Introduction
Lung cancer (LC) remains the leading cancer type in
morbidity and mortality worldwide (1), with non-small cell LC (NSCLC)
representing the most prevalent subtype, accounting for ~85% of all
LC cases (2). The aberrant
activation of the epidermal growth factor receptor (EGFR) signaling
pathway is a key driver of NSCLC (2). EGFR-tyrosine kinase inhibitors (TKIs)
targeted therapy has demonstrated significant benefits in patients
with NSCLC harboring activating EGFR mutation (3). Moreover, although the response rate to
EGFR-TKIs is substantially lower in patients with NSCLC with
wild-type EGFR compared with those with mutated EGFR (4), clinical trials indicate that
EGFR-TKIs, when used as consolidation and maintenance treatment
following first-line chemotherapy, can improve outcomes in patients
with NSCLC with wild-type EGFR (5).
Nevertheless, the efficacy of EGFR-TKIs is considerably constrained
by intrinsic and acquired resistance, and the underlying mechanisms
of EGFR-TKIs resistance are complicated and heterogeneous,
encompassing secondary mutation in the target kinase domain,
activation of alternative signal pathways, concurrent alteration in
other oncogenes, histological transformation, and tumor
microenvironment (6), which remains
incompletely understood.
Primary cilia are ubiquitous, hair-like organelles
that projected from cell surface, originating from the basal body
and composed of an axoneme with a 9+0 microtubule doublet
configuration (7). The organelle
functions as distinctive sensory structure and signaling hub,
sensing and transducing extracellular chemical and physical stimuli
into intracellular responses by coordinating multiple signaling
pathways, including Hedgehog (HH), G protein-coupled receptor,
receptor tyrosine kinase (RTK), and WNT pathways (8). Alterations in ciliation and ciliary
signaling pathway status are significantly associated with various
cancer types, but the role of cilia varies depending on the cancer
type, stage and microenvironment (9). Notably, primary cilia have been
implicated in resistance to chemotherapeutic agents (10–12),
ionizing radiation (12,13), and protein kinase targeted
inhibitors (14,15), suggesting a regulatory role in
cellular response and resistance to cancer therapies.
It has been previously reported that the EGFR-TKI
gefitinib (Gef) remarkably restores ciliogenesis in NSCLC A549
cells, as well as in kidney cancer UMRC2, breast cancer SUM159, and
pancreatic cancer L3.6 cells (16).
Furthermore, a notable increase in cilia frequency, length and
fragmentation has been observed in NSCLC HCC4006, H2228, A549 and
H23 cells that have developed resistance to kinase inhibitors, and
ablation of cilia or inhibition of the cilia-dependent HH signaling
pathway is sufficient to overcome both acquired and de novo
kinase inhibitor resistance in NSCLC cells (14). These findings highlight the
involvement of primary cilia in promoting EGFR-TKIs resistance in
NSCLC, but the precise role and underlying mechanism are still
largely unexplored.
The adenylate cyclase (AC) superfamily, which
comprises nine membrane-bound subtypes (AC1-AC9) and one soluble
subtype, plays critical roles in development, progression and
therapy resistance across various cancer types by modulating the
cyclic adenosine monophosphate (cAMP) signaling pathway (17). This pathway's activation and
transduction are often orchestrated by primary cilia (18). AC3 is commonly localized to primary
cilia in mammals and is involved in the regulation of cilia
elongation and HH signaling (19,20).
While emerging evidence has linked AC3 to cell proliferation,
migration and invasion in gastric and pancreatic cancers (21,22),
its role in EGFR-TKIs' resistance mechanisms in NSCLC remains to be
fully elucidated.
In the present study, the prevalence of primary
cilia in EGFR-TKIs' insensitive and sensitive NSCLC cells, as well
as the dynamic alterations in ciliogenesis following EGFR-TKIs
treatment were investigated. The functional roles of primary cilia
and AC3 in modulating cellular responses to EGFR-TKIs were further
studied, which may help to provide a novel strategy for overcoming
drug resistance in NSCLC cells.
Materials and methods
Reagents
The first and second generation of EGFR-TKIs
gefitinib (Gef; cat. no. HY-50895) and dacomitinib (Dac; cat. no.
HY-13272) were purchased from MedChemExpress. Dimethyl sulfoxide
(DMSO; cat. no. 472301) and propidium iodide (PI; 1.0 µg/ml; cat.
no. P4864) were obtained from Sigma-Aldrich; Merck KGaA. For
immunoblotting experiment, the primary antibodies of ERK (1:1,000;
cat. no. ab184699) and phosphorylated-ERK (p-ERK; 1:1,000; cat. no.
ab201015) were purchased from Abcam; primary antibodies of BCL2
(1:2,000; cat. no. 12789-1-AP), α-tubulin (α-tub; 1:40,000; cat.
no. 11224-1-AP), IFT88 (1:1,000; cat. no. 60227-1-Ig), AC3
(1:1,000; cat. no. 19492-1-AP), Cyclin A2 (CCNA; 1:1,000; cat. no.
18202-1-AP), Cyclin B1 (CCNB; 1:1,000; cat. no. 55004-1-AP), Cyclin
D1 (CCND; 1:3,000; cat. no. 60186-1-Ig), Cyclin E1 (CCNE; 1:1,000;
cat. no. 11554-1-AP), CDK1 (1:1,000; cat. no. 19532-1-AP), CDK2
(1:1,000; cat. no. 10122-1-AP), p21 (1:1,000; cat. no. 10355-1-AP),
p16 (1:1,000; cat. no. 10883-1-AP), GAPDH (1:50,000; cat. no.
60004-1-Ig), and the secondary antibodies including HRP-conjugated
goat anti-mouse IgG (1:5,000; cat. no. SA00001-1), HRP-conjugated
goat anti-rabbit IgG (1:5,000; cat. no. SA00001-2) were supplied by
Proteintech Group, Inc. For immunofluorescence staining, the
primary antibodies of ADP ribosylation factor such as GTPase 13B
(ARL13B; 1:500; cat. no. 17711-1-AP/66739-1-Ig), AC3 (1:500; cat.
no. 19492-1-AP) and γ-tubulin (γ-tub; 1:500; cat. no. T6557) were
purchased from Proteintech Group, Inc. and Sigma-Aldrich; Merck
KGaA respectively. The secondary antibodies including Alexa Fluor
594-conjugated goat anti-mouse IgG, Alexa Fluor 594-conjugated goat
anti-mouse IgG (1:500; cat. no. A-11005), Alexa Fluor
488-conjugated goat anti-mouse IgG (1:500; cat. no. A-11001), Alexa
Fluor Plus 555-conjugated goat anti-rabbit IgG (1:500; cat. no.
A32732), and Alexa Fluor Plus 488-conjugated goat anti-rabbit IgG
(1:500; cat. no. A32731) were purchased from Thermo Fisher
Scientific, Inc. 4′,6-diamidino-2-phenylindole (DAPI) was obtained
from Molecular Probes; Thermo Fisher Scientific, Inc.
Cell culture
The human NSCLC cell lines A549 (CSTR:
19375.09.3101HUMSCSP503), NCI-H23 (H23; CSTR:
19375.09.3101HUMSCSP581), PC9 (CSTR: 19375.09.3101HUMSCSP5085) and
HCC827 (CSTR: 19375.09.3101HUMSCSP538) were purchased from the Cell
Bank/Stem Cell Bank, Chinese Academy of Sciences. A549 cells were
cultured in Dulbecco's Modified Eagle Medium/F-12 (Gibco; Thermo
Fisher Scientific, Inc.). H23, PC9 and HCC827 cells were maintained
in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.). The
medium was supplemented with 10% fetal bovine serum (Shanghai
ExCell Biology, Inc.), 100 units/ml penicillin and 100 µg/ml
streptomycin (Beijing Solarbio Science & Technology Co., Ltd.).
Cells were cultured in a humidified atmosphere at 37°C under 5%
CO2, and tested negative for mycoplasma.
Cell viability assay
NSCLC cells (1×105) were planted in
6-well plates and incubated for 24 h, followed by treated with
indicated concentrations of Gef or Dac for 48 h. In addition, NSCLC
cells (1×105) were seeded in 6 well-plates for 24 h
before transfected with siIFT88, siARL13B, siAC3, or NC. After
transfection, Gef, Dac, or DMSO was immediately added into the
medium and the cells were continuously cultured for 48 h. The cell
number was counted using a Coulter counter (Z2; Beckman Coulter
Inc.), the cell viability related to control group was determined
and the half maximal inhibitory concentration (IC50) of
Gef or Dac was calculated.
Immunofluorescence staining
Cells were fixed with 4% paraformaldehyde (Beijing
Solarbio Science & Technology Co., Ltd.) for 10 min at room
temperature following the pre-chilling methanol at −20°C for 20
min. The fixed cells were permeabilized with 0.5% Triton X-100 for
10 min, then blocked with 5% immunostaining blocking buffer
(Shanghai Yeasen Biotechnology Co., Ltd.) for 1 h at room
temperature. The primary antibodies of ARL13B and γ-tub were used
to visualize primary cilia and basal bodies, respectively. The
primary antibody of AC3 was used to visualize its localization. The
primary antibodies were incubated for 2 h at room temperature and
then cells were incubated with the secondary antibodies for 1 h.
The cell nuclei were counterstained with DAPI. The images were
captured under an RVL-100-G fluorescence microscope (ECHO). For
cilia frequency analysis, confidence level (α): 95%, Z-score: 1.96,
margin of error (e): 5%, population proportion (p): 1–60%, when
p=60% the population size (n)=369, and more than 500 cells for each
experiment were calculated. For AC3 positive cilia analysis,
confidence level (α): 95%, Z-score: 1.96, margin of error (e): 5%,
population proportion (p): 0–20%, when p=20% the population size
(n)=246, and more than 200 cilia were calculated for each group.
The length of the primary cilium (n>50) was measured using
ImageJ software (Version 1.52; National Institutes of Health).
Western blotting
Cells were lysed with RIPA buffer (Beyotime
Institute of Biotechnology). The total cellular proteins (20 µg)
were separated by 10% SDS-PAGE and transferred to a
methanol-activated PVDF membrane (Merck KGaA). The membrane was
blocked with 5% protein-free rapid blocking buffer (Wuhan
Servicebio Technology Co., Ltd.) for 2 h at room temperature and
probed with the primary antibodies for 2 h at room temperature.
After incubation with HRP-conjugated secondary antibody, the
protein bands were detected with chemiluminescence reagents (Merck
KGaA), and the images were captured by using Alliance LD4 gel
imaging system (UVITEC) and analyzed using ImageJ software.
Cell cycle distribution analysis
For the cell cycle assay, cells treated with Gef or
Dac for 1–3 days were harvested and fixed in −20°C pre-chilled 70%
alcohol overnight, then stained with 20 µg/ml PI for 15 min at room
temperature. Flow cytometric analysis was performed using Amnis
imaging flow cytometer (Merck KGaA), and at least 10,000 gated
events were acquired from each sample. The data were analyzed with
Amnis IDEAS Application v6.0 (Merck KGaA).
PI staining
The control cells or cells transfected with small
interfering RNAs (siRNAs) were treated with Gef or Dac for
indicated time, then incubated with 20 µg/ml PI for 15 min to stain
the dead cells. The images of bright field (BF) and fluorescence
field were captured by using a DMI6000 fluorescence microscope
(Leica Microsystems GmbH), and at least 500 cells were counted for
each group.
RNA interference
Two individual stealth siRNAs targeting IFT88
(siIFT88-1/2, 25 nM, Thermo Fisher Scientific, Inc.) or ARL13B
(siARL13B-1/2; 50 nM; Guangzhou RiboBio Co., Ltd.) were introduced
into the cells by using Lipofectamine 2000 reagent (Thermo Fisher
Scientific, Inc.) to ablate primary cilia. Three individual siRNAs
against AC3 (Guangzhou RiboBio Co., Ltd.) were employed to
knockdown the target gene expression at a final concentration of 50
nM. A negative control siRNA (NC; siN0000001-1-5; Guangzhou RiboBio
Co., Ltd.) was used as the control group. Cells were transfected
with siRNAs for 5 h at 37°C and then the medium was replaced with
fresh culture medium. The interference efficiency of siRNAs was
evaluated by immunoblotting analysis. The sequences of siRNAs are
as follows: siIFT88-1: 5′-CCAAAGCCAUUAAAUUCUACCGAAU-3′; siIFT88-2:
5′-GAAGAAAGCUGUAUUGCCAAUAGUU-3′; siARL13B-1:
5′-GTGTCAGATAGAACCATGT-3′; siARL13B-2: 5′-GAACCATGTTCAGCAATCT-3′;
siAC3-1: 5′-GCACCAGCTTCCTCAAAGT-3′; siAC3-2:
5′-GGACTGGTGTTGGACATCA-3′; siAC3-3: 5′-GACGTATGATGAGATTGGA-3′. The
sequences of NC were not provided for the commercial patent
security protection.
Cell senescence assay
The cellular senescence was measured by
Senescence-Tracker (Sen-Tra) assay as previously reported (23). Briefly, cells were exposed to DMSO
or Gef (25 µM) for 1–3 days, followed by incubation with Sen-Tra
fluorescence probe (10 µM; cat. no. C0603; Beyotime Institute of
Biotechnology) overnight at 37°C; then, the cells were washed with
PBS for three times before imaging. The fluorescence images of
senescent cells were captured on a DMI6000 fluorescence microscope.
Quantification of images obtained from cells was performed using
ImageJ software, and >500 cells were counted for each group.
Statistical analysis
All the experiments were repeated at least three
times, and the data are presented as the mean ± standard deviation
(SD). Statistical analysis was performed with Origin 2019
(OriginLab Corporation) using paired Student's t-test for two
groups and one-way ANOVA followed by Tukey's post hoc test for
multiple comparisons. *P<0.05 was considered to indicate a
statistically significant difference.
Results
EGFR-TKIs' insensitive and sensitive
NSCLC cells present different ciliation status
To determine whether primary cilia are associated
with cellular resistance to EGFR-TKIs in NSCLC, the
drug-insensitive A549 and H23 cells (EGFR wild-type) and the
drug-sensitive HCC827 and PC9 cells (EGFR mutant) were treated with
increasing concentrations of Gef and Dac for 48 h to measure the
IC50. The results showed that the IC50 values
for A549 and H23 to Gef/Dac are ~20.3/3.9 and 15.5/1.9 µM
respectively (Fig. 1A and B;
Fig. S1A and B), and that for
HCC827/PC9 are significantly lower than 0.05/0.01 and 0.05/0.01 µM
respectively (Fig. 1C and D;
Fig. S1C and D). To further
corroborate the distinct inhibitory effectiveness of EGFR-TKI on
the drug-insensitive and drug-sensitive cells, the activation of
ERK, a key EGFR downstream signaling factor (24), was detected in A549 and HCC827 cells
challenged with different doses of Gef. As demonstrated, the
expression level of activated ERK (p-ERK) exhibited a significant
decrease in HCC827 but not in A549 cells (Fig. 1E and F), indicating that HCC827 is
distinctly more sensitive to Gef and Dac than A549. Next, the
immunofluorescence staining assay with the cilia and basal body
markers ARL13B and γ-tub was performed, and typical primary cilia
structure was observed in A549, H23 and HCC827 cells cultured in
full serum-containing medium (Figs.
1G and S2A), but that was
completely lacking in PC9 cells (Fig.
S2C). The cilia frequency is significantly higher in A549
(19.9±3.5%) and H23 (~1.5%) cells compared with that in HCC827
(<0.05%) and PC9 (none) cells (Figs.
1G and S2), suggesting a
negative association between cilia incidence and cellular
sensitivity to EGFR-TKIs.
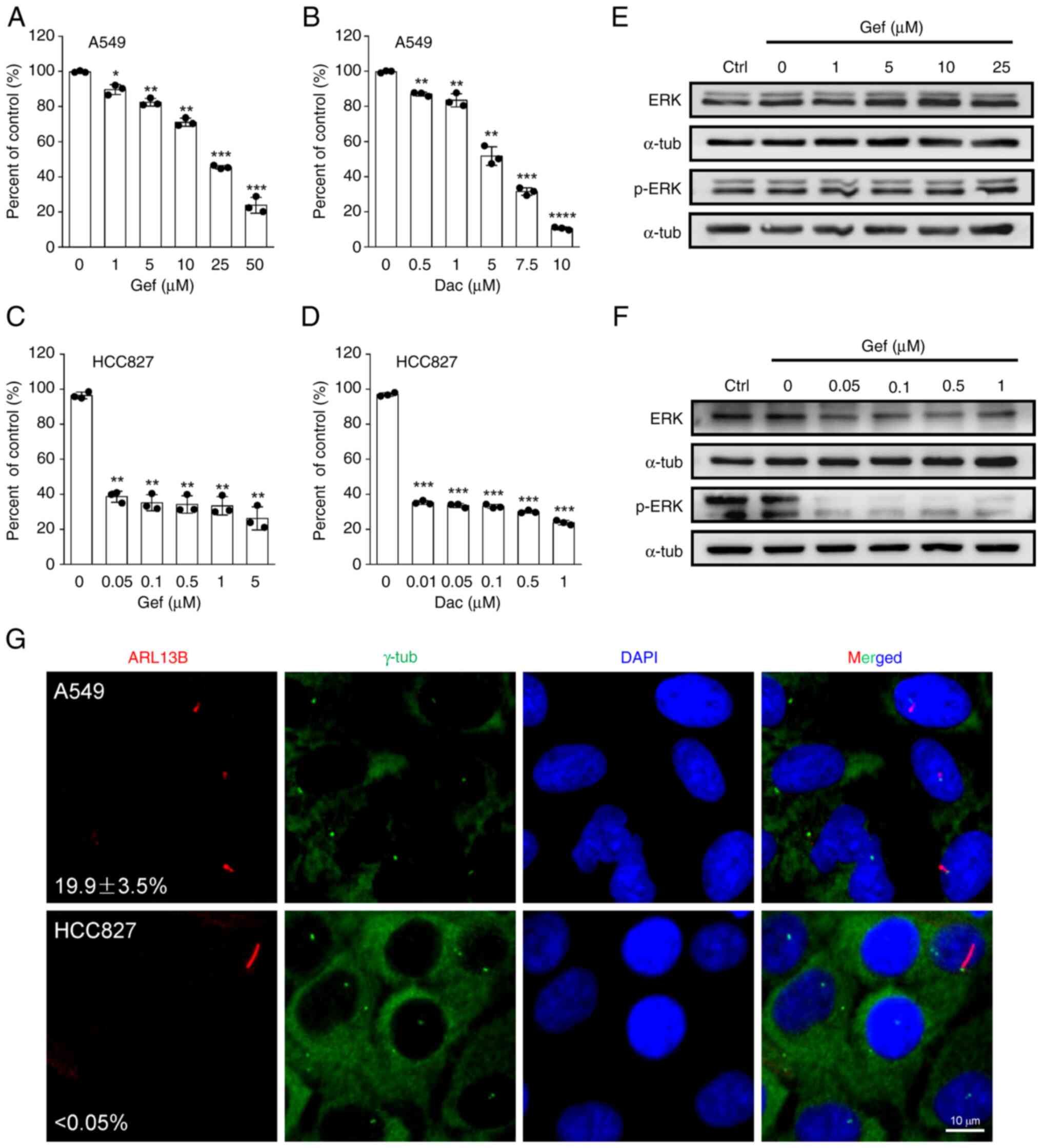 | Figure 1.Epidermal growth factor
receptor-tyrosine kinase inhibitor-insensitive A549 cells harbor
high cilia incidence. (A and B) A549 cells were treated with
increasing concentrations of (A) Gef or (B) Dac, and the cell
viability related to untreated control (Ctrl) was measured at 48 h
post-treatment. (C and D) HCC827 cells were treated with increasing
concentrations of (C) Gef or (D) Dac, and the cell viability
related to untreated control was measured at 48 h post-treatment.
(E and F) A549 and HCC827 cells were exposed to increasing
concentrations of Gef for 48 h, and the immunoblotting analysis was
performed to measure the protein expression levels of ERK and p-ERK
at Thr202/Tyr204. α-tubulin (α-tub) was used as a loading control.
(G) Representative images of primary cilia labeled with ARL13B
(red) and basal bodies labeled with γ-tub (green) in A549 and
HCC827 cells, and the cilia incidence was calculated. The nuclei
were counterstained with DAPI (blue). Scale bar, 10 µm. All
quantitative data were obtained from three replicates and shown as
the mean ± SD. Error bars, ± SD. *P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001 compared with 0 µM (0.1% DMSO).
Gef, gefitinib; Dac, dacomitinib; p-, phosphorylated; SD, standard
deviation. |
EGFR-TKIs facilitate primary cilia
formation and elongation in A549 and H23 but not HCC827 and PC9
cells
Increased cilia frequency and length have been
reported in TKI-resistant NSCLC cells, such as erlotinib-resistant
HCC4006 (14) and
dasatinib-resistant A549 cells (25), but no significant alterations in
afatinib- and erlotinib-resistant PC9 cells compared with the
parental cells (14). To
investigate the effects of EGFR-TKIs on primary ciliogenesis in
NSCLC cells, the A549, H23, HCC827 and PC9 cells were treated with
multiple doses of Gef and Dac. As revealed in Fig. 2A-E, administration of Gef and Dac
resulted in remarkable cilia formation and elongation in A549 cells
in a dose-dependent manner. The proportion of ciliated cells was
raised from ~20% in DMSO (0 µM Gef)-treated cells to nearly 60% in
5 µM Gef-treated cells, and to nearly 50% in 1 µM Dac-treated
cells. Similar alterations were observed in H23 cells treated with
5 µM Gef or 1 µM Dac (Fig. S2A and
B). As the drug concentration increased, the cilia incidence
and length were gradually decreased, and exposure to 50 µM Gef or
10 µM Dac even lead to deciliation in A549 cells. However, Gef and
Dac were not able to induce cilia formation in HCC827 and PC9 cells
(Fig. 2F and G; Fig. S2C). Together, these data suggested
that the ciliogenesis responding to EGF-TKIs maybe contribute to
drug resistance in NSCLC cells.
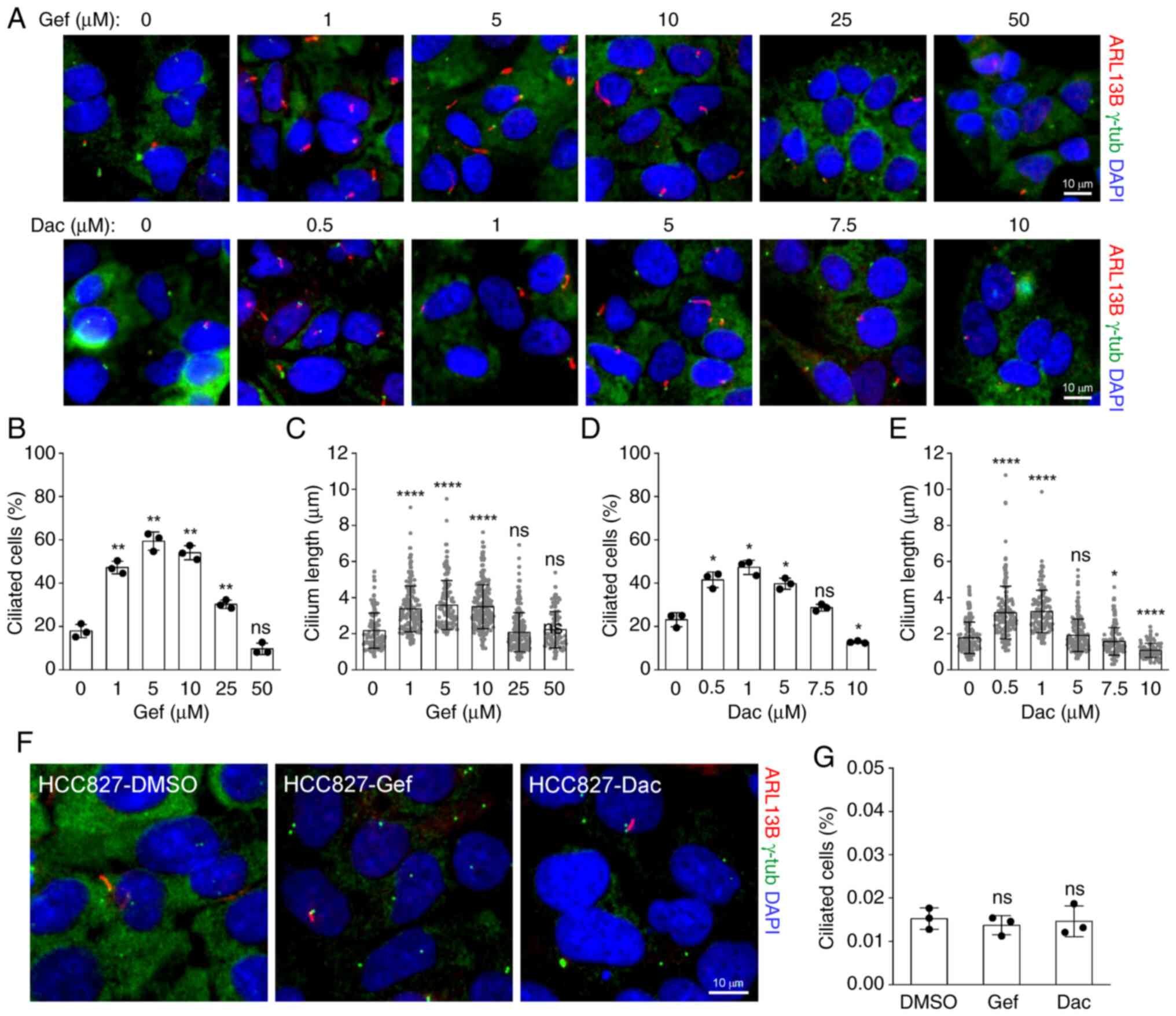 | Figure 2.Epidermal growth factor
receptor-tyrosine kinase inhibitors promote ciliogenesis in
dose-dependent manner in A549 but not HCC827 cells. (A)
Representative images of primary cilia labeled with ARL13B (red)
and basal bodies labeled with γ-tub (green) in A549 cells treated
with increasing doses of Gef or Dac for 48 h. (B and C)
Quantification of (B) ciliated cells and the (C) cilium length of
A549 cells treated with 0 (n=117), 1 (n=176), 5 (n=133), 10
(n=216), 25 (n=210), 50 (n=95) µM Gef for 48 h. **P<0.01 and
****P<0.0001 compared with 0 µM; ns, not significant. (D and E)
Quantification of (D) ciliated cells and the (E) cilium length of
A549 cells treated with 0 (n=133), 0.5 (n=153), 1 (n=138), 5
(n=163), 7.5 (n=130), 10 (n=80) µM Dac for 48 h. *P<0.05 and
****P<0.0001 compared with 0 µM; ns, not significant. (F)
Representative images of primary cilia labeled with ARL13B (red)
and basal bodies labeled with γ-tub (green) in HCC827 cells treated
with 0.01 µM Gef or 0.001 µM Dac for 48 h. (G) Quantification of
ciliated cells in HCC827 cells in (F). ns, not significant compared
with DMSO. The nuclei were counterstained with DAPI (blue). Scale
bar, 10 µm. All quantitative data were obtained from three
replicates and shown as the mean ± SD. Error bars, ± SD. Gef,
gefitinib; Dac, dacomitinib; p-, phosphorylated; SD, standard
deviation; ns, not significant. |
EGFR-TKIs induce cytostatic but not
cytotoxic effects
Next, the effects of EGF-TKIs on the cell cycle and
cell fate were investigated. Upon Gef (25 µM) treatment, A549 cells
were predominantly blocked in G0/G1 phase in a time-dependent
manner (Fig. 3A and B). Similar
phenomenon was observed in HCC827 exposed to 0.05 µM Gef (Fig. 3C and D). Moreover, the expression
levels of G1 cyclins (CCND and CCNE) and cyclin-dependent kinase
inhibitor (p16 and p21) were consistently upregulated following Gef
and Dac treatment in A549 cells, while the expression levels of S
and G2/M cyclins (CCNA and CCNB) were suppressed, and the
cyclin-dependent kinase CDK1 and CDK2 showed slight changes
(Fig. 3E), supporting a G1 phase
arrest induced by EGFR-TKIs. Notably, Gef treatment resulted in
little cell death in A549 cells; <5% dead cells were detected at
3 days post-treatment (Fig. 3F).
The expression levels of p-ERK in Gef-treated cells showed modest
reduction compared with the DMSO-treated group, but the positive
ciliogenesis modulator IFT88 and the negative apoptosis regulator
BCL2 were gradually elevated (Fig.
3G), indicating enhanced capacities of ciliogenesis and
anti-apoptosis. Strikingly, increased senescent cells were detected
by the Sen-Tra labeling assay in A549 cells exposed to 25 µM Gef
(Fig. 3H), and the proportion of
senescent cells reached ~60% at 3 days after Gef exposure (Fig. 3I). Collectively, these data
demonstrated that EGFR-TKIs induce cytostatic but not cytotoxic
effects in NSCLC cells.
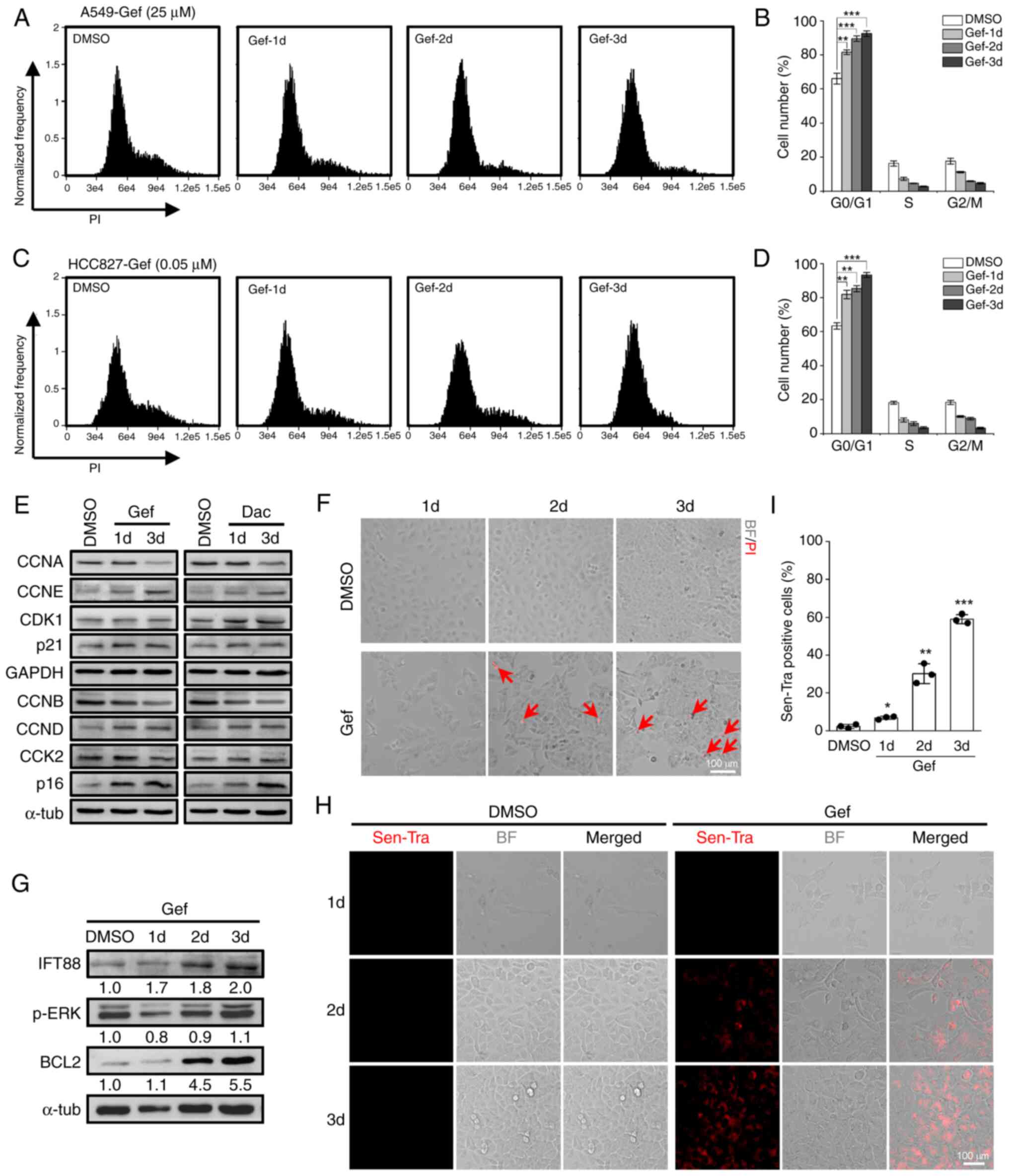 | Figure 3.Gef treatment leads to G1 phase
arrest and senescence. (A and B) Cell cycle distribution assay of
A549 cells treated with DMSO (0.1%) or 25 µM Gef for 1, 2, and 3
days (d) by flow cytometry with (A) PI staining; (B) quantification
data. (C and D) Cell cycle distribution assay of HCC827 cells
treated with DMSO (0.1%) or 0.05 µM Gef for 1, 2, and 3 days by
flow cytometry with (C) PI staining; (D) quantification data. (E)
Immunoblotting analysis of the expression levels of CCNA, CCNB,
CCND, CCNE, CDK1, CDK2, p16 and p21 in A549 cells treated with
DMSO, Gef (25 µM), or Dac (5 µM) for 1 and 3 days. α-tubulin
(α-tub) and GAPDH were used as loading controls. (F) Representative
PI staining images of A549 cells exposed to DMSO (0.1%) or Gef (25
µM) for 1, 2, 3 days, the dead cells (red) were stained with PI and
indicated with red arrows. Scale bar, 100 µm. (G) Immunoblotting
analysis of the expression levels of IFT88, p-ERK (Thr202/Tyr204)
and BCL2 in A549 cells treated with DMSO (0.1%) or Gef (25 µM) for
1, 2, 3 days. α-tubulin (α-tub) was used as a loading control. The
values under the immunoblot bands indicate the quantitative
densitometry value measured using ImageJ software. (H and I) The
(H) cellular senescence assay of A549 cells treated with DMSO
(0.1%) or Gef (25 µM) for 1–3 d labeling by senescence-tracker
(Sen-Tra) fluorescence probe, and the (I) quantification data.
Scale bar, 100 µm. Data are expressed as the mean ± SD. Error bars,
± SD. *P<0.05, **P<0.01 and ***P<0.001 compared with DMSO.
Gef, gefitinib; PI, propidium iodide; CCNA, Cyclin A2; CCNB, Cyclin
B1; CCND, Cyclin D1; CCNE, Cyclin E1; Dac, dacomitinib; BF, bright
field; p-, phosphorylated; SD, standard deviation. |
Disruption of ciliogenesis attenuates
cellular resistance to EGFR-TKIs
Given that Gef and Dac can promote primary cilia
formation in A549 and H23 cells (Figs.
2A and S2A), it is likely that
the primary cilia may play important roles in responding to
EGFR-TKIs in the insensitive NSCLC cells. To determine this
conjecture, the siRNAs targeting IFT88 (siIFT88-1/2) or ARL13B
(siARL13B-1/2) were introduced into A549 cells. The results showed
that transfection of siIFT88 or siARL13B prominently suppressed the
protein expression levels of IFT88 or ARL13B in A549 cells
(Fig. 4A). Moreover, knockdown of
IFT88 or ARL13B by siRNAs resulted in significant decreases in the
cilia frequency and length in A549 cells exposed to Gef as expected
(Fig. 4B-D). Importantly, the
ablation of primary cilia by siIFT88 and siARL13B profoundly
improved the inhibitory efficacy of Gef and Dac against A549 cells
(Fig. 4E and F). In addition, the
PI staining assay revealed that the abrogation of ciliogenesis by
IFT88 or ARL13b silencing strongly provoked the cell death in A549
cells exposed to 25 µM Gef (Fig.
4G), and the proportion of dead cells rose to ~30% at 3 days
after treatment (Fig. 4H). These
results indicated that the inhibition of ciliogenesis sensitizes
A549 cells to EGFR-TKIs by switching senescence to cell death.
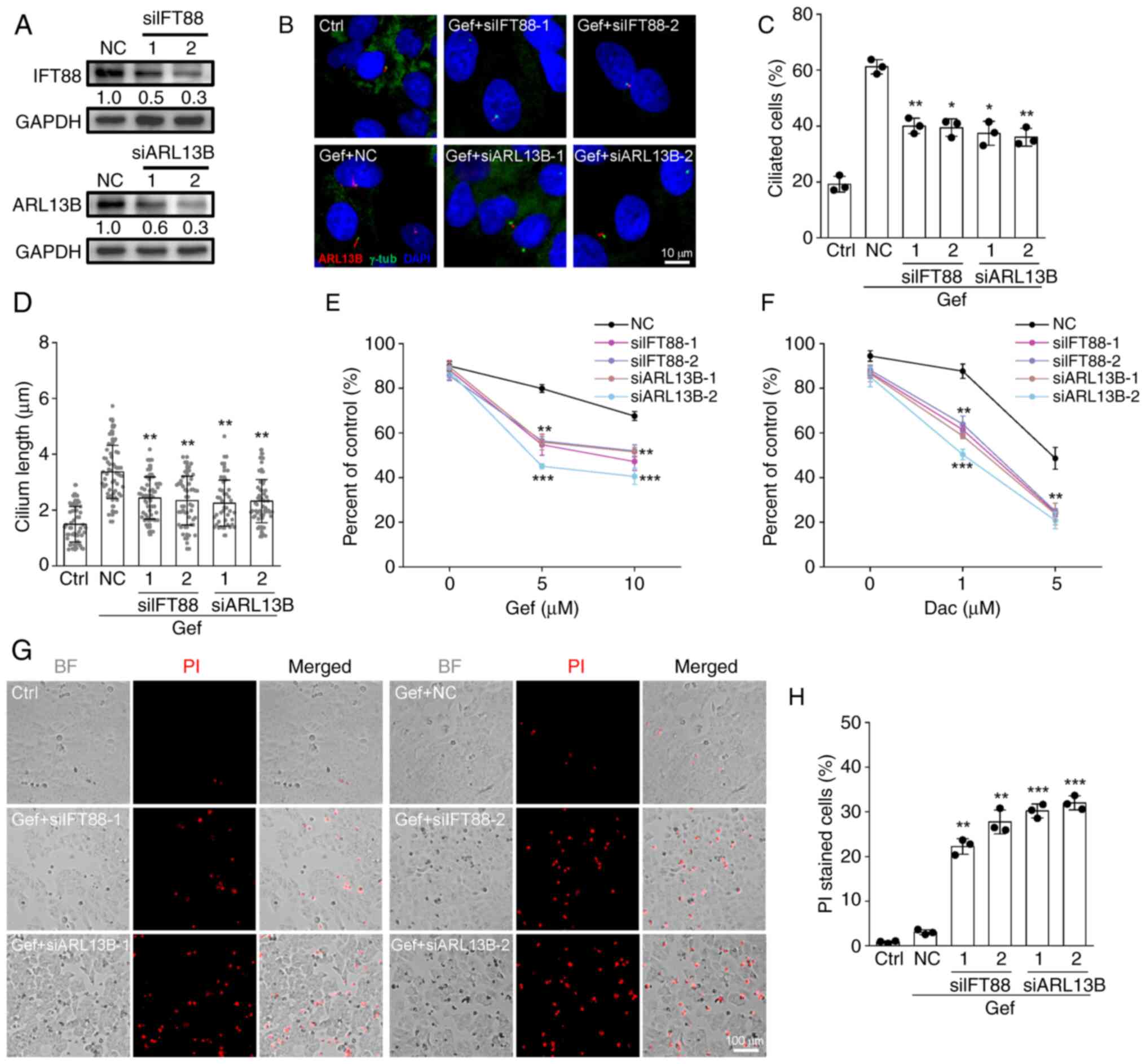 | Figure 4.Inhibition of ciliogenesis sensitizes
A549 cells to epidermal growth factor receptor-tyrosine kinase
inhibitors. (A) Immunoblotting analysis of the protein expression
levels of IFT88 and ARL13B in A549 cells transfected with siRNAs
against IFT88 (siIFT88-1/2), ARL13B (siARL13B-1/2), or NC. GAPDH
was employed as a loading control. The values under the immunoblot
bands indicate the quantitative densitometry value measured using
ImageJ software. (B-D) Immunofluorescence labeling of primary cilia
(ARL13B, red) and basal bodies (γ-tub, green) in A549 control cells
(Ctrl) and cells transfected with siIFT88-1/2, siARL13B-1/2, or NC
following treated with 10 µM Gef for 48 h, (C) the proportion of
ciliated cells and (D) cilium length in each group. Ctrl, n=52;
Gef-NC, n=59; Gef-siIFT88-1, n=57; Gef-siIFT88-2, n=64;
Gef-siARL13B-1, n=56; Gef-siARL13B-2, n=69. The nuclei were stained
with DAPI (blue). (E and F) A549 cells transfected with NC,
siIFT88-1/2, or ARL13B-1/2 siRNAs were exposed to (E) Gef (0, 5, 10
µM) or (F) Dac (0, 1, 5 µM) for 48 h, the cell viability related to
untreated control was measured. (G and H) A549 cells transfected
with indicated siRNAs were exposed 10 µM Gef for 48 h and stained
with PI to detect the dead cells and quantification data for each
group. Scale bar, 100 µm. Data are expressed as the mean ± SD.
Error bars, ± SD. *P<0.05, **P<0.01 and ***P<0.001
compared with NC. siRNA, small interfering RNA; NC, negative
control; Gef, gefitinib; Dac, dacomitinib; BF, bright field; SD,
standard deviation. |
EGFR-TKIs promote AC3 expression and
ciliary localization
To gain insight into the role for AC3 in primary
cilia-mediated cellular resistance to EGFR-TKIs in NSCLC cells, the
expression of AC3 in responding to Gef and Dac stresses in A549
cells were detected. As demonstrated in Fig. 5A, both Gef and Dac treatment leads
to persistent upregulation of AC3 protein expression, which is in
accordance with ciliary gene ARL13B protein expression. Previously,
it has been reported that AC3 presents on primary cilia in multiple
cell types such as synoviocyte (20) osteocyte (26) and astrocyte (27). To determine AC3 subcellular
localization in NSCLC cells exposed to EGFR-TKIs, A549 cells were
treated with DMSO (0.1%), Gef (10 µM) and Dac (5 µM) for 3 days
followed by visualization of the primary cilia with
immunofluorescence staining of antibodies against ARL13B, and the
results identified that the proportion of AC3 positive cilia
increased to >20% in Gef- and Dar-treated cells while that was
merely found in DMSO-treated cells (Fig. 5B and C). However, EGFR-TKIs
treatment slightly altered ARL13B expression levels and even
decreased AC3 expression levels in HCC827 cells (Fig. 5D), indicating different responses of
ciliogenesis and AC3 expression to EGFR-TKI stress between A549 and
HCC827 cells. Furthermore, it was found that Gef-induced AC3
expression and ciliary localization were mitigated by disruption of
primary cilia (Fig. 5E and F).
Collectively, these results indicated that EGFR-TKI-induced AC3
expression and localization to primary cilia may contribute to
cellular drug resistance.
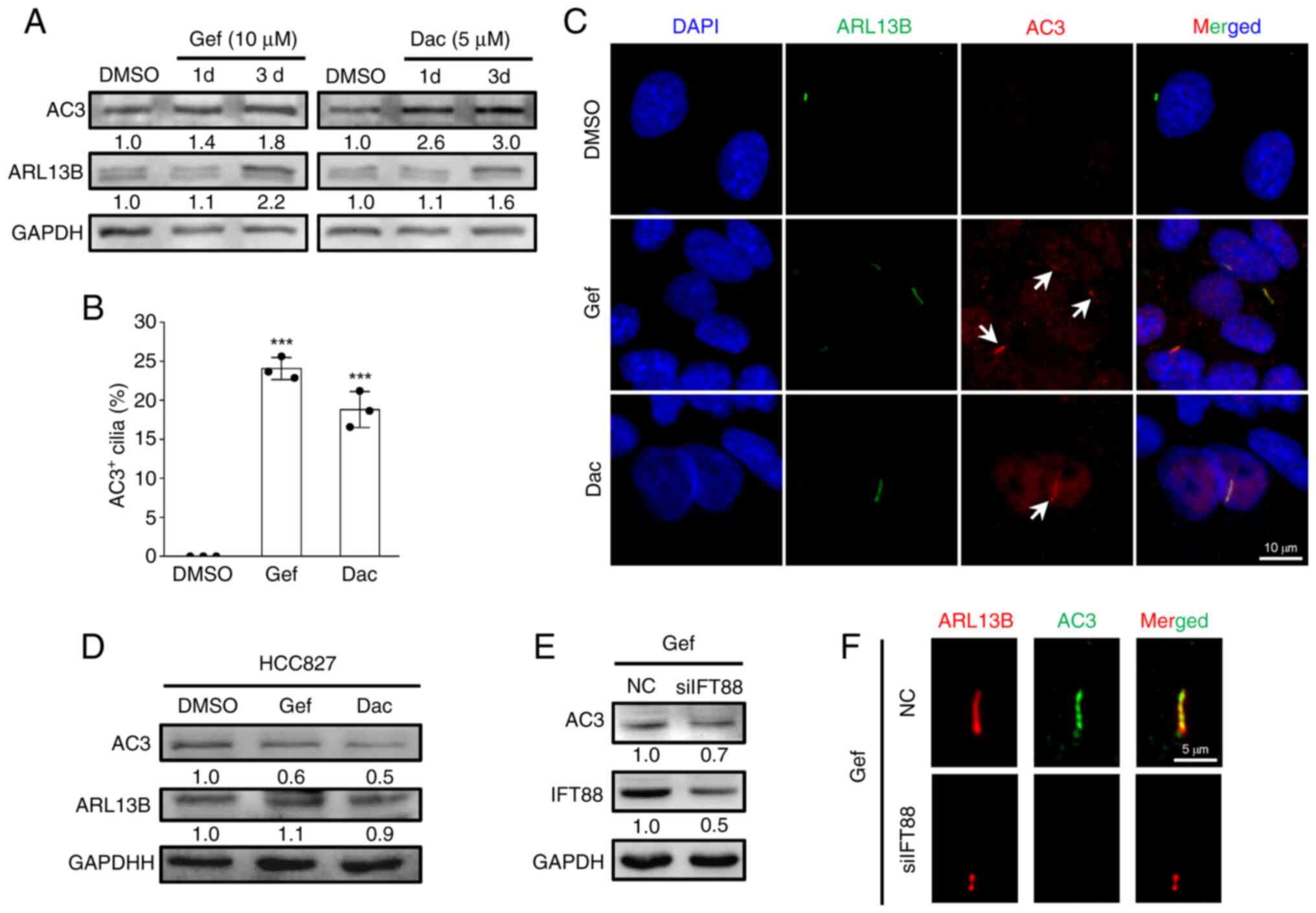 | Figure 5.Epidermal growth factor
receptor-tyrosine kinase inhibitors induce AC3 expression and
ciliary localization. (A) Immunoblotting analysis of the expression
levels of AC3 and ARL13B in A549 cells exposed to DMSO (0.1%), Gef
(10 µM) and Dac (5 µM) for 1 and 3 days. (B and C)
Immunofluorescence labeling of primary cilia (Arl13b, green) and
AC3 (red, indicated by white arrows) in A549 cells administrated
with DMSO (0.1%), Gef (10 µM), and Dac (5 µM) for 3 days, and
quantification of AC3 positive (AC3+) cilia in each
group. The nuclei were stained with DAPI (blue). Scale bar, 10 µm.
Data are expressed as the mean ± SD. Error bars, ± SD.
***P<0.001 compared with DMSO. (D) Immunoblotting analysis of
the expression levels of AC3 and ARL13B in HCC827 cells exposed to
DMSO (0.1%), Gef (0.05 µM) and Dac (0.01 µM) for 3 days. (E)
Immunoblotting analysis of the protein expression levels of AC3 and
IFT88 in Gef-treated A549 cells transfected with siIFT88-1
(siIFT88) or NC siRNA for 3 days. (F) Immunofluorescence labeling
of primary cilia (Arl13b, red) and AC3 (green) in cells in (E).
Scale bar, 5 µm. GAPDH was used as a loading control; the values
under the immunoblot bands indicate the quantitative densitometry
value measured using ImageJ software. Gef, gefitinib; Dac,
dacomitinib; SD, standard deviation; si-, small interfering; NC,
negative control; AC3, adenylate cyclase 3. |
Suppression of AC3 expression is
sufficient to sensitize A549 cells to Gef
Given that EGFR-TKIs promote AC3 expression and
presence on primary cilia, the role of AC3 in cellular EGFR-TKI
sensitivity was investigated. Three individual siRNAs specifically
against AC3 (siAC3-1/2/3) were introduced to suppress its protein
expression (Fig. 6A), whereas the
expression of ARL13b was merely affected upon siAC3 treatment.
Subsequently, the cell viability was measured and the results
showed that the cellular sensitivity was significantly enhanced in
A549 cells transfected with siAC3 compared with that transfected
with NC siRNA following Gef combination treatment at 0, 5 and 10 µM
for 48 h (Fig. 6B), indicating a
positive association between AC3, cell proliferation and drug
resistance ability. Additionally, inhibition of AC3 could induce or
reduce cilia elongation in different cell types (20,26),
suggesting a feedback regulation of AC3 for ciliogenesis. To
address this issue, the cilia frequency of A549 cells transfected
with siAC3-1 or NC was detected following 10 µM Gef administrations
for 48 h. However, it was found that the proportion of ciliated
cells was barely altered upon AC3 suppression (Fig. 6C and D), indicating that
EGFR-TKIs-induced cilia formation is not associated with AC3
expression and cilia localization.
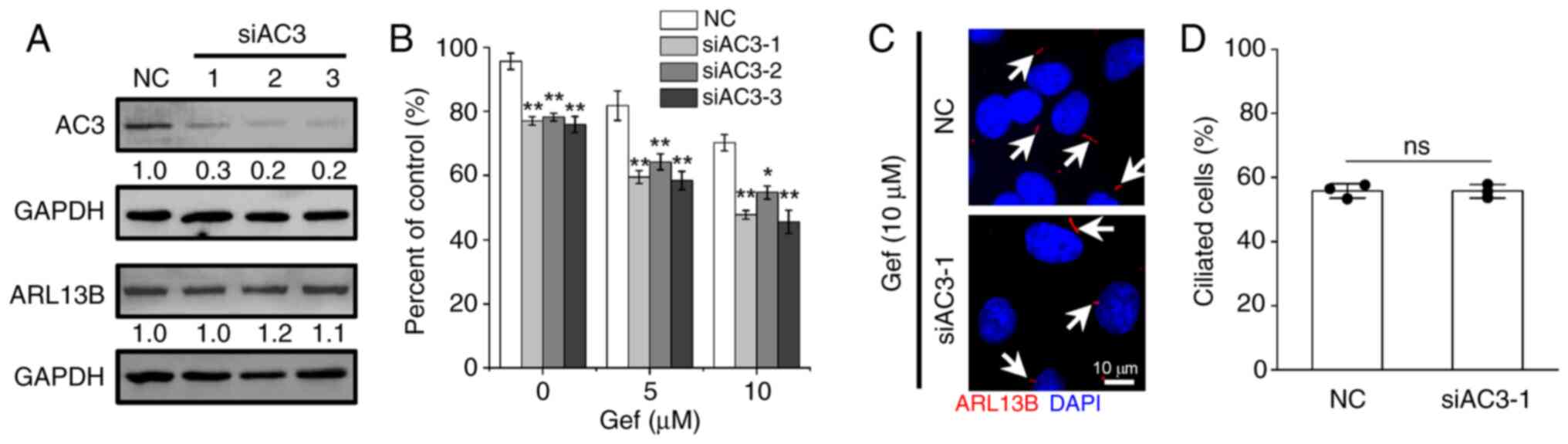 | Figure 6.Silencing AC3 expression enhances
cellular sensitivity to Gef. (A) Immunoblotting analysis of the
expression levels of AC3 and ARL13B in A549 cells transfected with
siRNAs targeting AC3 (siAC3-1/2/3) or NC for 48 h. GAPDH was used
as a loading control. The values under the immunoblot bands
indicate the quantitative densitometry value measured using ImageJ
software. (B) A549 cells transfected NC or siAC3-1/2/3 were exposed
to Gef (0, 5, 10 µM) for 48 h, the cell viability related to
untreated control was measured. (C and D) Immunofluorescence
labeling of primary cilia (Arl13b, red, indicated by white arrows)
in A549 cells transfected with NC and siAC3-1 following
administrated with Gef (10 µM) for 3 days, and quantification of
ciliated cells in each group. The nuclei were stained with DAPI
(blue). Scale bar, 10 µm. Data are expressed as the mean ± SD.
Error bars, ± SD. *P<0.05 and **P<0.01 compared with NC. AC3,
adenylate cyclase 3; Gef, gefitinib; si-, small interfering; NC,
negative control; SD, standard deviation; ns, not significant. |
Discussion
In the present study, it was demonstrated that
EGFR-TKIs facilitate primary ciliogenesis in the drug-insensitive
A549 and H23 cells but not in the drug-sensitive HCC827 and PC9
cells. Specifically, it was revealed that the disruption of primary
cilia reduces cellular resistance to EGFR-TKIs, potentially linked
to AC3 expression and its ciliary localization. Together, these
results suggested a connection between the primary cilia-AC3 axis
and cellular sensitivity to EGFR-TKIs in NSCLC.
Primary cilia, functioning as antennae on cell
surface, play a crucial role in responding to extracellular signals
by coordinate various signaling pathways that regulate cell
proliferation, division, migration, metabolism and physiology
(28). Despite the frequent loss or
repression of primary cilia in numerous solid tumor types (9), an increasing number of small molecular
anticancer drugs, including diverse protein kinase inhibitors, have
been identified that can restore ciliogenesis in cancer cells
(14,16,29,30).
This restoration suppresses cancer progression and alters drug
sensitivity simultaneously (31).
In the EGFR-mutant NSCLS cell line HCC4006, which completely lacks
primary cilia, sustained administration of the EGFR-TKI erlotinib
results in acquired drug resistance and increased ciliogenesis
(14). In the present study, the
results revealed that EGFR-TKI de novo-resistant NSCLC A549
cells maintain a significantly larger population of ciliated cells
than drug-sensitive HCC827 cells (Fig.
1), and short-term exposure to Gef and Dac further enhances
primary cilia formation in A549 and H23 cells but not in HCC827 and
PC9 cells (Figs. 2 and S2), suggesting that primary cilia may be
one of the EGFR-TKI resistance mechanisms in EGFR wild-type NSCLC
compared with EGFR mutant type. Thus, these results robustly
confirm the involvement of primary cilia in both the innate and
adaptive resistance of NSCLC to EGFR-TKIs.
The molecular mechanisms underlying acquired
resistance to EGFR-TKIs in NSCLC predominantly involve
reactivation, bypass and indifference of the EGFR pathway (6). However, it remains limited in cancer
types lacking mutations of the drug target, hindering the
development and application of novel therapeutic strategies. As a
distinctive sensory organelle, primary cilia play crucial role in
cell fate determination in responding to anticancer drugs by
regulating receptors located on cilia (32). Notably, EGFR has been shown to
localize to primary cilia (33),
where its signaling activity is restrict appropriately (33). Additionally, the aberrant activation
of EGFR signaling leads to the loss of primary cilia via Aurora A
in oral mucosa carcinoma and human retinal epithelia RPE1 cells
(34,35), suggesting a negative feedback loop
between primary cilia and EGFR.
Activation of the EGFR pathway drives cell
proliferation, migration and metabolism through downstream
effectors including the PI3K-AKT, JAK-STAT and RAS-RAF-MEK-ERK
pathways (6). A recent study has
uncovered the role of primary cilia in facilitating AKT and ERK
activation induced by type A platelet-derived growth factor during
Leydig cell development (36).
Additionally, treatment with EGFR-TKIs significantly reduces
proliferation without causing cell death in various cancer cells,
with a process linked to YAP-mediated ciliogenesis (37,38).
The present findings also demonstrated that A549 cells experienced
strong G1 cell cycle arrest (Fig.
3A), formed cilia (Fig. 2A),
and subsequently entered senescence (Fig. 3H) rather than undergoing cell death
(Fig. 3F) after Gef and Dac
treatment, implicating a positive relationship between primary
cilia and senescence induced by EGFR-TKIs in NSCLC.
Accumulating evidence has highlighted the functional
role of primary cilia in cell fate determination. Loss of primary
cilia in thyroid cancer cells leads to apoptogenic stimuli and
subsequent apoptotic cell death both in vitro and in
vivo (39). Genotoxic
stress-induced ciliogenesis drives cell senescence initiation and
maintenance (13,40,41),
the removal of cilia increases cellular sensitivity to ionizing
radiation by inducing apoptosis in senescent cells (13). In the present study, results also
revealed that the suppression of primary cilia by knockdown of
ciliary gene IFT88 or ARL13B significantly enhanced the efficacy of
EGFR-TKIs against NSCLC (Fig. 4E and
F), potentially due to increased cell death (Fig. 4G and H). However, the specific type
of cell death remains unconfirmed in the present study. Further
investigation into the role of primary cilia in cell fate
determination and the underlying mechanisms may facilitate the
identification of novel therapeutic avenue to sensitize NSCLC to
EGFR-TKIs.
Accumulating evidence indicates a reciprocal
relationship between primary cilia and cellular senescence.
Increased ciliogenesis have been observed in senescent cells
triggered by various stimuli (13,42,43).
Conversely, it has been demonstrated that primary ciliogenesis is
an essential cellular process in senescence induction induced by
etoposide (43) or ionizing
radiation (13), mediated through
autophagy or HH signaling activation in tumor cells. Furthermore,
Ma et al (41) demonstrated
that stress-induced transient primary ciliogenesis is required for
senescence initiation in human fetal lung fibroblast IMR-90 cells
exposed to ionizing radiation, oxidative stress, or inflammatory
stimuli, and that these cilia disassemble after senescence entry
(41). However, a recent study by
the authors revealed persistent primary ciliogenesis in senescent
tumor cells following ionizing radiation, which critically
contributes to both the initiation and maintenance of senescence,
and targeting either primary cilia or the senescence-associated
BCL2 pathway with ABT-263 significantly enhanced the lethal effects
of radiation (44). Therefore,
targeting the crosstalk between primary cilia and senescence may
represent an effective strategy for improving treatment
efficacy.
The AC family numbers play a pivotal role in
physiological and biological processes by modulating cAMP-related
signaling pathways, primarily involving protein kinase A (PKA) and
cAMP-activated guanine exchange factor (EPAC) (17,45).
The upregulation of cAMP by AC1 has been shown to reduce apoptosis
induced by DNA damage by promoting DNA damage repair mechanisms,
thereby increasing cellular resistance to chemotherapy and
radiotherapy (46,47). AC3, recognized as a marker for
cilia, is localized within primary cilia in a cell type-dependent
manner (19). In the present study,
it was reported for the first time to the best of our knowledge,
that EGFR-TKIs provoke the expression and ciliary localization of
AC3 in NSCLC cells with wild-type EGFR that are insensitive to the
drugs (Fig. 5A-C). Furthermore, the
inhibition of AC3 expression was found to enhance the cytotoxic
effects of EGFR-TKIs (Fig. 6).
Although the downstream signaling pathways of AC3 were not
investigated, it is imperative to focus on cAMP-PKA/EPAC pathways,
as they are closely associated with senescence and survival
(45). Understanding these factors
is crucial for elucidating the mechanism of EGFR-TKI resistance
mediated by the cilia-AC3 axis.
Although the present results suggest a potential
functional interplay between primary cilia and cellular resistance
to EGFR-TKIs in wild-type EGFR NSCLC, several limitations and open
questions remain: i) While the present study discloses a novel
association between primary cilia and intrinsic resistance to
EGFR-TKIs, the absence of drug-resistant EGFR-mutant NSCLC cell
models precludes conclusions about their role in acquired
resistance; ii) neither alternative RTK pathway activation nor
aberrant EGFR downstream pathway activation, two crucial resistance
mechanisms (24), were examined, as
well as their relationship to primary cilia; iii) increased cell
death was observed in cilia-disrupted A549 cells treated with Gef
using PI staining, a method that cannot distinguish type of cell
death; iv) the study lacks in vivo validation using
tumor-bearing animal models or patient-derived organoids to
substantiate the functional role of primary cilia; and v) it was
found that EGFR-TKI treatment promotes cilia formation (Fig. 2A-E) and AC3 ciliary localization
(Fig. 5B and C), and the disruption
of ciliogenesis and interference of AC3 expression increases cell
sensitivity to EGFR-TKIs in A549 cells, whereas the involvement of
AC3 downstream signaling, particularly the cAMP-PKA/EPAC pathway
(45), in response to EGFR-TKI and
its contribution to drug resistance remain unexplored. Further
research is therefore required to elucidate the mechanisms by which
the primary cilia-AC3 axis modulates EGFR-TKI resistance in
NSCLC.
In conclusion, the present study revealed that the
primary cilia-AC3 axis contributes to cellular resistance to
EGFR-TKIs in NSCLC and underscores its potential as a sensitizing
strategy for combination therapy in NSCLC.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Dan Xu and Dr
Qingfeng Wu (Biomedical Platform of the Public Technology Center,
Institute of Modern Physics, Chinese Academy of Sciences) for
providing technical assistance in flow cytometric analysis.
Funding
The present study was supported by the Non-profit Central
Research Institute Fund of Chinese Academy of Medical Sciences
(grant no. 2019PT320005), the Science and Technology Research
Project of Gansu (grant nos. 23JRRA533, 24JRRA952 and 25JRRA1204),
the National Natural Science Foundation of China (grant no.
12375355) and the Youth Innovation Promotion Association CAS (grant
no. 2021415).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
LJ, LW and JHu conducted investigation, curated
data, developed methodology, acquired funding and wrote the
original draft. RZ and JC conducted investigation and developed
methodology. JHe and YY conceptualized and supervised the study,
wrote, reviewed and edited the manuscript, and acquired funding.
All authors read and approved the final version of the manuscript.
JHe and YY confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263.
2024.PubMed/NCBI
|
|
2
|
Hendriks LEL, Remon J, Faivre-Finn C,
Garassino MC, Heymach JV, Kerr KM, Tan DSW, Veronesi G and Reck M:
Non-small-cell lung cancer. Nat Rev Dis Primers. 10:712024.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pao W and Chmielecki J: Rational,
biologically based treatment of EGFR-mutant non-small-cell lung
cancer. Nat Rev Cancer. 10:760–774. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Inoue A, Suzuki T, Fukuhara T, Maemondo M,
Kimura Y, Morikawa N, Watanabe H, Saijo Y and Nukiwa T: Prospective
phase II study of gefitinib for chemotherapy-naive patients with
advanced non-small-cell lung cancer with epidermal growth factor
receptor gene mutations. J Clin Oncol. 24:3340–3346. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cappuzzo F, Ciuleanu T, Stelmakh L,
Cicenas S, Szczésna A, Juhász E, Esteban E, Molinier O, Brugger W,
Melezínek I, et al: Erlotinib as maintenance treatment in advanced
non-small-cell lung cancer: A multicentre, randomised,
placebo-controlled phase 3 study. Lancet Oncol. 11:521–529. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Du Z, Kan H, Sun J, Liu Y, Gu J,
Akemujiang S, Zou Y, Jiang L, Wang Q, Li C, et al: Molecular
mechanisms of acquired resistance to EGFR tyrosine kinase
inhibitors in non-small cell lung cancer. Drug Resist Updat.
82:1012662025. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Satir P, Pedersen LB and Christensen ST:
The primary cilium at a glance. J Cell Sci. 123:499–503. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Anvarian Z, Mykytyn K, Mukhopadhyay S,
Pedersen LB and Christensen ST: Cellular signalling by primary
cilia in development, organ function and disease. Nat Rev Nephrol.
15:199–219. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kiseleva AA, Nikonova AS and Golemis EA:
Patterns of ciliation and ciliary signaling in cancer. Rev Physiol
Biochem Pharmacol. 185:87–105. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shireman JM, Atashi F, Lee G, Ali ES,
Saathoff MR, Park CH, Savchuk S, Baisiwala S, Miska J, Lesniak MS,
et al: De novo purine biosynthesis is a major driver of
chemoresistance in glioblastoma. Brain. 144:1230–1246. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chao YY, Huang BM, Peng IC, Lee PR, Lai
YS, Chiu WT, Lin YS, Lin SC, Chang JH, Chen PS, et al: ATM- and
ATR-induced primary ciliogenesis promotes cisplatin resistance in
pancreatic ductal adenocarcinoma. J Cell Physiol. 237:4487–4503.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wei L, Ma W, Cai H, Peng SP, Tian HB, Wang
JF, Gao L and He JP: Inhibition of ciliogenesis enhances the
cellular sensitivity to temozolomide and ionizing radiation in
human glioblastoma cells. Biomed Environ Sci. 35:419–436.
2022.PubMed/NCBI
|
|
13
|
Ma W, Wei L, Jin L, Ma Q, Zhang T, Zhao Y,
Hua J, Zhang Y, Wei W, Ding N, et al: YAP/Aurora A-mediated
ciliogenesis regulates ionizing radiation-induced senescence via
Hedgehog pathway in tumor cells. Biochim Biophys Acta Mol Basis
Dis. 1870:1670622024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jenks AD, Vyse S, Wong JP, Kostaras E,
Keller D, Burgoyne T, Shoemark A, Tsalikis A, de la Roche M,
Michaelis M, et al: Primary cilia mediate diverse kinase inhibitor
resistance mechanisms in cancer. Cell Rep. 23:3042–3055. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee C, Yi J, Park J, Ahn B, Won YW, Jeon
J, Lee BJ, Cho WJ and Park JW: Hedgehog signalling is involved in
acquired resistance to KRASG12C inhibitors in lung
cancer cells. Cell Death Dis. 15:562024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Khan NA, Willemarck N, Talebi A, Marchand
A, Binda MM, Dehairs J, Rueda-Rincon N, Daniels VW, Bagadi M,
Thimiri Govinda Raj DB, et al: Identification of drugs that restore
primary cilium expression in cancer cells. Oncotarget. 7:9975–9992.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo R, Liu T, Shasaltaneh MD, Wang X,
Imani S and Wen Q: Targeting adenylate cyclase family: New concept
of targeted cancer therapy. Front Oncol. 12:8292122022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Johnson JLF and Leroux MR: cAMP and cGMP
signaling: Sensory systems with prokaryotic roots adopted by
eukaryotic cilia. Trends Cell Biol. 20:435–444. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brewer KK, Brewer KM, Terry TT, Caspary T,
Vaisse C and Berbari NF: Postnatal dynamic ciliary ARL13B and ADCY3
localization in the mouse brain. Cells. 13:2592024. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ou Y, Ruan Y, Cheng M, Moser JJ, Rattner
JB and van der Hoorn FA: Adenylate cyclase regulates elongation of
mammalian primary cilia. Exp Cell Res. 315:2802–2817. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hong SH, Goh SH, Lee SJ, Hwang JA, Lee J,
Choi IJ, Seo H, Park JH, Suzuki H, Yamamoto E, et al: Upregulation
of adenylate cyclase 3 (ADCY3) increases the tumorigenic potential
of cells by activating the CREB pathway. Oncotarget. 4:1791–1803.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Quinn SN, Graves SH, Dains-McGahee C,
Friedman EM, Hassan H, Witkowski P and Sabbatini ME: Adenylyl
cyclase 3/adenylyl cyclase-associated protein 1 (CAP1) complex
mediates the anti-migratory effect of forskolin in pancreatic
cancer cells. Mol Carcinog. 56:1344–1360. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hu L, Dong C, Wang Z, He S, Yang Y, Zi M,
Li H, Zhang Y, Chen C, Zheng R, et al: A rationally designed
fluorescence probe achieves highly specific and long-term detection
of senescence in vitro and in vivo. Aging Cell. 22:e138962023.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang Y, Li S, Wang Y, Zhao Y and Li Q:
Protein tyrosine kinase inhibitor resistance in malignant tumors:
Molecular mechanisms and future perspective. Signal Transduct
Target Ther. 7:3292022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim SO, Kim BY and Lee KH: Synergistic
effect of anticancer drug resistance and Wnt3a on primary
ciliogenesis in A549 cell-derived anticancer drug-resistant subcell
lines. Biochem Biophys Res Commun. 635:1–11. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Duffy MP, Sup ME and Guo XE: Adenylyl
cyclase 3 regulates osteocyte mechanotransduction and primary
cilium. Biochem Biophys Res Commun. 573:145–150. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sterpka A and Chen X: Neuronal and
astrocytic primary cilia in the mature brain. Pharmacol Res.
137:114–121. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hilgendorf KI, Myers BR and Reiter JF:
Emerging mechanistic understanding of cilia function in cellular
signalling. Nat Rev Mol Cell Biol. 25:555–573. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kiseleva AA, Korobeynikov VA, Nikonova AS,
Zhang P, Makhov P, Deneka AY, Einarson MB, Serebriiskii IG, Liu H,
Peterson JR and Golemis EA: Unexpected activities in regulating
ciliation contribute to off-target effects of targeted drugs. Clin
Cancer Res. 25:4179–4193. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guen VJ and Prigent C: Targeting Primary
ciliogenesis with small-molecule inhibitors. Cell Chem Biol.
27:1224–1228. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Collinson R and Tanos B: Primary cilia and
cancer: A tale of many faces. Oncogene. 44:1551–1566. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Saito M, Otsu W, Miyadera K and Nishimura
Y: Recent advances in the understanding of cilia mechanisms and
their applications as therapeutic targets. Front Mol Biosci.
10:12321882023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pant K, Richard S, Peixoto E, Baral S,
Yang R, Ren Y, Masyuk TV, LaRusso NF and Gradilone SA:
Cholangiocyte ciliary defects induce sustained epidermal growth
factor receptor signaling. Hepatology. 81:1132–1145. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yin F, Chen Q, Shi Y, Xu H, Huang J, Qing
M, Zhong L, Li J, Xie L and Zeng X: Activation of EGFR-Aurora A
induces loss of primary cilia in oral squamous cell carcinoma. Oral
Dis. 28:621–630. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kasahara K, Aoki H, Kiyono T, Wang S,
Kagiwada H, Yuge M, Tanaka T, Nishimura Y, Mizoguchi A, Goshima N
and Inagaki M: EGF receptor kinase suppresses ciliogenesis through
activation of USP8 deubiquitinase. Nat Commun. 9:7582018.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tsai YC, Kuo TN, Chao YY, Lee PR, Lin RC,
Xiao XY, Huang BM and Wang CY: PDGF-AA activates AKT and ERK
signaling for testicular interstitial Leydig cell growth via
primary cilia. J Cell Biochem. 124:89–102. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee JE, Park HS, Lee D, Yoo G, Kim T, Jeon
H, Yeo MK, Lee CS, Moon JY, Jung SS, et al: Hippo pathway effector
YAP inhibition restores the sensitivity of EGFR-TKI in lung
adenocarcinoma having primary or acquired EGFR-TKI resistance.
Biochem Biophys Res Commun. 474:154–160. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Guo Y, Dupart M, Irondelle M, Peraldi P,
Bost F and Mazure NM: YAP1 modulation of primary cilia-mediated
ciliogenesis in 2D and 3D prostate cancer models. FEBS Lett.
598:3071–3086. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee J, Park KC, Sul HJ, Hong HJ, Kim KH,
Kero J and Shong M: Loss of primary cilia promotes
mitochondria-dependent apoptosis in thyroid cancer. Sci Rep.
11:41812021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jeffries EP, Di Filippo M and Galbiati F:
Failure to reabsorb the primary cilium induces cellular senescence.
FASEB J. 33:4866–4882. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ma X, Zhang Y, Zhang Y, Zhang X, Huang Y,
He K, Chen C, Hao J, Zhao D, LeBrasseur NK, et al: A stress-induced
cilium-to-PML-NB route drives senescence initiation. Nat Commun.
14:18402023. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Breslin L, Prosser SL, Cuffe S and
Morrison CG: Ciliary abnormalities in senescent human fibroblasts
impair proliferative capacity. Cell Cycle. 13:2773–2779. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Teng YN, Chang HC, Chao YY, Cheng HL, Lien
WC and Wang CY: Etoposide triggers cellular senescence by inducing
multiple centrosomes and primary cilia in adrenocortical tumor
cells. Cells. 10:14662021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu X, Wei L, Zhang R, Chen J, Zhang T,
Hua J, Wang J, He J and Xie X: The DNA-PKcs-primary cilia axis
maintains ionizing radiation-induced senescence in tumor cells.
Acta Biochim Biophys Sin. 2025.Doi: 10.3724/abbs.2025168.
View Article : Google Scholar
|
|
45
|
Zhang H, Liu Y, Liu J, Chen J, Wang J, Hua
H and Jiang Y: cAMP-PKA/EPAC signaling and cancer: The interplay in
tumor microenvironment. J Hematol Oncol. 17:52024. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ginsberg G, Angle K, Guyton K and Sonawane
B: Polymorphism in the DNA repair enzyme XRCC1: Utility of current
database and implications for human health risk assessment. Mutat
Res. 727:1–15. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zou T, Liu J, She L, Chen J, Zhu T, Yin J,
Li X, Li X, Zhou H and Liu Z: A perspective profile of ADCY1 in
cAMP signaling with drug-resistance in lung cancer. J Cancer.
10:6848–6857. 2019. View Article : Google Scholar : PubMed/NCBI
|














