Introduction
Alternative splicing (AS) enables individual genes
to produce multiple mRNA isoforms, substantially expanding
eukaryotic proteomic diversity and functional complexity (1,2).
Occurring in ~95% of multi-exon genes, AS generates tissue- and
context-specific variants that regulate key processes such as
proliferation, apoptosis and differentiation (1,3,4). AS is
a critical mechanism for regulating gene expression (5). Its biological importance is also
highlighted by genomic studies and ~18% of cancer-associated
single-nucleotide polymorphisms in the COSMIC database are located
within splice-regulatory motifs (6). These mutations compromise exon-intron
recognition, often causing the production of aberrant transcripts
that support oncogenesis and resistance to therapy. In cancer,
aberrant AS causes transcriptomic rewiring that supports tumor
heterogeneity and adaptive evolution; for example, splice variants
such as BRCA1-Δ11q (1) and
BCL2L12 (3) have been
identified that interfere with apoptotic signalling, and variants
of CD44v, which facilitate epithelial-mesenchymal transition (EMT)
and metastasis. Pan-cancer analyses have allowed for the
identification of more than 15,000 cancer-specific splice variants,
numerous of which are associated with chemoresistance and stemness
(7,8). Such alterations are often the result
of mutations affecting spliceosomal components (for example,
SF3B1) or dysregulated splicing factors (for example,
QKI and PTBP1), which can rewire tumor phenotypes
allowing them to be more resilient to cell stresses and to survive
(8,9).
Despite significant progress in chemotherapy and
targeted therapies, drug resistance continues to pose a major
challenge in oncology and is responsible for more than 50% of
cancer-related deaths, with relapse rates exceeding 70% in
aggressive cancers such as pancreatic ductal adenocarcinoma (PDAC)
and glioblastoma (3,8). Although numerous established
resistance mechanisms involve secondary genetic mutations,
including reversion mutations that restore homologous recombination
in BRCA1/2-mutant cancers, increasing evidence underscores
the clinical relevance of dysregulated RNA splicing. For instance,
in BRCA1-mutant ovarian cancers, secondary splice-site
mutations (SSMs) that cause exon skipping (AS) and produce
resistant BRCA1 isoforms are detected in roughly 10% of patients
following PARP inhibitor therapy, a considerable increase from the
pretreatment frequency of ~1% (10). Pan-cancer analyses further reveal
that AS produces diverse tumor-specific neoantigens and
drug-resistant isoforms that promote immune evasion and treatment
failure across cancer types. Although the exact proportion of
splicing-mediated resistance has yet to be fully quantified and
likely varies by cancer type and regimen, splicing dysregulation
constitutes a major non-mutational pathway contributing
significantly to the overall resistance burden, second only to
genetic alterations (11,12).
AS-mediated mechanisms span diverse therapies:
BRCA1-Δ11q confers resistance to PARP inhibitors and
cisplatin in breast cancer (1),
while c-Mpl-del splice variants promote chemoresistance in acute
megakaryocytic leukemia (13).
Under hypoxic conditions, LUCAT1 recruits PTBP1 to alter
PARP3 splicing and foster oxaliplatin resistance in
colorectal cancer (CRC) (9),
paralleled by FBXO7 stabilization of RBFOX2 to drive
pro-survival FoxM1 and POSTN splicing in
temozolomide-resistant glioblastoma (3).
Key splicing factors act as molecular hubs:
QKI establishes basal-like splicing programs supporting PDAC
chemoresistance (8), while SNHG
family lncRNAs employ intron retention (IR) to suppress snoRNAs and
enhance hepatoblastoma drug tolerance (14). Although hypoxia-induced LUCAT1 binds
PTBP1 to modulate DNA damage response genes (9), the hierarchical organization of these
regulatory networks remains undefined. Elucidating these mechanisms
will require integrated approaches to identify master regulators
and advance isoform-directed therapeutic strategies.
Molecular architecture of AS in cancer
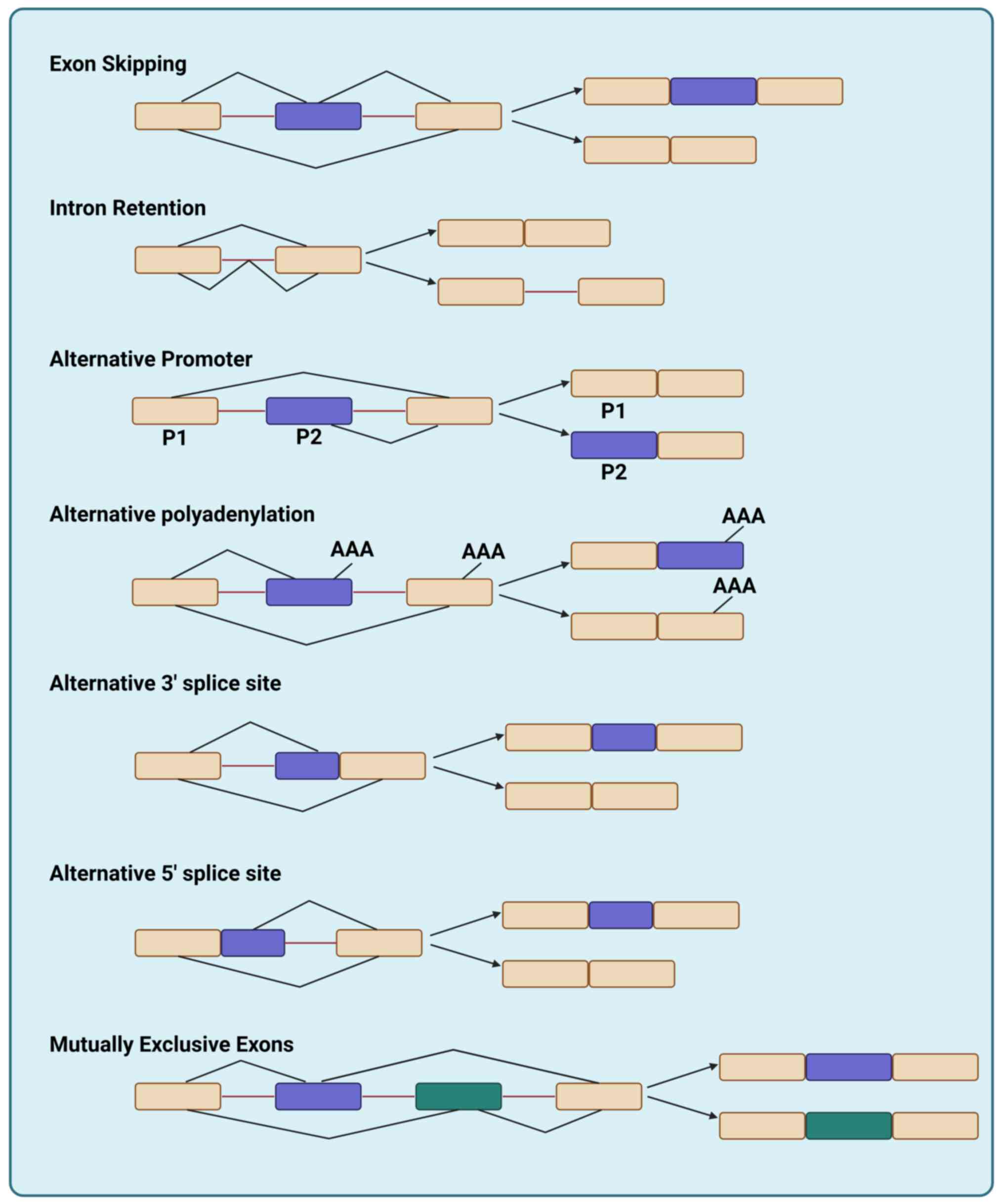
Classification of AS events
Canonical AS classification and the splicing
resistance map framework
Transcripts from nearly all human protein-coding
genes undergo one or more modes of AS, such as inclusion or
skipping of individual ‘cassette’ exons, selection between
alternative 5′ and 3′ splice sites, differential retention of
introns, mutually exclusive splicing of adjacent exons, and other,
more complex patterns of splicing (15). These diverse outcomes are broadly
classified into classical patterns of AS, such as ES, IR,
alternative promoter (AP) usage, alternative polyadenylation (APA),
alternative 3′ splice site, alternative 5′ splice site and mutually
exclusive exons (MXE) (Fig. 1).
These canonical splicing patterns constitute the foundational
elements of the ‘splicing resistance map’, a unified landscape of
AS events that collectively underlie therapeutic resistance across
diverse cancers. In The Cancer Genome Atlas (TCGA) pan-cancer
analysis, AP and ES were the most prevalent splicing patterns;
importantly, these frequencies were derived from bulk tumor
transcriptomes and therefore reflect composite signals from
malignant and stromal/immune cells (16) (Fig.
S1).
ES is reported to be one of the most common AS
event, characterized by loss of functional domains/sites or a shift
in the open reading frame, leading to a variety of human diseases
(17), which involves the exclusion
of specific exons during mRNA maturation, often modifying protein
domains critical for tumor progression (18–20).
ES events constitute a prominent component of the splicing
resistance map, yielding isoforms that circumvent targeted
therapies. For example, in triple-negative breast cancer (TNBC),
PRMT5 inhibition induces ES in AURKB transcripts, resulting
in mitotic catastrophe and chemoresistance by disrupting chromatin
stability (21). In the mesenchymal
subtype of CRC (CMS4), retention of exon 17 in the MYOF gene
and skipping of exon 11 in the ENAH gene underlie poor
prognosis and enhanced chemoresistance by promoting EMT and
augmenting tumor invasiveness (22). IR, once considered rare, is now
recognized as a hallmark of aggressive malignancies (23–26).
IR events represent a major axis of the splicing resistance map,
particularly in advanced and therapy-refractory malignancies. In
prostate cancer, IR induces the accumulation of aberrant
transcripts (for example, SNHG19), which fosters castration
resistance and stemness by impairing DNA repair pathways, enhancing
chemoresistance, and activating oncogenic cascades, thereby
nominating the spliceosome as a promising therapeutic target whose
inhibitors reverse IR-mediated aggressive phenotypes (27,28).
APs, involve the use of different transcription
start sites within a gene, thereby altering the 5′-untranslated
regions (UTRs) sequences to generate distinct variants. Promoters
not only determine when a gene is active and to what extent, but
also regulate which gene isoforms are expressed (29). AP-mediated isoform switching
constitutes a central mechanism within the splicing resistance map,
facilitating adaptive transcriptional reprogramming under
therapeutic pressure. APs enable tissue-specific isoform
expression, as observed in MET exon 14 skipping in non-small cell
lung cancer (NSCLC), where splice site mutations produce oncogenic
isoforms that evade kinase inhibitor targeting (30). In prostate cancer, distal APs
(P2/P3) supersede the proximal promoter (P1) to drive
UGT2B17 transcription, generating n2/n3 isoforms, which
contain additional 5′ exons (for example, 1b/1c) through AS and
encode full-length proteins, that dominate the androgen
inactivation pathway in localized and metastatic tumors (31). In liver cancer, selective DNA
methylation loss in CpG-poor regions increases chromatin
accessibility, thereby enabling the binding of tissue-specific
transcription factors and activation of APs, which drive
tumor-specific transcription (32).
These canonical AS patterns are systematically cataloged in
large-scale projects such as TCGA, which demonstrated that >90%
of cancer-related genes exhibit at least one AS event (33). Computational analysis of these
splicing events employs specialized tools including the SpliceSeq
package, which aligns RNA-Seq reads to a gene-specific exon
junction graph and computes Percent Spliced In values to
systematically quantify exon or splice junction inclusion across
numerous tumor and normal samples (34). Long-read sequencing has recently
revealed that 30% of isoforms in breast cancer contain unannotated
ES or IR events, numerous of which are associated with
HER2+ or TNBC subtypes (21,33).
Other common splicing classes include MXE, exemplified by CD44
variants that promote ovarian cancer metastasis, and alternative
3′/5′ splice site selection that alters EGFR kinase domains to
confer tyrosine kinase inhibitor resistance (35).
Functionally classified isoforms in
therapy resistance
New classification systems focus on AS events that
directly influence therapeutic response, particularly those
enabling drug resistance. Understanding these mechanisms requires
precise definition of ‘therapy-resistance-associated isoforms’,
alternatively spliced mRNAs experimentally demonstrated to confer
resistance by modifying drug targets, activating compensatory
pathways, or enhancing survival (10–12).
To qualify, isoforms must satisfy three criteria: i) functional
validation through drug sensitivity assays; ii) association with a
specific treatment; and iii) identification in human tumors or
clinically relevant models.
Numerous examples demonstrate this concept. In PARP
inhibitor-resistant ovarian cancer, BRCA1 exon 11 skipping
generates Δ11q isoforms that restore homologous recombination
repair (HRR) capacity, circumventing nonsense-mediated decay
(10). Similarly, NT5C2 exon
6a inclusion yields a hyperactive nucleotidase conferring
6-mercaptopurine resistance in leukemia (36). NSCLC with KRAS G12 mutations
exhibits MET exon 14 skipping that activates RAS/ERK
signaling to evade MET inhibitor effects (30). Splicing alterations also perturb
epigenetic regulation: PRMT5 inhibition in CDK4/6
inhibitor-resistant breast cancer induces FUS-dependent IR,
impairing DNA synthesis proteins such as PCNA and MCM2 (37). Given their clinical relevance, these
mechanisms are emerging as therapeutic targets themselves, for
instance, CRISPR/dCasRx-mediated exclusion of TIMP1 exons
counteracts SRSF1-driven splicing and attenuates metastasis in CRC
(38). Such subclassifications are
transforming precision oncology, with clinical trials currently
evaluating splice-switching antisense oligonucleotides (ASOs)
targeting PTBP1 in glioblastoma (39). Additionally, APA events in
BCL2L1 produce anti-apoptotic isoforms in hepatocellular
carcinoma (HCC), while poison exon inclusion in MAP3K7
disrupts TGF-β signaling to foster chemoresistance (27,35).
Isoforms are excluded from this category if evidence is purely
computational or derived from correlative expression without direct
functional proof, or if the resistance mechanism is indirect (for
example, via general proliferation effects) (10–12).
Representative isoforms that satisfy these criteria,
highlighting their drug contexts, functional validation methods,
and clinical evidence levels are presented in Table I.
 | Table I.Exemplary
therapy-resistance-associated isoforms. |
Table I.
Exemplary
therapy-resistance-associated isoforms.
| First author/s,
year | Isoform | Gene | Drug context | Functional
assay | Clinical evidence
level | (Refs.) |
|---|
| Nesic et al,
2024 | Δ11q | BRCA1 | PARP inhibitors
(olaparib, rucaparib) | RNA-seq, minigene
assays, CRISPR/Cas9 | Patient biopsies,
PDX models | (10) |
| Bradley and
Anczuków, 2023 | BQ323636.1 | NCOR2 | Tamoxifen,
aromatase inhibitors (anastrozole) | RNA-seq,
immunohistochemistry, siRNA knockdown | Patient tissue
microarrays, cell lines | (11) |
| Sciarrillo et
al, 2020 | AR-V7 | AR | Androgen receptor
inhibitors (enzalutamide, abiraterone) | RT-qPCR,
RNA-sequencing, circulating tumor cell analysis | Patient circulating
tumor cells | (40) |
| Kahles et
al, 2018 | PKM2 | PKM | Metabolic
inhibitors (for example, glycolysis inhibitors) | siRNA knockdown,
CRISPR screening | Pan-cancer TCGA
data, cell lines | (12) |
| Sciarrillo et
al, 2020 | HER2Δ16 | ERBB2 | Trastuzumab | Immunoblotting,
colony formation assays | Breast cancer
patient biopsies | (40) |
|
| BRAF p61 | BRAF | Vemurafenib | RNA-seq, protein
immunoprecipitation | Melanoma
biopsies |
|
|
| CD44v6 | CD44 | 5-FU, radiation
therapy | Antibody blockade,
RNA interference | Patient tumor
samples, cell lines |
|
|
| MENAINV | ENAH | Paclitaxel | Invasion assays,
isoform-specific antibodies | Breast cancer
metastases |
|
|
| BARD1β | BARD1 | PARP
inhibitors | Immunoblotting,
homologous | Colon cancer cell
lines |
|
|
|
|
|
| recombination
assays |
|
|
|
| KLF6-SV1 | KLF6 | Sorafenib,
cisplatin | Apoptosis assays,
xenograft models | Hepatocellular
carcinoma samples |
|
|
| CHK1-S | CHEK1 | DNA-damaging
agents | Kinase activity
assays, survival analysis | Patient tumor
tissues |
|
|
|
|
| (for example,
topotecan) |
|
|
|
|
| RonΔ165 | MST1R | MET inhibitors
(crizotinib) | Migration assays,
immunoblotting | PDX models, NSCLC
cell lines |
|
|
| FGFR1β | FGFR1 | FGFR inhibitors
(erdafitinib) | Proliferation
assays, reverse transcription | Bladder cancer
models |
|
|
|
|
|
| PCR |
|
|
|
| IL-6R soluble | IL6R | Tocilizumab | ELISA, signaling
pathway analysis | Patient serum
samples |
|
|
| CD19e2Δ | CD19 | CAR-T therapy | Flow cytometry,
sequencing | B-ALL patient
samples at relapse |
|
|
| CD22e2Δ | CD22 | CAR-T therapy | Isoform-specific
PCR, cytotoxicity assays | B-cell malignancy
models |
|
|
| USP5s | USP5 | Gemcitabine | Splicing
modulation, drug sensitivity tests | Glioma cell
lines |
|
|
| MDM4-S | MDM4 | MDM2 inhibitors
(nutlin-3) | Apoptosis assays,
xenograft studies | Patient tumor
tissues |
|
|
| SRSF3-S | SRSF3 | Paclitaxel | Colony formation,
ASO treatment | Oral squamous cell
carcinoma |
|
|
| circCDYL2 | CDYL2 | Cisplatin,
radiation | RNAi, miRNA
sponging assays | Patient plasma,
xenograft models |
|
|
| VEGFxxxb | VEGFA | Anti-angiogenic
therapies | Splicing reporter
assays, tube formation | Patient serum and
tumor tissues |
|
|
|
|
| (bevacizumab) | assays |
|
|
This systematic compilation underscores the
diversity of splicing-mediated resistance mechanisms, which often
coexist with genetic mutations and contribute substantially to
treatment failure. Functional validation through targeted assays
(for example, CRISPR and ASOs) is essential to establish causality,
while clinical evidence ranges from preclinical models to direct
patient data, highlighting the translational relevance of these
isoforms (10,12,40).
Cis-regulatory elements and trans-acting
factors
Cis-regulatory elements and
trans-acting factors form the molecular scaffold of AS regulation
in cancer
Cis-acting elements, including exonic/intronic
splicing enhancers (ESE/ISE) and silencers (ESS/ISS), are
nucleotide sequences within pre-mRNA that recruit trans-acting
regulators (for example, SR proteins, hnRNPs) to govern splice site
selection (41,42), Cell-type specific gene expression
patterns are regulated by cis-regulatory elements such as promoters
and enhancers (43) (Fig. 2). Moreover, the regulation of AS
entails recognition of cis-acting RNA elements in the pre-mRNA by
trans-acting protein splicing factors (44). These elements operate
combinatorially to enhance or repress spliceosome assembly, with
mutations or dysregulated expression commonly observed in cancer
(12). For example, BCL2L1
ESE mutations drive pro-survival BCL2L1-L isoform
expression, conferring apoptosis resistance (40). Genome-wide analyses indicate that
~15% of cancer-associated mutations map to splicing regulatory
elements, with ESE/ESS regions in oncogenes for example,
MET) exhibiting recurrent alterations (12,45).
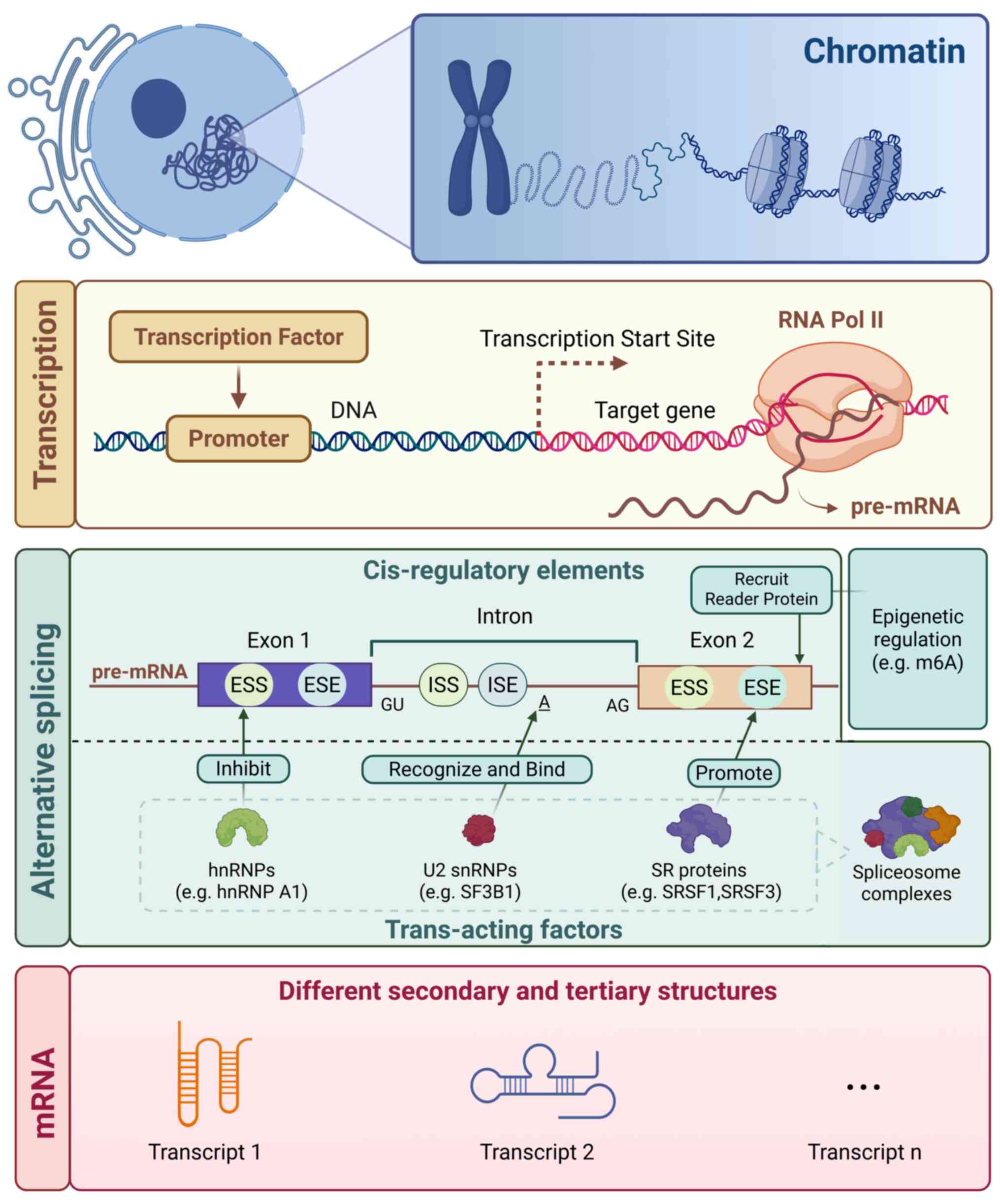 | Figure 2.Alternative splicing mechanism and
spliceosome complex proteins with their functions. Pre-mRNA
splicing is the process of removal of intronic regions and joining
the exons to form mature mRNA. Trans-acting factors consist of
splicing factors which recognize conserved premRNA sequences
(cis-regulatory elements) and recruit the core spliceosome
machinery which coordinates and executes the excision of introns.
U2 snRNP (for example, SF3B1) recognizes and binds to branch point
A, initiating spliceosome assembly and splicing reactions; SR
proteins (for example, SRSF1) primarily bind to ESE elements (with
partial assistance from ISE elements) to promote splicing; hnRNPs
(for example, hnRNP A1) inhibit splicing by binding to ESS/ISS
elements; epigenetic regulation (for example, m6A modification)
acts on exons through recruiting reader proteins (for example,
YTHDC1). These factors collectively act on pre-mRNA, regulating
alternative splicing and ultimately generating distinct mRNA
transcripts. ESE, exonic splicing enhancer; ISE, intronic splicing
enhancer; ESS, exonic splicing silencer; ISS, intronic splicing
silencer. |
Integrated cis-trans-epigenetic
mechanisms enable adaptive therapy resistance
Chronic myeloid leukemia (CML) cells exhibit ESS
defects in SRSF1-targeted transcripts (for example, PRKCH),
sustaining oncogenic signaling despite BCR-ABL1 inhibition
(46). Glioblastoma-associated
SF3B1 inhibition alters BCL2L1 splicing, favoring
anti-apoptotic isoform expression (47). High-throughput studies identify
recurrent ESE/ESS mutations in drug resistance genes (for example,
ABCB1), which disrupt repressive elements to circumvent
therapy (44). For example,
aberrant ESS motifs in TP53 exon 6 promote ES, generating
dominant-negative isoforms that compromise chemotherapy-induced
apoptosis (40). SR proteins (for
example, SRSF1) and hnRNPs (for example, hnRNP A1) are frequently
overexpressed in therapy-resistant cancers. In CML, SRSF1
upregulation enhances BCL2L1 exon 2 inclusion, elevating
pro-survival BCL2L1-S isoforms and undermining imatinib-induced
apoptosis (46). SF3B1 dysfunction
in glioblastoma drives BCL2L1-L overexpression via preferential 3′
splice site selection, facilitating temozolomide evasion (47). Post-transcriptional modifications
further modulate splicing factor activity: METTL3-mediated m6A
methylation stabilizes SRSF1 mRNA in pancreatic cancer,
potentiating cisplatin resistance (48). Epigenetic regulation, encompassing
DNA methylation and RNA epitranscriptomics (for example, m6A),
modulates splicing by altering RNA-protein interactions (49–51).
In gastric cancer, SNORA37 promotes CMTR1-ELAVL1
interaction to drive CD44 AS and tumor progression (52). Dysregulation of the epigenome drives
aberrant transcriptional programs that promote cancer onset and
progression (53,54). METTL3 deposits m6A on
QSOX1 exon 14, recruiting YTHDC1 to enhance exon inclusion
and β-catenin activation, thereby conferring gefitinib resistance
in NSCLC (55). Conversely, ALKBH5
erases m6A from FOXO1 mRNA, enabling SRSF3 binding and
anti-apoptotic isoform generation (55). Chromatin remodelers (for example,
SWI/SNF) also influence splicing: ARID1A loss induces
BRD9 ES, activating Wnt signaling in ovarian cancer
(44). These findings collectively
define a dynamic ‘splicing code’ that integrates cis-regulatory
mutations, trans-factor dysregulation and epigenetic modifications,
a multilayered system that enables tumor cells to rapidly adapt and
develop resistance to targeted therapies, chemotherapy, and
epigenetic drugs (Fig. 2).
Splicing dysregulation: Protein
alterations and cis-element mutations
Altered spliceosome proteins due to somatic
mutations, overexpression or post-translational modifications are
key drivers of aberrant splicing that promotes tumour survival and
therapy resistance. Mutations in SF3B1 in glioblastoma lead
to ES such as BRCA1-Δ11q that results in a hypomorphic
isoform that provides resistance to PARP inhibition (12). In CML, SRSF1 is overexpressed in
leukemic blasts and IL-3 maintains SRSF1 expression following
BCR-ABL1 inhibition, leading to imatinib resistance via PKC-
and PLC-dependent mechanisms (46).
These events promote proliferation and inhibit apoptosis providing
a selective advantage in cancer.
DNA cis-element mutations (for example, in exonic
splicing silencers) disrupt trans-factor binding, leading to
aberrant splicing and drug resistance. In acute lymphoblastic
leukemia (ALL), NT5C2 exon 6a inclusion creates a
gain-of-function isoform with elevated nucleotidase activity,
driving resistance to 6-mercaptopurine (36). These mutations are enriched in
relapse samples and represent promising targets for ASOs due to
their sequence-specific nature; ASOs can be designed to bind mutant
cis-elements or aberrant splice sites, blocking incorrect splicing
and restoring normal isoform expression. This re-sensitizes cells
to therapy, as demonstrated in leukemia models where ASOs reversed
chemoresistance by correcting NT5C2 splicing (Figs. 3B and 4) (36).
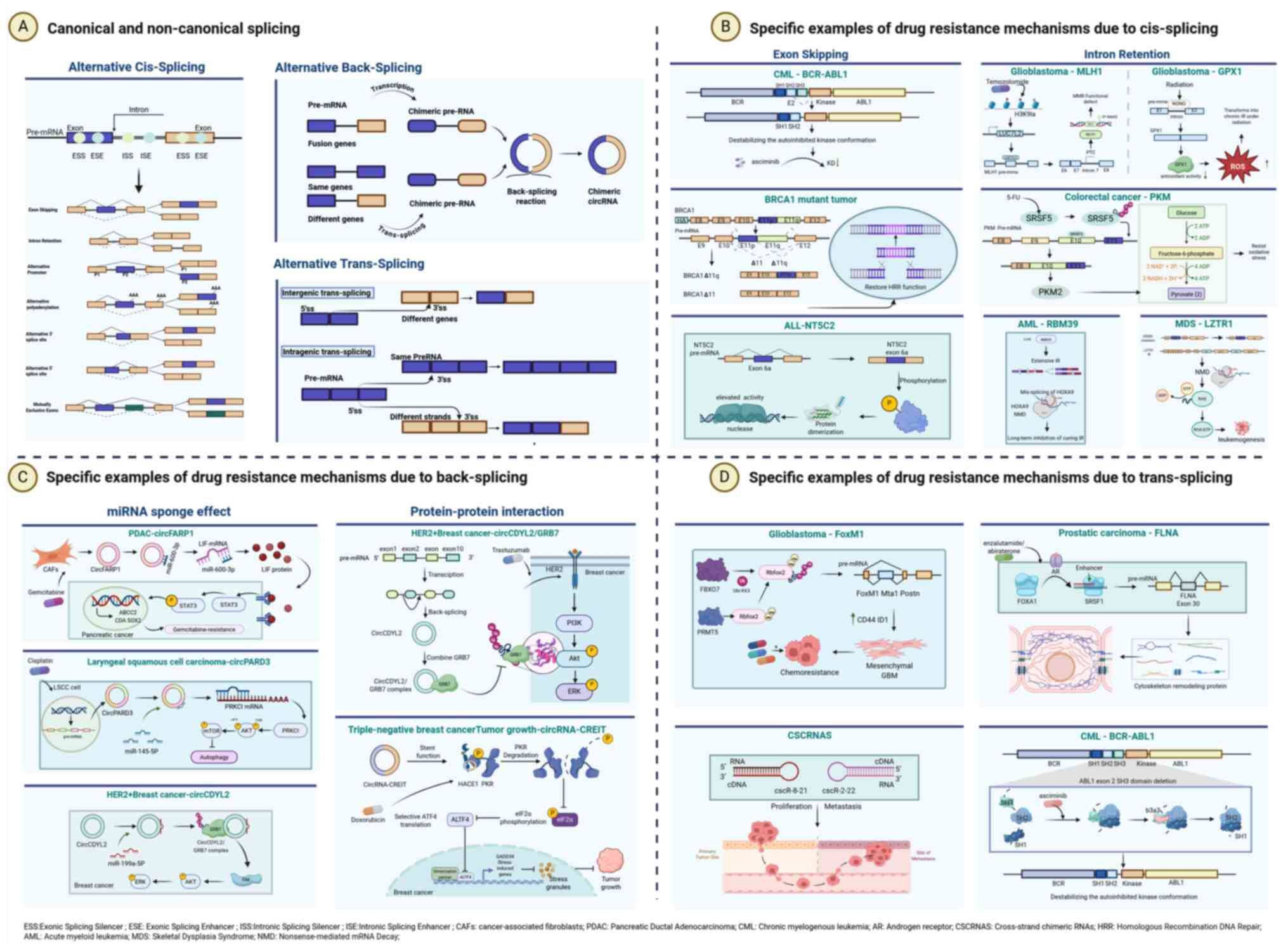 | Figure 3.Canonical vs. non-canonical splicing
events and their associated resistance mechanisms. (A) The AS can
be classified into two categories, canonical splicing events: ACS
and non-canonical splicing events: ATS and ABS. There are seven
subtypes of ACS, consistent with the content in Fig. 1. ABS is a non-canonical RNA
processing mechanism distinct from linear splicing, wherein a
downstream splice donor site covalently joins an upstream splice
acceptor site, forming a closed-loop RNA structure called circular
RNA (circRNA). ATS connects fragments from two distinct primary RNA
transcripts generating chimeric RNAs. ATS can be classified into
two types based on the origin of the primary RNA transcripts,
involving intragenic and intergenic trans-splicing. (B) Specific
examples of drug resistance mechanisms due to cis-splicing. Exon
skipping (left panel) confers resistance through alterations in
protein structure and function. Examples include: deletion of the
SH3 domain in BCR-ABL1, which reduces asciminib binding in chronic
myeloid leukemia (CML); skipping of exon 11 in BRCA1, which
restores homologous recombination repair (HRR) and leads to PARP
inhibitor (PARPi) resistance; and inclusion of exon 6a in NT5C2,
which promotes dimerization and enhances enzymatic activity in
acute lymphoblastic leukemia (ALL). Intron retention (right panel)
facilitates resistance via rapid stress response and regulation of
gene expression. Key instances are: temozolomide-induced detained
introns in MLH1, resulting in mismatch repair (MMR) deficiency in
glioblastoma; radiation-induced intron retention in GPX1, mediated
by NONO, that impairs its antioxidant function, elevates reactive
oxygen species (ROS), and promotes radiation resistance in
glioblastoma; SRSF5-mediated retention of intron 9 in PKM,
upregulating PKM2 to enhance glycolysis and confer oxidative stress
resistance in colorectal cancer; widespread intron retention
following RBM39 loss, which triggers nonsense-mediated decay (NMD)
of oncogenic transcripts in acute myeloid leukemia (AML); and
finally, intron retention in LZTR1 in myelodysplastic syndromes
(MDS) that evades NMD, thereby activating the RAS pathway and
driving leukemogenesis. (C) Specific examples of drug resistance
mechanisms due to back-splicing. The miRNA sponge mechanism (left
panel) is illustrated by the following: exosomal circFARP1 derived
from cancer-associated fibroblasts sequesters miR-660-3p, leading
to upregulation of LIF and activation of STAT3 signaling, thereby
promoting stemness and gemcitabine resistance in pancreatic ductal
adenocarcinoma; circPARD3 acts as a sponge for miR-145-5p,
activating the PRKCI/AKT/mTOR pathway to suppress autophagy and
induce cisplatin resistance in laryngeal squamous cell carcinoma;
and circCDYL2 sequesters miR-199a-5p, resulting in elevated GRB7
expression and subsequent FAK signaling activation, which
contributes to trastuzumab resistance in HER2-positive breast
cancer. Protein-protein interactions (right panels) are
demonstrated through two examples: circCDYL2 directly binds to and
stabilizes GRB7, sustaining HER2/AKT/ERK signaling; and
circRNA-CREIT scaffolds the HACE1-mediated degradation of PKR,
which disrupts stress granule formation and promotes doxorubicin
resistance in triple-negative breast cancer. (D) Specific examples
of drug resistance mechanisms due to trans-splicing. In
glioblastoma, Rbfox2 stabilized by FBXO7 promotes trans-splicing,
generating FoxM1 variants that upregulate stemness markers (for
example, CD44, ID1) and confer chemoresistance. In prostate cancer,
FOXA1 facilitates the inclusion of FLNA exon 30 via trans-splicing,
resulting in cytoskeletal remodeling and resistance to androgen
receptor inhibitors. Trans-splicing between sense and antisense
transcripts also produces cross-strand chimeric RNAs (such as
cscR-8-21 and cscR-2-22), which contribute to cancer cell survival
and have been implicated in therapy resistance. Furthermore, in
chronic myeloid leukemia, trans-splicing gives rise to truncated
BCR-ABL1 isoforms (for example, b3a3) that lack the SH3 domain,
leading to constitutive kinase activity and resistance to
allosteric inhibitors such as asciminib. ACS, alternative
cis-Splicing; ATS, alternative trans-splicing; ABS, alternative
back-splicing |
AS-driven drug resistance mechanisms
Cis splicing in drug resistance
Overview of cis splicing
In cancer, dysregulated cis splicing promotes tumor
progression and therapy resistance by altering protein function,
stability, or subcellular localization (56). For instance, aberrant splicing of
apoptosis regulators, oncogenic kinases and drug transporters
directly affects treatment efficacy and disease relapse (10). The clinical relevance of
cis-splicing is underscored by its association with poor prognosis
and resistance to targeted therapies, immunotherapies and
chemotherapy (57).
ES
AS-mediated exclusion of ABL1 exon 2 results
in an in-frame deletion of SH3 domain residues. This 31-amino-acid
deletion in BCR-ABL1 weakens asciminib binding by 18-fold and
impairs the autoinhibitory conformation required for drug efficacy,
conferring resistance in CML (58).
In BRCA1, secondary SSMs induce exon 11
skipping, producing truncated Δ11 and Δ11q isoforms that restore
HRR capacity. These isoforms allow tumor cells to bypass
PARPi-induced synthetic lethality (10).
In relapsed ALL, the inclusion of exon 6a introduces
a novel phosphorylation site that promotes NT5C2
dimerization, leading to a 15-fold increase in enzymatic activity
of the protein compared with the wild-type protein (36).
IR
IR and the more recent concept of ‘detained introns’
(DIs), a type of IR, are critical splicing events that drive
resistance to cancer therapies (59,60).
DIs are introns that are retained in polyadenylated transcripts
owing to a stalled co-transcriptional splicing process. These
splicing events act as rapid and reversible regulators of gene
expression, whereas classical IR, is defined by persistent IR in
mature mRNAs.
In glioblastoma, multiple of splicing mechanisms
cooperate to mediate both acute and chronic therapy resistance.
Chemoradiation with agents such as temozolomide stalls RNA
polymerase II elongation and induces DIs in MLH1 through
H3K9 lactylation-dependent upregulation of LUC7L2 (62), which introduces a frameshift
mutation truncating the protein and leading to mismatch repair
(MMR) dysfunction. The functional loss of MMR leads to acute
chemoresistance. These DIs are capable of acting as rapid stress
sensors, such that inhibition of lactylation rescues MLH1
splicing and re-sensitizes the tumors to treatment (63). In addition, NONO-induced IR in
GPX1 results in the loss of antioxidant activity and an
increase in reactive oxygen species which leads to apoptosis
(63). Radiation further stalls
splicing progression, converting the acute DI response into chronic
IR in an epigenetically dependent manner, highlighting how stress
duration mediates resistance plasticity (61).
In CRC, SRSF5-induced IR in PKM intron 9
leads to the upregulation of PKM2, which increases glycolysis and
enables survival under 5-fluorouracil-induced oxidative stress
(56).
In AML, loss of RBM39 leads to widespread IR
promoting the nonsense-mediated decay (NMD) of pro-leukemic
transcripts including HOXA9 targets (64). IR in spliceosome mutant cells
mediates their acute vulnerability to this mechanism, providing a
measurable biomarker of therapeutic response. Chronic RBM39
inhibition leads to the consolidation of the IR state promoting
continual NMD.
In myelodysplastic syndromes (MDS), loss of
ZRSR2 causes IR in a subset of minor introns in LZTR1
inducing NMD and subsequent RAS pathway activation, which drives
leukemogenesis in these patients (65). Taken together, chemoradiation stalls
co-transcriptional splicing, inducing DIs as an acute, reversible
response to therapy (61). Chronic
stress promotes the conversion of DIs into stable IR states,
allowing chronic resistance to be established. This dichotomy
explains how tumors adapt rapidly (via DIs) and persist (via IR)
under therapeutic pressure (Fig. 3A and
B).
Back-splicing and circRNA-driven drug
resistance.
Back-splicing: Mechanisms,
classification and functional roles in drug resistance
Back-splicing is a non-canonical RNA processing
mechanism distinct from linear splicing, wherein a downstream
splice donor site covalently joins an upstream splice acceptor
site, forming a closed-loop RNA structure called circular RNA
(circRNA) (66). Numerous
protein-coding genes in higher eukaryotes are capable of producing
circRNAs through back-splicing of exons (67–69).
This process competes with canonical splicing for splice sites
(70–72), resulting in exonuclease-resistant
circRNAs that lack 5′ caps and 3′ poly(A) tails, thereby enhancing
their stability and allowing secretion via extracellular vesicles
(73,74). Experimental evidence underscores the
remarkable stability of circRNAs; for instance, Enuka et al
(75) demonstrated, using metabolic
labeling that the median half-life of circRNAs in mammary cells
ranges from 18.8–23.7 h, significantly longer than the 4.0–7.4 h
observed for linear RNAs. Their exceptional longevity makes
circRNAs well-suited as biomarkers, a feature demonstrated across
multiple studies to stem from their covalently closed circular
architecture which confers intrinsic resistance to exonuclease
degradation (76–78). CircRNAs are broadly classified into
three subtypes: Exonic circRNAs (ecircRNAs), intronic circRNAs and
exon-intron circRNAs (EIciRNAs), with ecircRNAs being the most
prevalent and functionally characterized in cancer (79) (Fig.
3A). Their aberrant expression in cancers provides insights
into roles in tumorigenesis (80),
and they can act as miRNA sponges, protein scaffolds or peptide
templates (74,81). For example, circCDYL2 in
HER2+ breast cancer stabilizes GRB7 to sustain
HER2/AKT/ERK signaling and confers trastuzumab resistance (73), while circPARD3 in laryngeal squamous
cell carcinoma sponges miR-145-5p to activate PRKCI/AKT/mTOR
signaling and suppress autophagy, promoting chemoresistance
(79). The conservation of
back-splicing and its dysregulation in tumors highlight its role in
therapy evasion, with circRNAs like circRNA-CREIT in TNBC serving
as biomarkers for poor prognosis under doxorubicin treatment
(74).
Role of the circFARP1 miRNA sponge
effect in gemcitabine resistance through the miR-660-3p/ leukemia
inhibitory factor (LIF) axis
CircRNAs may act as competing endogenous RNAs to
sponge miRNAs and relieve repression of oncogenic targets. In PDAC,
cancer-associated fibroblasts secrete exosomal circFARP1, which
sponges miR-660-3p to upregulate LIF, activating STAT3 signaling to
induce stemness and gemcitabine resistance (81). In this way, circFARP1 may bind
miR-660-3p to derepress the cytokine LIF, which in turn promotes
IL-6/STAT3 survival signaling pathways (81). Likewise, circPARD3 acts as a sponge
for miR-145-5p, leading to upregulation of PRKCI to activate
AKT/mTOR signaling and inhibit autophagy to enhance cisplatin
resistance in laryngeal squamous cell carcinoma (79). Such circRNA/miRNA/protein signaling
axes are often amplified in tumor cells that are resistant to
therapy, for example, circCDYL2 sponges miR-199a-5p to upregulate
growth factor adaptor protein GRB7 and focal adhesion kinase
signaling in HER2+ breast cancer (73). CircRNA sponging can also be blocked
in tumor cells to sensitize them to chemotherapy such as with
anti-circFARP1 small interfering RNA (siRNA) to to restore miRNA
activity (Fig. 3C) (81).
CircRNA-protein interactions in therapy
resistance: CircCDYL2/growth factor receptor-bound protein 7
(GRB7) complex formation and circRNA-CREIT-mediated protein
kinase R (PKR) degradation. CircRNAs also participate in direct
protein interactions that modulate cellular function. In
HER2+ breast cancer, circCDYL2 binds GRB7,
protecting it from ubiquitination, and thereby maintaining
HER2/AKT/ERK signaling in the presence of trastuzumab (73). This leads to HER2+
tumors exhibiting resistance to HER2− targeted
therapies (73). In TNBC,
circRNA-CREIT acts as a molecular scaffold to bring HACE1 (HACE1
ubiquitin protein ligase) and PKR together, leading to degradation
of PKR and disruption of stress granules, and thereby allowing the
tumor to exhibit resistance to doxorubicin (74). These protein-interaction mechanisms
establish circRNAs as versatile regulators of therapeutic
resistance, suggesting that targeting such interfaces, for instance
through GRB7 inhibition, may represent a promising approach to
reestablish treatment sensitivity (Fig.
3C).
Trans-splicing. Definition, mechanisms
and classification of alternative trans-splicing (ATS)
ATS generates chimeric mRNAs by joining exons from
two distinct pre-mRNA transcripts through spliceosome-mediated
ligation (82,83). Unlike conventional cis-splicing, ATS
connects splice sites located on different RNA molecules, often
mediated by complementary base pairing or trans-acting factors
(4,84). The process involves coordinated
recruitment of spliceosomal components, including U1 and U2 snRNPs,
to donor and acceptor sites on distinct pre-mRNAs, enabling exon
joining via transesterification reactions (11). ATS events are classified as
intergenic when combining exons from different chromosomal loci or
intragenic when linking exons from alternative transcripts of the
same gene (4,12) (Fig.
3A). This mechanism is thus expected to substantially increase
transcript diversity in cancers and likely underlies oncogenic
adaptation in treatment-resistant tumors (84). Although fusion genes such as
ABL/BCR are widely considered to arise from translocations
such as the Philadelphia chromosome, ATS alone can generate similar
chimeric transcripts at the RNA level.
In CML, ATS gives rise to fusion transcripts such
as CLEC12A-MIR223HG, which has been found in both malignant and
normal cells, demonstrating its capacity to diversify the
transcriptome in the absence of DNA rearrangement (85). ATS variants of the BCR-ABL
oncogene may escape detection or cause resistance to imatinib due
to an altered configuration of the kinase domain (58). By augmenting the functional
transcriptome beyond what the genome can encode, ATS can increase
the adaptability of tumor cells for oncogenic adaptation under
therapeutic pressure (86,87).
Mechanisms of ATS in drug
resistance
ATS confers drug resistance through the production
of oncogenic chimeric transcripts or via the disruption of
drug-target interactions. In glioblastoma, FBXO7 stabilizes
RBFOX2 to induce trans-splicing events that produce FOXM1
variants, which further promote the expression of stemness-related
markers such as CD44 and ID1, thereby conferring chemotherapy
resistance (3) (Fig. 3D). Apart from resistance to
chemotherapy, ATS may also play a role in immune escape mechanisms.
Pan-cancer analyses have identified tumor-specific splicing
junctions, or ‘neojunctions’, that produce peptides not found in
normal tissues, and computational predictions suggest these
peptides may serve as ligands for MHC-I proteins and thereby
influence the response to immune checkpoint inhibitors (12). Trans-splicing between BCL2L1
and adjacent genes generates truncated BCL2 variants that impair
chemotherapy-induced apoptosis (11). These mechanisms highlight how ATS
diversifies the transcriptome to promote survival under therapeutic
pressure.
Case studies of ATS in drug-resistant
cancers
Specific ATS events are linked to therapy
resistance. In prostate cancer, FOXA1 orchestrates ATS by binding
enhancer regions of splicing factors such as SRSF1, leading to exon
30 inclusion in FLNA, which stabilizes isoforms promoting
cytoskeletal remodeling and resistance to androgen receptor (AR)
inhibitors (88). Additionally,
FBXO7-driven trans-splicing in glioblastoma stabilizes RBFOX2,
which coordinates splicing of mesenchymal genes such as
POSTN to augment temozolomide resistance through
integrin-mediated survival signaling (3). Furthermore, cross-strand chimeric RNAs
(cscRNAs) produced by ATS between sense and antisense transcripts
have been identified in multiple cancer cells. For instance, in
prostate cancer PC3 cells and HCC Huh7 cells, specific cscRNAs such
as cscR-8-21 and cscR-2-22 are generated through the fusion of
transcripts from opposite DNA strands. Knockdown of these cscRNAs
using siRNA significantly diminishes cell proliferation, colony
formation and migration, as validated by reverse transcription PCR,
Sanger sequencing, RNA fluorescence in situ hybridization,
and functional assays, supporting their role in promoting cancer
cell survival and potentially contributing to therapy resistance
(89). In CML, canonical splicing
of BCR-ABL produces p210 or p190 transcripts, but ATS can
generate truncated isoforms for example, BCR-ABL1/b3a3) that
lack ABL1 exon 2-encoded SH3 domain residues, leading to
constitutive kinase activity and resistance to allosteric
inhibitors such as asciminib by destabilizing the autoinhibited
kinase conformation (58).
Moreover, EZH2-mediated repression of splicing factors such as
CELF2 in CML results in aberrant splicing patterns that promote
stem cell-like properties and diminish sensitivity to tyrosine
kinase inhibitors (Fig. 3D)
(87). These ATS-derived variants
activate alternative signaling pathways, such as dynamic
phosphorylation events, thereby contributing to treatment failure
and relapse (86). These findings
demonstrate that ATS actively contributes to the development of
aggressive, therapy-resistant cancers.
ATS and its prominence compared with
chromosomal abnormalities
AS events including trans-splicing, are prevalent
somatic perturbations in cancer that promote tumor heterogeneity
and therapy resistance (90). By
contrast, hereditary cancer syndromes are frequently driven by
germline chromosomal abnormalities, including RB1 deletions
in retinoblastoma, TP53 mutations in Li-Fraumeni syndrome,
NF1 deletions in neurofibromatosis type 1, APC
mutations in familial adenomatous polyposis, and BRCA1/BRCA2
germline mutations in hereditary breast cancer (91–93).
AS predominantly occurs as a somatic event that broadly reprograms
gene networks, while chromosomal abnormalities generally underlie
inherited cancer syndromes with high penetrance. In
neurofibromatosis type 1, mis-splicing of NF1 exon 23a
disrupts neurofibromin function in high-grade gliomas, thereby
activating the Ras/MAPK pathway to drive tumor formation (94).
Similarly, retinoblastoma exhibits upregulated
spliceosome activity regulated by pRB/E2F3a under oncogenic stress
(90). Genomic studies establish AS
dysregulation as a nearly universal feature of tumorigenesis,
whereas chromosomal abnormalities primarily drive ~10–20% of
hereditary cancer syndromes (91–93).
This predominance establishes AS as a central mechanism in sporadic
cancers and a promising therapeutic target, as exemplified by
metabolic vulnerabilities conferred by TP53 splicing
variants in Li-Fraumeni syndrome (95).
Splicing regulation in cancer immunity,
computational discovery, and systems crosstalk
Splicing-derived neoantigens and
resistance to immunotherapy
Somatic mutations in core spliceosomal components
including SRSF2 and SF3B1 produce recurrent
mis-splicing events that create novel immunogenic peptides, these
splicing-derived neoantigens are presented by MHC class I molecules
and trigger CD8+ T-cell responses in myeloid
malignancies (96,97). While these neoantigens provide
effective targets for immunotherapy, tumors can develop resistance
through selection of low-immunogenicity splice variants or impaired
antigen presentation through mechanisms such as aberrant HLA-I
splicing (98). Pharmacological
spliceosome modulation with agents such as indisulam or MS-023
counteracts this resistance by restoring neoantigen presentation,
and exhibits synergistic effects with anti-PD-1 therapy to enhance
T-cell-mediated tumor clearance in preclinical models (97).
Mis-splicing of microexons, short exons typically
regulated in tissue-specific patterns, represents an important
disease mechanism that disrupts essential protein interaction
networks, in autism spectrum disorder, CPEB4 microexon mis-splicing
drives protein aggregation and functional impairment (99). While this mechanism has been
primarily characterized in neurological disorders, similar
processes likely operate in cancer biology, where microexon
alterations may influence oncogenic signaling pathways and immune
evasion capabilities, thereby creating opportunities for novel
biomarkers and therapeutic strategies (98,99).
These observations underscore splicing-derived neoantigens as
promising biomarkers of response to immune checkpoint blockade
response and as a basis for emerging approaches such as
personalized cancer vaccines and combination therapies that target
spliceosome activity is targeted (97,98).
Computational deconvolution and
single-cell dissection of splicing networks
Computational methods have been developed to enable
systematic study of splicing heterogeneity with both bulk and
single-cell RNA sequencing data (scRNA-seq). Deep learning models
such as APARENT-Perturb can predict splicing from sequence and
uncover key regulatory mechanisms including the CFIm complex, as
well as clinically relevant splicing signatures associated with
drug resistance (100).
Integrating these models with large-scale genomic repositories such
as TCGA enables systematic pan-cancer studies that associate
specific splicing patterns with clinical outcomes.
Single-cell technologies provide an unprecedented
resolution of splicing dynamics among cell populations in the tumor
microenvironment (TME). Reference atlases such as Tabula Sapiens
show cell-type-specific splicing of MYL6 and other genes,
and highlight the required resolution to resolve complex cellular
heterogeneity (101). In aplastic
anemia, scRNA-seq enables detection of aberrant splicing in the
rare population of hematopoietic stem cells, and can provide
mechanistic hypotheses for clonal evolution and treatment
resistance (102).
Recent work in computational biology has led to the
development of advanced frameworks for integration of multi-omics
data and the mapping of splicing regulatory networks. The model
APARENT-Perturb relies on deep neural networks to predict the
effect of genetic perturbations on splicing outcomes, and has
uncovered functional interactions between regulatory elements, as
well as sequence features influencing polyadenylation site
selection (100). Integration of
splicing data with other molecular measurements using tools such as
CellPhoneDB enables systematic investigation of networks of
cell-cell communication that underlie splicing heterogeneity in
tissue microenvironments (101).
Systemic crosstalk of pre-mRNA
splicing with metabolism, cellular stress and epitranscriptome
Splicing regulation can dynamically respond to
metabolic and stress signals through adaptive systems such as the
minor spliceosome (MiS), which is activated during cancer
progression. In prostate cancer, a progressively dysregulated MiS,
as evidenced by elevated U6atac snRNA levels, has been associated
with disease progression and splicing reprogramming under metabolic
stress (103). Cellular stressors,
including androgen deprivation therapy and AR pathway inhibition,
have been shown to further enhance the efficiency of minor intron
splicing, a clear indication of how treatment-related stress can
remodel the spliceosome to facilitate the development of
therapeutic resistance (103).
This dynamic regulation of splicing in turn selectively reprograms
the splicing network to express additional antiapoptotic isoforms,
firmly positioning MiS at the center of the adaptive response in
cancer.
The U12-type spliceosome functions as a specialized
regulatory system for processing stress and oncogenic signals via
minor intron splicing. The structural analyses of the MiS point to
MiS-specific components, such as SCNM110 and PPIL2, as key factors
that ensure a stable catalytic core required for the accurate
processing of genes involved in cell-cycle checkpoint and DNA
repair pathways (104,105). The evolutionary conservation of
this system and the mutational vulnerability of its key components
underscore the importance of MiS in preserving cellular homeostasis
under metabolic and therapeutic stress (103,104).
In conclusion, splicing serves as an integrator of
metabolic and stress signals, and the U12-type spliceosome offers a
compelling model of how an ancient system of gene regulation can
influence the tumor adaptive response. Further work is needed to
elucidate the direct links between metabolism and splicing as well
as the epitranscriptomic layer of regulation, both of which still
remain to be fully characterized in the current literature.
Therapeutic targeting of aberrant
splicing
Perturbations in splicing patterns are well
documented in cancer, and mutations in spliceosomal proteins and
cancer driver genes are enriched across malignancies (106). Aberrant AS contributes to tumor
progression and therapeutic resistance by generating tumor-specific
splice variants that evade conventional therapies. New therapeutic
modalities are under development to directly target dysregulated
splicing machinery or its downstream effects. These include small
molecules that inhibit the activity of specific splicing factors or
the assembly of the spliceosome, ASOs that block the selection of
pathogenic splice sites, and nanoparticle-delivered circRNAs that
leverage the endogenous splicing machinery.
Together these modalities aim to reestablish
physiologic splice patterns and counteract tumor-promoting isoforms
to overcome therapeutic resistance (Fig. 4). The development of these agents
reflects an increasing focus on splicing-targeted therapeutics that
integrate mechanistic insights into splicing dysregulation with
recent advances in drug design and delivery (Table II).
| Table II.Clinical trials of splicing-targeted
therapies in cancer. |
Table II.
Clinical trials of splicing-targeted
therapies in cancer.
| First author/s,
year | Therapeutic
agent | Target | Cancer type | Clinical trial
identifier and phase | Key Findings /
Status | (Refs.) |
|---|
| Steensma et
al, 2021; | H3B-8800 | SF3B1 |
Myelodysplastic | NCT02841540 | Established maximum
tolerated dose (MTD); reversible | (107,108) |
| Seiler et
al, 2018 |
| (Spliceosome) | syndromes,
leukemia | (Phase I) | QTcF prolongation
and gastrointestinal (GI) toxicity as common adverse events. Showed
antitumor activity in SF3B1-mutant patients. |
|
| Chi et al,
2017 | Custirsen | Clusterin (via
ASO) | Metastatic
castration-resistant prostate cancer | SYNERGY (Phase
III) | Failed to improve
overall survival when combined with docetaxel, highlighting
delivery challenges for antisense oligonucleotides (ASOs) in solid
tumors. | (109) |
Small molecule splicing
modulators
Small molecule splicing modulators are an emerging
class of therapeutics designed to correct or exploit dysregulated
pre-mRNA splicing in cancer. These molecules bind to core
components of the spliceosome, the macromolecular machinery that
mediates intron removal and exon ligation, thereby either directly
altering splice site selection or modulating the kinetics of
spliceosome assembly. One prominent target is the SF3b complex, a
subcomplex of the U2 small nuclear ribonucleoprotein (U2snRNP) that
is responsible for recognizing the branchpoint sequences during the
early stages of spliceosome assembly. Inhibitors of SF3b, such as
pladienolide derivatives including H3B-8800 and E7107, stabilize a
conformation that prevents its interaction with pre-mRNA, leading
to widespread IR or ES (108,110). The effect of these inhibitors is
amplified in cancer cells harboring mutations in spliceosomal genes
(for example, SF3B1, U2AF1, or SRSF2) since these cells
exhibit heightened dependence on already compromised spliceosome
function. Recent studies also highlight the role of nuclear
condensates, which are phase-separated compartment enriched in
splicing factors, in potentially concentrating splicing modulators
selectively in cancer cells (111). In addition to SF3b-targeting
molecules, several small molecules have been identified that
modulate other components of the splicing machinery, such as SR
proteins, or even target upstream RNA modification pathways, such
as METTL3, which mediates N6-methyladenosine (m6A)
methylation, a post-transcriptional modification known to regulate
splicing efficiency and pre-mRNA transcript stability (112,113). Consequently, the diversity of
mechanisms underlying the action of splicing modulators suggests
their potential efficacy across a wide range of splicing-driven
malignancies.
The therapeutic efficacy of SF3b inhibitors is
exemplified in MDS and chronic lymphocytic leukemia (CLL), where
recurrent SF3B1 mutations drive aberrant splicing and
oncogenesis. H3B-8800, an orally bioavailable pladienolide
derivative, selectively eliminates spliceosome-mutant cells by
causing retention of short GC-rich introns in genes that encode
spliceosome proteins and increase splicing stress (108). Preclinical studies in
SF3B1-mutated pancreatic cancer and leukemia models have
shown that H3B-8800 reduced viability by greater than 50% at
nanomolar concentrations, with only mild effects in wild-type
cells. Resistance to H3B-8800 was caused by mutations in other
components of SF3b, such as PHF5A-Y36C, indicating on-target
activity (108). In MDS, the
SF3B1 mutations lead to a hypersensitivity to splicing
perturbation, as these cells depend on residual activity of the
spliceosome to produce transcripts critical for the survival of
mutant cells (110). In early
clinical trials in MDS, H3B-8800 treatment led to a reduction in
blasts and an improvement in other hematologic parameters in
patients with SF3B1-mutated MDS (108).
However, the Phase I first-in-human dose-escalation
study of H3B-8800 (NCT02841540) completed dose escalation with
defined dose-limiting toxicities, including ocular toxicity
observed with prior pladienolide derivatives (for example, E7107).
The trial identified reversible QTcF prolongation and
gastrointestinal disturbances (diarrhea, nausea and vomiting) as
common treatment-emergent adverse events, with the maximum
tolerated dose established at 30 mg for a 5-days-on/9-days-off
schedule and 14 mg for a 21-days-on/7-days-off schedule (107). Despite safety considerations,
these findings validate SF3b as a therapeutically tractable node in
spliceosome-mutant cancers and provide a roadmap for targeting
splicing vulnerabilities in other malignancies.
Aberrant splicing also underlies resistance to
targeted therapies, as observed in BRAF-mutant melanomas
treated with vemurafenib. ~30% of resistant tumors express
BRAF splice variants (for example, BRAF3-9), which
lack the RAS-binding domain and dimerize independently of upstream
signals (113). A C-to-G mutation
in intron 8 of BRAF creates a cryptic branchpoint, promoting
BRAF3-9 expression through enhanced recognition by mutant
SF3B1 (113). The splicing
modulators, including spliceostatin A (SSA), reverse the shift in
isoform expression by inhibiting SF3b and thus re-establishing
sensitivity to BRAF inhibitors. In vemurafenib-resistant melanoma
xenografts, SSA treatment decreased BRAF3-9 levels by 60% and
slowed tumor growth by 40%, showing that splicing modulation can
overcome drug resistance (113).
The splicing modulators and BRAF inhibitors are enriched in nuclear
condensates containing MED1 and BRD4, which enhance the efficacy of
the combination therapy (111).
Parallel studies implicate RNA m6A methylation in regulating
BRAF splicing, as METTL3 knockdown reduces
BRAF3-9 expression and synergizes with vemurafenib (112). These insights highlight the dual
utility of splicing modulators: As monotherapies in
spliceosome-mutant cancers and as adjuvants to reverse adaptive
splicing in drug-resistant tumors.
ASOs
ASOs are short synthetic nucleic acids.
Specifically, ASOs are single-stranded, chemically modifiable short
nucleic acid sequences usually in the range of 18–25 nucleotides in
length (114). They are designed
to bind pre-mRNA and modulate splicing events by blocking
splice-site recognition or altering exon inclusion. These molecules
are chemically modified (for example, phosphorothioate backbones,
2′-MOE/cEt modifications) to enhance stability and binding
affinity. For example, in HCC, ASO1-cEt/DNA targeting PKM
pre-mRNA induced a splice switch from the oncogenic PKM2 isoform to
tumor-suppressive PKM1, suppressing glycolysis and tumor growth in
xenografts (115). Similarly, in
KRAS Q61K-mutant cancers, ASOs disrupting exonic splicing
enhancer motifs near cryptic splice sites induced ES, generating
non-functional KRAS transcripts and sensitizing tumors to
osimertinib (116). These studies
underscore ASOs' ability to target splicing-dependent oncogenic
drivers with high specificity.
A key design strategy is to target variants that
mediate resistance to therapy. In gallbladder cancer, PTBP3
mediates the skipping of IL-18 exon 3, leading to the
expression of a tumor-specific ΔIL-18 isoform that in turn
is responsible for the suppression of the tumour-fighting activity
of CD8+ T-cells. ASO4, designed to block the splice site
of the ΔIL-18 isoform, was shown to restore expression of
full-length IL-18 and also to induce improved tumor
clearance by T-cells in vivo (117). Likewise, in B-cell tumors, two
CD20 mRNA isoforms with different 5′-UTRs (V1/V3) have been shown
to control protein translation. In these isoforms, splice-switching
morpholinos selecting for the translation-competent V3 isoform
raised CD20 levels, the increased CD20 expression resensitized
rituximab-resistant cell lines (118). These examples demonstrate how ASOs
can reverse resistance by reprogramming splicing toward
nonpathogenic isoforms.
Clinical progress in solid tumors mirrors advances
observed with nusinersen (a spinal muscular atrophy therapy). In
ovarian cancer, BUD31 sustains antiapoptotic BCL2L12
expression by promoting exon 3 inclusion. BUD31-targeting ASOs
induced ES, triggering nonsense-mediated decay of BCL2L12
mRNA and apoptosis in xenograft models (119). Meanwhile, in HCC, systemic
delivery of a mouse-specific ASO (mASO3-cEt/DNA) redirected
PKM splicing, inhibiting tumorigenesis without toxicity
(115). Similarly, in lung cancer
models, the addition of the MERTK ASO to
XRT+anti-PD1 and XRT+ anti-CTLA4 profoundly
slowed the growth of both primary and secondary tumors and
significantly extended survival (120). Challenges remain in tumor-specific
delivery, but chemical conjugates (for example, GalNAc for hepatic
targeting) and lipid nanoparticles (LNPs) show promise.
Previous advances in delivery platforms have
significantly enhanced the in vivo efficacy of ASOs by
improving targeted delivery and splice-switching efficiency. GalNAc
conjugates, which facilitate hepatocyte-specific uptake through
asialoglycoprotein receptor-mediated endocytosis, have demonstrated
a 10-fold improvement in gene silencing potency in murine liver
models, highlighting their specificity for hepatic applications
(121). LNPs, engineered to
encapsulate splice-switching oligonucleotides, have achieved over
60% splice correction in tumor xenografts, as measured by
functional recovery of target genes, underscoring their utility in
oncology settings (122). Exosomes
allow delivery of nucleic acids to neurons by exosomes, which can
be functionalized with RVG peptides to target delivery to the
brain. It has led to ~50% reduction in models of neurodegenerative
disorders (123). These approaches
allow for delivery to be more precise, and for the systemic levels
of agents to be lower, reducing off-target effects and improving
therapeutic indices.
However, the translation of ASO medicines into the
clinic for the treatment of solid tumors is significantly hampered
by challenges that arise during systemic delivery. This includes
poor tissue penetration, limited uptake and poor transport of ASOs
into and within cells. A key clinical failure for systemic delivery
of ASOs was the SYNERGY Phase 3 trial of custirsen, a
clusterin-targeting ASO, in combination with docetaxel and
prednisone in patients with metastatic castration-resistant
prostate cancer. It was observed that custirsen had no impact on
overall survival in these patients, a finding that underscores the
need to overcome delivery barriers through means such as ligand
conjugates and nanocarriers (109).
circRNA-directed nanotherapeutics
Circular RNAs (circRNAs) are covalently closed RNA
molecules formed by back-splicing of precursor mRNAs. CircRNAs are
highly stable molecules that are resistant to exonucleases.
CircRNAs are involved in the development of cancers by regulating
the expression of target genes through microRNA sponging, protein
interactions and transcriptional regulation (124,125). CircRNA-directed nanotherapeutics
is a preclinical proof-of-concept approach that employs RNA
interference technology in combination with a delivery system that
integrates nanoparticles to target oncogenic circRNAs.
CircRNA-directed nanotherapeutics utilizes siRNAs or ASOs
encapsulated in biocompatible nanoparticles to enhance stability,
targeted delivery and mitigate off-target effects relative to free
RNA agents. This strategy holds promise to overcome issues such as
rapid renal clearance and enzymatic degradation of free RNA, and
has been shown to enable specific targeting of dysregulated
circRNAs in tumors (126,127).
Recent studies have provided proof-of-concept
evidence that circRNA-targeted nanomedicines can be effective in
different cancer types in mouse models. In gastric cancer,
lipid-polymer hybrid nanoparticles carrying siRNAs to knockdown
circ_0008315 resensitized tumors to cisplatin by disrupting
miR-3666/CPEB4 signaling. This nano-formulation exhibited
2.3-fold higher tumoral accumulation than free siRNA and
significantly decreased tumor volumes in a patient-derived
xenograft model (65.8 mm3 vs. 132.4 mm3 in
free siRNA-treated mice; with no effects on organ function)
(124). In HCC, poly(β-amino
ester)-based nanoparticles carrying circMDK siRNA achieved (78%
target gene knockdown) inhibited tumor growth by upregulating
ATG16L1 via an m6A-associated mechanism. The nanoparticles
were designed with pH-sensitive components, so that 92% of the
siRNA payload could be released specifically in the TME, suggesting
potential superiority over conventional chemotherapy treatment in
an orthotopic tumor model (126).
These examples underscore the potential, at a proof-of-concept
stage, for nanocarriers to improve the pharmacokinetic properties
of circRNA-targeting agents in a preclinical model.
There are other examples of preclinical validation
of circRNA nanotherapeutics, whereby multifunctional nanoparticle
designs provide supporting evidence (Table III). For instance, delivery of
circRHBDD1 siRNA in PLGA-PEG nanoparticles effectively
blocked the the IGF2BP2/PD-L1 axis and increased
infiltration of CD8+ T cells by 4.1-fold in murine
gastric cancer, achieving complete tumor regression in 40% of
treated animals when combined with anti-PD-1 antibody (125). PLGA nanoparticles were also used
to deliver si-cSERPINE2 to breast cancer cells in orthotopic
mouse models. This treatment attenuated IL-6-mediated
crosstalk between breast cancer cells and tumor-associated
macrophages, and inhibited lung metastasis by 72% (127). Current early-phase clinical trials
are beginning to validate such alternative strategies. For
instance, a Phase I clinical trial of LNP-formulated circRNA
inhibitors was conducted in solid tumors, and indicated a favorable
safety profile, but was undertaken as an exploratory study and was
not further validated in large animal studies.
| Table III.Summary of therapeutic strategies
targeting alternative splicing in cancer. |
Table III.
Summary of therapeutic strategies
targeting alternative splicing in cancer.
| First author/s,
year | Therapeutic
modality | Specific
agent/approach | Molecular
target/action | Resistance
mechanism addressed |
Proof-of-concept/model | Clinical
status | (Refs.) |
|---|
| Steensma et
al, 2021; Seiler et al, 2018 | Small molecule
inhibitors | H3B-8800 | SF3b complex
modulator | Induces lethal
splicing errors in spliceosomemutant cells | SF3B1-mutant
leukemia and pancreatic cancer models | Clinical Phase
I | (107,108) |
| Salton et
al, 2015 |
| Spliceostatin
A | SF3b complex
inhibitor | Reverses BRAF3-9
isoform expression |
Vemurafenib-resistant melanoma
xenografts | Preclinical | (113) |
| Xu et al,
2023 |
| NVP2 (SRPK1
inhibitor) | Inhibits SR protein
kinase | Normalizes VEGF
splicing | Pancreatic ductal
adenocarcinoma models | Preclinical | (128) |
| Ma et al,
2022 | ASOs | PKM-targeting
ASO | Switches PKM2 to
PKM1 isoform | Suppresses
oncogenic glycolysis | HCC xenografts | Preclinical | (115) |
| Zhao et al,
2024 |
| IL-18-targeting
ASO | Corrects pathogenic
IL-18 splicing | Restores
CD8+ T-cell function | Gallbladder cancer
models | Preclinical | (117) |
| Chi et al,
2017 |
| Custirsen | Clusterin mRNA
(ASO) | Aimed to inhibit
anti-apoptotic protein | Patients with
metastatic castration-resistant prostate cancer | Clinical Phase III
(Failed) | (109) |
| Sun et al,
2023 |
CRISPR/Splice-Switching | CRISPR/dCasRx | Targets TIMP1
pre-mRNA | Reverses
SRSF1-driven pro-metastatic splicing | Colorectal cancer
models | Preclinical | (38) |
| Fei et al,
2024; Li et al, 2024; Du et al, 2022 |
Nanotherapeutics | siRNA-loaded
nanoparticles | Targets oncogenic
circRNAs (for example, circ_0008315, circRHBDD1) | Reverses
chemoresistance or immune evasion | Gastric cancer, HCC
xenograft models | Preclinical | (124–126) |
Conclusions and future perspectives
The detailed association between dysregulation of
AS and cancer drug resistance is increasingly appreciated, wherein
numerous splicing events, including ES, IR, and selection of
alternative splice sites, can directly influence therapeutic
response. Recent advances in long-read sequencing methods,
particularly Oxford Nanopore Technologies and PacBio sequencing,
have enabled the resolution of full-length transcript isoforms,
allowing cancer-specific AS events to be precisely identified. For
instance, in spatial isoform transcriptomics of esophageal squamous
cell carcinoma (ESCC), the isoform switching of CD74 was found to
contribute to chemoresistance by promoting the interaction of
C1QC+ tumor associated macrophages and exhausted
CD8+ T-cells, which in turn fosters an immunosuppressive
TME (129). In PDAC, the circRNA
hsa_circ_0007919 was found to be involved in gemcitabine resistance
by recruiting the transcription factors FOXA1 and
TET1 to demethylate the LIG1 promoter, thereby
boosting DNA repair by regulating AS (128). These observations align with
splicing quantitative trait loci (sQTL) studies in CRC, in which a
genetic variant was found to impact the splicing of PRMT7,
which in turn was implicated in the MAPK signalling pathway and
chemotherapy resistance (27).
Importantly, recent structural analyses of the minor spliceosome
have identified several components specific to U11 or U12 snRNPs
(for example, 35K and 48K) and lactylated nucleolin (NCL) that are
required for stabilizing the recognition of U12-type introns, which
in turn drive the oncogenic splicing programs (104,130). Together, these findings highlight
the dual role of AS in both driving molecular diversity and
representing a potential vulnerability for therapeutic intervention
in cancer.
In the future, it will be essential to optimise
long-read sequencing protocols to improve their accuracy and
scalability in studying spatially resolved isoform diversity in the
metastatic niche. A key priority would be to study how the minor
spliceosome, responsible for excising a minority of spliceosomal
introns (U12, accounting for ~0.5% of all human genes) that often
include oncogenes and tumor suppressors, is regulated.
Cryo-electron microscopy studies have elucidated the structure of
the spliceosome responsible for processing U12-dependent introns,
which revealed that some of the components appear to be
functionally orthologous to the SF3a complex that stabilizes
branchpoint recognition in the major spliceosome, notably
SCNM1, and demonstrated that a U-box protein PPIL2 is
essential for the assembly of the catalytic core (104). On a more clinical note, it might
also be valuable to examine spatially resolved splicing that is sex
or tissue specific, as such splicing patterns could provide useful
biomarkers for precision medicine. Multi-ancestry sQTL studies have
shown that the spliceosome component PRPF8 is regulated in
an ancestry-specific manner in TNBC where DHX9 suppresses
circRNA-CREIT and induces chemoresistance (27,74).
Furthermore, combining single-cell isoform sequencing with spatial
transcriptomics in tumor microenvironments could be explored to
identify how subclonal splicing variants drive adaptive resistance.
Yang et al (130) showed
that in cholangiocarcinoma, lactate-driven NCL lactylation
reprograms the RNA splicing machinery via MADD isoform
switching, which could be a potential therapeutic target. Such
studies will require collaboration between basic scientists and
clinicians to standardize isoform annotation pipelines and
cross-validate key splicing nodes in multiple models.
Therapeutic strategies specifically targeting
AS-mediated drug resistance are rapidly evolving with promising
approaches being investigated in preclinical models that target the
cis-regulatory elements, trans-acting factors and various
spliceosome inhibitors. Small molecules such as the SRPK1
kinase inhibitor NVP2 act by normalizing VEGF splicing in
PDAC to resensitize tumour cells to cisplatin (128). ASOs targeting the pathogenic
isoform in ESCC, such as CD74v6, which is known to promote immune
evasion via macrophage apoptosis, demonstrated efficacy in
suppressing metastasis in preclinical trials (129). Inhibition of lactylation-dependent
activation of NCL by SGC3027 also abrogated oncogenic MADD
splicing in cholangiocarcinoma and synergized with chemotherapy to
reduce tumor burden (130).
Combinatorial approaches are also gaining traction; for instance,
pairing spliceosome modulators for example, pladienolide B) with
immune checkpoint blockers enhances T-cell infiltration in tumors
with high SF3B1 mutation burden (27). Structural information about the
minor spliceosome will also provide a framework for rational drug
design as has been recently demonstrated for the interaction of
PPIL2-U5 snRNA to inhibit tri-snRNP assembly (104). However, the main concern regarding
these drugs is mitigating off-target effects due to the ubiquitous
nature of the splicing machinery in normal cells. Clinical trials
should aim to develop isoform-specific therapies, and leveraging
CRISPR-based screens that identify synthetic lethal interactions
between splicing factors and chemotherapeutic drugs could be
employed. As the field progresses, integrating splicing-centric
therapies with conventional regimens will be pivotal for overcoming
the adaptive plasticity of cancer cells and improving patient
outcomes.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by Key Science and Technology
Tackling Program of Henan (grant no. 232102311077), the National
Natural Science Foundation of China (grant no. 82503868), the
medical science and technology Project of Henan (grant nos.
LHGJ20220198, LHGJ20240104 and LHGJ20240108), the Natural Science
Foundation of Henan (grant no. 242300420578), the Henan Provincial
Medical Education Research Project (grant no. Wjlx2020107), the
Graduate Education Reform Research and Curriculum Development
Program of the Academy of Medical Sciences, Zhengzhou University
(grant no. 040012023B062) and the National Key Clinical Discipline
Construction Project.
Availability of data and materials
Not applicable.
Authors' contributions
WZhu, ZW and CL conceptualized the study. WZhu
wrote the original draft. BB, WZha and GC wrote, reviewed and
edited the manuscript. HY and HA supervised the research work, and
contributed to the study methodology, investigation and data
curation. FL and ZL solicited for funding, contributed to project
administration, validation and provision of resources. All authors
read and approved the final version of the manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ALL
|
acute lymphoblastic leukemia
|
|
AP
|
alternative promoter
|
|
APA
|
alternative polyadenylation
|
|
AR
|
androgen receptor
|
|
AS
|
alternative splicing
|
|
ASO
|
antisense oligonucleotide
|
|
ATS
|
alternative trans-splicing
|
|
CML
|
chronic myeloid leukemia
|
|
circRNA
|
circular RNA
|
|
DIs
|
detained introns
|
|
ecircRNA
|
exonic circRNA
|
|
EIciRNA
|
exon-intron circRNA
|
|
EMT
|
epithelial-mesenchymal transition
|
|
ER
|
estrogen receptor
|
|
ESE
|
exonic splicing enhancer
|
|
ESS
|
exonic splicing silencer
|
|
ES
|
exon skipping
|
|
HCC
|
hepatocellular carcinoma
|
|
HRR
|
homologous recombination repair
|
|
IR
|
intron retention
|
|
ISE
|
intronic splicing enhancer
|
|
ISS
|
intronic splicing silencer
|
|
LNP
|
lipid nanoparticle
|
|
MDS
|
myelodysplastic syndromes
|
|
MiS
|
minor spliceosome
|
|
MXE
|
mutually exclusive exons
|
|
NMD
|
nonsense-mediated decay
|
|
NSCLC
|
non-small cell lung cancer
|
|
PDAC
|
pancreatic ductal adenocarcinoma
|
|
pre-mRNA
|
precursor mRNA
|
|
scRNA-seq
|
single-cell RNA sequencing
|
|
sQTL
|
splicing quantitative trait loci
|
|
SSM
|
splice-site mutation
|
|
TCGA
|
The Cancer Genome Atlas
|
|
TNBC
|
triple-negative breast cancer
|
|
UTR
|
untranslated region
|
References
|
1
|
Yang Q, Zhao J, Zhang W, Chen D and Wang
Y: Aberrant alternative splicing in breast cancer. J Mol Cell Biol.
11:920–929. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marasco LE and Kornblihtt AR: The
physiology of alternative splicing. Nat Rev Mol Cell Biol.
24:242–254. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li S, Chen Y, Xie Y, Zhan H, Zeng Y, Zeng
K, Wang L, Zhan Z, Li C, Zhao L, et al: FBXO7 confers mesenchymal
properties and chemoresistance in glioblastoma by controlling
Rbfox2-mediated alternative splicing. Adv Sci. 10:e23035612023.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu M, Zhang S, Zhou H, Hu X, Li J, Fu B,
Wei M, Huang H and Wu H: The interplay between non-coding RNAs and
alternative splicing: From regulatory mechanism to therapeutic
implications in cancer. Theranostics. 13:2616–2631. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bauer M, Schöbel CM, Wickenhauser C,
Seliger B and Jasinski-Bergner S: Deciphering the role of
alternative splicing in neoplastic diseases for immune-oncological
therapies. Front Immunol. 15:13869932024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu X and Hurst LD: Determinants of the
usage of splice-associated cis-motifs predict the distribution of
human pathogenic SNPs. Mol Biol Evol. 33:518–529. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Venkataramany AS, Schieffer KM, Lee K,
Cottrell CE, Wang PY, Mardis ER, Cripe TP and Chandler DS:
Alternative RNA splicing defects in pediatric cancers: New insights
in tumorigenesis and potential therapeutic vulnerabilities. Ann
Oncol. 33:578–592. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ruta V, Naro C, Pieraccioli M, Leccese A,
Archibugi L, Cesari E, Panzeri V, Allgöwer C, Arcidiacono PG,
Falconi M, et al: An alternative splicing signature defines the
basal-like phenotype and predicts worse clinical outcome in
pancreatic cancer. Cell Rep Med. 5:1014112024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huan L, Guo T, Wu Y, Xu L, Huang S, Xu Y,
Liang L and He X: Hypoxia induced LUCAT1/PTBP1 axis modulates
cancer cell viability and chemotherapy response. Mol Cancer.
19:112020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nesic K, Krais JJ, Wang Y, Vandenberg CJ,
Patel P, Cai KQ, Kwan T, Lieschke E, Ho GY, Barker HE, et al: BRCA1
secondary splice-site mutations drive exon-skipping and PARP
inhibitor resistance. Mol Cancer. 23:1582024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bradley RK and Anczuków O: RNA splicing
dysregulation and the hallmarks of cancer. Nat Rev Cancer.
23:135–155. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kahles A, Lehmann KV, Toussaint NC, Hüser
M, Stark SG, Sachsenberg T, Stegle O, Kohlbacher O and Sander C;
Cancer Genome Atlas Research Network; Rätsch G, : Comprehensive
analysis of alternative splicing across tumors from 8,705 patients.
Cancer Cell. 34:211–224.e6. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li F, Xiong Y, Yang M, Chen P, Zhang J,
Wang Q, Xu M, Wang Y, He Z, Zhao X, et al: c-mpl-del, a c-mpl
alternative splicing isoform, promotes AMKL progression and
chemoresistance. Cell Death Dis. 13:8692022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhen N, Zhu J, Mao S, Zhang Q, Gu S, Ma J,
Zhang Y, Yin M, Li H, Huang N, et al: Alternative splicing of
lncRNAs from SNHG family alters snoRNA expression and induces
chemoresistance in hepatoblastoma. Cell Mol Gastroenterol Hepatol.
16:735–755. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ule J and Blencowe BJ: Alternative
splicing regulatory networks: functions, mechanisms, and evolution.
Mol Cell. 76:329–345. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Duan C, Zhang Y, Li L, Liu K, Yao X, Wu X,
Li B, Mao X, Wu H, Liu H, et al: Identification of alternative
splicing associated with clinical features: From pan-cancers to
genitourinary tumors. Front Oncol. 13:12499322023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim P, Yang M, Yiya K, Zhao W and Zhou X:
ExonSkipDB: Functional annotation of exon skipping event in human.
Nucleic Acids Res. 48:D896–D907. 2020.PubMed/NCBI
|
|
18
|
Mazieres J, Paik PK, Garassino MC, Le X,
Sakai H, Veillon R, Smit EF, Cortot AB, Raskin J, Viteri S, et al:
Tepotinib treatment in patients with MET exon 14-skipping non-small
cell lung cancer: Long-term follow-up of the VISION phase 2
nonrandomized clinical trial. JAMA Oncol. 9:1260–1266. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mazieres J, Vioix H, Pfeiffer BM, Campden
RI, Chen Z, Heeg B and Cortot AB: MET exon 14 skipping in NSCLC: A
systematic literature review of epidemiology, clinical
characteristics, and outcomes. Clin Lung Cancer. 24:483–497. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen S, Yang C, Wang ZW, Hu JF, Pan JJ,
Liao CY, Zhang JQ, Chen JZ, Huang Y, Huang L, et al: CLK1/SRSF5
pathway induces aberrant exon skipping of METTL14 and cyclin L2 and
promotes growth and metastasis of pancreatic cancer. J Hematol
Oncol. 14:602021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang K, Wei J, Zhang S, Fei L, Guo L, Liu
X, Ji Y, Chen W, Ciamponi FE, Chen W, et al: A chemical screen
identifies PRMT5 as a therapeutic vulnerability for
paclitaxel-resistant triple-negative breast cancer. Cell Chem Biol.
31:1942–1957.e6. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ambeskovic A, McCall MN, Woodsmith J, Juhl
H and Land H: Exon-skipping-based subtyping of colorectal cancers.
Gastroenterology. 167:1358–1370.e12. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hsu TYT, Simon LM, Neill NJ, Marcotte R,
Sayad A, Bland CS, Echeverria GV, Sun T, Kurley SJ, Tyagi S, et al:
The spliceosome is a therapeutic vulnerability in MYC-driven
cancer. Nature. 525:384–388. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Koedoot E, van Steijn E, Vermeer M,
González-Prieto R, Vertegaal ACO, Martens JWM, Le Dévédec SE and
van de Water B: Splicing factors control triple-negative breast
cancer cell mitosis through SUN2 interaction and sororin intron
retention. J Exp Clin Cancer Res. 40:822021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Grabski DF, Broseus L, Kumari B, Rekosh D,
Hammarskjold M and Ritchie W: Intron retention and its impact on
gene expression and protein diversity: A review and a practical
guide. Wiley Interdiscip Rev RNA. 12:e16312021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Monteuuis G, Schmitz U, Petrova V, Kearney
PS and Rasko JEJ: Holding on to junk bonds: intron retention in
cancer and therapy. Cancer Res. 81:779–789. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang M, Chen C, Lu Z, Cai Y, Li Y, Zhang
F, Liu Y, Chen S, Zhang H, Yang S, et al: Genetic control of
alternative splicing and its distinct role in colorectal cancer
mechanisms. Gastroenterology. 165:1151–1167. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang D, Hu Q, Liu X, Ji Y, Chao HP, Liu
Y, Tracz A, Kirk J, Buonamici S, Zhu P, et al: Intron retention is
a hallmark and spliceosome represents a therapeutic vulnerability
in aggressive prostate cancer. Nat Commun. 11:20892020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Demircioğlu D, Cukuroglu E, Kindermans M,
Nandi T, Calabrese C, Fonseca NA, Kahles A, Lehmann KV, Stegle O,
Brazma A, et al: A pan-cancer transcriptome analysis reveals
pervasive regulation through alternative promoters. Cell.
178:1465–1477.e17. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Suzawa K, Offin M, Lu D, Kurzatkowski C,
Vojnic M, Smith RS, Sabari JK, Tai H, Mattar M, Khodos I, et al:
Activation of KRAS mediates resistance to targeted therapy in MET
exon 14-mutant non-small cell lung cancer. Clin Cancer Res.
25:1248–1260. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lévesque E, Labriet A, Hovington H, Allain
ÉP, Melo-Garcia L, Rouleau M, Brisson H, Turcotte V, Caron P,
Villeneuve L, et al: Alternative promoters control
UGT2B17-dependent androgen catabolism in prostate cancer and its
influence on progression. Br J Cancer. 122:1068–1076. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nepal C and Andersen JB: Alternative
promoters in CpG depleted regions are prevalently associated with
epigenetic misregulation of liver cancer transcriptomes. Nat
Commun. 14:27122023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Veiga DFT, Nesta A, Zhao Y, Mays AD, Huynh
R, Rossi R, Wu TC, Palucka K, Anczukow O, Beck CR and Banchereau J:
A comprehensive long-read isoform analysis platform and sequencing
resource for breast cancer. Sci Adv. 8:eabg67112022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ryan M, Wong WC, Brown R, Akbani R, Su X,
Broom B, Melott J and Weinstein J: TCGASpliceSeq a compendium of
alternative mRNA splicing in cancer. Nucleic Acids Res. 44((D1)):
D1018–D1022. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rogalska ME, Mancini E, Bonnal S, Gohr A,
Dunyak BM, Arecco N, Smith PG, Vaillancourt FH and Valcárcel J:
Transcriptome-wide splicing network reveals specialized regulatory
functions of the core spliceosome. Science. 386:551–560. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Torres-Diz M, Reglero C, Falkenstein CD,
Castro A, Hayer KE, Radens CM, Quesnel-Vallières M, Ang Z, Sehgal
P, Li MM, et al: An alternatively spliced gain-of-function NT5C2
isoform contributes to chemoresistance in acute lymphoblastic
leukemia. Cancer Res. 84:3327–3336. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lin CC, Chang TC, Wang Y, Guo L, Gao Y,
Bikorimana E, Lemoff A, Fang YV, Zhang H, Zhang Y, et al: PRMT5 is
an actionable therapeutic target in CDK4/6 inhibitor-resistant
ER+/RB-deficient breast cancer. Nat Commun. 15:22872024. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sun Q, Han Y, He J, Wang J, Ma X, Ning Q,
Zhao Q, Jin Q, Yang L, Li S, et al: Long-read sequencing reveals
the landscape of aberrant alternative splicing and novel
therapeutic target in colorectal cancer. Genome Med. 15:762023.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Song X, Tiek D, Miki S, Huang T, Lu M,
Goenka A, Iglesia R, Yu X, Wu R, Walker M, et al: RNA splicing
analysis deciphers developmental hierarchies and reveals
therapeutic targets in adult glioma. J Clin Invest.
134:e1737892024. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sciarrillo R, Wojtuszkiewicz A, Assaraf
YG, Jansen G, Kaspers GJL, Giovannetti E and Cloos J: The role of
alternative splicing in cancer: From oncogenesis to drug
resistance. Drug Resist Updat. 53:1007282020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Baralle FE and Giudice J: Alternative
splicing as a regulator of development and tissue identity. Nat Rev
Mol Cell Biol. 18:437–451. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dou Z, Zhao D, Chen X, Xu C, Jin X, Zhang
X, Wang Y, Xie X, Li Q, Di C and Zhang H: Aberrant bcl-x splicing
in cancer: From molecular mechanism to therapeutic modulation. J
Exp Clin Cancer Res. 40:1942021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Preissl S, Gaulton KJ and Ren B:
Characterizing cis-regulatory elements using single-cell
epigenomics. Nat Rev Genet. 24:21–43. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Soni K, Jagtap PKA, Martínez-Lumbreras S,
Bonnal S, Geerlof A, Stehle R, Simon B, Valcárcel J and Sattler M:
Structural basis for specific RNA recognition by the alternative
splicing factor RBM5. Nat Commun. 14:42332023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Dvinge H and Bradley RK: Widespread intron
retention diversifies most cancer transcriptomes. Genome Med.
7:452015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sinnakannu JR, Lee KL, Cheng S, Li J, Yu
M, Tan SP, Ong CCH, Li H, Than H, Anczuków-Camarda O, et al: SRSF1
mediates cytokine-induced impaired imatinib sensitivity in chronic
myeloid leukemia. Leukemia. 34:1787–1798. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fuentes-Fayos AC, Pérez-Gómez JM, G-García
ME, Jiménez-Vacas JM, Blanco-Acevedo C, Sánchez-Sánchez R, Solivera
J, Breunig JJ, Gahete MD, Castaño JP and Luque RM: SF3B1 inhibition
disrupts malignancy and prolongs survival in glioblastoma patients
through BCL2L1 splicing and mTOR/ß-catenin pathways imbalances. J
Exp Clin Cancer Res. 41:392022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Taketo K, Konno M, Asai A, Koseki J,
Toratani M, Satoh T, Doki Y, Mori M, Ishii H and Ogawa K: The
epitranscriptome m6A writer METTL3 promotes chemo- and
radioresistance in pancreatic cancer cells. Int J Oncol.
52:621–629. 2017.PubMed/NCBI
|
|
49
|
Wang N, Hu Y and Wang Z: Regulation of
alternative splicing: Functional interplay with epigenetic
modifications and its implication to cancer. Wiley Interdiscip Rev
RNA. 12:e18152023.PubMed/NCBI
|
|
50
|
Lei Y and Lai M: Epigenetic regulation and
therapeutic targeting of alternative splicing dysregulation in
cancer. Pharmaceuticals (Basel). 18:7132025. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ilango S, Paital B, Jayachandran P, Padma
PR and Nirmaladevi R: Epigenetic alterations in cancer. Front
Biosci (Landmark Ed). 25:1058–1109. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bao B, Tian M, Wang X, Yang C, Qu J, Zhou
S, Cheng Y, Tong Q and Zheng L: SNORA37/CMTR1/ELAVL1 feedback loop
drives gastric cancer progression via facilitating CD44 alternative
splicing. J Exp Clin Cancer Res. 44:152025. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sun L, Zhang H and Gao P: Metabolic
reprogramming and epigenetic modifications on the path to cancer.
Protein Cell. 13:877–919. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hogg SJ, Beavis PA, Dawson MA and
Johnstone RW: Targeting the epigenetic regulation of antitumour
immunity. Nat Rev Drug Discov. 19:776–800. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang Y, Wei J, Feng L, Li O, Huang L, Zhou
S, Xu Y, An K, Zhang Y, Chen R, et al: Aberrant m5C
hypermethylation mediates intrinsic resistance to gefitinib through
NSUN2/YBX1/QSOX1 axis in EGFR-mutant non-small-cell lung cancer.
Mol Cancer. 22:812023. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Bian Z, Yang F, Xu P, Gao G, Yang C, Cao
Y, Yao S, Wang X, Yin Y, Fei B and Huang Z: LINC01852 inhibits the
tumorigenesis and chemoresistance in colorectal cancer by
suppressing SRSF5-mediated alternative splicing of PKM. Mol Cancer.
23:232024. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wickland DP, McNinch C, Jessen E, Necela
B, Shreeder B, Lin Y, Knutson KL and Asmann YW: Comprehensive
profiling of cancer neoantigens from aberrant RNA splicing. J
Immunother Cancer. 12:e0089882024. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Leyte-Vidal A, DeFilippis R, Outhwaite IR,
Kwan I, Lee JY, Leavitt C, Miller KB, Rea D, Rangwala AM, Lou K, et
al: Absence of ABL1 exon 2-encoded SH3 residues in BCR::ABL1
destabilizes the autoinhibited kinase conformation and confers
resistance to asciminib. Leukemia. 38:2046–2050. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Monteuuis G, Wong JJL, Bailey CG, Schmitz
U and Rasko JEJ: The changing paradigm of intron retention:
Regulation, ramifications and recipes. Nucleic Acids Res.
47:11497–11513. 2019.PubMed/NCBI
|
|
60
|
Wong JJL and Schmitz U: Intron retention:
Importance, challenges, and opportunities. Trends Genet.
38:789–792. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Tan ZW, Fei G, Paulo JA, Bellaousov S,
Martin SES, Duveau DY, Thomas CJ, Gygi SP, Boutz PL and Walker S:
O-GlcNAc regulates gene expression by controlling detained intron
splicing. Nucleic Acids Res. 48:5656–5669. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yue Q, Wang Z, Shen Y, Lan Y, Zhong X, Luo
X, Yang T, Zhang M, Zuo B, Zeng T, et al: Histone H3K9 lactylation
confers temozolomide resistance in glioblastoma via LUC7L2-mediated
MLH1 intron retention. Adv Sci (Weinh). 11:23092902024. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wang X, Han M, Wang S, Sun Y, Zhao W, Xue
Z, Liang X, Huang B, Li G, Chen A, et al: Targeting the splicing
factor NONO inhibits GBM progression through GPX1 intron retention.
Theranostics. 12:5451–5469. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wang E, Lu SX, Pastore A, Chen X, Imig J,
Lee SCW, Hockemeyer K, Ghebrechristos YE, Yoshimi A, Inoue D, et
al: Targeting an RNA-binding protein network in acute myeloid
leukemia. Cancer Cell. 35:369–384.e7. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Inoue D, Polaski JT, Taylor J, Castel P,
Chen S, Kobayashi S, Hogg SJ, Hayashi Y, Pineda JMB, El Marabti E,
et al: Minor intron retention drives clonal hematopoietic disorders
and diverse cancer predisposition. Nat Genet. 53:707–718. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Xu Z, Chen Y, Ma L, Chen Y, Liu J, Guo Y,
Yu T, Zhang L, Zhu L and Shu Y: Role of exosomal non-coding RNAs
from tumor cells and tumor-associated macrophages in the tumor
microenvironment. Mol Ther. 30:3133–3154. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Chen LL: The expanding regulatory
mechanisms and cellular functions of circular RNAs. Nat Rev Mol
Cell Biol. 21:475–490. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Liu CX and Chen LL: Circular RNAs:
Characterization, cellular roles, and applications. Cell.
185:2016–2034. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Chen R, Wang SK, Belk JA, Amaya L, Li Z,
Cardenas A, Abe BT, Chen CK, Wender PA and Chang HY: Engineering
circular RNA for enhanced protein production. Nat Biotechnol.
41:262–272. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Pitolli C, Marini A, Sette C and
Pagliarini V: Non-canonical splicing and its implications in brain
physiology and cancer. Int J Mol Sci. 23:28112022. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Xue W, Ma XK and Yang L: Fast and furious:
insights of back splicing regulation during nascent RNA synthesis.
Sci China Life Sci. 64:1050–1061. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Xu B, Meng Y and Jin Y: RNA structures in
alternative splicing and back-splicing. Wiley Interdiscip Rev RNA.
12:e16262021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Ling Y, Liang G, Lin Q, Fang X, Luo Q, Cen
Y, Mehrpour M, Hamai A, Liu Z, Shi Y, et al: circCDYL2 promotes
trastuzumab resistance via sustaining HER2 downstream signaling in
breast cancer. Mol Cancer. 21:82022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Wang X, Chen T, Li C, Li W, Zhou X, Li Y,
Luo D, Zhang N, Chen B, Wang L, et al: CircRNA-CREIT inhibits
stress granule assembly and overcomes doxorubicin resistance in
TNBC by destabilizing PKR. J Hematol Oncol. 15:1222022. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Enuka Y, Lauriola M, Feldman ME, Sas-Chen
A, Ulitsky I and Yarden Y: Circular RNAs are long-lived and display
only minimal early alterations in response to a growth factor.
Nucleic Acids Res. 44:1370–1383. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kristensen LS, Andersen MS, Stagsted LVW,
Ebbesen KK, Hansen TB and Kjems J: The biogenesis, biology and
characterization of circular RNAs. Nat Rev Genet. 20:675–691. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Rybak-Wolf A, Stottmeister C, Glažar P,
Jens M, Pino N, Giusti S, Hanan M, Behm M, Bartok O, Ashwal-Fluss
R, et al: Circular RNAs in the mammalian brain are highly abundant,
conserved, and dynamically expressed. Mol Cell. 58:870–885. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Memczak S, Jens M, Elefsinioti A, Torti F,
Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer
M, et al: Circular RNAs are a large class of animal RNAs with
regulatory potency. Nature. 495:333–338. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Gao W, Guo H, Niu M, Zheng X, Zhang Y, Xue
X, Bo Y, Guan X, Li Z, Guo Y, et al: circPARD3 drives malignant
progression and chemoresistance of laryngeal squamous cell
carcinoma by inhibiting autophagy through the PRKCI-akt-mTOR
pathway. Mol Cancer. 19:1662020. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Chen L and Shan G: CircRNA in cancer:
Fundamental mechanism and clinical potential. Cancer Lett.
505:49–57. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Hu C, Xia R, Zhang X, Li T, Ye Y, Li G, He
R, Li Z, Lin Q, Zheng S and Chen R: circFARP1 enables
cancer-associated fibroblasts to promote gemcitabine resistance in
pancreatic cancer via the LIF/STAT3 axis. Mol Cancer. 21:242022.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Hong EM, Ingemarsdotter CK and Lever AML:
Therapeutic applications of trans -splicing. Br Med Bull. 136:4–20.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Berger A, Maire S, Gaillard M, Sahel J,
Hantraye P and Bemelmans A: mRNA trans-splicing in gene therapy for
genetic diseases. Wiley Interdiscip Rev RNA. 7:487–498. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Zhang J, Xu X, Deng H, Liu L, Xiang Y and
Feng J: Overcoming cancer drug-resistance calls for novel
strategies targeting abnormal alternative splicing. Pharmacol Ther.
261:1086972024. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Dhungel BP, Monteuuis G, Giardina C, Tabar
MS, Feng Y, Metierre C, Ho S, Nagarajah R, Fontaine ARM, Shah JS,
et al: The fusion of CLEC12A and MIR223HG arises from a
trans-splicing event in normal and transformed human cells. Int J
Mol Sci. 22:121782021. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Lee G and Muir TW: Distinct phases of
cellular signaling revealed by time-resolved protein synthesis. Nat
Chem Biol. 20:1353–1360. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Brunmeir R, Ying L, Yan J, Hee YT, Lin B,
Kaur H, Leong QZ, Teo WW, Choong G, Jen WY, et al: EZH2 modulates
mRNA splicing and exerts part of its oncogenic function through
repression of splicing factors in CML. Leukemia. 39:650–662. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Del Giudice M, Foster JG, Peirone S,
Rissone A, Caizzi L, Gaudino F, Parlato C, Anselmi F, Arkell R,
Guarrera S, et al: FOXA1 regulates alternative splicing in prostate
cancer. Cell Rep. 40:1114042022. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Wang Y, Zou Q, Li F, Zhao W, Xu H, Zhang
W, Deng H and Yang X: Identification of the cross-strand chimeric
RNAs generated by fusions of bi-directional transcripts. Nat
Commun. 12:46452021. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Tu J, Huo Z, Yu Y, Zhu D, Xu A, Huang MF,
Hu R, Wang R, Gingold JA, Chen YH, et al: Hereditary retinoblastoma
iPSC model reveals aberrant spliceosome function driving bone
malignancies. Proc Natl Acad Sci USA. 119:e21178571192022.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Allen I, Hassan H, Walburga Y, Huntley C,
Loong L, Rahman T, Allen S, Garrett A, Torr B, Bacon A, et al:
Second primary cancer risks after breast cancer in BRCA1 and BRCA2
pathogenic variant carriers. J Clin Oncol. 43:651–661. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Esplin ED, Hanson C, Wu S, Horning AM,
Barapour N, Nevins SA, Jiang L, Contrepois K, Lee H, Guha TK, et
al: Multiomic analysis of familial adenomatous polyposis reveals
molecular pathways associated with early tumorigenesis. Nat Cancer.
5:1737–1753. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Yadav S, Boddicker NJ, Na J, Polley EC, Hu
C, Hart SN, Gnanaolivu RD, Larson N, Holtegaard S, Huang H, et al:
Contralateral breast cancer risk among carriers of germline
pathogenic variants in ATM, BRCA1, BRCA2, CHEK2, and PALB2. J Clin
Oncol. 41:1703–1713. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Pacheco AG and Lou H: Alternative splicing
of exon 23a in neurofibromatosis type 1 pre-mRNA: Its contribution
to the protein structure and function of neurofibromin. Wiley
Interdiscip Rev RNA. 16:e700212025. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Choe JH, Kawase T, Xu A, Guzman A,
Obradovic AZ, Low-Calle AM, Alaghebandan B, Raghavan A, Long K,
Hwang PM, et al: Li-fraumeni syndrome-associated dimer-forming
mutant p53 promotes transactivation-independent mitochondrial cell
death. Cancer Discov. 13:1250–1273. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Kim WJ, Crosse EI, De Neef E, Etxeberria
I, Sabio EY, Wang E, Bewersdorf JP, Lin KT, Lu SX, Belleville A, et
al: Mis-splicing-derived neoantigens and cognate TCRs in splicing
factor mutant leukemias. Cell. 188:3422–3440.e24. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Lu SX, De Neef E, Thomas JD, Sabio E,
Rousseau B, Gigoux M, Knorr DA, Greenbaum B, Elhanati Y, Hogg SJ,
et al: Pharmacologic modulation of RNA splicing enhances antitumor
immunity. Cell. 184:4032–4047.e31. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Zhang Y, Qian J, Gu C and Yang Y:
Alternative splicing and cancer: A systematic review. Signal
Transduct Target Ther. 6:782021. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Garcia-Cabau C, Bartomeu A, Tesei G,
Cheung KC, Pose-Utrilla J, Picó S, Balaceanu A, Duran-Arqué B,
Fernández-Alfara M, Martín J, et al: Mis-splicing of a neuronal
microexon promotes CPEB4 aggregation in ASD. Nature. 637:496–503.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Kowalski MH, Wessels HH, Linder J,
Dalgarno C, Mascio I, Choudhary S, Hartman A, Hao Y, Kundaje A and
Satija R: Multiplexed single-cell characterization of alternative
polyadenylation regulators. Cell. 187:4408–4425.e23. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
The Tabula Sapiens Consortium, . Jones RC,
Karkanias J, Krasnow MA, Pisco AO, Quake SR, Salzman J, Yosef N,
Bulthaup B, Brown P, et al: The tabula sapiens: A multiple-organ,
single-cell transcriptomic atlas of humans. Science.
376:eabl48962022. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Zhu C, Lian Y, Wang C, Wu P, Li X, Gao Y,
Fan S, Ai L, Fang L, Pan H, et al: Single-cell transcriptomics
dissects hematopoietic cell destruction and T-cell engagement in
aplastic anemia. Blood. 138:23–33. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Augspach A, Drake KD, Roma L, Qian E, Lee
SR, Clarke D, Kumar S, Jaquet M, Gallon J, Bolis M, et al: Minor
intron splicing is critical for survival of lethal prostate cancer.
Mol Cell. 83:1983–2002.e11. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Bai R, Yuan M, Zhang P, Luo T, Shi Y and
Wan R: Structural basis of U12-type intron engagement by the fully
assembled human minor spliceosome. Science. 383:1245–1252. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Bai R, Wan R, Wang L, Xu K, Zhang Q, Lei J
and Shi Y: Structure of the activated human minor spliceosome.
Science. 371:eabg08792021. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Lee SCW and Abdel-Wahab O: Therapeutic
targeting of splicing in cancer. Nat Med. 22:976–986. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Steensma DP, Wermke M, Klimek VM,
Greenberg PL, Font P, Komrokji RS, Yang J, Brunner AM, Carraway HE,
Ades L, et al: Phase I first-in-human dose escalation study of the
oral SF3B1 modulator H3B-8800 in myeloid neoplasms. Leukemia.
35:3542–3550. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Seiler M, Yoshimi A, Darman R, Chan B,
Keaney G, Thomas M, Agrawal AA, Caleb B, Csibi A, Sean E, et al:
H3B-8800, an orally available small-molecule splicing modulator,
induces lethality in spliceosome-mutant cancers. Nat Med.
24:497–504. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Chi KN, Higano CS, Blumenstein B, Ferrero
JM, Reeves J, Feyerabend S, Gravis G, Merseburger AS, Stenzl A,
Bergman AM, et al: Custirsen in combination with docetaxel and
prednisone for patients with metastatic castration-resistant
prostate cancer (SYNERGY trial): A phase 3, multicentre,
open-label, randomised trial. Lancet Oncol. 18:473–485. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Aird D, Teng T, Huang CL, Pazolli E, Banka
D, Cheung-Ong K, Eifert C, Furman C, Wu ZJ, Seiler M, et al:
Sensitivity to splicing modulation of BCL2 family genes defines
cancer therapeutic strategies for splicing modulators. Nat Commun.
10:1372019. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Klein IA, Boija A, Afeyan LK, Hawken SW,
Fan M, Dall'Agnese A, Oksuz O, Henninger JE, Shrinivas K, Sabari
BR, et al: Partitioning of cancer therapeutics in nuclear
condensates. Science. 368:1386–1392. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Lan Q, Liu PY, Bell JL, Wang JY,
Hüttelmaier S, Zhang XD, Zhang L and Liu T: The emerging roles of
RNA m6A methylation and demethylation as critical regulators of
tumorigenesis, drug sensitivity, and resistance. Cancer Res.
81:3431–3440. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Salton M, Kasprzak WK, Voss T, Shapiro BA,
Poulikakos PI and Misteli T: Inhibition of vemurafenib-resistant
melanoma by interference with pre-mRNA splicing. Nat Commun.
6:71032015. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Ramasamy T, Ruttala HB, Munusamy S,
Chakraborty N and Kim JO: Nano drug delivery systems for antisense
oligonucleotides (ASO) therapeutics. J Control Release.
352:861–878. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Ma WK, Voss DM, Scharner J, Costa ASH, Lin
KT, Jeon HY, Wilkinson JE, Jackson M, Rigo F, Bennett CF and
Krainer AR: ASO-based PKM splice-switching therapy inhibits
hepatocellular carcinoma growth. Cancer Res. 82:900–915. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Kobayashi Y, Chhoeu C, Li J, Price KS,
Kiedrowski LA, Hutchins JL, Hardin AI, Wei Z, Hong F, Bahcall M, et
al: Silent mutations reveal therapeutic vulnerability in RAS Q61
cancers. Nature. 603:335–342. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Zhao C, Zhao J, Zhang Y, Zhu YD, Yang ZY,
Liu SL, Tang QY, Yang Y, Wang HK, Shu YJ, et al: PTBP3 mediates
IL-18 exon skipping to promote immune escape in gallbladder cancer.
Adv Sci. 11:24066332024. View Article : Google Scholar
|
|
118
|
Ang Z, Paruzzo L, Hayer KE, Schmidt C, Diz
MT, Xu F, Zankharia U, Zhang Y, Soldan S, Zheng S, et al:
Alternative splicing of its 5′ UTR limits CD20 mRNA translation and
enables resistance to CD20-directed immunotherapies. Blood.
142:1724–1739. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Wang Z, Wang S, Qin J, Zhang X, Lu G, Liu
H, Guo H, Wu L, Shender VO, Shao C, et al: Splicing factor BUD31
promotes ovarian cancer progression through sustaining the
expression of anti-apoptotic BCL2L12. Nat Commun. 13:62462022.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Hu Y, Revenko A, Barsoumian H, Bertolet G,
Fowlkes NW, Maazi H, Green MM, He K, Sezen D, Voss TA, et al:
Inhibition of MER proto-oncogene tyrosine kinase by an antisense
oligonucleotide enhances treatment efficacy of immunoradiotherapy.
J Exp Clin Cancer Res. 43:702024. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Prakash TP, Graham MJ, Yu J, Carty R, Low
A, Chappell A, Schmidt K, Zhao C, Aghajan M, Murray HF, et al: NAR
breakthrough article targeted delivery of antisense
oligonucleotides to hepatocytes using triantennary N-acetyl
galactosamine improves potency 10-fold in mice. Nucleic Acids Res.
42:8796–8807. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Ojansivu M, Barriga HMG, Holme MN, Morf S,
Doutch JJ, Andaloussi SE, Kjellman T, Johnsson M, Barauskas J and
Stevens MM: Formulation and characterization of novel ionizable and
cationic lipid nanoparticles for the delivery of splice-switching
oligonucleotides. Adv Mater. 37:e24195382025. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Alvarez-Erviti L, Seow Y, Yin H, Betts C,
Lakhal S and Wood MJ: Delivery of siRNA to the mouse brain by
systemic injection of targeted exosomes. Nat Biotechnol.
29:341–345. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Fei Y, Cao D, Li Y, Wang Z, Dong R, Zhu M,
Gao P, Wang X, Cai J and Zuo X: Circ_0008315 promotes tumorigenesis
and cisplatin resistance and acts as a nanotherapeutic target in
gastric cancer. J Nanobiotechnology. 22:5192024. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Li Y, Wang Z, Gao P, Cao D, Dong R, Zhu M,
Fei Y, Zuo X and Cai J: CircRHBDD1 promotes immune escape via
IGF2BP2/PD-L1 signaling and acts as a nanotherapeutic target in
gastric cancer. J Transl Med. 22:7042024. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Du A, Li S, Zhou Y, Disoma C, Liao Y,
Zhang Y, Chen Z, Yang Q, Liu P, Liu S, et al: M6A-mediated
upregulation of circMDK promotes tumorigenesis and acts as a
nanotherapeutic target in hepatocellular carcinoma. Mol Cancer.
21:1092022. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Zhou B, Mo Z, Lai G, Chen X, Li R, Wu R,
Zhu J and Zheng F: Targeting tumor exosomal circular RNA cSERPINE2
suppresses breast cancer progression by modulating MALT1-NF-κB-IL-6
axis of tumor-associated macrophages. J Exp Clin Cancer Res.
42:482023. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Xu L, Ma X, Zhang X, Zhang C, Zhang Y,
Gong S, Wu N, Zhang P, Feng X, Guo J, et al: hsa_circ_0007919
induces LIG1 transcription by binding to FOXA1/TET1 to enhance the
DNA damage response and promote gemcitabine resistance in
pancreatic ductal adenocarcinoma. Mol Cancer. 22:1952023.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Yin Y, Wang Y, Yu X, Li Y, Zhao Y, Wang Y
and Liu Z: Spatial isoforms reveal the mechanisms of metastasis.
Adv Sci. 11:24022422024. View Article : Google Scholar
|
|
130
|
Yang L, Niu K, Wang J, Shen W, Jiang R,
Liu L, Song W, Wang X, Zhang X, Zhang R, et al: Nucleolin
lactylation contributes to intrahepatic cholangiocarcinoma
pathogenesis via RNA splicing regulation of MADD. J Hepatol.
81:651–666. 2024. View Article : Google Scholar : PubMed/NCBI
|