Introduction
Cholangiocarcinoma (CCA), an aggressive cancer
originating from the biliary epithelium, is the 2nd most common
primary liver cancer after hepatocellular carcinoma (1–4).
Histologically, CCA is typically an adenocarcinoma characterized by
dense desmoplastic stroma and a complex tumor microenvironment
(TME) enriched with cancer-associated fibroblasts and
immunosuppressive cells, which collectively foster tumor
progression and resistance to therapy (5,6). Due
to the absence of early symptoms and lack of effective screening
tools, only 15–30% of patients with CCA are eligible for curative
resection at diagnosis (with >65% presenting with unresectable
or metastatic disease), and recurrence rates after curative-intent
surgery remain high (50–83%) (7–9). The
standard first-line chemotherapy regimen, gemcitabine combined with
cisplatin, confers limited benefit. Although targeted therapies
against FGFR2 and IDH1 mutations have shown promise
in subsets of patients, resistance frequently emerges (10). The combination of molecular
heterogeneity, immunosuppressive TME and intrinsic chemoresistance
results in poor outcomes, with a median survival of only 4–8 months
and a 5-year survival rate of <20% (11). Thus, there is an urgent need to
identify novel biomarkers and therapeutic targets tailored to the
molecular and anatomical diversity of CCA.
Targeting specific molecular pathways and harnessing
the mechanisms of regulated cell death (RCD) have emerged as
promising strategies for cancer suppression (12–15).
Recently, a distinct RCD form termed disulfidptosis has been
identified, characterized by cytoskeletal collapse caused by
excessive disulfide bonding in actin filaments under
glucose-deprived conditions (16,17).
In cells overexpressing SLC7A11, continuous cystine uptake depletes
NADPH, hindering disulfide bond reduction and promoting toxic
protein crosslinking. Notably, disulfidptosis is unresponsive to
classical RCD inhibitors and can occur independently of ATP levels.
Therapeutic strategies that enhance thiol oxidation or inhibit
glucose uptake exacerbate disulfide stress and trigger this cell
death pathway (16,17). Emerging studies implicate
disulfidptosis in digestive tract malignancies such as gastric and
hepatocellular carcinoma, where disulfidptosis-related gene (DRG)
expression is associated with immune activity and clinical
prognosis (18–26). However, its role in CCA remains
unexplored.
Notably, research has highlighted key features of
CCA biology that suggest a unique vulnerability to disulfidptosis.
First, CCA is dependent on glucose metabolism and undergoes
extensive metabolic reprogramming to survive in a nutrient-poor
TME, creating a state of chronic metabolic stress (5,27).
Second, the aggressive, invasive nature of CCA is dependent on the
dynamic reorganization of the actin cytoskeleton, and an abnormal
actin network is itself a driver of the malignant phenotype and
metastasis (28). Furthermore, the
emerging susceptibility of CCA to other forms of redox-dependent
cell death such as ferroptosis points towards a reliance on solute
carriers such as the cystine-glutamate transporter xCT, which
includes SLC7A11, to manage oxidative stress (29). These characteristics, glucose
dependency, cytoskeletal vulnerability and active SLC7A11-related
pathways, establish a rationale for investigating disulfidptosis, a
novel cell death pathway that is directly associated with all three
of these biological pillars.
To investigate this unexplored area, DRGs in CCA
were systematically evaluated using RNA sequencing data from The
Cancer Genome Atlas (TCGA) and European Molecular Biology
Laboratory-European Bioinformatics Institute (EMBL-EBI).
Differential expression analysis was performed to identify
candidate DRGs, then unsupervised clustering was used to define
patient subgroups with distinct clinical and molecular
characteristics. Building on these findings, a prognostic signature
was developed using least absolute shrinkage and selection operator
(LASSO) regression and validated through functional assays to
elucidate the ways key DRGs regulate disulfidptosis. To the best of
our knowledge, the present study revealed the first comprehensive
landscape of disulfidptosis in CCA, associating DRG activity to
patient survival, immune microenvironment remodeling and
therapeutic responsiveness. These findings not only offer a
prognostic tool, but also highlight actionable targets, opening new
avenues for treating this intractable malignancy. The workflow of
the present study is shown in Fig.
1.
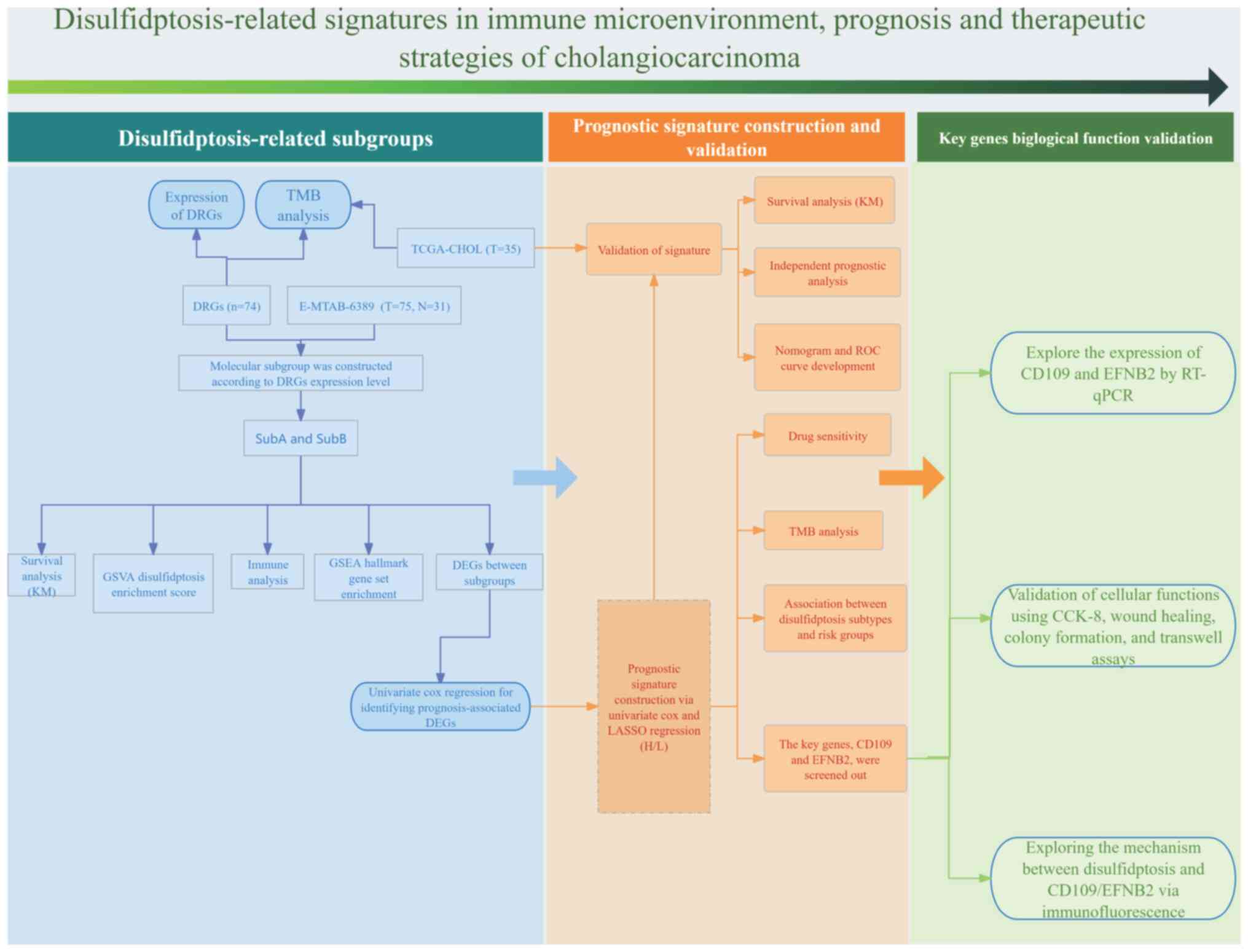 | Figure 1.Flow chart for the present study
analysis. DRGs (n=74) expression was assessed in the E-MTAB-6389
cohort (T=75, N=31), dividing patients into SubA and SubB based on
DRG levels. Molecular subtyping was followed by survival analysis
(KM), GSVA enrichment score, immune analysis and GSEA hallmark
enrichment. Prognosis-related DEGs were identified and used to
build a signature via univariate Cox and LASSO regression,
validated in the TCGA-CHOL cohort (T=35) with KM survival,
independent prognostic, nomogram, ROC, drug sensitivity and TMB
analyses. Disulfidptosis subtypes and risk groups were explored,
highlighting CD109 and EFNB2, validated by RT-qPCR and functional
assays. The disulfidptosis-CD109/EFNB2 mechanism was investigated
via immunofluorescence. DRGs, disulfidptosis-related genes; TCGA,
The Cancer Genome Atlas; GSVA, gene set variation analysis; GSEA,
Gene Set Enrichment Analysis; LASSO, least absolute shrinkage and
selection operator; KM, Kaplan-Meier; ROC, receiver operating
characteristic; TMB, tumor mutational burden; RT-qPCR, real-time
quantitative polymerase chain reaction; CCK-8, Cell Counting Kit-8;
TCGA-CHOL, cholangiocarcinoma cohort from The Cancer Genome Atlas
database; T, tumor; N, normal. |
Materials and methods
Data collection and processing
To construct and validate the prognostic signature,
independent training and validation cohorts were used. The present
study conducted a systematic search of public repositories,
including the Gene Expression Omnibus (GEO), The Cancer Genome
Atlas (TCGA) and ArrayExpress. For the training cohort, the
E-MTAB-6389 dataset (https://www.ebi.ac.uk/biostudies/studies/E-MTAB-6389)
was selected from the ArrayExpress database (https://www.ebi.ac.uk/arrayexpress/) after a
systematic search of public repositories. The dataset was chosen
due to its larger sample size (n=75) which provided greater
statistical power for the initial model construction. This dataset
contains gene expression data from 75 CCA tissues and 31 adjacent
normal tissues, generated on the Affymetrix Human Transcriptome
Array 2.0 microarray platform (GEO platform accession, GPL17585).
The raw data were processed using the robust multi-array average
algorithm and subsequently log2-transformed. For external
validation, TCGA-CHOL cohort was used, acquired from the Genomic
Data Commons portal (https://portal.gdc.cancer.gov/). This cohort consisted
of 35 patients with CCA and corresponding overall survival (OS)
data. Gene expression was quantified as Fragments Per Kilobase of
transcript per Million mapped reads (FPKM) on the Illumina HiSeq
2000 platform (Illumina, Inc.), and the values were
log2-transformed [log2(FPKM+1)] for subsequent analyses. Due to
incomplete clinical information in the training cohort, survival
modeling and mutation profiling were performed exclusively on TCGA
dataset. A total of 100 DRGs were identified through a systematic
literature review. The selection process began with core
mechanistic genes established in foundational studies, and was
expanded by incorporating DRGs from recent high-quality
bioinformatics and experimental studies that constructed prognostic
models or molecular subtypes (molecular subs; distinct tumor
subgroups characterized by specific disulfidptosis-related gene
expression profiles and clinical outcomes) across malignancies
(Table SI) (16–26).
All datasets are publicly available and adhere to ethical
standards. The harmonized clinicopathological characteristics for
subsequent analyses are elaborated in Table I.
 | Table I.Clinicopathological characteristics
of patients with cholangiocarcinoma in The Cancer Genome Atlas. |
Table I.
Clinicopathological characteristics
of patients with cholangiocarcinoma in The Cancer Genome Atlas.
| Clinicopathological
characteristics | TCGA cohort
(N=35) |
|---|
| Age (years), no.
(%) |
|
|
≤65 | 17 (48.6%) |
|
>65 | 18 (51.4%) |
| Gender, no.
(%) |
|
|
Female | 19 (54.3%) |
|
Male | 16 (45.7%) |
| Stage, no. (%) |
|
| I | 18 (51.4%) |
| II | 9 (25.7%) |
|
III | 1 (2.9%) |
| IV | 7 (20%) |
| T, no. (%) |
|
| 0 | 0 (0.0%) |
| 1 | 18 (51.4%) |
| 2 | 6 (17.1%) |
| 2a | 2 (5.7%) |
| 2b | 4 (11.5%) |
| 3 | 5 (14.3%) |
| 4 | 0 (0.0%) |
| N, no. (%) |
|
| N0 | 25 (71.4%) |
| N1 | 5 (14.3%) |
| NX | 5 (14.3%) |
| M, no. (%) |
|
| M0 | 27 (77.1%) |
| M1 | 5 (14.3%) |
| MX | 3 (8.6%) |
| Survival status,
no. (%) |
|
|
Alive | 17 (48.6%) |
|
Dead | 18 (51.4%) |
| Overall survival
time, days |
|
| Mean ±
SD | 742.54±548.58 |
|
Median | 640 |
Expression of DRGs and molecular
subtyping in CCA
An integrated analysis of 100 DRGs in CCA was
conducted. Expression profiles were extracted from the E-MTAB-6389
dataset, and differential expression was assessed using Wilcoxon
rank-sum tests (FDR <0.05). Significant DRGs were visualized
using boxplots. Pairwise co-expression relationships were evaluated
using Pearson's correlation analysis. All statistical and
bioinformatic analyses were performed using R version 4.4.1
(30). Somatic mutation landscapes
were analyzed using TCGA data and the ‘maftools’ package (31) (https://bioconductor.uib.no/packages/release/bioc/html/maftools.html)
version 2.18.0, with alterations depicted in waterfall plots. To
identify disulfidptosis-driven molecular Subs, unsupervised
consensus clustering was performed on tumor samples using the
‘ConsensusClusterPlus’ package (32) (https://bioconductor.org/packages/release/bioc/html/ConsensusClusterPlus.html)
version 1.54.0.
Prognostic, immune microenvironment
and mechanistic profiling of disulfidptosis-related molecular Subs
in CCA
To investigate the prognostic and biological
significance of disulfidptosis-related molecular Subs in CCA,
survival analysis was performed to compare OS across Subs.
Enrichment scores for disulfidptosis pathways were computed by
integrating gene sets with CCA expression matrices using the ‘GSVA’
R package, with Sub-specific differences assessed via Wilcoxon
tests. For immune microenvironment analysis, three complementary
approaches were used. Stromal and immune cell infiltration scores
were quantified using the Estimation of Stromal and Immune cells in
Malignant Tumor tissues using Expression data (ESTIMATE) algorithm
with the ‘estimate’ R package. Proportions of 22 immune cell types
were estimated through Cell-type Identification by Estimating
Relative Subsets of RNA Transcripts (CIBERSORT) deconvolution using
the ‘CIBERSORT’ R package. Immune cell-specific enrichment scores
were derived via single-sample (ss) Gene Set Enrichment Analysis
(GSEA) with the ‘GSVA’ R package. Subtype differences were
evaluated using Wilcoxon tests. Expression profiles of immune
checkpoint and human leukocyte antigen (HLA) family genes were
compared across Subs. Molecular pathway characterization was
performed using GSEA.
Identification of DRGs, and
construction and validation of the prognostic signature in CCA
Utilizing the ‘limma’ package in R, P-values and
log2FC were calculated for each gene, applying multiple testing
correction to obtain adjusted P-values (adj.P.Value). Genes with
adj.P.Value <0.05 and |log2FC|>0.585 were classified as
differentially expressed and disulfidptosis related. To identify
prognostic Sub-specific genes, univariate Cox regression analysis
was conducted using gene expression and survival data from CCA
samples. Genes with P<0.01 were deemed significant. A prognostic
model was then constructed using LASSO regression analysis using
the ‘lars’ package, incorporating 10-fold cross-validation for gene
selection. The risk score formula, RiskScore=β1X1 + β2X2 + ... +
βnXn, was employed, where β signifies LASSO regression coefficients
and X represents gene expression levels. Risk scores were
calculated for each sample in both the EMBL-EBI training set and
TCGA validation cohort, with patients stratified into high (H)-risk
and low (L)-risk groups based on an optimal risk score cut-off.
Kaplan-Meier assessed the association between risk groups and
survival outcomes. Univariate and multivariate Cox regression
analyses determined whether the model functioned as an independent
prognostic factor for CCA. Significant clinical features were
incorporated into a nomogram constructed with the ‘rms’ R package.
Calibration curves assessed model consistency, while receiver
operating characteristic (ROC) curves and clinical decision curves
evaluated the nomogram's predictive accuracy and practical
utility.
Drug sensitivity analysis of risk
groups
Drug sensitivity was analyzed between H- and L-risk
groups by employing predicted IC50 values sourced from
the Genomics of Drug Sensitivity in Cancer (https://www.cancerrxgene.org/). Utilizing the
‘pRRophetic’ R package, IC50 values were computed.
Significant disparities in drug sensitivity among the risk
subgroups were uncovered using Wilcoxon rank-sum tests,
highlighting therapeutic diversity.
Mutational landscape stratification by
risk groups
For the analysis of somatic variant profiles from
TCGA cohorts, the ‘maftools’ package was employed to investigate
oncogenic landscapes across risk subgroups. The varying mutation
frequencies among the top 20 driver genes were visualized using
‘oncoplots’. Additionally, the tumor mutational burden (TMB) was
quantified and evaluated using the Wilcoxon rank-sum test.
Association between disulfidptosis
Subs and risk groups
To explore the relationship between disulfidptosis
Subs and risk groups, the R ‘ggalluvial’ package was used to
generate Sankey diagrams. These diagrams visually represented the
distribution of samples across disulfidptosis Subs and H-/L-risk
groups, revealing the associations between these
classifications.
Cell culture
HuCCT1 cells (Procell Life Science & Technology
Co., Ltd.), primary human intrahepatic bile duct epithelial cells
(HiBEpiCs; passage 2; cat. no. CP-H042; Pricella Biotechnology),
RBE cells (The Cell Bank of Type Culture Collection of The Chinese
Academy of Sciences) and HuH-28 cells (cat. no. ZQ1030; Zhong Qiao
Xin Zhou Biotechnology Co., Ltd.) were cultured in RPMI-1640 medium
(HyClone; Cytiva) supplemented with 10% fetal bovine serum (FBS;
Gibco; Thermo Fisher Scientific, Inc.) at 37°C in a humidified 5%
CO2 incubator (Heal Force Bio-Meditech Holdings, Ltd.).
All cell handling procedures were performed in a biological safety
cabinet (BHC-1300IIA2; Suzhou Antai Air Technology Co., Ltd.). For
thawing, cells were rapidly thawed in a 37°C water bath (HWS-12;
Shanghai Yiheng Scientific Instrument Co., Ltd.) and gently
agitated. After centrifugation (200 × gp; 5 min; room temperature),
cells were resuspended in culture medium, seeded into dishes and
incubated at 37°C with 5% CO2. When cells achieved
80–90% confluency, they were trypsinized, centrifuged (200 × g; 5
min; room temperature) and re-seeded. For cryopreservation,
5×106 cells/ml were resuspended in Cell Freezing Medium
(cat. no. C0210; Beyotime Biotechnology), aliquoted into cryovials
and frozen at −80°C before storage in liquid nitrogen.
Small interfering (si)RNA
transfection
siRNA transfection was performed to knock down
CD109 and EFNB2 expression in HuCCT1 and RBE cells.
The specific siRNA sequences (listed in Table II) were synthesized by Sangon
Biotech Co., Ltd. A total of 5×105 cells/ml were seeded
in 2 ml culture medium in a 6-well plate and incubated for 24 h at
37°C in a 5% CO2 incubator. siRNAs [negative control
(NC) siRNA, CD109 siRNA and EFNB2 siRNA] were diluted to 100
pmol/µl. For transfection, 1 µl siRNA was mixed with 150 µl
Opti-MEM (Gibco; Thermo Fisher Scientific, Inc.) and incubated for
5 min at room temperature. Similarly, 5 µl
Lipofectamine™ RNAiMAX (cat. no. 13778150; Thermo Fisher
Scientific, Inc.) was mixed with 150 µl Opti-MEM. After 20 min of
incubation at room temperature, the transfection complex was added
dropwise to the cells. After 6 h at 37°C, the medium was replaced
with DMEM (cat. no. 11965-118; Gibco; Thermo Fisher Scientific
Inc.) containing 10% FBS, and cells were cultured for further
analysis. Transfection efficiency and plasmid construction were
validated using quantitative PCR (qPCR) and western blotting (WB)
24 h post-transfection.
| Table II.siRNA sequences about CD109, EFNB2
and si-NC. |
Table II.
siRNA sequences about CD109, EFNB2
and si-NC.
| siRNA | Sense, 5′-3′ | Antisense,
5′-3′ |
|---|
| si-NC | UUCUCCGAACGUGUCACGU
(dT dT) | ACGUGACACGUUCGGAGAA
(dT dT) |
| CD109-siRNA1 | AUCAAACCUCACUGUCUCU
(dT dT) | AGAGACAGUGAGGUUUGAU
(dT dT) |
| CD109-siRNA2 | ACACUUACUCUUCCAUCAC
(dT dT) | GUGAUGGAAGAGUAAGUGU
(dT dT) |
| CD109-siRNA3 | ACAAGCCAAAGCAAGAAGU
(dT dT) | ACUUCUUGCUUUGGCUUGU
(dT dT) |
| EFNB2-siRNA1 | ACCUGGACAAGGACUGGUA
(dT dT) | UACCAGUCCUUGUCCAGGU
(dT dT) |
| EFNB2-siRNA2 | GUUGGCCAGUAUGAAUAUU
(dT dT) | AAUAUUCAUACUGGCCAAC
(dT dT) |
| EFNB2-siRNA3 | GUGCCAAACCAGACCAAGA
(dT dT) | UCUUGGUCUGGUUUGGCAC
(dT dT) |
Reverse transcription (RT)-qPCR
Total RNA was extracted from normal intrahepatic
biliary epithelial cells (HIBEpiC) and CCA cell lines (HuCCT1, RBE,
and HuH-28) using 1 ml TRIzol® (Thermo Fisher
Scientific, Inc.), followed by chloroform separation, isopropanol
precipitation and ethanol washing. RNA concentration and purity
were assessed using a NanoDrop 2000. cDNA was synthesized from 1 µg
RNA using Reverse Transcriptase (Thermo Fisher Scientific, Inc.)
with Oligo-dT. The reaction was incubated at 70°C for 5 min, 37°C
for 5 min, 42°C for 60 min and 70°C for 5 min. qPCR was performed
to a 10 µl reaction containing 4 µl diluted cDNA, 5 µl
BeyoFast™ SYBR Green qPCR Mix (2X) (cat. no. D7260;
Beyotime Biotechnology), 0.4 µl forward and reverse primer (10 µM)
and 0.6 µl water on a CFX96 system. The thermocycling conditions
for qPCR were: 95°C for 5 min, 40 cycles of 95°C for 10 sec and
60°C for 20 sec, followed by a melting step and cooling at 40°C for
30 sec. Data were analyzed using the 2−ΔΔCq method
(33) with GAPDH as the
reference gene. Primer sequences are shown in Table III.
| Table III.Reverse transcription-quantitative
PCR primer sequences. |
Table III.
Reverse transcription-quantitative
PCR primer sequences.
| Gene | Sense, 5′-3′ | Antisense,
5′-3′ |
|---|
| CD109 |
AAGCCAGTGAAAGGAGACGTA |
CCAGGGGAAGATAGATCCAGG |
| EFNB2 |
TATGCAGAACTGCGATTTCCAA |
TGGGTATAGTACCAGTCCTTGTC |
| GAPDH |
GGAGCGAGATCCCTCCAAAAT |
GGCTGTTGTCATACTTCTCATGG |
WB
Total protein was collected from HuCCT1 and RBE
cells using RIPA lysis buffer (Beyotime Biotechnology) supplemented
with protease and phosphatase inhibitors. The lysate was collected
by centrifugation at 14,000 × g for 15 min at 4°C, and protein
concentration was measured using a BCA Protein Assay Kit (cat. no.
P0012; Beyotime Biotechnology). Samples were mixed with 4X Sample
Buffer (Beyotime Biotechnology) containing 100 mM dithiothreitol,
denatured at 95°C for 5 min and separated on 4–20% Bis-Tris gels
(cat. no. M00657; GenScript). Proteins were transferred to PVDF
membranes which were then blocked with 5% non-fat milk in TBS-Tween
[TBST; Tris-buffered saline containing 0.1% (V/V) Tween 20] for 2 h
at room temperature. The membranes were subsequently incubated with
primary antibodies diluted in TBST containing 5% bovine serum
albumin (Sigma-Aldrich; Merck KGaA). The specific primary
antibodies used were: CD109 (1:1,000; cat. no. A23787; ABclonal
Biotech Co., Ltd.) and Ephrin B2 Polyclonal antibody (1:1,500; cat.
no. 26533-1-AP; Proteintech Group, Inc.). The incubation was
performed overnight (12–16 h) at 4°C on a shaker. Following primary
antibody incubation, membranes were washed three times with TBST
(10 min each) and incubated with HRP-linked secondary antibody
(1:6,000; cat. no. 7074; Cell Signaling Technology Inc.) for 1 h at
room temperature for detection. For the loading control, membranes
were probed separately using GAPDH (D16H11) Rabbit mAb (HRP
Conjugate) (cat. no. 8884; Cell Signaling Technology Inc.), which
was detected directly. Chemiluminescent signals were captured with
a Tanon-4600 imaging system and analyzed using Quantity One
(version 4.6.6; Bio-Rad Laboratories, Inc.).
Cell Counting Kit-8 (CCK-8) assay
A total of 2×104 cells/ml were seeded in
100 µl DMEM with 10% FBS in 96-well plates and incubated for 24 h
at 37°C with 5% CO2. At 0, 24, 48 and 72 h, 10 µl CCK-8
solution (Beyotime Biotechnology) was added to each well, avoiding
bubbles. Plates were incubated for 1 h at 37°C, and absorbance was
measured at 450 nm using a microplate reader.
Cell cloning assay
Cells were serially diluted and seeded at 100 cells
per 60-mm dish. Dishes were gently rotated for even distribution
and incubated at 37°C with 5% CO2 for 2–3 weeks. When
visible clones appeared, the medium was removed and cells were
washed twice with 1X PBS, fixed with 4% paraformaldehyde for 15 min
at room temperature and stained with 0.1% crystal violet for 10–30
min at room temperature. After washing and air drying, clones were
imaged using a light microscope and imaging system (Olympus
Corporation) and quantified with ImageJ software (version 1.54;
National Institutes of Health).
Wound healing assay
A total of 5×105 HuCCT1 and RBE tumor
cells/well were seeded in 6-well plates and cultured for 16–24 h at
37°C until 90% confluence. A 10-µl pipette tip was used to create
3–5 linear scratches per well. Cells were washed three times with
PBS to remove debris and cultured in serum-free DMEM for 24 h at
37°C. Wound closure was imaged at 0 and 24 h using a light
microscope (Nikon Corporation) and analyzed using ImageJ software
(version 1.54; National Institutes of Health).
Transwell assay
Tumor cell migration and invasion were assessed
using 24-well Transwell inserts with an 8-µm pore size
polycarbonate membrane. For the migration assay, cells were
trypsinized using 0.25% Trypsin-EDTA, centrifuged at 300 × g for 5
min at room temperature, washed twice with PBS and resuspended in
serum-free RPMI-1640 medium with 1% FBS to a concentration of
5×105 cells/ml. Subsequently, 200 µl of the cell
suspension (1×105 cells) was seeded into the upper
chamber. For the invasion assay, Transwell inserts were pre-coated
with Matrigel Matrix and solidified for 1 h at 37°C before seeding
cells. RPMI-1640 medium containing 20% FBS was added to the lower
chamber as a chemoattractant. The plates were incubated for 24 h at
37°C in a humidified atmosphere containing 5% CO2.
Non-migrated cells on the upper surface were removed using cotton
swabs. Migrated or invaded cells on the lower surface were fixed
with 4% paraformaldehyde for 30 min at room temperature and stained
with 0.1% crystal violet solution for 20 min at room temperature.
The inserts were washed with PBS, and the stained cells were imaged
and counted using ImageJ software (version 1.54; National
Institutes of Health) for quantitative analysis.
F-actin immunofluorescence
staining
A total of 2×104 cells/ml were added in
500 µl medium in 6-well plates (Corning, Inc.) and cultured in DMEM
(cat. no. 11965-118; Gibco; Thermo Fisher Scientific, Inc.) for 24
h at 37°C with 5% CO2 until reaching 50% confluence. To
investigate the role of EFNB2 and CD109 in disulfidptosis, the
medium was replaced with glucose-free DMEM (cat. no. 11966-025;
Gibco; Thermo Fisher Scientific, Inc.) for glucose deprivation.
Cells were then treated with the GLUT1 inhibitor BAY-876 (10 µM;
cat. no. HY-100017, MedChemExpress) and/or the reducing agent,
Tris-(2-carboxyethyl)-phosphine hydrochloride (TCEP; 20 mM; cat.
no. A600974; Sangon Biotech Co., Ltd.) for 6 h at 37°C. Following
treatment, cells were washed twice with pre-warmed 1X PBS (pH 7.4;
Sangon Biotech Co., Ltd.). Cells were fixed with 4%
paraformaldehyde (Shanghai Lingfeng Chemical Reagent Co., Ltd.) in
PBS for 10 min at room temperature, followed by three PBS washes.
Permeabilization was performed with 0.5% Triton X-100 (Bio-Rad
Laboratories, Inc.) in PBS for 5 min at room temperature, followed
by PBS washes. Cells were then incubated with Alexa
Fluor™ 594 Phalloidin (1:400 dilution in 1% BSA/PBS;
cat. no. A12381, Thermo Fisher Scientific Inc.) for 30 min in a,
humidified chamber in the dark. After washing, cells were
counterstained with DAPI (100 nM; cat. no. HY-D0814,
MedChemExpress) for 30 sec at room temperature. Coverslips were
mounted on slides with AntiFade Mounting Medium (cat. no. HY-K1042;
MedChemExpress) and sealed. Fluorescence was observed using a ZEISS
LSM800 confocal microscope (ZEISS).
Statistical analysis
Statistical analysis was conducted using R (version
4.4.1; The R Foundation for Statistical Computing), SPSS (version
26.0; IBM Corp.) and GraphPad Prism (version 10.0; Dotmatics).
Intergroup comparisons were carried out using the Wilcoxon rank-sum
test, while chi-square tests assessed association between subgroups
and clinicopathological features. Survival curves were plotted
using the Kaplan-Meier method. Prognostic factors for CCA were
identified via LASSO regression, followed by univariate and
multivariate Cox regression to calculate hazard ratios (HRs) and
95% confidence intervals (CIs). P<0.05 was considered to
indicate a statistically significant difference with FDR <0.05.
Significance levels are denoted as *P<0.05, **P<0.01 and
***P<0.001. All experiments were performed in triplicate.
Results
Development of
disulfidptosis-regulated clusters and their characteristics in
CCA
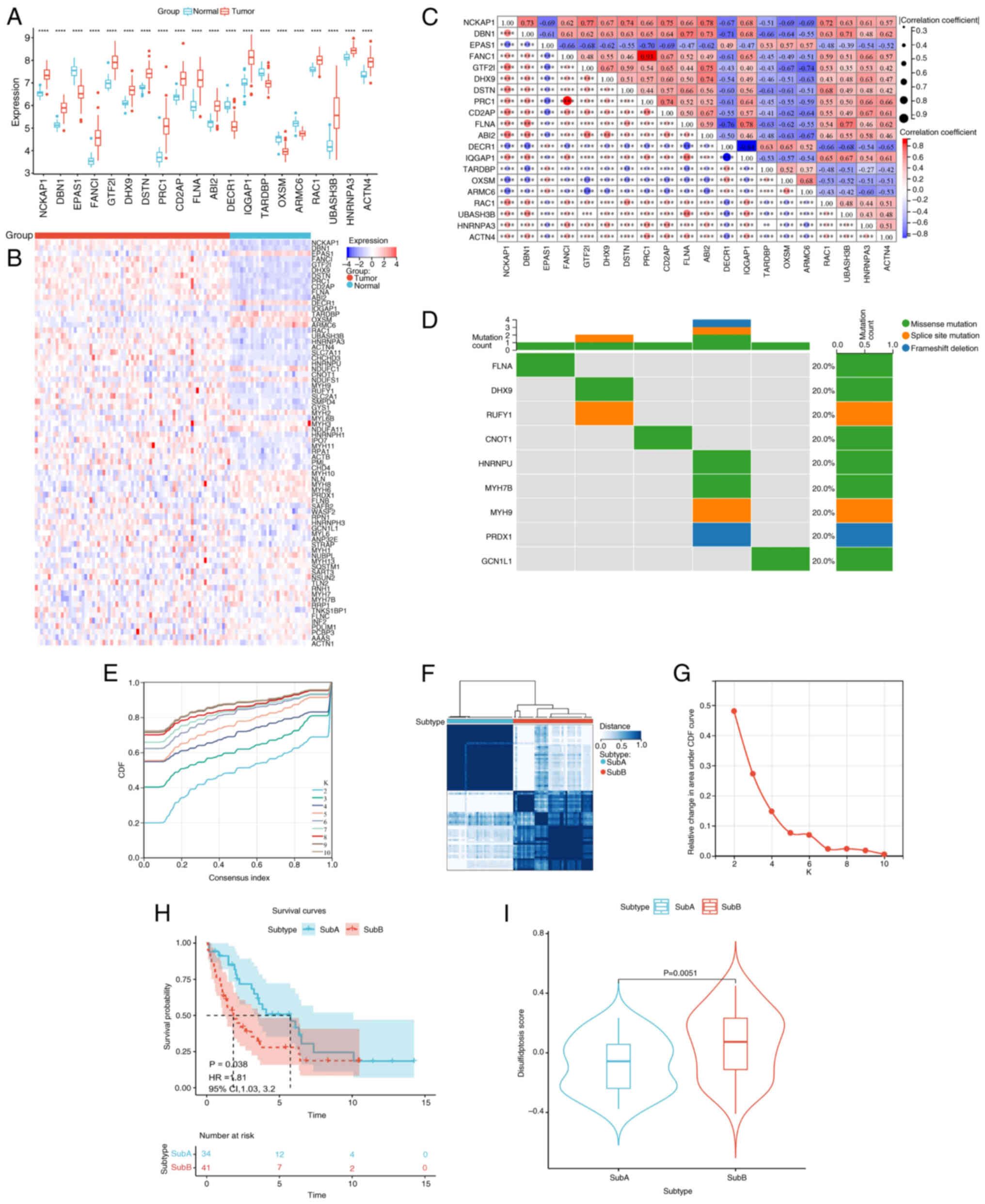
Differential expression analysis of 100 DRGs in CCA
vs. adjacent normal tissues identified 74 DRGs (|log2FC|>1; FDR
<0.05) using Wilcoxon rank-sum tests (Table SII). The top 20 DRGs are shown in
Fig. 2A and a correlation heatmap
of all 74 DRGs is shown in Fig. 2B.
Co-expression analysis of the top 20 DRGs revealed significant
intergenic correlations (Fig. 2C).
Somatic mutation analysis via TCGA data detected mutations in nine
DRGs (Fig. 2D). Unsupervised
consensus clustering (K=2; range, 2–10; Fig. 2E) classified 75 samples into SubA
(n=34) and SubB (n=41) Subs (Fig.
2F; Table SIII). Sub stability
was confirmed using cumulative distribution function curve analysis
(Fig. 2G). Kaplan-Meier analysis
indicated shorter OS for SubB (P=0.038; Fig. 2H). GSVA revealed elevated
disulfidptosis pathway activity in SubB (P=0.005; Fig. 2I; Table
SIV).
Immune landscape and checkpoint gene
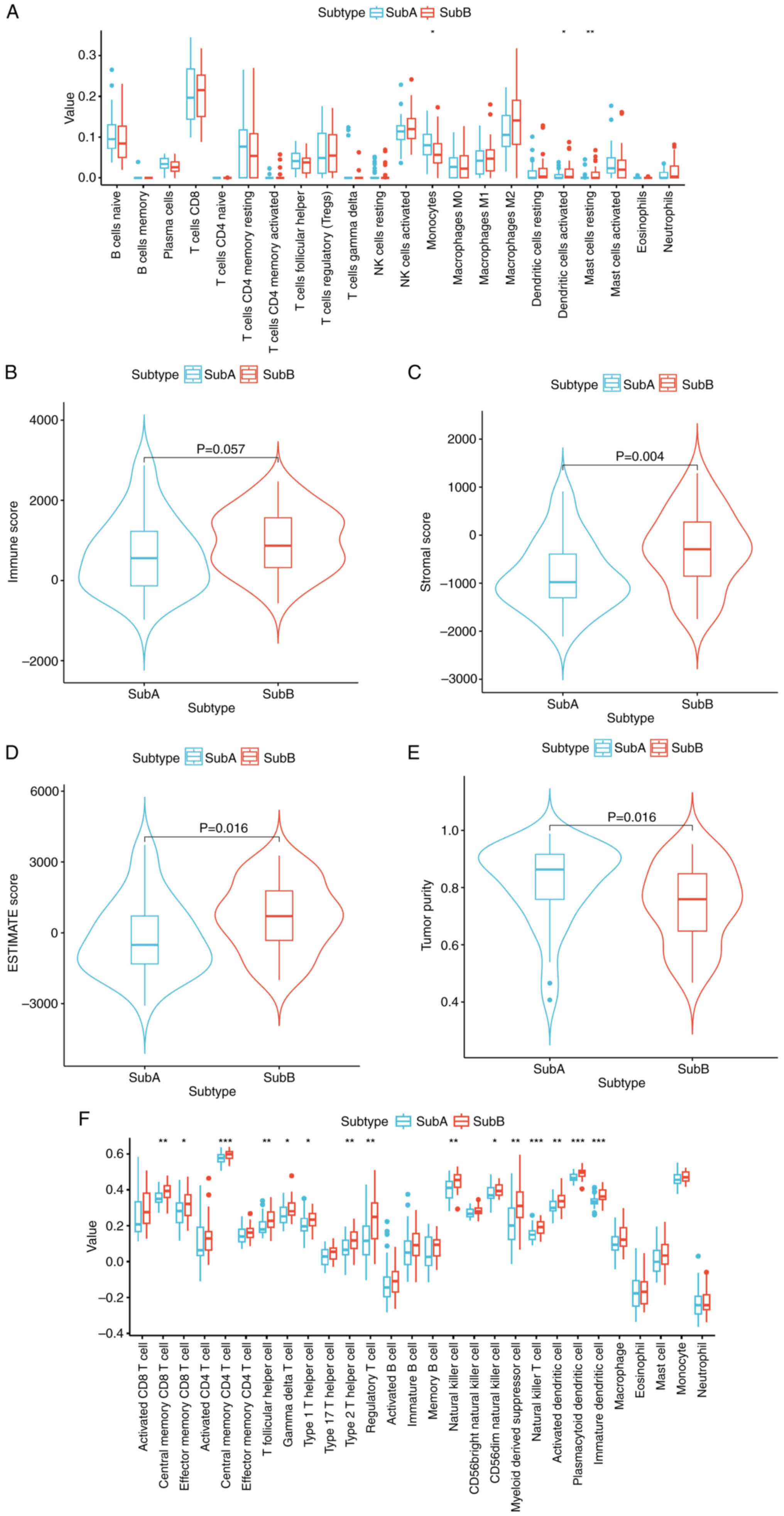
expression profiles in disulfidptosis Subs of CCA
To investigate the relationship between
disulfidptosis Subs and the immune microenvironment in CCA, the
CIBERSORT algorithm was applied to evaluate infiltration of 22
immune cell types. Comparative analysis (P<0.05) revealed
significantly lower monocyte levels in SubB compared with SubA,
with an increased number of activated dendritic and resting mast
cells in SubB (Fig. 3A). ESTIMATE
analysis indicated higher stromal and ESTIMATE scores in SubB
(P<0.05), while SubA exhibited greater tumor purity (Fig. 3B-E). Immune scores trended higher in
SubB. ssGSEA analysis revealed significantly enhanced effector
immune cell activity in SubB, including CD8+ T cells,
CD4+ T cells and natural killer cells (P<0.05),
indicating robust antitumor immunity (Fig. 3F). By contrast, SubA exhibited
elevated levels of γδ T cells and dendritic cells, associated with
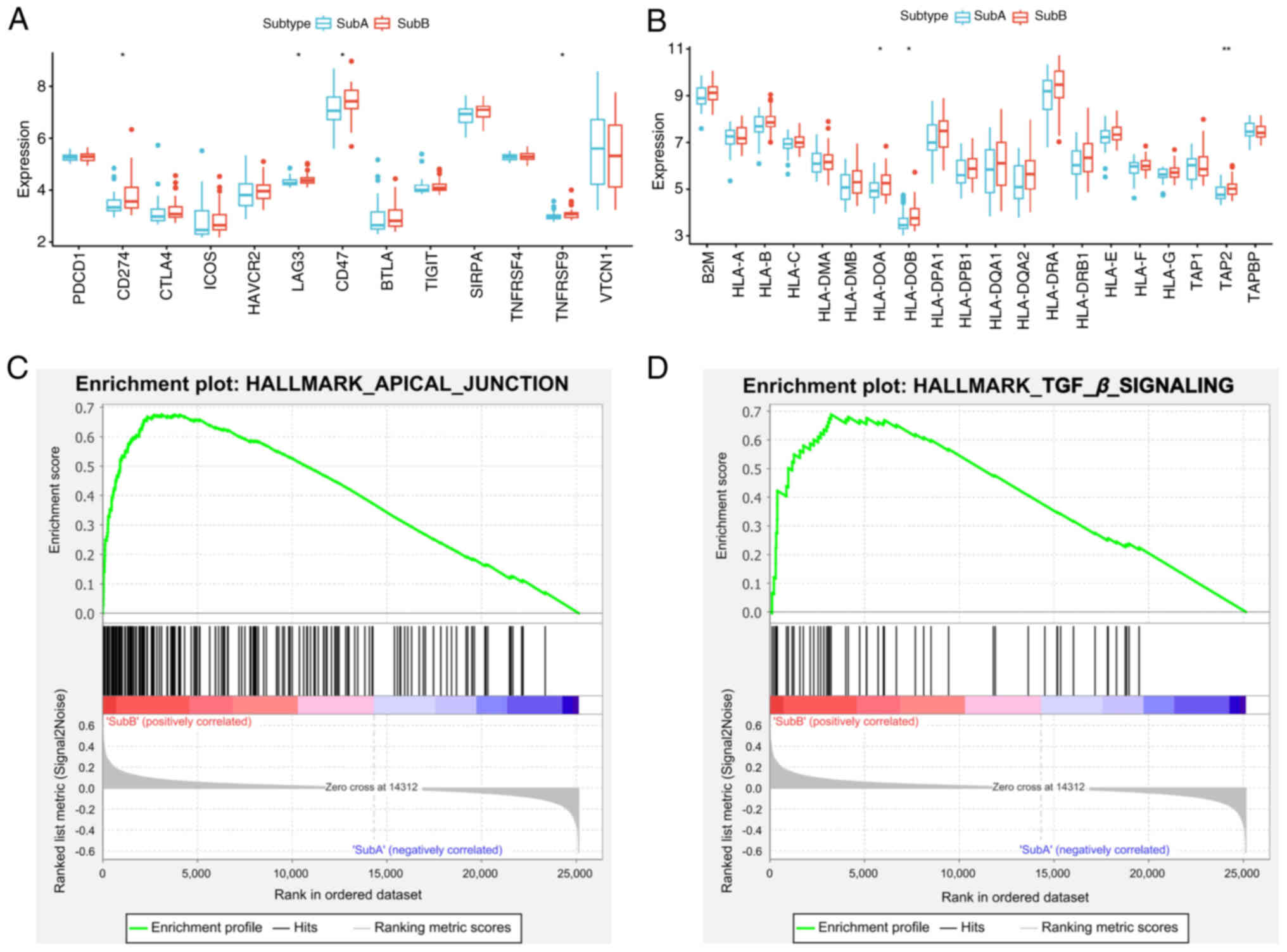
adaptive immunity and antigen presentation. Immune checkpoint genes
(PD-L1, LAG3, CD47 and TNFRSF9) and HLA antigen
presentation genes (HLA-DOA, HLA-DOB and TAP2) were
significantly upregulated in SubB (P<0.05; Fig. 4A and B). GSEA analysis of the
E-MTAB-6389 dataset revealed SubB enrichment in APICAL_JUNCTION and
TGF_β_SIGNALING pathways (Fig. 4C and
D; FDR <0.05), suggesting roles in immune evasion and
structural remodeling.
Identifying DRGs, construction and
validation of the disulfidptosis-related predictive signature in
CCA
Differentially expressed gene (DEG) analysis between
the DRG subgroups identified 190 upregulated genes and 173
downregulated genes (Fig. 5A;
Table SV). Univariate Cox
regression analysis of the 363 DEGs revealed seven
prognosis-related genes (ADAMTS12, SLC2A1, CD109, CALB2, EFNB2,
KLK6 and KRT6A) with P<0.01 and HRs ranging from 1.5
(95% CI, 1.1–1.9) to 2.2 (95% CI, 1.5–3.4; Fig. 5B). LASSO regression selected a
four-gene prognostic signature (EFNB2, CD109, KLK6 and
ADAMTS12) with positive coefficients (Fig. 5C and D; Table SVI). The risk score was calculated
as follows: RiskScore=0.3589 × Exp (EFNB2) + 0.2604 × Exp
(CD109) +0.2382 × Exp (ADAMTS12) + 0.1120 × Exp
(KLK6).
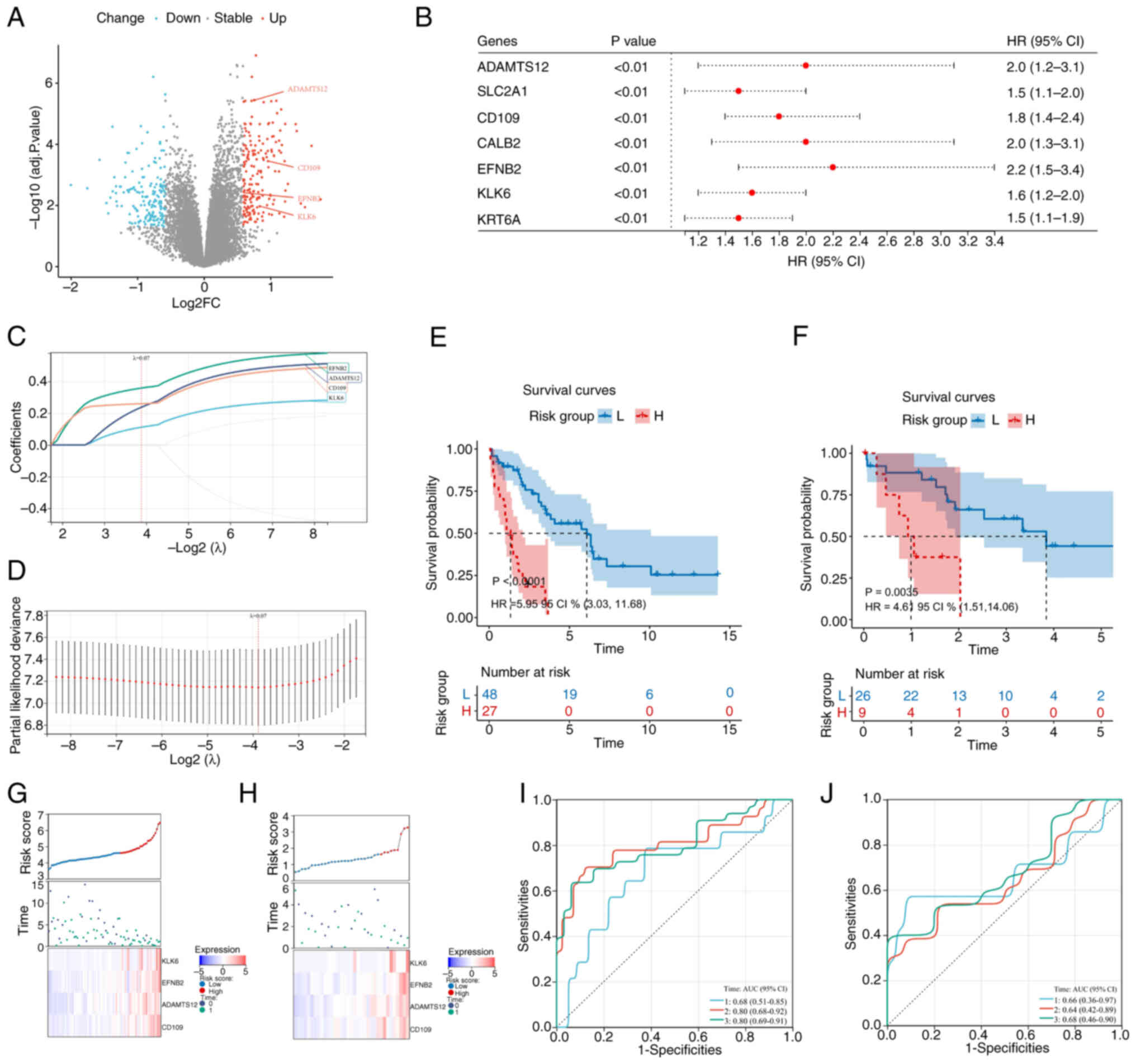 | Figure 5.Prognostic gene signature and risk
score analysis in cholangiocarcinoma. (A) Volcano plot of
differentially expressed genes (blue, downregulated; red,
upregulated); (B) Univariate Cox regression forest plot of seven
prognostic genes; (C) LASSO coefficient distribution profile; (D)
likelihood deviance for LASSO coefficients, with vertical dashed
lines indicating λ. min (left) and λ.1se (right); (E) Kaplan-Meier
survival curves for patients stratified by the 4-gene signature in
the training cohort (Risk group; P<0.0001; HR=5.95; 95% CI
3.03–11.68); (F) Kaplan-Meier survival curves for patients
stratified by the 4-gene signature in the validation cohort; (G)
training cohort: Risk score distribution (top), survival status
(middle) and gene expression heatmap (bottom); (H) validation
cohort: Risk score distribution (top), survival status (middle) and
gene expression heatmap (bottom); (I) time-dependent ROC curves
(1-, 2-, 3-year) in the training cohort; (J) time-dependent ROC
curves (1-, 2-, 3-year) in the validation cohort. LASSO, least
absolute shrinkage and selection operator; HR, hazard ratio; AUC,
area under the curve. |
Using optimal cut-offs (EMBL-EBI; 4.595; TCGA,
1.608), patients with CCA were classified into H- and L-risk
groups. Kaplan-Meier analysis revealed significant survival
differences in both the EMBL-EBI (P=0.0001; HR, 5.95; 95% CI,
3.03–11.68) and TCGA (P=0.0035; HR, 4.61; 95% CI, 1.51–14.06)
datasets (Fig. 5E and F). Heatmaps
showed increased expression of the signature genes in H-risk
patients, consistent with positive coefficients (Fig. 5G and H). Time-dependent ROC analysis
demonstrated area under the curves (AUCs) of 0.680, 0.800 and 0.800
(EMBL-EBI) and 0.660, 0.640 and 0.680 (TCGA; Fig. 5I and J) at 1, 2 and 3 years,
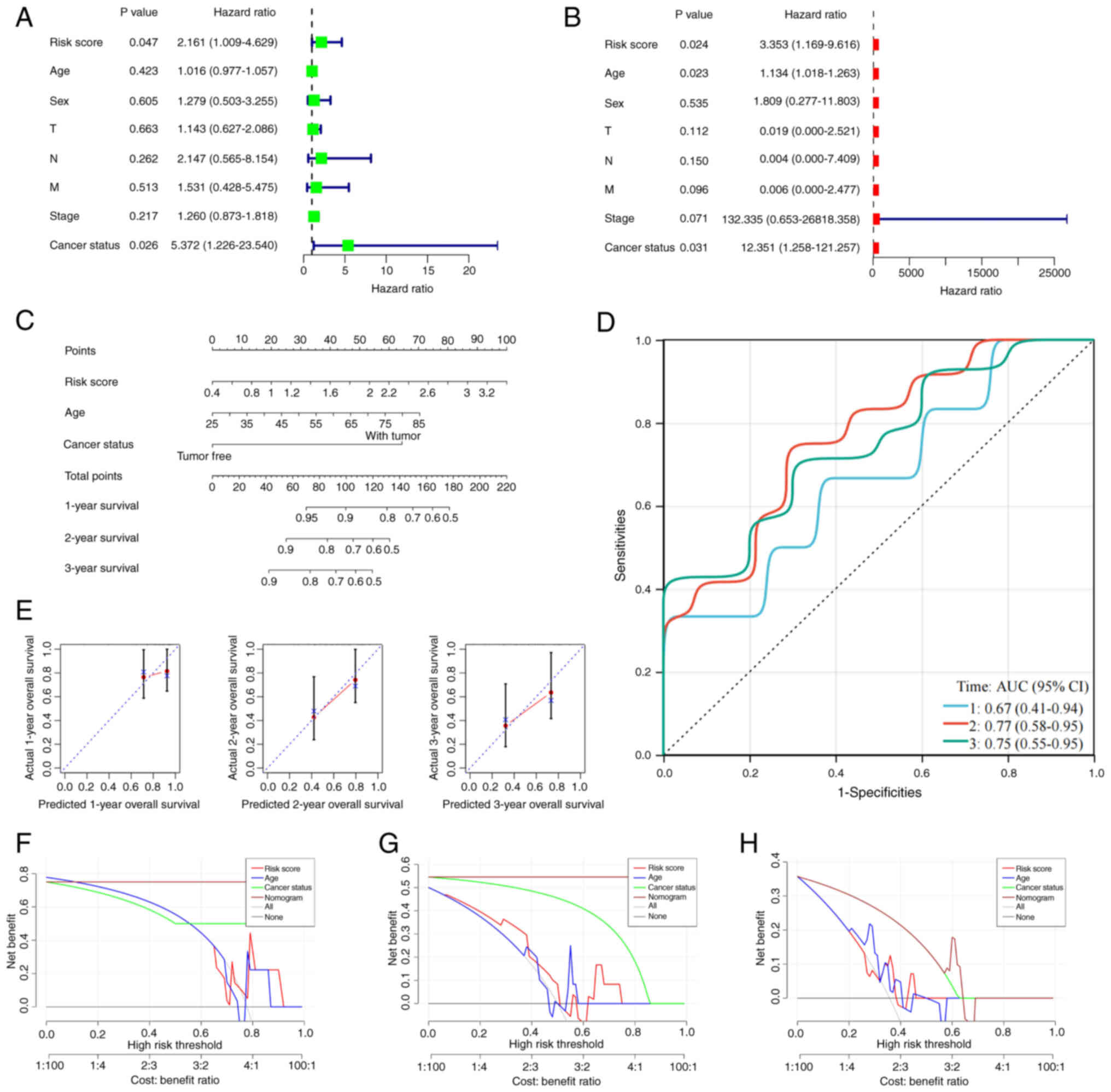
respectively. Univariate and multivariate Cox regression confirmed
that the risk score was an independent prognostic factor (P=0.024;
HR, 3.353; 95% CI, 1.169–9.616) and multivariate analysis,
including age (P=0.023; HR, 1.134; 95% CI, 1.018–1.263) and cancer
status (P=0.031; HR, 12.351; 95% CI, 1.258–121.257), further
validated its independence (Fig. 6A and
B; Table SVII). A nomogram
integrating the risk score, age and cancer status achieved AUCs of
0.670, 0.770 and 0.750 for 1-, 2- and 3-year predictions (Fig. 6C and D; Table SVIII). Calibration curves indicated
strong agreement between predicted and observed outcomes (Fig. 6E). Decision curve analysis for the
1-, 2- and 3-year outcomes showed that the risk score model
provided superior net benefit over age or cancer status alone,
especially at medium-to-high thresholds, confirming its clinical
utility in risk stratification and treatment decision-making for
CCA (Fig. 6F-H).
To elucidate the biological underpinnings of this
signature, the genes with the strongest contribution to the risk
score were prioritized for functional validation. Given that
EFNB2 and CD109 possessed the two highest risk
coefficients in the LASSO model, they were selected for subsequent
in vitro experiments.
Integrated profiling of drug
sensitivity, TMB and molecular Subs in the prognostic risk
model
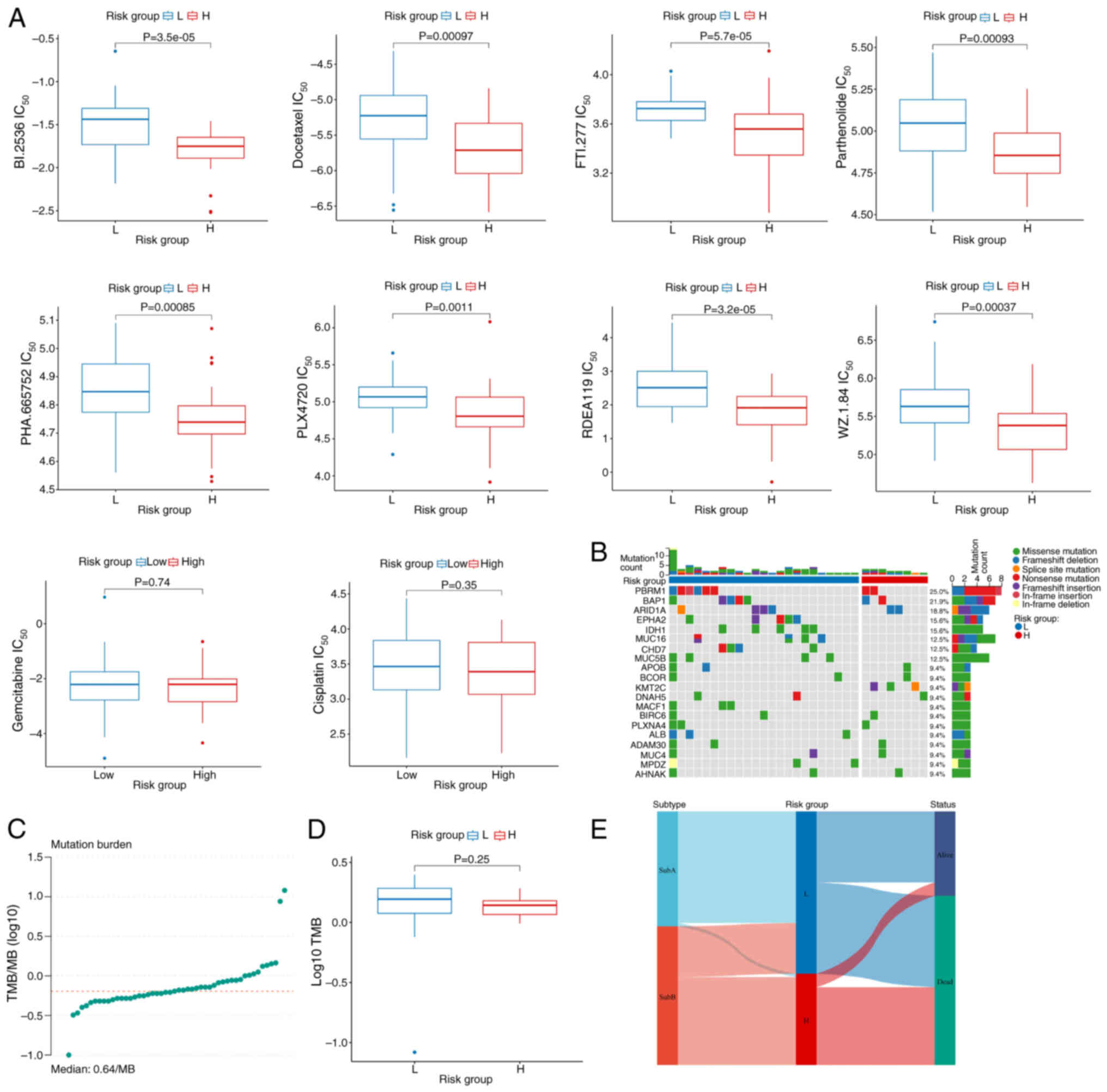
Chemotherapeutic drug sensitivity analysis
demonstrated that eight antineoplastic agents, PLX-4720, WZ-1-84,
refametinib, PHA-665752, parthenolide, FTI-277, docetaxel and
BI2536, exhibited significantly lower IC50 values in the
H-risk group compared with those in the L-risk group, suggesting
heightened chemosensitivity in H-risk patients with CCA. However,
no significant difference in efficacy was observed between risk
groups for the standard first-line regimen, gemcitabine plus
cisplatin (P<0.05; Fig. 7A;
Table SIX). Mutation analysis
identified PBRM1 as the most frequently altered gene among
the top 20 mutated genes (Fig. 7B).
TMB analysis using TCGA-CHOL data showed similar distributions
across risk groups, with no significant differences in TMB values
(Fig. 7C and D). Sankey diagram
analysis revealed a predominant clustering of H-risk cases within
the SubB disulfidptosis Sub (Fig.
7E), consistent with the association of the Sub with worse
clinical outcomes.
Establishment of CD109 and EFNB2
knockdown CCA cell model
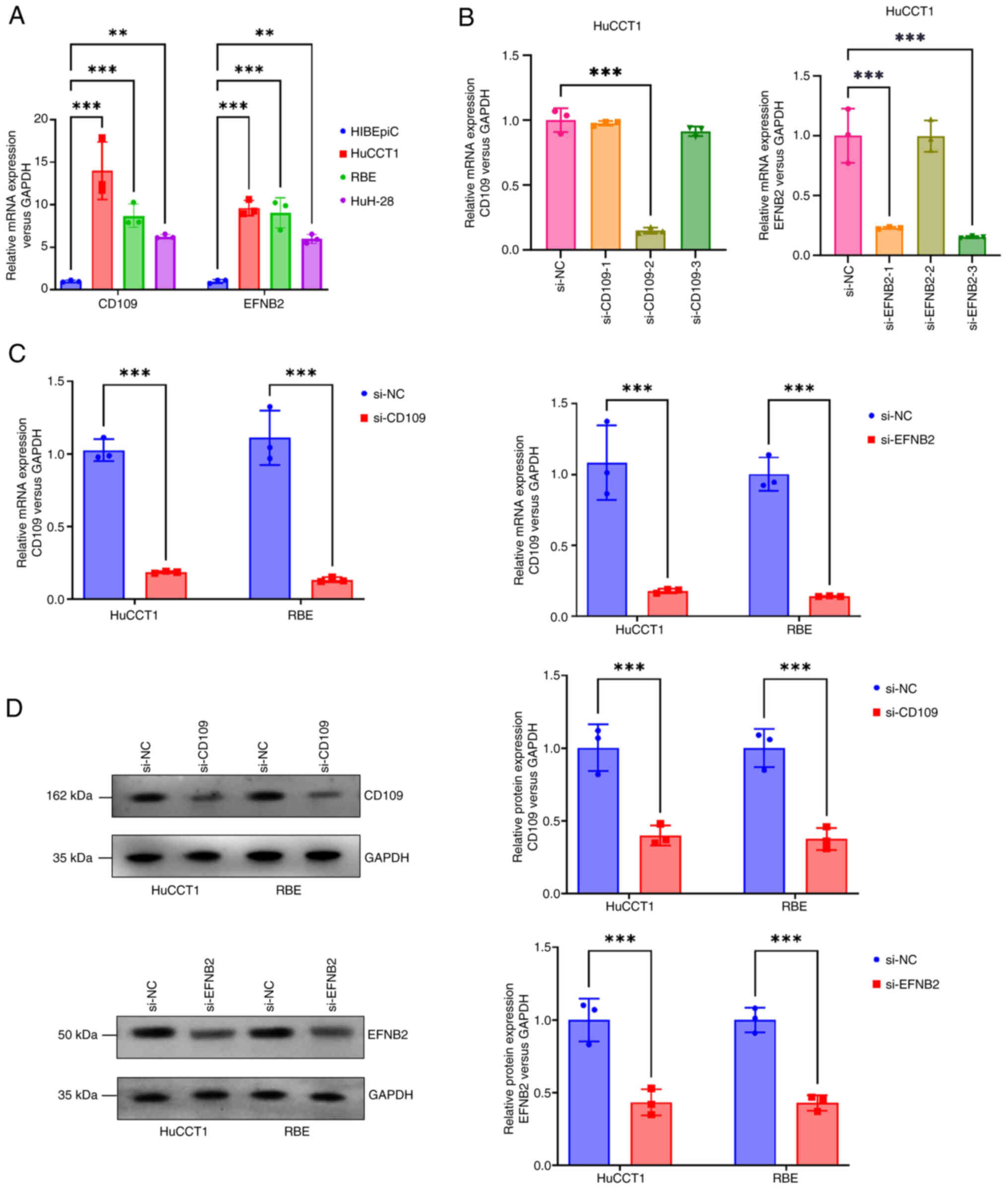
RT-qPCR was performed to assess mRNA expression of
CD109 and EFNB2 in three CCA cell lines (HuCCT1, RBE
and HuH-28) and in normal human intrahepatic biliary epithelial
cells (HiBEpiCs). Compared with HiBEpiCs, CD109 and
EFNB2 expression was significantly upregulated in HuCCT1 and
RBE cells (P<0.001), and moderately upregulated in HuH-28 cells
(P<0.01; Fig. 8A). Based on
these results, HuCCT1 and RBE cells were selected for
siRNA-mediated knockdown of CD109 and EFNB2. Three
siRNAs were designed for each gene, and their mRNA suppression
efficiency was evaluated by RT-qPCR. In HuCCT1 cells, si-CD109-2
and si-EFNB2-3 showed the most significant downregulation
(P<0.001; Fig. 8B), and were
selected for subsequent experiments. After transfecting si-CD109
and si-EFNB2 into HuCCT1 and RBE cells, RT-qPCR confirmed
significant reductions in CD109 and EFNB2 mRNA levels
(P<0.001; Fig. 8C), indicating
efficient transfection. WB further revealed significantly reduced
CD109 and EFNB2 protein levels in both cell lines (P<0.001;
Fig. 8D), consistent with mRNA
changes.
Functional assays for CD109 and EFNB2
knockdown in CCA cells
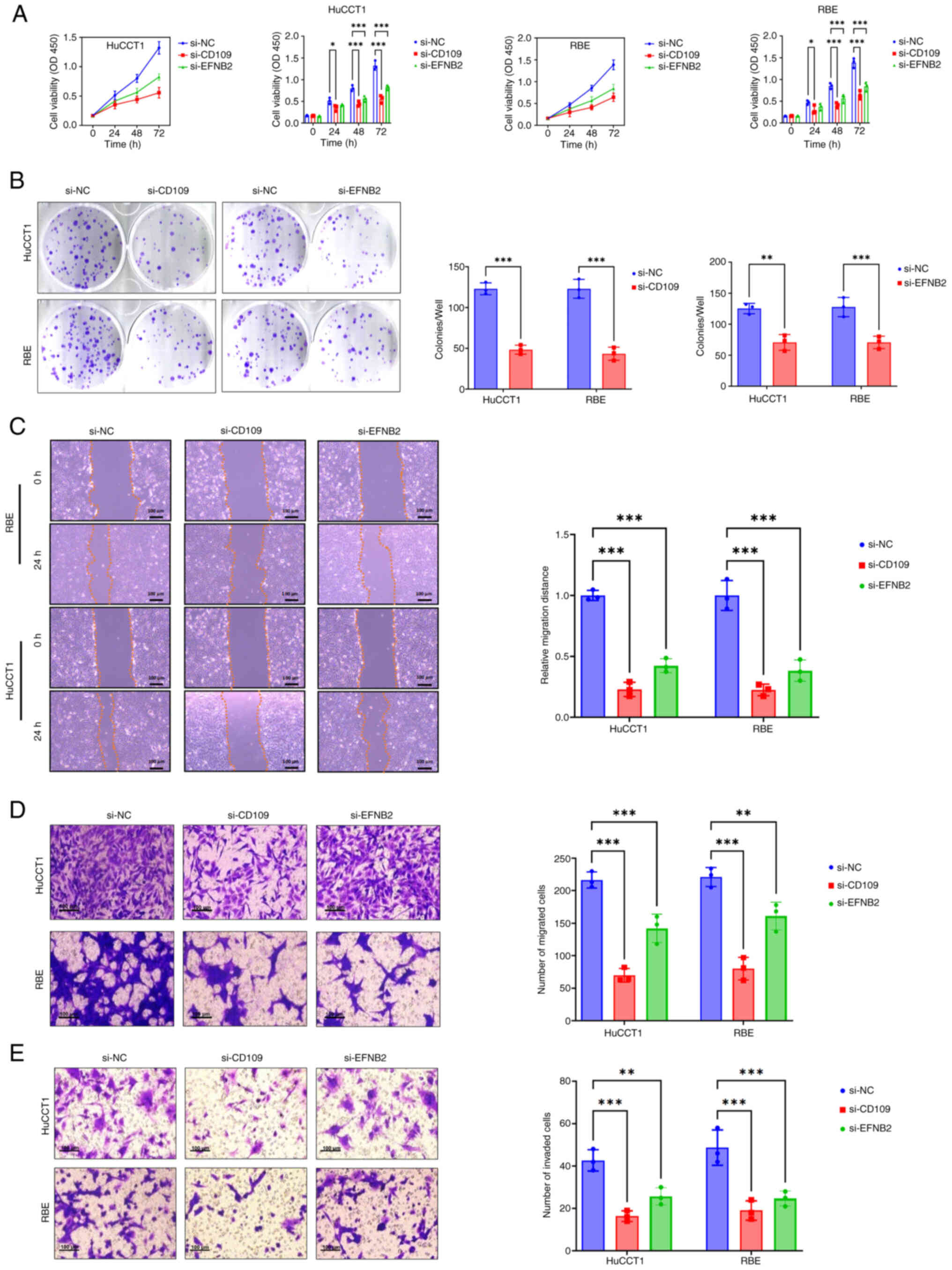
To evaluate the effects of CD109 and
EFNB2 knockdown on CCA cell behavior, cell proliferation was
assessed using the CCK-8 assay at 0, 24, 48 and 72 h in HuCCT1 and
RBE cells transfected with si-CD109, si-EFNB2 or si-NC (control).
Compared with the si-NC group, knockdown of CD109 or
EFNB2 significantly reduced cell proliferation, with
pronounced effects at 48 and 72 h (P<0.001); notably, si-CD109
exhibited stronger inhibition compared with si-EFNB2 (Fig. 9A). Clonogenic assays were performed
to assess colony formation. HuCCT1 and RBE cells transfected with
si-CD109 or si-EFNB2 formed significantly fewer colonies compared
with the si-NC group (P<0.001; Fig.
9B). Wound healing assays evaluated cell migration. Compared
with the si-NC group, the si-CD109 and si-EFNB2 groups exhibited
significantly reduced wound closure in HuCCT1 and RBE cells, with
si-CD109 showing greater inhibition (P<0.001; Fig. 9C). Transwell assays assessed
migration and invasion. Migration assays showed significantly fewer
migrating cells in si-CD109 and si-EFNB2 groups compared with si-NC
(P<0.001; Fig. 9D). Invasion
assays revealed similarly reduced invasive cell numbers in both
knockdown groups (P<0.001; Fig.
9E). These findings indicate that CD109 and EFNB2
knockdown significantly inhibits proliferation, colony formation,
migration and invasion in HuCCT1 and RBE cells, suggesting their
key roles in CCA progression.
Role of EFNB2 and CD109 in
disulfidptosis
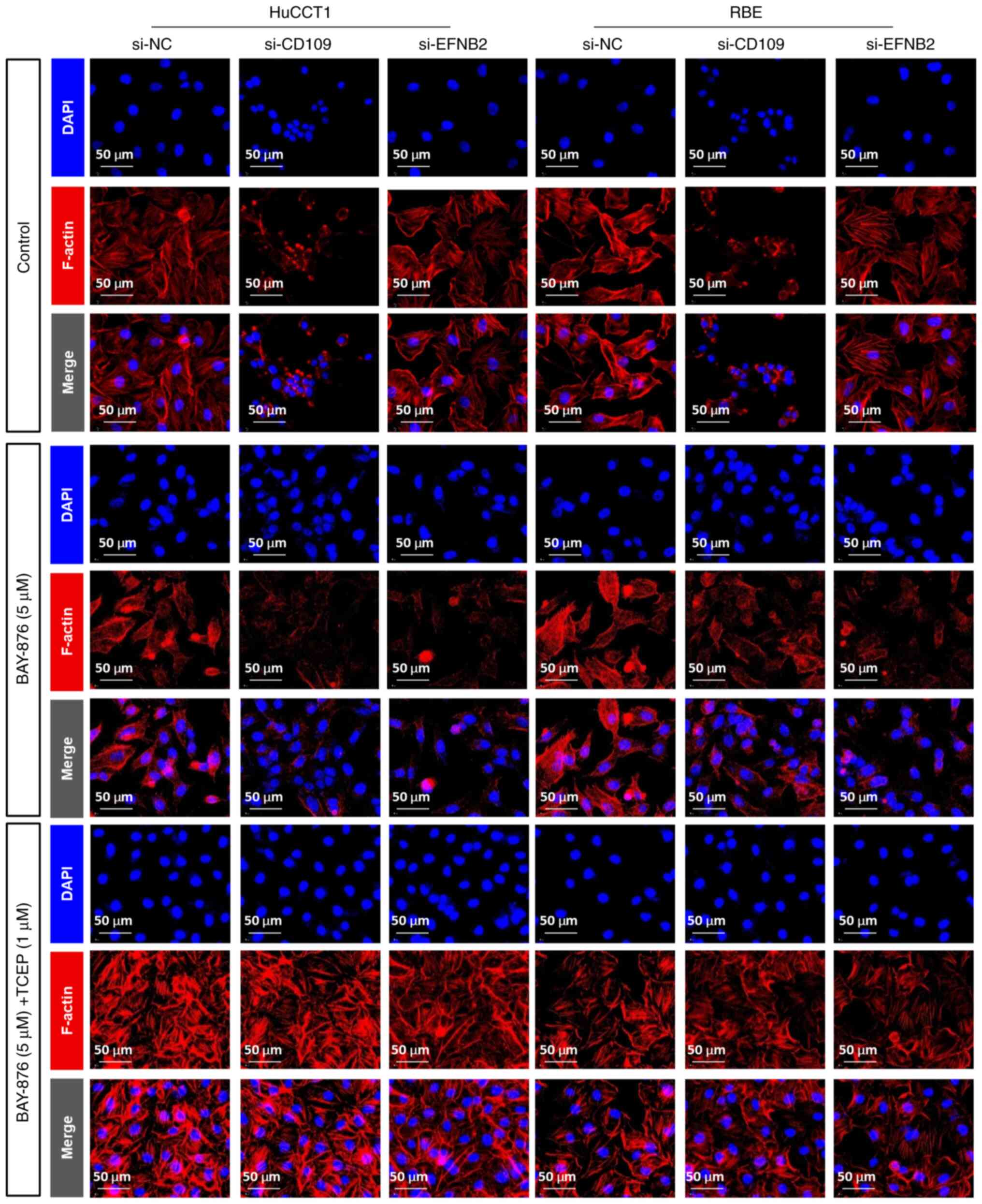
To investigate the role of EFNB2 and CD109 in
disulfidptosis, immunofluorescence staining was carried out. The
results (Fig. 10) showed that
under glucose deprivation conditions, knocking down EFNB2 or
CD109 in HuCCT1 and RBE cells induced cell shrinkage and
abnormal F-actin aggregation. Treatment with the GLUT1 inhibitor
BAY-876 exacerbated these morphological changes, with more
pronounced actin depolymerization observed in the si-CD109 group,
suggesting that CD109 may regulate cytoskeletal dynamics during
disulfide bond accumulation. Intervention with the reducing agent
TCEP largely alleviated F-actin aggregation in the si-CD109 group,
but only partially in the si-EFNB2 group, indicating that EFNB2 may
carry out a more key role in maintaining cytoskeletal stability.
These findings suggest that the knockout of EFNB2 and
CD109 enhances BAY-876-induced disulfidptosis, with TCEP
preventing these effects.
Discussion
In the present study, the first comprehensive
analysis of disulfidptosis in CCA, a malignancy characterized by
metabolic dysregulation and oxidative stress, was presented
(1). By integrating transcriptomic
profiling, molecular subtyping and prognostic modeling, its
potential as a novel therapeutic target was unveiled. Through
unsupervised clustering of 74 DRGs, two distinct molecular Subs
(SubA and SubB) with significant differences in prognosis and the
TME were identified. The more clinically aggressive SubB exhibited
heightened disulfidptosis pathway activity. This finding mirrors
observations in ferroptosis, where sublethal stress can foster
tumor adaptation. It is hypothesized that under metabolic stress
such as glucose deprivation CCA cells in the SubB adaptively
leverage components of the disulfidptosis machinery, potentially
through stress-activated pathways such as PI3K/AKT or MAPK/ERK
(34–39), could enhance invasive potential.
Leveraging these Sub distinctions, a robust
four-gene prognostic signature (CD109, EFNB2, KLK6 and
ADAMTS12) was developed and validated using LASSO Cox
regression. This model effectively stratified patients into H- and
L-risk groups, with the H-risk group showing significantly lower OS
in both the training and validation cohorts. The strong predictive
accuracy of the model for 1-, 2- and 3-year survival and its status
as an independent prognostic factor highlight its clinical
potential. The interplay between disulfidptosis and the TME is a
key, yet underexplored, determinant of CCA progression. The
findings of the present study reveal distinct immune profiles
between the molecular Subs, with the aggressive SubB harboring an
immunogenic yet profoundly immunosuppressive TME. CIBERSORT and
ssGSEA analyses demonstrated that SubB, despite enhanced
infiltration of effector immune cells such as CD8+ T
cells, also exhibited significant upregulation of immune checkpoint
genes (PD-L1, LAG3, CD47 and TNFRSF9) and HLA-related
genes. This ‘activated-exhausted’ phenotype where cytotoxic
lymphocytes co-express inhibitory checkpoint molecules, points
toward a compromised antitumor response and aligns with the worse
survival outcomes of the Sub (40–43).
GSEA provided a compelling mechanistic association
for this phenotype, revealing a significant enrichment of the TGF-β
signaling pathway in SubB. Mechanistically, it is hypothesized that
sustained TGF-β signaling orchestrates this immunosuppressive
landscape. While TGF-β can initially act as a chemoattractant for
effector immune cells such as CD8+ T cells, its chronic
presence drives extensive extracellular matrix (ECM) remodeling and
induces the expression of multiple immune checkpoints on both tumor
and stromal cells (44,45). Within this remodeled TME,
infiltrating CD8+ T cells rapidly transition from an
activated to an exhausted state, characterized by functional
impairment and high inhibitory receptor expression. This
‘activation-to-exhaustion’ shift effectively aborts antitumor
immunity and promotes malignant progression, suggesting that
targeting the TGF-β axis could be key to reversing
immunosuppression in these patients.
The prognostic signature of the present study is
anchored by four genes: CD109, EFNB2, KLK6 and
ADAMTS12. Among these, CD109 and EFNB2 possess
the highest risk coefficients and are intrinsically associated with
this TGF-β driven malignancy. CD109 functions as a negative
co-receptor for TGF-β, acting as a rheostat that suppresses
canonical tumor-suppressive signals while enhancing pro-tumorigenic
outputs such as EMT (46–48). EFNB2, through Ephrin receptor
signaling, activates downstream pathways to promote the dynamic
cytoskeletal remodeling essential for invasion and metastasis
(49–51). In vitro validation confirmed
their roles as key drivers of CCA malignancy. Silencing either
CD109 or EFNB2 markedly suppressed proliferation,
migration and invasion. Mechanistically, it was demonstrated that
these genes protect CCA cells from glucose starvation-induced
disulfidptosis, an effect that was reversed by the reducing agent
TCEP. Immunofluorescence analysis revealed that silencing these
genes led to F-actin cytoskeletal collapse under metabolic stress.
It is therefore hypothesized that CD109 and EFNB2 confer resistance
to disulfidptosis by preserving cytoskeletal integrity through
redox modulation, possibly by enhancing NADPH regeneration or
regulating SLC7A11-dependent cystine metabolism to maintain the
glutathione pool (52).
Concurrently, they might restrain the excessive actin network
polymerization seen in disulfidptosis by influencing key regulatory
nodes, such as the Rac1-WAVE pathway that governs actin dynamics.
Future investigations employing metabolomics, live-cell redox
imaging and non-reducing protein electrophoresis will be essential
to systematically assess the way CD109 or EFNB2 depletion alters
intracellular NADPH pools, thioredoxin system activity and actin
disulfide cross-linking.
Beyond the experimentally validated markers, the
prognostic signature of the present study incorporates KLK6
and ADAMTS12. The inclusion of these genes is justified by
robust bioinformatic and literature-based evidence. Both genes are
significantly upregulated in CCA tumors and are associated with
worse survival, aligning with their positive risk coefficients from
LASSO regression. KLK6, a serine protease, exerts dual roles in
inflammation and tumor progression (53,54).
In pancreatic ductal adenocarcinoma (PDAC), KLK6 promotes
invasiveness, indicating potential therapeutic relevance for CCA
(54). ADAMTS12, a
metalloproteinase, modulates ECM remodeling in inflammation,
fibrosis and cancer. In PDAC, ADAMTS12 enhances cell migration,
while in hepatocellular carcinoma, it is associated with poor
differentiation and recurrence (55,56).
By collectively shaping a microenvironment conducive to tumor
invasion and metastasis, these two genes serve as rational and key
components of the H-risk prognostic model of the present study.
Elucidating their direct functional roles in the disulfidptosis
pathway remains an important direction for future
investigation.
Despite the novel insights provided by the present
study, several limitations should be acknowledged. First, the
reliance on public databases, particularly TCGA-CHOL cohort with
its limited sample size (n=35), may constrain the statistical power
and generalizability of the prognostic model and limits its utility
for more detailed investigations, such as Sub analyses. A key
limitation is that the TMB analysis was confined to TCGA cohort, as
comprehensive mutation data were unavailable in other datasets,
thereby precluding independent validation of these findings.
Furthermore, the prognostic model is derived from bioinformatic
approaches and awaits validation with prospective clinical data.
Therefore, large-scale, multi-center prospective studies are
essential to confirm its clinical robustness. Second, while the
functional roles of CD109 and EFNB2 were established,
the contributions of the other two signature genes KLK6 and
ADAMTS12 to disulfidptosis and CCA progression remain to be
experimentally elucidated, which is a direction for future
research. Third, the drug sensitivity analysis was based solely on
computational predictions, which may not fully capture the
complexity of the TME. Experimental validation in clinically
relevant models such as patient-derived organoids or xenografts is
necessary to assess the therapeutic potential of the predicted
agents. Finally, regarding clinical translation, although measuring
the expression of signature genes such as CD109 and
EFNB2 in patient samples for example via
immunohistochemistry on FFPE tissues is technically feasible,
establishing standardized and reproducible protocols is a key
prerequisite for future clinical application.
In conclusion, the DRG risk model developed in the
present study provides a valuable tool for survival prediction in
patients with CCA, reinforcing the central role of disulfidptosis
in CCA pathogenesis. Specifically, CD109 and EFNB2,
by collaboratively regulating cytoskeletal stability, metabolic
reprogramming and immune suppression, hold promise as potential
therapeutic targets for reversing tumor progression and overcoming
treatment resistance. This not only offers important insights for
personalized treatment strategies, but also lays the theoretical
foundation for the development of disulfidptosis-based precision
therapies in CCA.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was financially supported by the Changzhou Sci
& Tech Program (grant no. CJ20220064, CJ20243007), the 2023
Changzhou Health Commission Science and Technology Project (grant
no. QY202301), Youth Talent Science and Technology Project of
Changzhou Health Commission (grant no. QN202434), Science and
Technology Project of Changzhou Health Commission (grant no.
ZD202428) and Top Talent of Changzhou ‘The 14th Five-Year Plan’
High-level Health Personnel Training Project (grant no.
2022260).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YC, JWu, DZha and WH contributed to the conception
and design of the study. YC, WH, DZhu, LJ and JWa were involved in
the acquisition, analysis and interpretation of the data. YC and WH
drafted the manuscript. DZhu, LJ and JWa critically revised the
manuscript for important intellectual content. JWu, DZha and WH
supervised the study. YC and WH confirm the authenticity of all the
raw data. All authors read and approved the final manuscript and
agreed to take responsibility for all aspects of the work.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CCA
|
cholangiocarcinoma
|
|
EMBL-EBI
|
European Molecular Biology
Laboratory-European Bioinformatics Institute
|
|
TCGA
|
The Cancer Genome Atlas
|
|
DRGs
|
Disulfidptosis-related genes
|
|
KM
|
Kaplan-Meier
|
|
GSVA
|
gene set enrichment analysis
|
|
DEGs
|
differentially expressed genes
|
|
ssGSEA
|
single-sample gene set enrichment
analysis
|
|
GSEA
|
gene set enrichment analysis
|
|
LASSO
|
least absolute shrinkage and
selection operator
|
|
OS
|
overall survival
|
|
ROC
|
receiver operating characteristic
|
|
DCA
|
clinical decision curves
|
|
EFNB2
|
ephrin-B2
|
|
TME
|
tumor microenvironment
|
|
TMB
|
tumor mutational burden
|
|
ADAMTS12
|
A disintegrin and metalloproteinase
with thrombospondin motifs 12
|
|
KLK6
|
Kallikrein-related peptidase 6
|
|
RCD
|
regulated cell death
|
|
ESTIMATE
|
Estimation of Stromal and Immune
cells in Malignant Tumor tissues using Expression
|
|
CIBERSORT
|
Cell-type Identification by
Estimating Relative Subsets of RNA Transcripts
|
|
RT-qPCR
|
reverse transcription-quantitative
PCR
|
|
WB
|
western blot analysis
|
|
CCK-8
|
Cell Counting Kit-8
|
|
IF
|
immunofluorescence
|
|
EMT
|
epithelial-mesenchymal transition
|
References
|
1
|
Brindley PJ, Bachini M, Ilyas SI, Khan SA,
Loukas A, Sirica AE, Teh BT, Wongkham S and Gores GJ:
Cholangiocarcinoma. Nat Rev Dis Primers. 7:652021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cardinale V: Classifications and
misclassification in cholangiocarcinoma. Liver Int. 39:260–212.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Banales JM, Cardinale V, Carpino G,
Marzioni M, Andersen JB, Invernizzi P, Lind GE, Folseraas T, Forbes
SJ, Fouassier L, et al: Expert consensus document:
Cholangiocarcinoma: Current knowledge and future perspectives
consensus statement from the European network for the study of
cholangiocarcinoma (ENS-CCA). Nat Rev Gastroenterol Hepatol.
13:261–280. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Clements O, Eliahoo J, Kim JU,
Taylor-Robinson SD and Khan SA: Risk factors for intrahepatic and
extrahepatic cholangiocarcinoma: A systematic review and
meta-analysis. J Hepatol. 72:95–103. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sirica AE, Gores GJ, Groopman JD, Selaru
FM, Strazzabosco M, Wei Wang X and Zhu AX: Intrahepatic
cholangiocarcinoma: Continuing challenges and translational
advances. Hepatology. 69:1803–1815. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fabris L, Perugorria MJ, Mertens J,
Björkström NK, Cramer T, Lleo A, Solinas A, Sänger H, Lukacs-Kornek
V, Moncsek A, et al: The tumour microenvironment and immune milieu
of cholangiocarcinoma. Liver Int. 39 (Suppl 1):S63–S78. 2019.
View Article : Google Scholar
|
|
7
|
Arrivé L and Djelouah M: Refining
prognosis in intrahepatic cholangiocarcinoma: The expanding role of
imaging. Radiol Imaging Cancer. 7:e2503832025. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
PDQ Adult Treatment Editorial Board. Bile
duct cancer (cholangiocarcinoma) treatment (PDQ®), . Health
Professional Version. PDQ Cancer Information Summaries. National
Cancer Institute; Bethesda, MD: 2002
|
|
9
|
Lockie EB, Sylivris A, Pandanaboyana S,
Zalcberg J, Skandarajah A and Loveday BP: Relationship between
pancreatic cancer resection rate and survival at population level:
Systematic review. BJS Open. 9:zraf0072025. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Valle JW, Lamarca A, Goyal L, Barriuso J
and Zhu AX: New horizons for precision medicine in biliary tract
cancers. Cancer Discov. 7:943–962. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Banales JM, Marin JJG, Lamarca A,
Rodrigues PM, Khan SA, Roberts LR, Cardinale V, Carpino G, Andersen
JB, Braconi C, et al: Cholangiocarcinoma 2020: The next horizon in
mechanisms and management. Nat Rev Gastroenterol Hepatol.
17:557–588. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kerr JF, Wyllie AH and Currie AR:
Apoptosis: A basic biological phenomenon with wide-ranging
implications in tissue kinetics. Br J Cancer. 26:239–257. 1972.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tang D, Kang R, Berghe TV, Vandenabeele P
and Kroemer G: The molecular machinery of regulated cell death.
Cell Res. 29:347–364. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shi Y, Wang Y, Niu K, Zhang W, Lv Q and
Zhang Y: How CLSPN could demystify its prognostic value and
potential molecular mechanism for hepatocellular carcinoma: A
crosstalk study. Comput Biol Med. 172:1082602024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shi Y, Wang Y, Niu K and Zhang Y: A
commentary on ‘A bibliometric analysis of gastric cancer liver
metastases: Advances in mechanisms of occurrence and treatment
options’. Int J Surg. 110:5897–5898. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu X, Nie L, Zhang Y, Yan Y, Wang C,
Colic M, Olszewski K, Horbath A, Chen X, Lei G, et al: Actin
cytoskeleton vulnerability to disulfide stress mediates
disulfidptosis. Nat Cell Biol. 25:404–414. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu X, Zhuang L and Gan B: Disulfidptosis:
Disulfide stress-induced cell death. Trends Cell Biol. 34:327–337.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zheng T, Liu Q, Xing F, Zeng C and Wang W:
Disulfidptosis: A new form of programmed cell death. J Exp Clin
Cancer Res. 42:1372023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tang J, Peng X, Xiao D, Liu S, Tao Y and
Shu L: Disulfidptosis-related signature predicts prognosis and
characterizes the immune microenvironment in hepatocellular
carcinoma. Cancer Cell Int. 24:192024. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang HB, Pan JY and Zhu T: A
disulfidptosis-related lncRNA prognostic model to predict survival
and response to immunotherapy in lung adenocarcinoma. Front
Pharmacol. 14:12541192023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang J, Zhang J, Zhang F, Lu S, Guo S,
Shi R, Zhai Y, Gao Y, Tao X, Jin Z, et al: Identification of a
disulfidptosis-related genes signature for prognostic implication
in lung adenocarcinoma. Comput Biol Med. 165:107402023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen Y, Xue W, Zhang Y, Gao Y and Wang Y:
A novel disulfidptosis-related immune checkpoint genes signature:
Forecasting the prognosis of hepatocellular carcinoma. J Cancer Res
Clin Oncol. 149:12843–12854. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dong X, Liao P, Liu X, Yang Z, Wang Y,
Zhong W and Wang B: Construction and validation of a reliable
disulfidptosis-related LncRNAs signature of the subtype,
prognostic, and immune landscape in colon cancer. Int J Mol Sci.
24:129152023. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kang K, Li X, Peng Y and Zhou Y:
Comprehensive analysis of disulfidptosis-related LncRNAs in
molecular classification, immune microenvironment characterization
and prognosis of gastric cancer. Biomedicines. 11:31652023.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Feng Z, Zhao Q, Ding Y, Xu Y, Sun X, Chen
Q, Zhang Y, Miao J and Zhu J: Identification a unique
disulfidptosis classification regarding prognosis and immune
landscapes in thyroid carcinoma and providing therapeutic
strategies. J Cancer Res Clin Oncol. 149:11157–11170. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Qi C, Ma J, Sun J, Wu X and Ding J: The
role of molecular subtypes and immune infiltration characteristics
based on disulfidptosis-associated genes in lung adenocarcinoma.
Aging (Albany NY). 15:5075–5095. 2023.PubMed/NCBI
|
|
27
|
Raggi C, Taddei ML, Rae C, Braconi C and
Marra F: Metabolic reprogramming in cholangiocarcinoma. J Hepatol.
77:849–864. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Duwe L, Fouassier L, Lafuente-Barquero J
and Andersen JB: Unraveling the actin cytoskeleton in the malignant
transformation of cholangiocyte biology. Transl Oncol.
26:1015312022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhao X, Zhang M, He J, Li X and Zhuang X:
Emerging insights into ferroptosis in cholangiocarcinoma (review).
Oncol Lett. 28:6062024. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
R Core Team, . R: A language and
environment for statistical computing. R Foundation for Statistical
Computing; Vienna: 2024, URL. https://www.R–project.org/
|
|
31
|
Mayakonda A, Lin DC, Assenov Y, Plass C
and Koeffler HP: Maftools: Efficient and comprehensive analysis of
somatic variants in cancer. Genome Res. 28:1747–1756. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wilkerson MD and Hayes DN:
ConsensusClusterPlus: A class discovery tool with confidence
assessments and item tracking. Bioinformatics. 26:1572–1573. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang ZJ, Huang YP, Li XX, Liu ZT, Liu K,
Deng XF, Xiong L, Zou H and Wen Y: A novel ferroptosis-related
4-gene prognostic signature for cholangiocarcinoma and photodynamic
therapy. Front Oncol. 11:7474452021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Amontailak S, Titapun A, Jusakul A, Thanan
R, Kimawaha P, Jamnongkan W, Thanee M, Sirithawat P and Techasen A:
Prognostic values of ferroptosis-related proteins ACSL4, SLC7A11,
and CHAC1 in cholangiocarcinoma. Biomedicines. 12:20912024.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wan S, Liang C, Wu C, Wang S, Wang J, Xu
L, Zhang X, Hou Y, Xia Y, Xu L and Huang X: Disulfidptosis in tumor
progression. Cell Death Discov. 11:2052025. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hu F and Lito P: Insights into how
adeno-squamous transition drives KRAS inhibitor resistance. Cancer
Cell. 42:330–332. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mi T, Kong X, Chen M, Guo P and He D:
Inducing disulfidptosis in tumors: Potential pathways and
significance. MedComm (2020). 5:e7912024. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xiao Y, Li ZZ, Zhong NN, Cao LM, Liu B and
Bu LL: Charting new frontiers: Co-inhibitory immune checkpoint
proteins in therapeutics, biomarkers, and drug delivery systems in
cancer care. Transl Oncol. 38:1017942023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cruz D, Rodríguez-Romanos R,
González-Bartulos M, García-Cadenas I, de la Cámara R, Heras I,
Buño I, Santos N, Lloveras N, Velarde P, et al: LAG3 genotype of
the donor and clinical outcome after allogeneic transplantation
from HLA-identical sibling donors. Front Immunol. 14:10663932023.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Perea F, Sánchez-Palencia A, Gómez-Morales
M, Bernal M, Concha Á, García MM, González-Ramírez AR, Kerick M,
Martin J, Garrido F, et al: HLA class I loss and PD-L1 expression
in lung cancer: Impact on T-cell infiltration and immune escape.
Oncotarget. 9:4120–4133. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Saigí M, Mate JL, Carcereny E,
Martínez-Cardús A, Esteve A, Andreo F, Centeno C, Cucurull M, Mesia
R, Pros E and Sanchez-Cespedes M: HLA-I levels correlate with
survival outcomes in response to immune checkpoint inhibitors in
non-small cell lung cancer. Lung Cancer. 189:1075022024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Itatani Y, Kawada K and Sakai Y:
Transforming growth Factor-β signaling pathway in colorectal cancer
and its tumor microenvironment. Int J Mol Sci. 20:58222019.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hao Y, Baker D and Ten Dijke P:
TGF-β-mediated epithelial-mesenchymal transition and cancer
metastasis. Int J Mol Sci. 20:27672019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Naoi H, Suzuki Y, Miyagi A, Horiguchi R,
Aono Y, Inoue Y, Yasui H, Hozumi H, Karayama M, Furuhashi K, et al:
CD109 attenuates bleomycin-induced pulmonary fibrosis by inhibiting
TGF-β signaling. J Immunol. 212:1221–1231. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Taki T, Shiraki Y, Enomoto A, Weng L, Chen
C, Asai N, Murakumo Y, Yokoi K, Takahashi M and Mii S: CD109
regulates in vivo tumor invasion in lung adenocarcinoma through
TGF-β signaling. Cancer Sci. 111:4616–4628. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Litvinov IV, Bizet AA, Binamer Y, Jones
DA, Sasseville D and Philip A: CD109 release from the cell surface
in human keratinocytes regulates TGF-β receptor expression, TGF-β
signalling and STAT3 activation: Relevance to psoriasis. Exp
Dermatol. 20:627–632. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zeng X, Hunt A, Jin SC, Duran D, Gaillard
J and Kahle KT: EphrinB2-EphB4-RASA1 signaling in human
cerebrovascular development and disease. Trends Mol Med.
25:265–286. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhu F, Dai SN, Xu DL, Hou CQ, Liu TT, Chen
QY, Wu JL and Miao Y: EFNB2 facilitates cell proliferation,
migration, and invasion in pancreatic ductal adenocarcinoma via the
p53/p21 pathway and EMT. Biomed Pharmacother. 125:1099722020.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Xu C, Gu L, Kuerbanjiang M, Jiang C, Hu L,
Liu Y, Xue H, Li J, Zhang Z and Xu Q: Adaptive activation of
EFNB2/EPHB4 axis promotes post-metastatic growth of colorectal
cancer liver metastases by LDLR-mediated cholesterol uptake.
Oncogene. 42:99–112. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Koppula P, Zhang Y, Zhuang L and Gan B:
Amino acid transporter SLC7A11/xCT at the crossroads of regulating
redox homeostasis and nutrient dependency of cancer. Cancer Commun
(Lond). 38:122018.PubMed/NCBI
|
|
53
|
Hwang YS, Cho HJ, Park ES, Lim J, Yoon HR,
Kim JT, Yoon SR, Jung H, Choe YK, Kim YH, et al: KLK6/PAR1 axis
promotes tumor growth and metastasis by regulating cross-talk
between tumor cells and macrophages. Cells. 11:41012022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang L, Lovell S, De Vita E, Jagtap PKA,
Lucy D, Goya Grocin A, Kjær S, Borg A, Hennig J, Miller AK and Tate
EW: A KLK6 activity-based probe reveals a role for KLK6 activity in
pancreatic cancer cell invasion. J Am Chem Soc. 144:22493–22504.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
He RZ, Zheng JH, Yao HF, Xu DP, Yang MW,
Liu DJ, Sun YW and Huo YM: ADAMTS12 promotes migration and
epithelial-mesenchymal transition and predicts poor prognosis for
pancreatic cancer. Hepatobiliary Pancreat Dis Int. 22:169–178.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Dekky B, Azar F, Bonnier D, Monseur C,
Kalebić C, Arpigny E, Colige A, Legagneux V and Théret N: ADAMTS12
is a stromal modulator in chronic liver disease. FASEB J.
37:e232372023. View Article : Google Scholar : PubMed/NCBI
|