Introduction
The major cellular components and mediators of the
tumor microenvironment (TME), including cancer cells, immune cells
[such as T cells, B cells, dendritic cells (DCs), myeloid-derived
suppressor cells (MDSCs) and tumor-associated macrophages (TAMs)],
stromal cells [cancer-associated fibroblasts (CAFs) and
tumor-associated endothelial cells], cytokines and chemokines,
tumor vasculature, lymphoid tissue, as well as adipocytes and
exosomes, serve a critical role in cancer initiation, progression,
spread and metastasis (1–3). Interactions between the tumor stroma,
especially those mediated by CAFs and other stromal components,
actively promote immune evasion and malignant growth in a variety
of solid tumors, including lung and breast cancer as well as other
epithelial malignancies (2).
Tumor-infiltrating lymphocytes are a crucial part of antitumor
immunity among the immune cells found in tumors and a recent study
has demonstrated that immunotherapies can alter TIL activity as
well as the interactions between immune and stromal cells in the
TME. The TME usually contains signals that inhibit the immune
response, such as regulatory T cells (Tregs) and myeloid-derived
suppressor cells, which prevent the infiltration and killing
function of immune cells (3).
Breaking through this immunosuppression has long been the key to
immunotherapy.
In non-small cell lung cancer (NSCLC) with epidermal
growth factor receptor (EGFR) mutations, the TME manifests as an
immunosuppressive phenotype. Compared with EGFR wild-type (wt)
cancer, these tumors usually have a much lower tumor mutational
burden, which results in a limited development of neoantigens that
can successfully trigger immune recognition. Insufficient
neoantigen presentation weakens T-cell priming and activation,
resulting in a relatively ‘immune-cold’ environment. The blunting
of antitumor immune activation under such conditions is primarily
responsible for the lower therapeutic benefit observed with immune
checkpoint inhibitors (ICIs) in this population (4,5).
However, the TME in NSCLC can be affected by chemotherapy,
radiotherapy or EGFR-tyrosine kinase inhibitors (TKIs). Short-term
TKI treatment excels in tumor clearance and immunological
upregulation, improving the overall survival and quality of life of
patients (6). Nevertheless, the
immunosuppressive TME gradually emerges with time, and EGFR-TKI
resistance is unavoidable (7–9). This
process reveals the dynamic evolution in tumor immune
microenvironment induced by EGFR-TKI treatment (10).
Mechanisms of resistance to EGFR-TKIs
In Asian populations, over half of patients with
NSCLC had EGFR-activating mutations (11). For individuals with locally
progressed or metastatic NSCLC, EGFR-TKIs are now the standard
therapy regimen (12,13). Their principal mechanism is
competitive binding to the ATP-binding site inside the EGFR kinase
domain, which inhibits kinase autophosphorylation and downstream
signaling cascades. This effect inhibits tumor cell development,
proliferation and metastasis (14).
With improvements in medical research, numerous EGFR-TKIs from the
first to third generations are now clinically available, markedly
improving patient survival rates. However, despite initial success,
nearly all patients develop acquired resistance to EGFR-TKI
therapy, with a median progression-free survival (mPFS) of ~1 year
(15,16). Resistance to EGFR-TKIs can be
divided into two categories: Primary and acquired (16). Of patients with EGFR mutations, ~30%
demonstrate primary resistance at the start of initial treatment,
indicating no objective response to TKI therapy; however, the
mechanisms causing this resistance remain unknown. However,
acquired resistance refers to disease progression that occurs
following an initial response to treatment. Its causes are
complicated and diverse, consisting mostly of EGFR-dependent
resistance, non-EGFR-dependent resistance (induced by activation of
EGFR bypass or downstream signaling pathways) and histological or
phenotypic alteration (17).
EGFR-dependent drug resistance
The T790M mutation accounts for 50–60% of acquired
resistance in patients treated with first- and second-generation
EGFR-TKIs (18). This mutation
replaces a bulky methionine (M) with a threonine (T) at position
790 in exon 20 of the EGFR gene. This substitution creates steric
hindrance between the aniline moiety of EGFR-TKIs and the
drug-binding site within the ATP pocket of EGFR, thereby weakening
drug-binding affinity. Additional mechanisms include a marked
increase in ATP binding affinity for EGFR T790M, alterations in the
catalytic domain and changes in overall conformational dynamics,
collectively contributing to acquired resistance to TKIs (18,19).
With the widespread usage of the third-generation
EGFR-TKI osimertinib, the C797S mutation has emerged as the
predominant mode of resistance. The EGFR C797S mutation, located at
position 797 in exon 20 of the EGFR gene, is a missense mutation in
which serine replaces cysteine. It accounts for 10–26% of
second-line osimertinib resistance cases and 7% of first-line
resistance cases (20). This
mutation damages the ATP-binding pocket, preventing
third-generation TKIs from making covalent connections with the
ATP-binding domain and so losing their inhibitory function
(21). The C797S mutation commonly
coexists with EGFR T790M in two structural forms: Cis (EGFR T790M
and C797S occur on the same allele) and trans (EGFR T790M and C797S
occur on different alleles). In ~85% of instances, the EGFR
C797S/T790M mutation is in the cis configuration, with ~10% of
patients having the C797S/T790M trans configuration. Whether the
C797S and T790M mutations form a cis structure has important
biological implications since it influences the therapeutic
efficacy of subsequent TKIs (17,22).
In addition to the most prevalent C797S mutation, C797G is another
missense mutation at the same location that causes drug resistance
by affecting the binding of the drug to the residue.
Aside from the classic T790M and C797S mutations,
uncommon mutations at other places within the EGFR kinase domain
can also cause resistance to osimertinib, mostly through
interference with drug-kinase binding. Mutations at the L718 site
(such as L718Q and L718V) and the adjacent G719A mutation primarily
interfere spatially, preventing the critical alanine ring in the
osimertinib molecule from forming a stable bond with its binding
pocket. L792 mutations (most commonly and notably L792H) directly
disrupt the tight binding of osimertinib to the kinase domain.
Mutations at the G796 site sterically clash with the solvent-front
aromatic ring of osimertinib, preventing kinase domain binding. The
G724S mutation is located in the ATP-binding region and may
interfere with osimertinib binding to its target by a variety of
mechanisms, including generating protein structural changes,
increasing ATP affinity or maintaining the kinase activation state,
resulting in drug resistance. Another form of resistance is target
upregulation caused by EGFR gene amplification (17,20,23).
EGFR-independent drug resistance
Activation of bypasses and downstream pathways, such
as HER2, HER3, MET, KRAS, NRAS proto-oncogene, GTPase (NRAS), BRAF,
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α
(PIK3CA), AXL receptor tyrosine kinase (AXL) and insulin-like
growth factor-1 receptor (IGF-1R), were often classified as
‘bypass’ mechanisms of resistance. These pathways enable tumor
cells to activate alternative routes that engage key EGFR effectors
essential for tumor cell growth and survival. The most common
bypass mechanism of resistance to first- and second-generation EGFR
TKIs is HER2 amplification (24,25).
Downstream signaling pathways activated following receptor
phosphorylation included the mitogen-activated protein kinase
pathway, phosphatidylinositol 3-kinase (PI3K)/Akt pathway,
phospholipase Cγ1, protein kinase C and various transcriptional
regulators that modulate gene expression (26).
The MET gene encoded a receptor tyrosine kinase
known as c-Met, or hepatocyte growth factor receptor, which
supports cancer cell growth and survival by activating the
HER3-PI3K/AKT and RAS/RAF/MEK/ERK signaling pathways, thereby
bypassing the inhibitory effects of EGFR receptor signaling
(18,27). The Ras-Raf-MEK-ERK pathway is
crucial for regulating cell cycle progression and proliferation.
KRAS and NRAS, both members of the Ras family, could activate MEK
and ERK through this signaling pathway, contributing to drug
resistance (28,29). BRAF mutations, although rare in
EGFR-mutant (mt) NSCLC, involve kinases located downstream of RAS
in the Ras-Raf-MEK-ERK pathway and may contribute to the activation
of the GFR/RAS/RAF signaling pathway (30). PIK3CA, a catalytic subunit of the
PI3K family of lipid kinases, serves an oncogenic role in lung
adenocarcinoma by activating the PI3K/AKT/mTOR pathway. This
signaling pathway regulates key cellular functions, including
growth, proliferation, metabolism, angiogenesis and metastasis, and
is frequently mutated or hyperactivated in various cancers.
Patients with PIK3CA mutations generally exhibit a worse prognosis
and shorter median survival compared with those without these
mutations (31,32). IGF-1R and AXL serve key roles in
regulating cell growth, differentiation, apoptosis, transformation
and other critical physiological processes through the downstream
PI3K/AKT signaling pathway, thereby contributing to the development
of secondary drug resistance (33,34).
Histology and phenotypic
conversion
Another mechanism of drug resistance involves
histological transformation within the tumor itself. Specifically,
some EGFR-mt lung adenocarcinomas develop into small cell lung
cancer (SCLC), accounting for 3–14% of acquired resistance cases.
This transformation is confirmed by histopathological biopsy, and
transformed tumors typically respond to standard SCLC treatment
regimens (35–38). Research indicates that the
co-deletion of Rb1 and TP53 genes constitutes the key molecular
basis driving such transformation (16,39).
Tumor cells can also undergo epithelial-mesenchymal
transition (EMT) and develop treatment resistance. During this
process, epithelial markers are downregulated and mesenchymal
markers are upregulated, resulting in increased proliferation,
invasion, migration and metastatic potential. However, the exact
processes underpinning EMT are unknown; this process may be driven
by AXL through activation of the PI3K/AKT signaling pathway
(40,41) (Fig.
1).
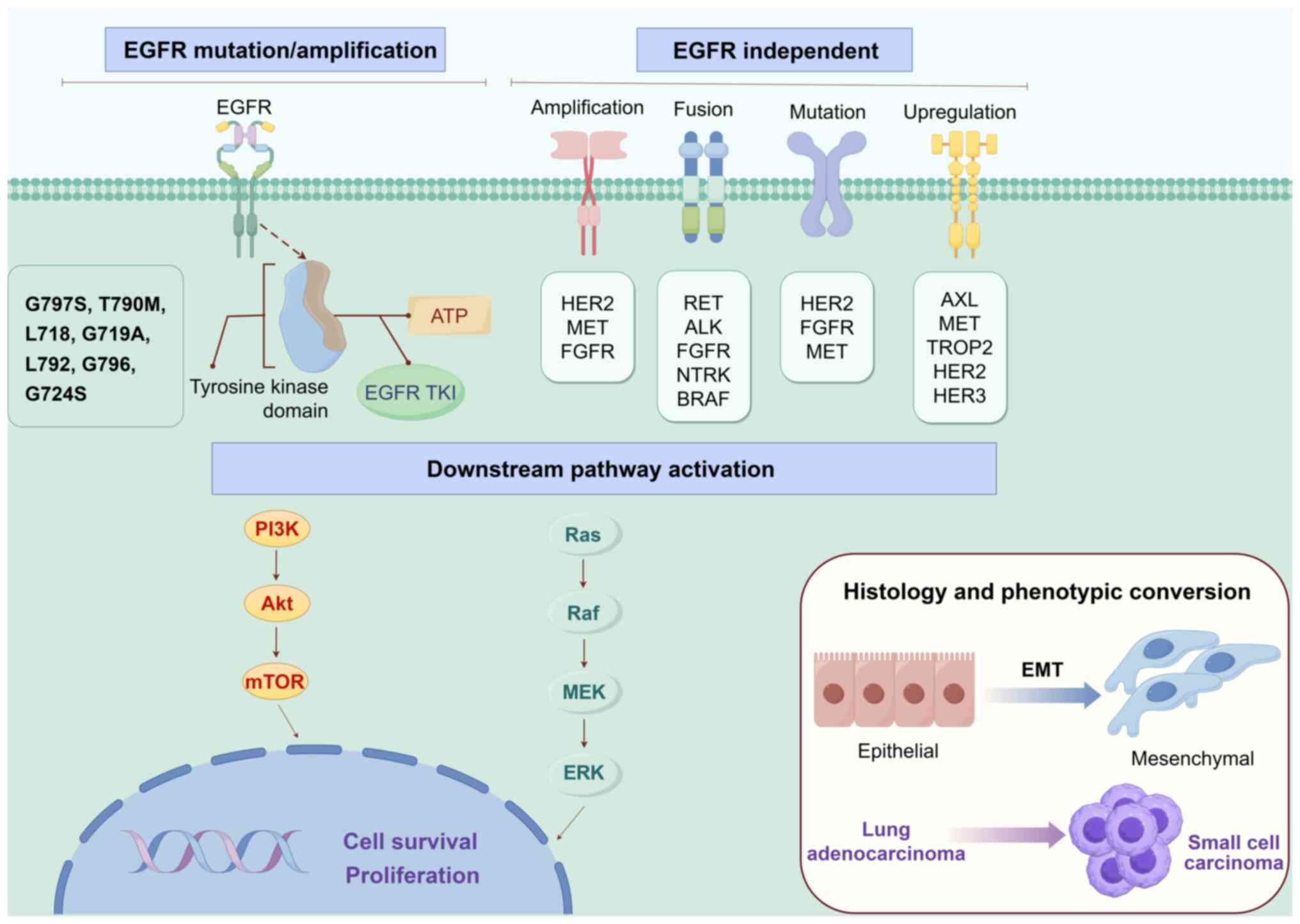 | Figure 1.The illustration outlines the key
mechanisms of resistance to EGFR-TKIs in EGFR-mutant NSCLC.
EGFR-dependent resistance arises from evolution of the target
itself (e.g., T790M/C797S mutations or gene amplification), which
restores EGFR kinase activity and re-activates the downstream
PI3K/Akt/mTOR and Ras/Raf/MEK/ERK pathways. EGFR independent
resistance occurs via bypass activation or downstream nodal
mutations, circumventing EGFR inhibition and ultimately converging
on the same core pathways. In addition, tumors may evade therapy
through histology or phenotypic transformation, such as
transformation to SCLC or EMT. EGFR, epidermal growth factor
receptor; TKIs, tyrosine kinase inhibitors; NSCLC, non-small cell
lung cancer; SCLC, small cell lung cancer; EMT,
epithelial-mesenchymal transition. |
Tumor-infiltrating immune cells
CD4+/CD8+ T
cells/Treg cells
Infiltration of CD8+ T cells is lower in
EGFR-mt lung adenocarcinoma (LA) compared with in EGFR-wt LA, and T
cell proliferation is inhibited (42,43).
It has been shown that after TKI treatment, immune cell
infiltration is increased and the antitumor response is enhanced in
EGFR-mt patient samples (44).
However, changes in the TME by TKI treatment appear
to be dynamic. TKI treatment induces the recruitment of
CD8+ and CD4+ T cells (45). With short-term TKI treatment, the
expression levels of CD8 and granzyme B (GB) in EGFR-TKI-treated T
cells co-cultured with tumor cells initially increases and then
decreases with the duration of treatment, and T cells infiltration
shifts from enhanced to suppressed (10). Evidence has shown that following TKI
resistance, the tumor can transform into a ‘hot’ tumor with
increased immune cell infiltration, with notable effector and T
cell infiltration (46).
In the early stages of treatment, sensitive
EGFR-TKIs can increase the number of cytotoxic CD8+ T
cells, while short-term EGFR inhibition reduces the proportion of
Foxp3+ Tregs. However, these changes in the TME, which
favor immune-mediated combination therapies for cancer, may
gradually diminish with continued treatment (47).
TAMs
TAMs serve an important role in tumor progression in
EGFR-mt NSCLC, and their enrichment in human NSCLC is associated
with poor clinical outcomes (48).
How M1 and M2 type macrophages are distinguished may be influenced
by the TME and macrophages may exhibit a mixed phenotype of both
types of markers.
EGFR mutations promote the expansion of alveolar
macrophages (AM), but TKI treatment markedly reduces AM numbers in
tumor-bearing mice (48). In
EGFR-mt cells, M2 macrophages increase, and while short-term TKI
treatment reduces M2 infiltration, the number of M2 macrophages
rise again with the onset of TKI resistance (10,45).
DCs
EGFR-mt lung cancer (LC) drives the immune phenotype
of tumor-infiltrating DCs (TIDC) toward an immunosuppressive
direction and is closely associated with exosomes. Tumor-derived
exosomes (TEXs) bridge the interaction between tumor cells and
immune cells, thereby altering antitumor immune responses. Research
has shown that TEXs can inhibit myeloid differentiation, promoting
the transformation of monocytes from immune-stimulating to
immune-suppressive cells, thereby supporting immune evasion
(49). There is also evidence that
EGFR activation by TEXs weakens the innate immunity of the host
(50) and promotes tumor metastasis
(51).
Compared with EGFR-wt tumor-bearing mice, TIDCs and
lymph node (LN) DCs isolated from EGFR-19del tumor-bearing mice
produced markedly less IL-12p40. This finding suggests that
EGFR-19del Lewis LC tumors drive the process of immunosuppression
by affecting DCs in both the tumor and LN. Furthermore, EGFR-mt LC
cells may influence DC function by secreting exosomes in
vitro. These exosomes not only accelerate tumor growth but also
induce immune suppression (43).
However, short-term TKI treatment markedly increases DC
infiltration within the TME (47).
Natural killer (NK) cells
In EGFR-mt tumors, both the innate and adaptive
lymphocyte compartments exhibit signs of functional exhaustion. The
proportion of cytotoxic NK cells is reduced, whereas NK T cell
(NKT) subsets with low cytotoxic potential are markedly increased
(52,53). These NKT subsets display diminished
expression of activation and cytotoxicity-related genes, indicating
weakened innate immune cytotoxicity within the EGFR-mt TME.
In EGFR-mt LC, TKI therapy increases NK cell
infiltration and cytotoxicity (44,54).
Elevated IL-6 levels are linked to decreased immune cell
infiltration during short-term TKI treatment. On the other hand, NK
cell activity is decreased in EGFR-mt NSCLC with acquired TKI
resistance due to increased upregulation of IL-6 (55).
B cells
Tertiary lymphoid structures (TLS) are primarily
composed of B lymphocytes, which are essential to their development
(53). TLS are associated with a
higher response to immunotherapies and increased survival. In the
TME of EGFR-negative LUAD, B cells build the TLS (56). Tumor-infiltrating B cells and
antigen presentation-related markers were notably lower in EGFR-mt
tumors compared with in EGFR-wt tumors, according to single-cell
transcriptome analysis, which resulted in decreased T-cell
activation (52). Immune
infiltration, including B cells and CD8+ T cells, is
increased in TKI-responsive samples but not in resistant ones after
EGFR-TKI treatment, indicating that combining ICIs may be more
advantageous prior to the development of TKI resistance (44).
Immunomodulatory molecules
Programmed death ligand 1 (PD-L1)
PD-L1 is a critical immune checkpoint protein that
is broadly expressed on the surface of tumor cells and
tumor-infiltrating immune cells. It inhibits anticancer immune
responses by binding to programmed cell death protein-1 (PD-1) on T
cells, B cells, DCs and NK T cells, and is an indicator of poor
survival in most advanced cancers (57–59).
It is regulated in NSCLC by two distinct mechanisms: Driver genetic
alterations and inflammation. Previous research has reported the
dynamic relationship between EGFR mutation status and PD-L1
expression before and after EGFR-TKI, but the exact relationship
remains controversial. Meanwhile, it is worth noting that there may
be an association between the levels of PD-L1 expression and the
efficacy of EGFR-TKI therapy (60).
Compared with EGFR-wt, EGFR-mt typically results in
upregulation of PD-L1 (59,61–65), a
process that is closely associated with activation of the
IL-6/JAK/STAT3 and p-ERK1/2/p-c-Jun signaling pathways (66,67).
However, one clinical study found that EGFR-mt patients exhibit
relatively low PD-L1 expression (16%; PD-L1 ≥5%) before TKI
treatment (68). An analysis
involving 18 studies and 3,969 patients revealed that, compared
with EGFR-wt tumors, EGFR-mt NSCLC is less likely to display PD-L1
positivity (65).
EGFR-TKI not only directly inhibits the activity of
tumor cells, but also indirectly enhances the antitumor immune
response by downregulating PD-L1 (61,66).
After short-term TKI treatment, PD-L1 expression is markedly
reduced (10), which can be
monitored by immuno-positron emission tomography imaging (69–71).
This downregulation of PD-L1 may be partly dependent on the
activation of the NF-kB pathway or AKT-STAT3 pathway (62,63).
However, a previous study suggested the opposite conclusion,
proposing that TKI treatment increases PD-L1 expression in patients
with EGFR-mt NSCLC (72). As
reported in one study (73), PD-L1
expression showed marked changes following EGFR-TKI treatment, with
the proportion of patients with PD-L1 strong positive tumors [tumor
proportion score (TPS) ≥50%] increasing from 14% at baseline to 28%
after TKI treatment. With subsequent ICI treatment, patients with
high PD-L1 expression (TPS ≥50%) achieved a longer mPFS compared
with those with low PD-L1 expression [TPS <50%; 7.1 vs. 1.7
months; HR=0.18 (0.04–0.56); P=0.0033]. The PD-1 signaling pathway
is activated in EGFR-TKI resistant tumors (74). After long-term TKI treatment, the
expression levels of PD-1 typically increase (10,72).
As EGFR-TKI resistance develops, the proportion of patients with
PD-L1 strong positive tumors increases from baseline (73). A retrospective analysis showed that
PD-L1 expression levels changed during the development of
resistance in 16 patients (28%), of which 12 patients had higher
PD-L1 expression levels after resistance (68). In addition, changes in PD-L1
expression levels in tumor-infiltrating immune cells between
baseline and the development of EGFR-TKI resistance were also of
interest, with the proportion of patients with ≥10% PD-L1
expression in tumor-infiltrating immune cells increasing from 11%
at baseline to 25% after TKI treatment.
Evidence suggests that patients with EGFR-TKI
resistance, particularly those who are T790M negative, may derive
greater benefit from PD-1 inhibitors, in part due to their higher
PD-L1 expression levels (75). This
suggests that further research is needed to determine whether
patients with EGFR-TKI resistance can benefit from PD-1 inhibitor
therapy. Due to the potential efficacy of immunotherapy in patients
with active PD-1 pathways, investigating the combined effects of
immunotherapy and its interactions with TKIs after TKI resistance
may help to optimize treatment strategies for these patients.
MHC-II, CD40, CD80 and CD86
LN DC and AM in EGFR-mt tumor-bearing mice exhibit
an immunosuppressive phenotype compared with EGFR-wt tumor-bearing
mice, characterized by downregulation of MHC-II and the
costimulatory molecules CD40, CD80, and CD86 (43,48).
CD47
Integrin-associated protein (CD47) is a cell surface
immunoglobulin-like molecule that inhibits phagocytosis by
interacting with signal regulatory protein α on phagocytes. CD47 is
selectively upregulated in patients with EGFR mutations. After
treatment with TKI, CD47 expression is downregulated, which
promotes DC phagocytosis of NSCLC cells. However, after the
development of resistance in vitro, CD47 expression is
upregulated (76).
Cytokines and chemokines
Cytokines and chemokines in the TME are also
regulated by EGFR-TKIs. According to molecular research, cytokines
can modulate immunological signaling by causing target-cell
receptors to undergo structural or functional changes, such as
receptor breakage or shedding (77–79).
In patients with EGFR-mt NSCLC, the pro-inflammatory cytokine
IL-17A was highly expressed, along with markedly increased
expression of transforming growth factor-β (TGF-β), which are
closely associated with cell proliferation and induction of TKI
resistance (80,81). Compared with AM in control mice, AM
in hormone mice produced more cytokines (such as IL-1α and TNF-α)
and chemokines [such as CXC motif chemokine ligand (CXCL) 1 and
CXCL2] (48). In addition, the
chemokine CXCL2 secreted by CAFs is considered to induce the
expression of PD-L1 in LA cells, thereby indirectly modulating
tumor immunity (82).
TKI treatment promotes the transformation of EGFR-mt
NSCLC from a ‘cold’ to a ‘hot’ tumor. After EGFR-TKI treatment, the
level of type I interferon (IFN) is notably increased in EGFR-mt
human LC cell lines, and the expression of the chemokines CXCL9,
CXCL10 and CXCL11 is also markedly upregulated. Meanwhile, the
CXCL10/CXCR3 pathway is activated in the EGFR-mt LC transgenic
mouse model (10,42,55).
Furthermore, an analysis of serum IFN-γ levels in 20 patients with
NSCLC treated with EGFR-TKI revealed that serum IFN-γ levels were
markedly higher compared with baseline levels in treated patients
(66).
In EGFR-TKI-resistant patients, the TME remodels
from a non-inflammatory to an inflammatory state. Studies have
shown that acquired resistance to TKI therapy can promote an
inflammatory response, with the IFN-γ pathway becoming markedly
enriched after TKI resistance, along with higher levels of granzyme
A expression (46). Type I IFN
levels are decreased in resistant cell lines, whereas expression of
the pro-inflammatory cytokines IL-6 and TGF-β1 is notably increased
(10,81,83,84).
CD24 and lymphocyte-activation gene 3
(LAG-3)
In a related study (85), the expression of the innate immune
checkpoint CD24 was found to be upregulated in EGFR-mt cells in
vitro following EGFR-TKI treatment, which is consistent with
the observation after TKI resistance. These findings suggest that
EGFR inhibition in EGFR-mt NSCLC cells promotes the development of
a TME conducive to immune escape. Furthermore, the expression of
the checkpoint protein LAG-3 is markedly elevated after EGFR-TKI
treatment (86).
Immunotherapy clinical trials in EGFR-mt
advanced NSCLC
Immunological monotherapy or dual
therapy
A pair of meta-analyses (87,88),
which included studies such as CheckMate057, KEYNOTE-010, OAK and
POPLAR, demonstrated that the efficacy of immunotherapy monotherapy
was inferior compared with that of docetaxel monotherapy in
patients with EGFR-mt LC. As a result, patients with EGFR-mt NSCLC
are less likely to derive notable benefits from immunotherapy
monotherapy. The BIRCH study (89),
a phase II, single-arm, multicenter clinical trial evaluating the
efficacy and safety of atezolizumab monotherapy in advanced
PD-L1-expressing NSCLC, included 45 patients with EGFR mutations.
The results showed that atezolizumab exhibited antitumor activity
regardless of EGFR status and was particularly effective in the
high PD-L1-expressing subgroup (TC3 or IC3). However, even among
EGFR-mt patients with high PD-L1 expression, the objective response
rate (ORR) and median overall survival (mOS) remained notably lower
compared with those in EGFR-wt patients. The ATLANTIC (90) study was a single-arm, open-label,
phase II trial evaluating durvalumab (an anti-PD-L1 monoclonal
antibody) as a third-line or later treatment for advanced NSCLC,
enrolling a total of 77 patients with EGFR mutations. Subgroup
analysis revealed that while the ORR in patients with EGFR-mt NSCLC
with PD-L1 expression ≥25% was higher compared with those with low
PD-L1 expression [12.2% (9/74) vs. 3.6% (1/28)], it was still lower
compared with EGFR-wt patients [16.4% (24/146) vs. 7.5%
(7/93)].
In one cohort of the KEYNOTE-021 study (91), a total of 11 patients were enrolled.
The combination of ipilimumab [anti-cytotoxic T-lymphocyte
associated protein 4 (CTLA-4) antibody] and pembrolizumab showed
limited efficacy in patients with EGFR-mt NSCLC, with an ORR of
only 10%, substantially lower than the 30% observed in EGFR-wt
patients. Overall, the data suggest that while the combination
therapy exhibits some antitumor activity in patients with advanced
NSCLC who had received multiple lines of therapy, it was less
effective and more toxic in EGFR-mt patients, with a 64% incidence
of treatment-emergent adverse events (AEs), including 29% of grade
3–5 severe AEs. The study suggests that dual immunotherapy has
limited effectiveness in EGFR-mt patients and must be chosen with
caution, especially when PD-L1 expression levels are low. In
conclusion, these clinical trial data indicate that the overall
response rate to immunotherapy is lower in patients with EGFR-mt
NSCLC compared with those with EGFR-wt. Nevertheless, antitumor
activity was observed in EGFR-mt patients with high PD-L1
expression, suggesting that high PD-L1 expression may serve as an
important predictor of potential benefit from immunotherapy in this
subgroup.
Immunotherapy combined with targeted
therapy
CAURAL (92) is a
phase II clinical trial investigating the combination of
osimertinib (a third-generation EGFR-TKI) and durvalumab in
patients with advanced NSCLC with EGFR T790M mutations who had
progressed after prior EGFR-TKI treatment. The primary objective of
the trial was to evaluate safety, with efficacy assessed as an
exploratory endpoint. The results demonstrated an ORR of 64% in the
combination therapy arm, lower than the 80% observed in the
osimertinib monotherapy arm, failing to show an advantage over
single-agent therapy. Nonetheless, the trial was terminated early
due to the elevated risk of interstitial lung disease observed in
related studies. This highlights that safety issues need to be
considered when combining an EGFR-TKI with an ICI.
Immunotherapy combined with
chemotherapy
CT18 (93) is a
multicenter, single-arm, phase II clinical trial evaluating the
efficacy and safety of toripalimab (anti-PD-1 antibody) in
combination with carboplatin and pemetrexed in patients (n=40) with
advanced EGFR-mt NSCLC who had failed EGFR-TKI therapy and lacked
T790M mutations. The results showed an ORR of 50.0% (95% CI,
33.8–66.2), and a disease control rate (DCR) of 87.5% (95% CI,
73.2–95.8). The mPFS was 7.0 months (95% CI, 4.8–8.4), while the
mOS reached 23.5 months (95% CI, 18.0 to NR months). The overall
safety profile of the treatment was manageable, with 97.5% of
patients experiencing treatment-related AEs (TRAEs), of which 65.0%
were grade 3 or higher, most commonly bone marrow suppression,
elevated transaminases and nausea. Immune-related AEs occurred in
40% of patients, with only 5.0% being grade 3 or higher.
CheckMate722 (94)
is a phase III clinical trial designed for patients with advanced
NSCLC characterized by EGFR mutations and resistance to TKI therapy
(n=294). This investigation sought to compare the efficacy of
nivolumab combined with chemotherapy against chemotherapy alone.
While the combination therapy group showed a modest improvement in
mPFS (5.6 vs. 5.4 months; HR=0.75), the difference fell short of
statistical significance. Likewise, the combination treatment did
not notably extend mOS (19.4 vs. 15.9 months; HR=0.82). The ORR was
31.3 and 26.7% in the combination and chemotherapy groups,
respectively. Post-hoc subgroup analyses revealed a potential trend
of enhanced PFS in specific subpopulations, including those with
sensitizing EGFR mutations or patients treated exclusively with
first-line TKI therapy, with HRs of 0.72 and 0.64,
respectively.
The KEYNOTE-789 study (95), a randomized, double-blind phase III
trial in 492 patients with TKI-resistant, EGFR-mt advanced NSCLC,
demonstrated that pembrolizumab in combination with chemotherapy
only slightly improved mPFS (5.6 vs. 5.5 months; HR=0.80; P=0.0122)
or mOS (15.9 vs. 14.7 months; HR=0.84; P=0.0362) compared with
chemotherapy alone. Neither the CheckMate722 nor the KEYNOTE-789
trial met their primary endpoints, suggesting that merely combining
anti-PD-1 ICIs with chemotherapy may not provide meaningful
clinical benefits for this patient population. Consequently, it is
essential to further investigate potential biomarkers and refine
combination treatment strategies to enhance therapeutic
outcomes.
Immunotherapy combined with
chemotherapy + anti-angiogenic drugs
IMpower150 (96,97) is
a phase III clinical trial that enrolled 123 patients with EGFR-mt
NSCLC to assess the efficacy of three regimens of atezolizumab in
combination with carboplatin and paclitaxel (ACP), bevacizumab plus
carboplatin and paclitaxel (BCP) and atezolizumab plus bevacizumab
plus carboplatin and paclitaxel (ABCP). The results showed that
patients with EGFR mutations or ALK translocations in the ABCP
group demonstrated a longer mPFS compared with those in the BCP
group, at 9.7 vs. 6.1 months, respectively (HR=0.59; 95% CI,
0.37–0.94; P=0.025). The mOS for the ABCP group was 29.4 months,
which was notably superior to the 18.1 months observed in the BCP
group (HR=0.60; 95% CI, 0.31–1.14), while the ACP group (19.0
months) was similar to the BCP group (HR=1.00; 95% CI, 0.57–1.74).
In terms of safety, 100% of patients in the ABCP group experienced
TRAEs, of which 66.7% were grade 3/4, although no grade 5 events
were reported. The incidence of TRAE in the ACP and BCP groups was
88.6% (with 56.8% grade 3/4) and 95.3% (55.8% grade 3/4),
respectively. In summary, the IMpower150 study demonstrated that
the ABCP regimen substantially prolonged OS in patients with
EGFR-mt NSCLC with controllable AEs and had a favorable safety and
tolerability profile.
ORIENT-31 (98) is a
phase III clinical trial designed to assess the efficacy of
sintilimab ± IBI305 combined with chemotherapy (pemetrexed +
cisplatin) in patients with locally advanced or metastatic EGFR-mt
non-squamous NSCLC who have progressed after EGFR-TKI treatment.
The randomized trial with 476 patients indicated that sintilimab
combined with chemotherapy significantly improved mPFS, with 5.5
vs. 4.3 months for chemotherapy alone (HR=0.72; 95% CI, 0.55–0.94;
P=0.016). Sintilimab plus IBI305 and chemotherapy extended mPFS to
7.2 months, compared with 4.3 months for chemotherapy alone
(HR=0.51; 95% CI, 0.39–0.67; P<0.0001), demonstrating
significant benefit. Regarding mOS, the sintilimab plus IBI305
combination chemotherapy group had a mOS of 21.1 months (95% CI,
17.5–23.9), which was similar to the 19.2 months observed in the
chemotherapy alone group (HR=0.98; 95% CI, 0.72–1.34; P=0.8883),
with no statistically significant difference. The mOS for the
sintilimab plus chemotherapy group was 20.5 months (95% CI,
15.8–25.3), which was comparable to the chemotherapy alone group at
19.2 months (HR=0.97; 95% CI, 0.71–1.32; P=0.8202). In terms of
safety, 56% of patients (88/158) in the sintilimab plus IBI305 plus
chemotherapy group developed a grade 3 or higher TRAE, which was
notably higher compared with in the sintilimab plus chemotherapy
(41%) and chemotherapy alone (49%) groups. Despite immunotherapy
side effects (such as immune pneumonitis and rash) were more
common, overall safety and tolerability were satisfactory and most
AEs could be mitigated with appropriate management.
IMpower151 (99) is
a phase III study that included 305 patients with advanced
non-squamous NSCLC. The trial assessed the efficacy of atezolizumab
in combination with bevacizumab (an anti-VEGF monoclonal antibody)
and chemotherapy (carboplatin + pemetrexed) as a first-line
treatment. Preliminary findings indicated that the combination
therapy (ABCP) showed limited benefits in mPFS and mOS compared
with the control group (BCP), without reaching statistical
significance. The mPFS was 9.5 and 7.1 months (HR=0.84; 95% CI,
0.65–1.09; P=0.18) and the mOS was 20.7 and 18.7 months (HR=0.93;
95% CI, 0.67–1.28) for the two groups, respectively. These results
suggest that although the combination of immunotherapy and
chemotherapy provided a slight survival benefit for patients with
EGFR-mt NSCLC, the effect was modest compared with other NSCLC
subtypes and did not substantially alter the prognosis. Safety
analyses revealed a high incidence of AEs in both groups and no new
safety signals. The rates of all-cause AEs were 99.3% in the ABCP
group (with 66.4% being grade 3/4) and 100% in the BCP group (61.4%
grade 3/4).
ATTLAS (100) is a
phase III study performed in Korea that enrolled 225 patients with
stage IV NSCLC diagnosed with EGFR sensitizing mutations or ALK
translocations, including the ABCP group (n=151) and the PC group
(n=74). All patients had disease progression or intolerance to one
or more EGFR or ALK TKIs. A total of 168 patients with EGFR-mt
NSCLC who were resistant to EGFR-TKI were enrolled in the study,
109 in the ABCP arm and 59 in the PC arm. The results demonstrated
that the ABPC arm significantly improved PFS compared with the PC
arm, with mPFS of 8.48 vs. 5.62 months, respectively [HR=0.62 (95%
CI, 0.45–0.86); P=0.004]. Subgroup analysis of patients with
EGFR-TKI-resistant EGFR-mt NSCLC revealed similar findings, with
mPFS of 8.7 months in the ABCP arm and 5.6 months in the PC arm
(HR=0.60; 95% CI, 0.43–0.84; P=0.002). However, no notable OS
benefit was observed in either group. In terms of safety, the
incidence of grade 3 or higher TRAEs was higher in the ABCP arm
compared with in the PC arm, primarily related to cytotoxic
chemotherapy. Nevertheless, bevacizumab-related TRAEs were
generally manageable with appropriate supportive care.
HARMONi-A (101,102) is a phase III clinical trial
performed in China, enrolling 322 patients with advanced or
metastatic EGFR-mt NSCLC who experienced disease progression during
EGFR-TKI treatment. This study evaluated the efficacy of
ivonescimab in combination with chemotherapy compared with
chemotherapy alone. In the trial, eligible patients were randomized
1:1 to receive ivonescimab plus chemotherapy (pemetrexed and
carboplatin) or placebo plus chemotherapy. Results showed that
ivonescimab plus chemotherapy resulted in a significant improvement
in mPFS compared with the chemotherapy group at 7.1 and 4.8 months,
respectively [HR=0.46 (95% CI; 0.34–0.62); P<0.001]. The ORRs of
the two groups were 50.6 and 35.4%, respectively, and the DCRs were
93.1 and 83.2%, respectively. In terms of safety, the incidence of
AEs was 99.4 and 97.5% in the two groups, and the incidence of
grade 3 or higher treatment-emergent AEs was 61.5 and 49.1%,
respectively.
In addition, two ongoing phases II clinical trials
(103,104) have shown preliminary efficacy. A
single-arm phase II study enrolled 64 patients with EGFR-mt NSCLC
who had progressed following EGFR-TKI treatment and received
combination chemotherapy with PM8002/BNT327, a bispecific antibody
targeting PD-L1 and VEGF-A. The results revealed an overall ORR of
54.7% (95% CI, 41.8–67.2). In patients with a TPS ≥50%, the ORR
reached 92.3% (95% CI, 64.0–99.8), indicating that the antitumor
activity of PM8002/BNT327 treatment was positively associated with
tumor PD-L1 expression levels. Additionally, cohort 5
(EGFR-TKI-resistant cohort) of the DUBHE-L-201 study (104) included 31 patients. The cohort
received QL1706 + carboplatin + pemetrexed + bevacizumab
administered intravenously on day 1 of a 21-day cycle for four
cycles. Maintenance therapy was QL1706 + pemetrexed + bevacizumab.
QL1706 is a bifunctional MabPair product containing anti-PD-1 and
anti-CTLA-4 antibodies. The primary endpoint of the study was
safety and secondary endpoints include confirmed ORR,
investigator-assessed duration of remission (DoR), PFS and OS.
Preliminary results showed a median DoR of 11.3 months (95% CI,
4.2–19.9), PFS of 8.5 months (95% CI, 5.7–13.3) and OS of 26.5
months (95% CI, 12.8-not evaluable) (Table I).
 | Table I.ICI-based immunotherapy combinations
for EGFR-mutant advanced NSCLC. |
Table I.
ICI-based immunotherapy combinations
for EGFR-mutant advanced NSCLC.
| A, Immunotherapy
combined with targeted therapy |
|---|
|
|---|
| NCT | Clinical trial | Phase | Intervention | Result | (Refs.) |
|---|
| NCT02454933 | CAURAL | III | Osi + Durva
(n=14) | ORR:64% vs.
80% | (92) |
|
|
|
| Osi (n=15) |
|
|
|
| B, Immunotherapy
combined with chemotherapy |
|
| NCT03513666 | CT18 | II | Tori + Carbo +
Pem | ORR=50.0% (95% CI,
33.8–66.2) | (93) |
|
|
|
| (n=40) | DCR=87.5% (95%CI,
73.2–95.8) |
|
|
|
|
|
| mPFS=7.0 m (95% CI,
4.8–8.4) |
|
|
|
|
|
| mOS=23.5 m (95% CI,
18.0-NR) |
|
| NCT02864251 | CheckMate722 | III | Nivo + Chemo | mPFS:5.6 m vs. 5.4
m (HR=0.75; |
|
|
|
|
| (n=144) | 95% CI, 0.56–1.00)
mOS:19.4 m | (94) |
|
|
|
| Chemo (n=150) | vs. 15.9 m
(HR=0.82; 95% CI, 0.61–1.10) |
|
| NCT03515837 | KEYNOTE-789 | III | Pembro + Chemo | mPFS:5.6 mvs5.5 m
(HR=0.80; | (95) |
|
|
|
| (n=245) | 95% CI,
0.65–0.97) |
|
|
|
|
| Placebo +
Chemo | mOS:15.9 vs. 14.7
(HR=0.84; |
|
|
|
|
| (n=247) | 95% CI,
0.69–1.02) |
|
|
| C, Immunotherapy
combined with chemotherapy + anti-angiogenic drugs |
|
| NCT02366143 | IMpower150 | III | ABCP: Atezo + | mPFS: ABCP vs.
BCP:9.70 m | (96,97) |
|
|
|
| Bev + Carbo+ | vs. 6.1 m (HR=0.59;
95% CI, |
|
|
|
|
| Pac (n=34) | 0.37–0.94) |
|
|
|
|
| BCP: (n=44) | mOS: ABCP vs.
BCP:26.1 m |
|
|
|
|
| ACP: (n=45) | vs. 20.3 m
(HR=0.91; 95% CI, |
|
|
|
|
|
| 0.53–1.59) |
|
|
|
|
|
| ACP vs. BCP: 21.4 m
vs. 20.3 m |
|
|
|
|
|
| (HR=1.16; 95% CI,
0.71–1.89) |
|
| NCT03802240 | ORIENT-31 | III | SIC: Sinti + | mPFS: SIC vs. C:7.2
m vs. 4.3 m | (98) |
|
|
|
| IBI305 + Chemo | (HR=0.51; 95% CI,
0.39–0.67) |
|
|
|
|
| (n=158) | SC vs. C: 5.5 m vs.
4.3 m |
|
|
|
|
| SC: Sinti +
Chemo | (HR=0.72; 95% CI,
0.55–0.94) |
|
|
|
|
| (n=158) | mOS: SIC: 21.1 m
(95% CI) |
|
|
|
|
| C: Chemov
(n=160) | 17.5–23.9) SC:20.5
m (95% CI) |
|
|
|
|
|
| 15.8–25.3) C:19.2 m
(95% CI |
|
|
|
|
|
| (15.8–22.4) |
|
| NCT04194203 | IMpower151 | III | ABCP: (n=152) | mPFS: 9.5 m vs. 7.1
m (HR=0.84; | (99) |
|
|
|
| BCP: (n=153) | 95% CI, 0.65–1.09;
P=0.18) |
|
|
|
|
|
| mOS: 20.7 m vs.
18.7 m |
|
|
|
|
|
| (HR=0.93; 95% CI,
0.67–1.28) |
|
| NCT03991403 | ATTLAS | III | ABPC: Atezo + | mPFS: ABCP vs.
PC:8.71 m | (100) |
|
|
|
| Bev + Pac + | vs. 5.62 m
(HR=0.60; 95% CI, |
|
|
|
|
| Carbo (n=109) | 0.43–0.84) |
|
|
|
|
| PC: (n=59) |
|
|
| NCT05184712 | HARMONi-A | III | ivonescimab + Chemo
(n=161) | mPFS: 7.1 m vs. 4.8
m (HR=0.46; 95% CI, 0.34–0.62) | (101,102) |
|
|
|
| Chemo (n=161) |
|
|
| NCT05756972 | - | II | PM8002/BNT327
+ | ORR:54.7% | (103) |
|
|
|
| Carbo + Pem
(n=64) |
|
|
| NCT05329025 | DUBHE-L-201 | II | QL1706+ Carbo + Pem
+ Bev (n=31) | mDoR, PFS, and OS
were 11.3 (95% CI, 4.2–19.9), 8.5 (5.7–13.3), and 26.5 months
(12.8-not evaluable), respectively | (104) |
Discussion and future perspectives
In summary, TKI-treated and TKI-resistant TMEs show
a shift towards a ‘hot’ tumor with increased immune cell
infiltration (Fig. 2), with an
increase in the immune-activating components of the TME, including
increased numbers or upregulation of tumor-infiltrating immune
cells, immunomodulatory molecules, cytokines or chemokines and a
reduction or impairment of immunosuppressive components. Notably,
acquired EGFR-TKI resistance promotes immune escape in lung cancer
by upregulating PD-L1 expression. Detailed studies have shown that
changes in the TME following TKI treatment are dynamic, with
short-term effects suggesting a potential therapeutic window during
which TKI treatment could more effectively leverage the immune
system to target tumors (Fig. 3).
However, the long-term effects of TKI therapy are more complex and
may be influenced by various factors, such as treatment cycles,
drug sensitivity and the specific treatment regimen employed.
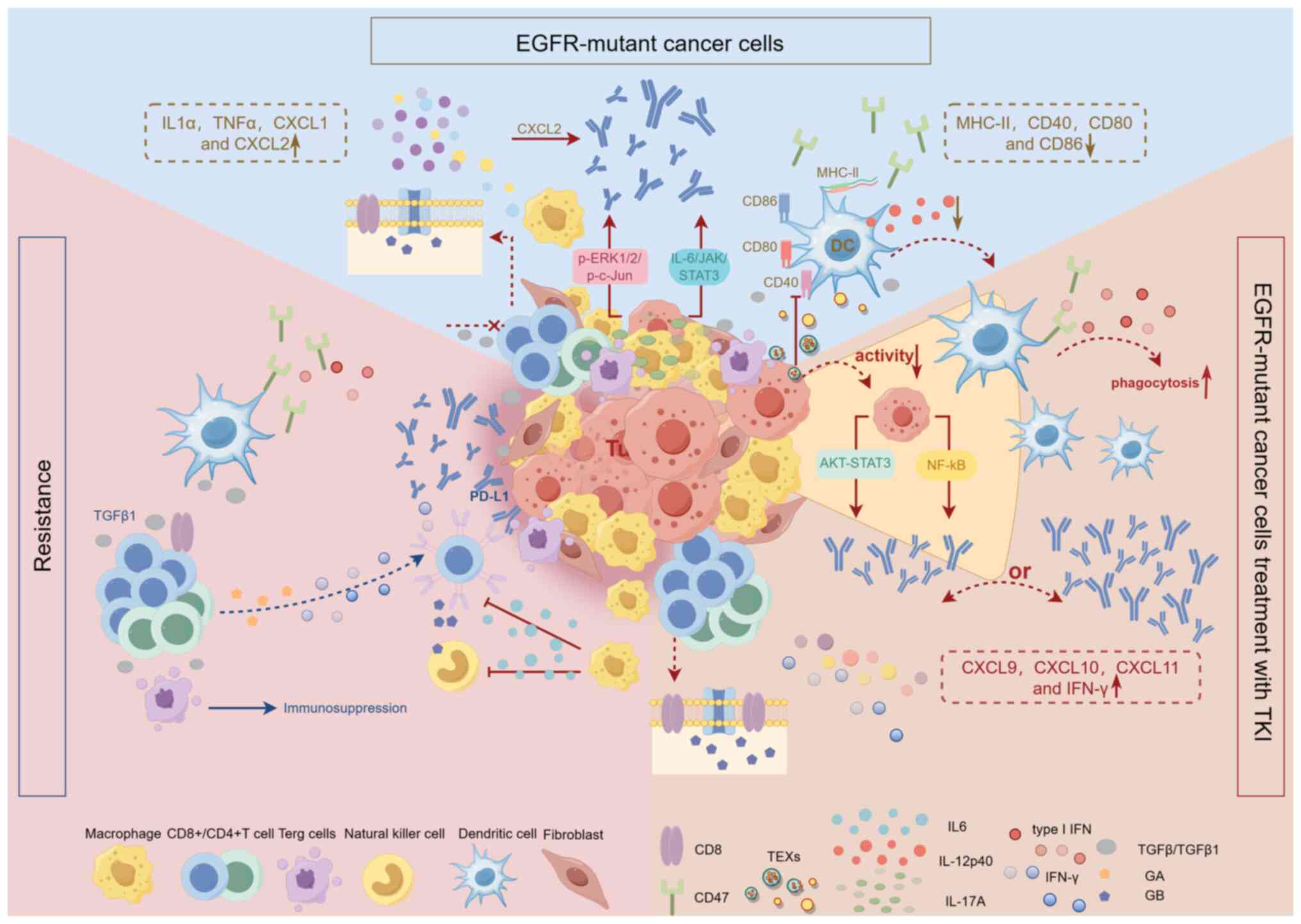 | Figure 2.TME in EGFR-mutant NSCLC and its
dynamics during short-term TKI treatment and resistance. The TME of
EGFR-mutant NSCLC is immunosuppressive. Low CD8+ T-cell
infiltration and inhibition of their proliferation and activation
by the cytokine TGF-β; EGFR mutation promotes AM proliferation;
TEXs inhibit DC function and reduce IL-12p40 production; MHC-II and
co-stimulatory molecules of DCs and AMs (CD40, CD80, CD86)
expression were downregulated; CD47 was selectively overexpressed;
EGFR mutations upregulated PD-L1 via IL-6/JAK/STAT3 and
p-ERK1/2/p-c-Jun pathways; high expression of pro-inflammatory
cytokine IL-17A; increased expression of cytokines TGF-β, IL1α and
TNFα and chemokines CXCL1 and CXCL2; and CAFs release of CXCL2
induced PD-L1 expression. EGFR-TKI can directly inhibit the
activity of tumor cells. Short-term TKI treatment transformed ‘cold
tumors’ into ‘hot tumors’, with an increase in T-cell and DC
infiltration, an increase in CD8 and GB expression levels Compared
with pre-treatment and a marked decrease in TAM infiltration and
Foxp3+ Tregs; CD47 is downregulated, enhancing
DC-mediated phagocytosis; PD-L1 expression could be reduced or
upregulated, and the reduced PL-L1 was considered to be associated
with the NF-κB and AKT-STAT3 pathways; the level of type I IFNs
(IFN-α, IFN-β) was increased and the expression of IFN-γ and the
chemokines CXCL9, CXCL10, and CXCL11 was upregulated. Upon TKI
resistance, the TME was remodeled towards inflammation, with
enrichment of inflammatory and IFN-γ pathway; CD8 and GB expression
decreased and granzyme A expression increased; TAM numbers rise;
CD47 was re-upregulated; the PD-1 pathway was activated and the
expression level of PD-L1 was increased; type I IFN decreased and
the expression of IL-6 and TGF-β1 was increased; the increased
expression of IL-6 suppressed T cell and NK cell cytotoxicity and
GB marker expression. TME, tumor microenvironment; EGFR, epidermal
growth factor receptor; NSCLC, non-small cell lung cancer; TGF-β,
transforming growth factor-β; AM, alveolar macrophages; CXCL, CXC
motif chemokine ligand; TEXs, tumor-derived exosomes; p-,
phosphorylated; IL, interleukin; TNF, tumor necrosis factor; CAFs,
cancer-associated fibroblasts; GB, granzyme B; IFN, interferon;
Tregs, regulatory T cells; DC, dendritic cell; TAMs,
tumor-associated macrophages; NK, natural killer; PD-L1, programmed
death ligand 1; TKI, tyrosine kinase inhibitor. |
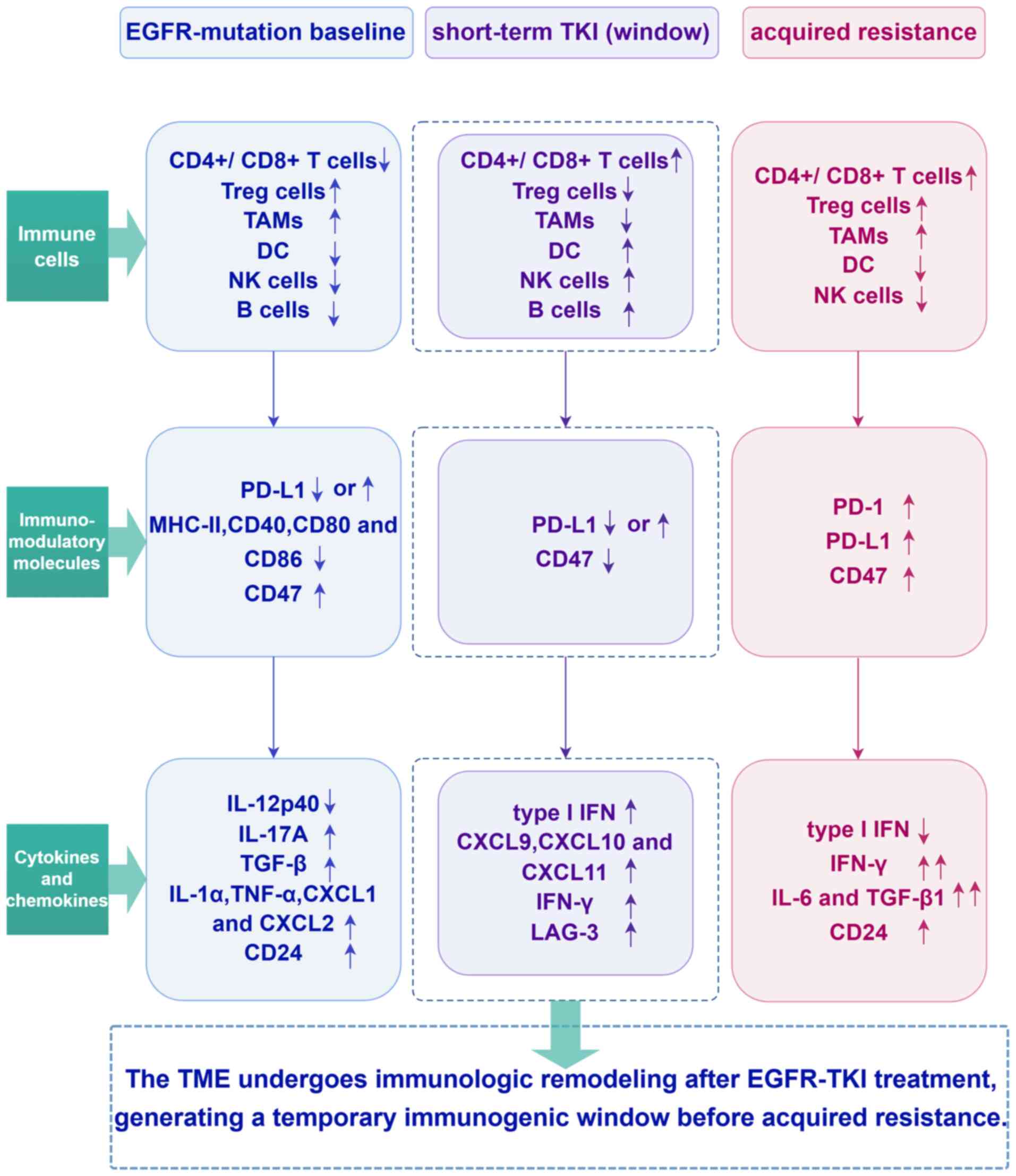 | Figure 3.A timeline schematic illustrating
dynamic changes in the TME from the EGFR-mutant baseline state
through short-term TKI exposure to the development of acquired
resistance. TME, tumor microenvironment; EGFR, epidermal growth
factor receptor; TKI, tyrosine kinase inhibitor; Treg cells,
regulatory T cells; TAMs, tumor-associated macrophages; DC,
dendritic cell; NK, natural killer; PD-L1, programmed death ligand
1; IL, interleukin; TGF-β, transforming growth factor-β; TNF, tumor
necrosis factor; CXCL, CXC motif chemokine ligand; IFN, interferon;
LAG-3, lymphocyte-activation gene 3; PD-1, programmed cell death
protein-1. |
Previous reports have shown that patients with high
PD-L1 expression have a greater survival benefit following
treatment with first-generation EGFR-TKI. However, the subsequent
FLAURA trial (105) reported
conflicting results, suggesting a shorter PFS in patients with high
PD-L1 expression. With second and third-generation TKI therapies,
high PD-L1 expression has been associated with worse survival and
higher rates of drug resistance. These findings suggest a strong
relationship between PD-L1 expression levels and TKI efficacy.
Previous cases are described in Table
II (106–117). However, discrepancies between
studies may be due to differences in drug generations, PD-L1
detection methods and thresholds for defining PD-L1 expression
levels. Most existing studies are retrospective, with limited
prospective studies and notable regional variability. There is an
urgent need for larger, multicenter, prospective clinical trials to
validate the relationship between PD-L1 expression and clinical
outcomes. Standardization of PD-L1 detection methods is also
critical to identifying the optimal population for ICI treatments,
ultimately providing greater clinical benefit to patients.
| Table II.Association between PD-L1 expression
and treatment efficacy of EGFR-TKI in EGFR-mutant NSCLC
patients. |
Table II.
Association between PD-L1 expression
and treatment efficacy of EGFR-TKI in EGFR-mutant NSCLC
patients.
| First
author/s,year | Gen. | EGFR-TKI | Country | EGFR-mutant (PD-L1
Testing) Patients (n) | PD-L1 expression in
EGFR-mutant/after TKIs | Associated clinical
outcomes | (Refs.) |
|---|
| D'Incecco et
al, 2015 | I | Gefitinib or
erlotinib | Italian | 95/123 | PD-L1 positivity
(2+ or 3+) in >5% of TCs: 55.3%/- | PD-L1+: higher RR
(61.2% vs. 34.8%, P=0.001), longer TTP (11.7 m vs. 5.7 m,
P<0.0001), and longer OS (21.9m vs. 12.5 m, P=0.09) | (64) |
| Lin et al,
2015 |
| Gefitinib or
erlotinib | China | 56 | Positive in 53.6%
of tumor specimens/- | PD-L1+: higher DCR
(93.3% vs. 61.5%, P=0.004), longer mPFS (16.5 vs. 8.6 m, P=0.001)
and mOS (35.3 vs. 19.8 m, P=0.004) | (106) |
| Soo et al,
2017 | I/II | Erlotinib,
gefitinib, or dacomitinib | South Korea | 90 | ≥1% positive: ICs
44%, TCs 59%/no association | PD-L1+: shorter PFS
[HR=1.008 (1.001–1.015), P=0.017] | (107) |
| Yoneshima et
al, 2018 |
| - | Japan | 71 | TPS of ≥1%:
42.3%/- | TPS ≥1% vs. <1%:
shorter mPFS (9 vs. 14 m, P=0.016) | (108) |
| Su et al,
2018 |
| - | China | 101 | Positive (TC3/IC3,
TC1-2/IC1-2): 35.6%/- | TC3/IC3 vs.
TC1-2/IC1-2 vs. TC0/IC0: lower ORR (35.7% vs. 63.2% vs. 67.3%,
P=0.002), shorter mPFS (3.8 vs. 6.0 vs. 9.5m, P<0.001), and
higher primary resistance rates (66.7% vs. 30.2%) P=0.009) | (109) |
| Hsu et al,
2019 |
| Gefitinib,
erlotinib, or afatinib | China | 123 | TPS of ≥1%:
30.1%/- | TPS ≥1% vs. <1%:
shorter mPFS (2.1 vs. 7.3 m, P<0.001) and mOS (11.2 vs. 38.2,
P=0.002), higher primary resistance rates (OR=5.95, 95% CI
2.35–15.05, P<0.001) | (110) |
| Yang et al,
2020 |
| Gefitinib,
erlotinib, or afatinib | China | 153 | TPS 1–49%: 25.5%,
TPS ≥50%: 11.8%/Stable in 60% (9/15), increased in 40% upon
progression | TPS 0% vs. 1–49%
vs. ≥50%: ORR (65.6% vs. 56.4% vs. 38.9%, P<0.001), DCR (93.8%
vs. 97.4% vs. 55.6%, P<0.001), mPFS (2.5 vs. 12.8 vs. 5.9 m,
P=0.027), primary resistance rates (6.3% vs. 2.6% vs. 44.4%,
P<0.001) | (60) |
| Isomoto et
al, 2020 | I/II/III | Gefitinib,
erlotinib, afatinib, dacomitinib or osimertinib | Japan | 134 | TPS of ≥50%:
14%/TPS of ≥50%: 28% | - | (73) |
| Brown et al,
2018 | I/III | Osimertinib vs.
gefitinib/erlotinib | Global | 128 | TC ≥1%: 51%/- | TC ≥1% vs. <1%:
Osimertinib group (n=54): similar mPFS (18.4 vs. 18.9 m);
gefitinib/erlotinib group (n=52): shorter mPFS (6.9 vs. 10.9
m) | (105) |
| Sakata et
al, 2021 | III | Osimertinib | Japan | 538 | TPS ≥50%: 11.9%,
1–49%: 31.6%, <1%: 30%, Unknown: 26.6%/- | mPFS: TPS ≥50%:
11.1 m (95% CI 8.3-NR) [HR=2.24 (1.17–4.30), P=0.015], TPS
1–49%:14.7 m (95% CI 13.6–20.5) [HR=1.66 (1.05–2.63), P=0.029], TPS
<1%: NR (95% CI 20.7-NR) | (111) |
| Alves et al,
2022 | I/III | Gefitinib,
erlotinib or osimertinib | Brazil | 278/188 | TC ≥1%:
36.7%/- | Higher PD-L1 (TPS
≥50% vs. 1–49% vs. <1%): not associated with mOS (37.5 vs. 46.5
vs. 50.4 m, P=0.48), and decreased event-free survival (median:9
vs. 34 vs. 26 m, P=0.014) | (112) |
| Papazyan et
al, 2024 | III | Osimertinib | France | 96 | TPS ≥50%:
20.8%/decreased in 4/15 patients at relapse | TPS ≥50% vs.
<50%: shorter mPFS (9.3 vs. 17.5 m, P=0.044), and mOS (14.3 vs.
26.0 m, P=0.025), ORR (53.3% vs. 46.5%, P>0.9) | (113) |
| Lakkunarajah et
al, 2023 |
| Osimertinib | Canada | 231 | TPS ≥50%:
29.4%/- | TPS ≥50% vs.
<1%: shorter PFS (HR=1.59, 95% CI 1.07–2.36, P=0.023), and OS
(HR=1.82, 95% CI 1.10–2.99, P=0.019) | (114) |
| Yoshimura et
al, 2021 |
| Osimertinib | Japan | 71 | TPS ≥50%:
21.1%/- | TPS ≥50% vs.
<50%: lower ORR (53.3% vs. 81.1%, P=0.043) and DCR (73.3% vs.
98.1%, P=0.007), shorter mPFS (5.0 vs. 17.4 m, P<0.001); TPS
≥50% vs. 1–49% vs. <1%: higher primary resistance rates (33.33%
vs. 3.85% vs3.45%, P=0.006) | (115) |
| Hsu et al,
2022 |
| Osimertinib | China | 85/71 | TPS ≥50%:
9.4%/- | TPS ≥50% vs.
<50%: shorter mPFS [9.7 vs. 26.5 m, aHR=0.19 (0.06–0.67),
P=0.009], shorter mOS [25.4 vs. NR, aHR=0.09 (0.01–0.70),
P=0.021] | (116) |
| Hamakawa et
al, 2023 |
| Osimertinib | Japan | 64 | TPS ≥20%:
34.4%/- | TPS ≥20% vs.
<20%: shorter mPFS (9.1 vs. 28.1 m, log-rank P=0.013), with
PD-L1 TPS ≥20% associated with early resistance to osimertinib | (117) |
Recent research has focused on patients with high
PD-L1 expression. Available evidence suggests that immunotherapy
can offer notable benefits to patients, both during short-term TKI
therapy and after the development of resistance. However, the
effect of short-course TKI therapy on PD-L1 expression levels has
long been a topic of debate. The timing of immunotherapy
interventions and the monitoring of their efficacy need to be
further confirmed. On the one hand, the immunological
characteristics of the TME, such as inflammatory or
immunosuppressive status, as well as the level of PD-L1 expression
should guide the choice of treatment. On the other hand,
identifying reliable biomarkers, such as PD-L1 and cytokine levels,
is crucial for accurately predicting treatment response. It is
particularly critical and urgent to conduct large-scale,
multicenter clinical trials to validate the role of these
biomarkers and to promote their standardized application in
clinical practice.
The growing importance of immunotherapy is further
underscored by the emergence of resistance to TKI therapy. In
patients with high PD-L1 expression and EGFR-TKI resistance,
immunotherapy may provide a notable survival benefit (73,103).
Conversely, in patients with low PD-L1 expression and EGFR-TKI
resistance, immunotherapy interventions do not yield the desired
therapeutic outcomes. Fourth-generation EGFR-TKI and antibody-drug
conjugates (ADCs) offer novel therapeutic strategies for patients
who have failed conventional TKI therapy and exhibit low PD-L1
expression. Fourth-generation TKIs are designed to overcome the
limitations of third-generation TKIs in the face of drug
resistance. ADCs targeting trophoblast surface antigen 2 have shown
notable therapeutic efficacy in immunotherapy non-responders.
Future studies should focus on elucidating the underlying
mechanisms of dynamic immune changes in the TME, as well as
performing comprehensive and systematic evaluations of combination
therapies that include TKIs, ICIs, ADCs and anti-angiogenic drugs.
Moreover, actively exploring the optimal therapeutic window and
addressing the safety issues associated with combination therapies
will greatly advance the field of oncology and bring new hope to
more patients.
Acknowledgements
No applicable.
Funding
The present study was supported by the Renxin Medical Research
Program of the Beijing Weiai Public Welfare Foundation (grant no.
RXYS2025-0200630118), the Scientific Research Program of the
Tianjin Municipal Education Commission (grant no. 2025ZD061), and
the Scientific Research Program of the Hebei Administration of
Traditional Chinese Medicine (grant no. T2026091).
Availability of data and materials
Not applicable.
Authors' contributions
All authors contributed to the study conception and
design. HM and LL conceived the study and wrote and revised the
manuscript. CJ, YC and JH wrote the manuscript. HM, LL, QT and CJ
wrote, reviewed and edited the manuscript. DY contributed to the
manuscript preparation. YZ supervised the study. Data
authentication is not applicable. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Osipov A, Saung MT, Zheng L and Murphy AG:
Small molecule immunomodulation: The tumor microenvironment and
overcoming immune escape. J Immunother Cancer. 7:2242019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vuletic A, Mirjacic Martinovic K and
Jurisic V: The role of tumor microenvironment in triple-negative
breast cancer and its therapeutic targeting. Cells. 14:13532025.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kraja FP, Jurisic VB, Hromić-Jahjefendić
A, Rossopoulou N, Katsila T, Mirjacic Martinovic K, De Las Rivas J,
Diaconu CC and Szöőr Á: Tumor-infiltrating lymphocytes in cancer
immunotherapy: From chemotactic recruitment to translational
modeling. Front Immunol. 16:16017732025. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vryza P, Fischer T, Mistakidi E and
Zaravinos A: Tumor mutation burden in the prognosis and response of
lung cancer patients to immune-checkpoint inhibition therapies.
Transl Oncol. 38:1017882023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
To KKW, Fong W and Cho WCS: Immunotherapy
in treating EGFR-mutant lung cancer: Current challenges and new
strategies. Front Oncol. 11:6350072021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jurisic V, Vukovic V, Obradovic J,
Gulyaeva LF, Kushlinskii NE and Djordjević N: EGFR polymorphism and
survival of NSCLC patients treated with TKIs: A systematic review
and meta-analysis. J Oncol. 2020:19732412020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li K, Quan L, Huang F, Li Y and Shen Z:
ADAM12 promotes the resistance of lung adenocarcinoma cells to
EGFR-TKI and regulates the immune microenvironment by activating
PI3K/Akt/mTOR and RAS signaling pathways. Int Immunopharmacol.
122:1105802023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jeong HO, Lee H, Kim H, Jang J, Kim S,
Hwang T, Choi DW, Kim HS, Lee N, Lee YM, et al: Cellular plasticity
and immune microenvironment of malignant pleural effusion are
associated with EGFR-TKI resistance in non-small-cell lung
carcinoma. iScience. 25:1053582022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu L, Wang C, Li S, Bai H and Wang J:
Tumor immune microenvironment in epidermal growth factor
receptor-mutated non-small cell lung cancer before and after
epidermal growth factor receptor tyrosine kinase inhibitor
treatment: A narrative review. Transl Lung Cancer Res.
10:3823–3839. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lu C, Gao Z, Wu D, Zheng J, Hu C, Huang D,
He C, Liu Y, Lin C, Peng T, et al: Understanding the dynamics of
TKI-induced changes in the tumor immune microenvironment for
improved therapeutic effect. J Immunother Cancer. 12:e0091652024.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Corvaja C, Passaro A, Attili I, Aliaga PT,
Spitaleri G, Signore ED and De Marinis F: Advancements in
fourth-generation EGFR TKIs in EGFR-mutant NSCLC: Bridging
biological insights and therapeutic development. Cancer Treat Rev.
130:1028242024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Owen DH, Ismaila N, Ahluwalia A, Feldman
J, Gadgeel S, Mullane M, Naidoo J, Presley CJ, Reuss JE, Singhi EK
and Patel JD: Therapy for stage IV non-small cell lung cancer with
driver alterations: ASCO living guideline, version 2024.3. J Clin
Oncol. 43:e2–e16. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Obradović J, Niševic-Lazović J, Sekeruš V,
Milašin J, Perin B and Jurisic V: Investigating the frequencies of
EGFR mutations and EGFR single nucleotide polymorphisms genotypes
and their predictive role in NSCLC patients in Republic of Serbia.
Mol Biol Rep. 52:3502025. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yamaoka T, Ohba M and Ohmori T:
Molecular-targeted therapies for epidermal growth factor receptor
and its resistance mechanisms. Int J Mol Sci. 18:24202017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu HA, Arcila ME, Rekhtman N, Sima CS,
Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M and Riely GJ:
Analysis of tumor specimens at the time of acquired resistance to
EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers.
Clin Cancer Res. 19:2240–2247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu SG and Shih JY: Management of acquired
resistance to EGFR TKI-targeted therapy in advanced non-small cell
lung cancer. Mol Cancer. 17:382018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zalaquett Z, Catherine Rita Hachem M,
Kassis Y, Hachem S, Eid R, Raphael Kourie H and Planchard D:
Acquired resistance mechanisms to osimertinib: The constant battle.
Cancer Treat Rev. 116:1025572023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Westover D, Zugazagoitia J, Cho BC, Lovly
CM and Paz-Ares L: Mechanisms of acquired resistance to first- and
second-generation EGFR tyrosine kinase inhibitors. Ann Oncol. 29
(Suppl 1):i10–i19. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ko B, Paucar D and Halmos B: EGFR T790M:
Revealing the secrets of a gatekeeper. Lung Cancer (Auckl).
8:147–159. 2017.PubMed/NCBI
|
|
20
|
Leonetti A, Sharma S, Minari R, Perego P,
Giovannetti E and Tiseo M: Resistance mechanisms to osimertinib in
EGFR-mutated non-small cell lung cancer. Br J Cancer. 121:725–737.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Thress KS, Paweletz CP, Felip E, Cho BC,
Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, et
al: Acquired EGFR C797S mutation mediates resistance to AZD9291 in
non-small cell lung cancer harboring EGFR T790M. Nat Med.
21:560–562. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Niederst MJ, Hu H, Mulvey HE, Lockerman
EL, Garcia AR, Piotrowska Z, Sequist LV and Engelman JA: The
allelic context of the C797S mutation acquired upon treatment with
third-generation EGFR inhibitors impacts sensitivity to subsequent
treatment strategies. Clin Cancer Res. 21:3924–3933. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Passaro A, Jänne PA, Mok T and Peters S:
Overcoming therapy resistance in EGFR-mutant lung cancer. Nat
Cancer. 2:377–391. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Proto C, Lo Russo G, Corrao G, Ganzinelli
M, Facchinetti F, Minari R, Tiseo M and Garassino MC: Treatment in
EGFR-mutated non-small cell lung cancer: How to block the receptor
and overcome resistance mechanisms. Tumori. 103:325–337. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Du X, Yang B, An Q, Assaraf YG, Cao X and
Xia J: Acquired resistance to third-generation EGFR-TKIs and
emerging next-generation EGFR inhibitors. Innovation (Camb).
2:1001032021.PubMed/NCBI
|
|
26
|
Roy V and Perez EA: Beyond trastuzumab:
Small molecule tyrosine kinase inhibitors in HER-2-positive breast
cancer. Oncologist. 14:1061–1069. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Punekar SR, Velcheti V, Neel BG and Wong
KK: The current state of the art and future trends in RAS-targeted
cancer therapies. Nat Rev Clin Oncol. 19:637–655. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ohashi K, Sequist LV, Arcila ME, Lovly CM,
Chen X, Rudin CM, Moran T, Camidge DR, Vnencak-Jones CL, Berry L,
et al: Characteristics of lung cancers harboring NRAS mutations.
Clin Cancer Res. 19:2584–2591. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Simanshu DK, Nissley DV and McCormick F:
RAS proteins and their regulators in human disease. Cell.
170:17–33. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Polivka J and Janku F: Molecular targets
for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol Ther.
142:164–175. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Eng J, Woo KM, Sima CS, Plodkowski A,
Hellmann MD, Chaft JE, Kris MG, Arcila ME, Ladanyi M and Drilon A:
Impact of concurrent PIK3CA mutations on response to EGFR tyrosine
kinase inhibition in EGFR-mutant lung cancers and on prognosis in
oncogene-driven lung adenocarcinomas. J Thorac Oncol. 10:1713–1719.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pollak M: Insulin and insulin-like growth
factor signalling in neoplasia. Nat Rev Cancer. 8:915–928. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Namba K, Shien K, Takahashi Y, Torigoe H,
Sato H, Yoshioka T, Takeda T, Kurihara E, Ogoshi Y, Yamamoto H, et
al: Activation of AXL as a preclinical acquired resistance
mechanism against osimertinib treatment in EGFR-mutant non-small
Cell Lung Cancer Cells. Mol Cancer Res. 17:499–507. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sequist LV, Waltman BA, Dias-Santagata D,
Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger
S, Cosper AK, et al: Genotypic and histological evolution of lung
cancers acquiring resistance to EGFR inhibitors. Sci Transl Med.
3:75ra262011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Oser MG, Niederst MJ, Sequist LV and
Engelman JA: Transformation from non-small-cell lung cancer to
small-cell lung cancer: Molecular drivers and cells of origin.
Lancet Oncol. 16:e165–e172. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yin X, Li Y, Wang H, Jia T, Wang E, Luo Y,
Wei Y, Qin Z and Ma X: Small cell lung cancer transformation: From
pathogenesis to treatment. Semin Cancer Biol. 86:595–606. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li Y, Xie T, Wang S, Yang L, Hao X, Wang
Y, Hu X, Wang L, Li J, Ying J and Xing P: Mechanism exploration and
model construction for small cell transformation in EGFR-mutant
lung adenocarcinomas. Signal Transduct Target Ther. 9:2612024.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee JK, Lee J, Kim S, Kim S, Youk J, Park
S, An Y, Keam B, Kim DW, Heo DS, et al: Clonal history and genetic
predictors of transformation into small-cell carcinomas from lung
adenocarcinomas. J Clin Oncol. 35:3065–3074. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huang L and Fu L: Mechanisms of resistance
to EGFR tyrosine kinase inhibitors. Acta Pharm Sin B. 5:390–401.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Neel DS and Bivona TG: Secrets of drug
resistance in NSCLC exposed by new molecular definition of EMT.
Clin Cancer Res. 19:3–5. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sumimoto H, Takano A, Igarashi T, Hanaoka
J, Teramoto K and Daigo Y: Oncogenic epidermal growth factor
receptor signal-induced histone deacetylation suppresses chemokine
gene expression in human lung adenocarcinoma. Sci Rep. 13:50872023.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yu S, Sha H, Qin X, Chen Y, Li X, Shi M
and Feng J: EGFR E746-A750 deletion in lung cancer represses
antitumor immunity through the exosome-mediated inhibition of
dendritic cells. Oncogene. 39:2643–2657. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fang Y, Wang Y, Zeng D, Zhi S, Shu T,
Huang N, Zheng S, Wu J, Liu Y, Huang G, et al: Comprehensive
analyses reveal TKI-induced remodeling of the tumor immune
microenvironment in EGFR/ALK-positive non-small-cell lung cancer.
Oncoimmunology. 10:19510192021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lin Z, Wang Q, Jiang T, Wang W and Zhao
JJ: Targeting tumor-associated macrophages with STING agonism
improves the antitumor efficacy of osimertinib in a mouse model of
EGFR-mutant lung cancer. Front Immunol. 14:10772032023. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen Q, Xia L, Wang J, Zhu S, Wang J, Li
X, Yu Y, Li Z, Wang Y, Zhu G and Lu S: EGFR-mutant NSCLC may
remodel TME from non-inflamed to inflamed through acquiring
resistance to EGFR-TKI treatment. Lung Cancer. 192:1078152024.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jia Y, Li X, Jiang T, Zhao S, Zhao C,
Zhang L, Liu X, Shi J, Qiao M, Luo J, et al: EGFR-targeted therapy
alters the tumor microenvironment in EGFR-driven lung tumors:
Implications for combination therapies. Int J Cancer.
145:1432–1444. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang DH, Lee HS, Yoon D, Berry G, Wheeler
TM, Sugarbaker DJ, Kheradmand F, Engleman E and Burt BM:
Progression of EGFR-mutant lung adenocarcinoma is driven by
alveolar macrophages. Clin Cancer Res. 23:778–788. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Valenti R, Huber V, Iero M, Filipazzi P,
Parmiani G and Rivoltini L: Tumor-released microvesicles as
vehicles of immunosuppression. Cancer Res. 67:2912–2915. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Gao L, Wang L, Dai T, Jin K, Zhang Z, Wang
S, Xie F, Fang P, Yang B, Huang H, et al: Tumor-derived exosomes
antagonize innate antiviral immunity. Nat Immunol. 19:233–245.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang H, Deng T, Liu R, Bai M, Zhou L,
Wang X, Li S, Wang X, Yang H, Li J, et al: Exosome-delivered EGFR
regulates liver microenvironment to promote gastric cancer liver
metastasis. Nat Commun. 8:150162017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Cho JW, Park S, Kim G, Han H, Shim HS,
Shin S, Bae YS, Park SY, Ha SJ, Lee I and Kim HR: Dysregulation of
TFH-B-TRM lymphocyte cooperation is
associated with unfavorable anti-PD-1 responses in EGFR-mutant lung
cancer. Nat Commun. 12:60682021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhao J, Lu Y, Wang Z, Wang H, Zhang D, Cai
J, Zhang B, Zhang J, Huang M, Pircher A, et al: Tumor immune
microenvironment analysis of non-small cell lung cancer development
through multiplex immunofluorescence. Transl Lung Cancer Res.
13:2395–2410. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
He S, Yin T, Li D, Gao X, Wan Y, Ma X, Ye
T, Guo F, Sun J, Lin Z and Wang Y: Enhanced interaction between
natural killer cells and lung cancer cells: Involvement in
gefitinib-mediated immunoregulation. J Transl Med. 11:1862013.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Patel SA, Nilsson MB, Yang Y, Le X, Tran
HT, Elamin YY, Yu X, Zhang F, Poteete A, Ren X, et al: IL6 Mediates
suppression of T- and NK-cell function in EMT-associated
TKI-resistant EGFR-mutant NSCLC. Clin Cancer Res. 29:1292–1304.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yang L, He YT, Dong S, Wei XW, Chen ZH,
Zhang B, Chen WD, Yang XR, Wang F, Shang XM, et al: Single-cell
transcriptome analysis revealed a suppressive tumor immune
microenvironment in EGFR mutant lung adenocarcinoma. J Immunother
Cancer. 10:e0035342022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chen J, Jiang CC, Jin L and Zhang XD:
Regulation of PD-L1: A novel role of pro-survival signalling in
cancer. Ann Oncol. 27:409–416. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang Y, Wang L, Li Y, Pan Y, Wang R, Hu
H, Li H, Luo X, Ye T, Sun Y and Chen H: Protein expression of
programmed death 1 ligand 1 and ligand 2 independently predict poor
prognosis in surgically resected lung adenocarcinoma. Onco Targets
Ther. 7:567–573. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Azuma K, Ota K, Kawahara A, Hattori S,
Iwama E, Harada T, Matsumoto K, Takayama K, Takamori S, Kage M, et
al: Association of PD-L1 overexpression with activating EGFR
mutations in surgically resected nonsmall-cell lung cancer. Ann
Oncol. 25:1935–1940. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yang CY, Liao WY, Ho CC, Chen KY, Tsai TH,
Hsu CL, Su KY, Chang YL, Wu CT, Hsu CC, et al: Association between
programmed death-ligand 1 expression, immune microenvironments, and
clinical outcomes in epidermal growth factor receptor mutant lung
adenocarcinoma patients treated with tyrosine kinase inhibitors.
Eur J Cancer. 124:110–122. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Akbay EA, Koyama S, Carretero J, Altabef
A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp
EM, Pugh TJ, et al: Activation of the PD-1 pathway contributes to
immune escape in EGFR-driven lung tumors. Cancer Discov.
3:1355–1363. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lin K, Cheng J, Yang T, Li Y and Zhu B:
EGFR-TKI down-regulates PD-L1 in EGFR mutant NSCLC through
inhibiting NF-κB. Biochem Biophys Res Commun. 463:95–101. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Abdelhamed S, Ogura K, Yokoyama S, Saiki I
and Hayakawa Y: AKT-STAT3 pathway as a downstream target of EGFR
signaling to regulate PD-L1 expression on NSCLC cells. J Cancer.
7:1579–1586. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
D'Incecco A, Andreozzi M, Ludovini V,
Rossi E, Capodanno A, Landi L, Tibaldi C, Minuti G, Salvini J,
Coppi E, et al: PD-1 and PD-L1 expression in molecularly selected
non-small-cell lung cancer patients. Br J Cancer. 112:95–102. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Soo RA, Lim SM, Syn NL, Teng R, Soong R,
Mok TSK and Cho BC: Immune checkpoint inhibitors in epidermal
growth factor receptor mutant non-small cell lung cancer: Current
controversies and future directions. Lung Cancer. 115:12–20. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chen N, Fang W, Zhan J, Hong S, Tang Y,
Kang S, Zhang Y, He X, Zhou T, Qin T, et al: Upregulation of PD-L1
by EGFR activation mediates the immune escape in EGFR-driven NSCLC:
Implication for optional immune targeted therapy for NSCLC patients
with EGFR mutation. J Thorac Oncol. 10:910–923. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zhang N, Zeng Y, Du W, Zhu J, Shen D, Liu
Z and Huang JA: The EGFR pathway is involved in the regulation of
PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in
EGFR-mutated non-small cell lung cancer. Int J Oncol. 49:1360–1368.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Gainor JF, Shaw AT, Sequist LV, Fu X,
Azzoli CG, Piotrowska Z, Huynh TG, Zhao L, Fulton L, Schultz KR, et
al: EGFR mutations and ALK rearrangements are associated with low
response rates to PD-1 pathway blockade in non-small cell lung
cancer: A retrospective analysis. Clin Cancer Res. 22:4585–4593.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Li D, Zou S, Cheng S, Song S, Wang P and
Zhu X: Monitoring the response of PD-L1 expression to epidermal
growth factor receptor tyrosine kinase inhibitors in nonsmall-cell
lung cancer xenografts by immuno-PET imaging. Mol Pharm.
16:3469–3476. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
He H, Qi X, Fu H, Xu J, Zheng Q, Chen L,
Zhang Y, Hua H, Xu W, Xu Z, et al: Imaging diagnosis and efficacy
monitoring by [89Zr]Zr-DFO-KN035 immunoPET in patients
with PD-L1-positive solid malignancies. Theranostics. 14:392–405.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Huang W, Zhou J, Liu Y, Yang Y, Saladin
RJ, Hsu JC, Cai W and Kang L: Advances in immunoPET/SPECT imaging:
The role of Fab and F(ab')2 fragments in theranostics.
Acta Pharm Sin B. 15:3888–3924. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Xing S, Hu K and Wang Y: Tumor immune
microenvironment and immunotherapy in non-small cell lung cancer:
Update and new challenges. Aging Dis. 13:1615–1632. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Isomoto K, Haratani K, Hayashi H, Shimizu
S, Tomida S, Niwa T, Yokoyama T, Fukuda Y, Chiba Y, Kato R, et al:
Impact of EGFR-TKI treatment on the tumor immune microenvironment
in EGFR mutation-positive non-small cell lung cancer. Clin Cancer
Res. 26:2037–2046. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Wang S, Rong R, Yang DM, Fujimoto J,
Bishop JA, Yan S, Cai L, Behrens C, Berry LD, Wilhelm C, et al:
Features of tumor-microenvironment images predict targeted therapy
survival benefit in patients with EGFR-mutant lung cancer. J Clin
Invest. 133:e1603302023. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Haratani K, Hayashi H, Tanaka T, Kaneda H,
Togashi Y, Sakai K, Hayashi K, Tomida S, Chiba Y, Yonesaka K, et
al: Tumor immune microenvironment and nivolumab efficacy in EGFR
mutation-positive non-small-cell lung cancer based on T790M status
after disease progression during EGFR-TKI treatment. Ann Oncol.
28:1532–1539. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Nigro A, Ricciardi L, Salvato I, Sabbatino
F, Vitale M, Crescenzi MA, Montico B, Triggiani M, Pepe S, Stellato
C, et al: Enhanced expression of CD47 is associated with off-target
resistance to tyrosine kinase inhibitor gefitinib in NSCLC. Front
Immunol. 10:31352020. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Jurisic V: Multiomic analysis of cytokines
in immuno-oncology. Expert Rev Proteomics. 17:663–674. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Jurisić V, Bogdanovic G, Srdic T, Jakimov
D, Mrdjanovic J, Baltic M and Baltic VV: Modulation of TNF-alpha
activity in tumor PC cells using anti-CD45 and anti-CD95 monoclonal
antibodies. Cancer Lett. 214:55–61. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Jurisic V, Srdic-Rajic T, Konjevic G,
Bogdanovic G and Colic M: TNF-α induced apoptosis is accompanied
with rapid CD30 and slower CD45 shedding from K-562 cells. J Membr
Biol. 239:115–122. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Lee KL, Lai TC, Lee WJ, Chen YC, Ho KH,
Hung WY, Yang YC, Chan MH, Hsieh FK, Chung CL, et al: Sustaining
the activation of EGFR signal by inflammatory cytokine IL17A
prompts cell proliferation and EGFR-TKI resistance in lung cancer.
Cancers (Basel). 15:32882023. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Huang H, Zhu X, Yu Y, Li Z, Yang Y, Xia L
and Lu S: EGFR mutations induce the suppression of CD8+
T cell and anti-PD-1 resistance via ERK1/2-p90RSK-TGF-β axis in
non-small cell lung cancer. J Transl Med. 22:6532024. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Hong SH, Kang N, Kim O, Hong SA, Park J,
Kim J, Lee MA and Kang J: EGFR-tyrosine kinase inhibitors induced
activation of the autocrine CXCL10/CXCR3 pathway through crosstalk
between the tumor and the microenvironment in EGFR-mutant lung
cancer. Cancers (Basel). 15:1242022. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Mirjačić Martinović K, Vuletić A, Tišma
Miletić N, Besu Žižak I, Milovanović J, Matković S and Jurišić V:
Circulating cytokine dynamics as potential biomarker of response to
anti-PD-1 immunotherapy in BRAFwt MM patients. Transl Oncol.
38:1017992023. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Inoue C, Miki Y, Saito R, Hata S, Abe J,
Sato I, Okada Y and Sasano H: PD-L1 induction by cancer-associated
fibroblast-derived factors in lung adenocarcinoma cells. Cancers
(Basel). 11:12572019. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Shiiya A, Noguchi T, Tomaru U, Ariga S,
Takashima Y, Ohhara Y, Taguchi J, Takeuchi S, Shimizu Y, Kinoshita
I, et al: EGFR inhibition in EGFR-mutant lung cancer cells perturbs
innate immune signaling pathways in the tumor microenvironment.
Cancer Sci. 114:1270–1283. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Zhou J, Yu X, Hou L, Zhao J, Zhou F, Chu
X, Wu Y, Zhou C and Su C: Epidermal growth factor receptor tyrosine
kinase inhibitor remodels tumor microenvironment by upregulating
LAG-3 in advanced non-small-cell lung cancer. Lung Cancer.
153:143–149. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Lee CK, Man J, Lord S, Cooper W, Links M,
Gebski V, Herbst RS, Gralla RJ, Mok T and Yang JC: Clinical and
molecular characteristics associated with survival among patients
treated with checkpoint inhibitors for advanced non-small cell lung
carcinoma: A systematic review and meta-analysis. JAMA Oncol.
4:210–216. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Lee CK, Man J, Lord S, Links M, Gebski V,
Mok T and Yang JCH: Checkpoint inhibitors in metastatic
EGFR-mutated non-small cell lung cancer-a meta-analysis. J Thorac
Oncol. 12:403–407. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Peters S, Gettinger S, Johnson ML, Jänne
PA, Garassino MC, Christoph D, Toh CK, Rizvi NA, Chaft JE,
Carcereny Costa E, et al: Phase II trial of atezolizumab as
first-line or subsequent therapy for patients with programmed
death-ligand 1-selected advanced non-small-cell lung cancer
(BIRCH). J Clin Oncol. 35:2781–2789. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Garassino MC, Cho BC, Kim JH, Mazières J,
Vansteenkiste J, Lena H, Corral Jaime J, Gray JE, Powderly J,
Chouaid C, et al: Durvalumab as third-line or later treatment for
advanced non-small-cell lung cancer (ATLANTIC): An open-label,
single-arm, phase 2 study. Lancet Oncol. 19:521–536. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Gubens MA, Sequist LV, Stevenson JP,
Powell SF, Villaruz LC, Gadgeel SM, Langer CJ, Patnaik A, Borghaei
H, Jalal SI, et al: Pembrolizumab in combination with ipilimumab as
second-line or later therapy for advanced non-small-cell lung
cancer: KEYNOTE-021 cohorts D and H. Lung Cancer. 130:59–66. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Yang JCH, Shepherd FA, Kim DW, Lee GW, Lee
JS, Chang GC, Lee SS, Wei YF, Lee YG, Laus G, et al: Osimertinib
Plus durvalumab versus osimertinib monotherapy in EGFR
T790M-positive NSCLC following previous EGFR TKI therapy: CAURAL
brief report. J Thorac Oncol. 14:933–939. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Jiang T, Wang P, Zhang J, Zhao Y, Zhou J,
Fan Y, Shu Y, Liu X, Zhang H, He J, et al: Toripalimab plus
chemotherapy as second-line treatment in previously EGFR-TKI
treated patients with EGFR-mutant-advanced NSCLC: A multicenter
phase-II trial. Signal Transduct Target Ther. 6:3552021. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Mok T, Nakagawa K, Park K, Ohe Y, Girard
N, Kim HR, Wu YL, Gainor J, Lee SH, Chiu CH, et al: Nivolumab plus
chemotherapy in epidermal growth factor receptor-mutated metastatic
non-small-cell lung cancer after disease progression on epidermal
growth factor receptor tyrosine kinase inhibitors: Final results of
CheckMate 722. J Clin Oncol. 42:1252–1264. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Yang JCH, Lee DH, Lee JS, Fan Y, de
Marinis F, Iwama E, Inoue T, Rodríguez-Cid J, Zhang L, Yang CT, et
al: Phase III KEYNOTE-789 study of pemetrexed and platinum with or
without pembrolizumab for tyrosine kinase inhibitor-resistant,
EGFR-mutant, metastatic nonsquamous non-small cell lung cancer. J
Clin Oncol. 42:4029–4039. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Socinski MA, Jotte RM, Cappuzzo F, Orlandi
F, Stroyakovskiy D, Nogami N, Rodríguez-Abreu D, Moro-Sibilot D,
Thomas CA, Barlesi F, et al: Atezolizumab for first-line treatment
of metastatic nonsquamous NSCLC. N Engl J Med. 378:2288–2301. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Nogami N, Barlesi F, Socinski MA, Reck M,
Thomas CA, Cappuzzo F, Mok TSK, Finley G, Aerts JG, Orlandi F, et
al: IMpower150 final exploratory analyses for atezolizumab plus
bevacizumab and chemotherapy in key NSCLC patient subgroups with
EGFR mutations or metastases in the liver or brain. J Thorac Oncol.
17:309–323. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Lu S, Wu L, Jian H, Cheng Y, Wang Q, Fang
J, Wang Z, Hu Y, Han L, Sun M, et al: Sintilimab plus chemotherapy
for patients with EGFR-mutated non-squamous non-small-cell lung
cancer with disease progression after EGFR tyrosine-kinase
inhibitor therapy (ORIENT-31): Second interim analysis from a
double-blind, randomised, placebo-controlled, phase 3 trial. Lancet
Respir Med. 11:624–636. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Zhou C, Dong X, Chen G, Wang Z, Wu X, Yao
Y, Zhang Y, Cheng Y, Pan H, Zhang X, et al: OA09.06 IMpower151:
Phase III Study of Atezolizumab + bevacizumab + chemotherapy in 1L
metastatic nonsquamous NSCLC. J Thorac Oncol. 18 (Suppl):S64–S65.
2023. View Article : Google Scholar
|
|
100
|
Park S, Kim TM, Han JY, Lee GW, Shim BY,
Lee YG, Kim SW, Kim IH, Lee S, Kim YJ, et al: Phase III, randomized
study of atezolizumab plus bevacizumab and chemotherapy in patients
with EGFR- or ALK-mutated non-small-cell lung cancer (ATTLAS,
KCSG-LU19-04). J Clin Oncol. 42:1241–1251. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
HARMONi-A Study Investigators, Fang W,
Zhao Y, Luo Y, Yang R, Huang Y, He Z, Zhao H, Li M, Li K, et al:
Ivonescimab plus chemotherapy in non-small cell lung cancer with
EGFR variant: A randomized clinical trial. JAMA. 332:561–570. 2024.
View Article : Google Scholar
|
|
102
|
Li Z, Fang W, Zhao Y, Luo Y, Yang R, Huang
Y, He Z, Zhao H, Li M, Li K, et al: Ivonescimab combined with
chemotherapy in patients with EGFR-mutant non-squamous non-small
cell lung cancer who progressed on EGFR tyrosine-kinase inhibitor
treatment (HARMONi-A): A randomized, double-blind, multi-center,
phase 3 trial. J Clin Oncol. 42 (16 Suppl):S85082024. View Article : Google Scholar
|
|
103
|
Wu YL, Wang Z, Cheng Y, Fang J, Meng X,
Pan Y, Zhao H, Zhao Y, Su H, Sun M, et al: 1255MO A phase II safety
and efficacy study of PM8002/BNT327 in combination with
chemotherapy in patients with EGFR-mutated non-small cell lung
cancer (NSCLC). Ann Oncol. 35 (Suppl 2):S8042024. View Article : Google Scholar
|
|
104
|
Fang WF, Yang Y, Zhao Y, Huang Y, Zhao H,
Zhou N, Zhang Y, Chen L, Zhou T, Chen G, et al: 646P Iparomlimab
and tuvonralimab (QL1706) plus chemotherapy and bevacizumab for
epidermal growth factor receptor inhibitor (EGFRi)-resistant,
EGFR-mutant, advanced non-small cell lung cancer (NSCLC): Updated
results from Cohort 5 in the DUBHE-L-201 study. Ann Oncol. 35
(Suppl 4):S16462024. View Article : Google Scholar
|
|
105
|
Brown H, Vansteenkiste J, Nakagawa K, Cobo
M, John T, Barker C, Kohlmann A, Todd A, Saggese M, Chmielecki J,
et al: Programmed cell death ligand 1 expression in untreated EGFR
mutated advanced NSCLC and response to osimertinib versus
comparator in FLAURA. J Thorac Oncol. 15:138–143. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Lin C, Chen X, Li M, Liu J, Qi X, Yang W,
Zhang H, Cai Z, Dai Y and Ouyang X: Programmed death-ligand 1
expression predicts tyrosine kinase inhibitor response and better
prognosis in a cohort of patients with epidermal growth factor
receptor mutation-positive lung adenocarcinoma. Clin Lung Cancer.
16:e25–e35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Soo RA, Kim HR, Asuncion BR, Fazreen Z,
Omar MFM, Herrera MC, Yun Lim JS, Sia G, Soong R and Cho BC:
Significance of immune checkpoint proteins in EGFR-mutant non-small
cell lung cancer. Lung Cancer. 105:17–22. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Yoneshima Y, Ijichi K, Anai S, Ota K,
Otsubo K, Iwama E, Tanaka K, Oda Y, Nakanishi Y and Okamoto I:
PD-L1 expression in lung adenocarcinoma harboring EGFR mutations or
ALK rearrangements. Lung Cancer. 118:36–40. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Su S, Dong ZY, Xie Z, Yan LX, Li YF, Su J,
Liu SY, Yin K, Chen RL, Huang SM, et al: Strong programmed death
ligand 1 expression predicts poor response and de novo resistance
to EGFR tyrosine kinase inhibitors among NSCLC patients with EGFR
mutation. J Thorac Oncol. 13:1668–1675. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Hsu KH, Huang YH, Tseng JS, Chen KC, Ku
WH, Su KY, Chen JJW, Chen HW, Yu SL, Yang TY and Chang GC: High
PD-L1 expression correlates with primary resistance to EGFR-TKIs in
treatment naïve advanced EGFR-mutant lung adenocarcinoma patients.
Lung Cancer. 127:37–43. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Sakata Y, Sakata S, Oya Y, Tamiya M,
Suzuki H, Shibaki R, Okada A, Kobe H, Matsumoto H, Yokoi T, et al:
Osimertinib as first-line treatment for advanced epidermal growth
factor receptor mutation-positive non-small-cell lung cancer in a
real-world setting (OSI-FACT). Eur J Cancer. 159:144–153. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Alves Pinto I, de Oliveira Cavagna R,
Virginio da Silva AL, Dias JM, Santana IV, Souza LC, Ferreira da
Silva FA, Biazotto Fernandes MF, Junqueira Pinto GD, Negreiros IS,
et al: EGFR mutations and PD-L1 expression in early-stage non-small
cell lung cancer: A real-world data from a single center in Brazil.
Oncologist. 27:e899–e907. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Papazyan T, Denis MG, Sagan C, Raimbourg
J, Herbreteau G and Pons-Tostivint E: Impact of PD-L1 expression on
the overall survival of caucasian patients with advanced
EGFR-mutant NSCLC treated with frontline osimertinib. Target Oncol.
19:611–621. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Lakkunarajah S, Truong PT, Bone JN,
Hughesman C, Yip S, Alex D, Hart J, Pollock P, Egli S, Clarkson M,
et al: First-line osimertinib for patients with EGFR-mutated
advanced non-small cell lung cancer: efficacy and safety during the
COVID-19 pandemic. Transl Lung Cancer Res. 12:1454–1465. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Yoshimura A, Yamada T, Okuma Y, Fukuda A,
Watanabe S, Nishioka N, Takeda T, Chihara Y, Takemoto S, Harada T,
et al: Impact of tumor programmed death ligand-1 expression on
osimertinib efficacy in untreated EGFR-mutated advanced non-small
cell lung cancer: A prospective observational study. Transl Lung
Cancer Res. 10:3582–3593. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Hsu KH, Tseng JS, Yang TY, Chen KC, Su KY,
Yu SL, Chen JJW, Huang YH and Chang GC: PD-L1 strong expressions
affect the clinical outcomes of osimertinib in treatment naïve
advanced EGFR-mutant non-small cell lung cancer patients. Sci Rep.
12:97532022. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Hamakawa Y, Agemi Y, Shiba A, Ikeda T,
Higashi Y, Aga M, Miyazaki K, Taniguchi Y, Misumi Y, Nakamura Y, et
al: Association of PD-L1 tumor proportion score ≥20% with early
resistance to osimertinib in patients with EGFR-mutated NSCLC.
Cancer Med. 12:17788–17797. 2023. View Article : Google Scholar : PubMed/NCBI
|














