Introduction
Gliomas are the most prevalent primary malignant
tumors of the central nervous system (CNS) in adults (1), with diffuse midline glioma (DMG)
standing out for its high invasiveness and abysmal prognosis.
Driven by the histone H3 lysine 27 (H3K27)M mutation, DMG exhibits
aggressive behavior and marked resistance to treatment, making it a
formidable challenge in neuro-oncology.
DMG predominantly affects children and adolescents,
whereas adult cases are uncommon. Clinically, numerous pediatric
DMGs present as diffuse intrinsic pontine glioma (DIPG), a
radiographic entity centered in the pons. Epidemiologic estimates
suggest 20–30 new DIPG cases annually in the UK (2) and 100–400 annually in the US (2,3) and a
Canadian population-based cohort identified 143 pediatric DIPG
cases over a 10-year period (~14 cases per year) (2). DMG remains highly lethal, with a
median overall survival (OS) of ~11 months (4) and 1-, 2- and 5-year survival rates of
42.3, 9.6 and 2.2% in a 1,008-case registry cohort (5).
DMGs are invasive tumors affecting midline
structures such as the thalamus, pons, brainstem and spinal cord.
Clinical manifestations vary by location, including cranial nerve
dysfunction, ataxia and raised intracranial pressure. MRI is key
for diagnosis, showing heterogeneous features such as diffuse or
distinct boundaries, variable contrast enhancement and similarities
to low-grade astrocytomas (6).
Pons-involved DMGs often infiltrate >50% of the pons and may
encase the basilar artery, with minimal T1 contrast enhancement and
uneven T1/T2 signal intensities, often with necrosis or hemorrhage
(7,8), reflecting their histopathological
heterogeneity.
Molecular pathological studies of DMG have revealed
its unique epigenetic characteristics, focusing on the pivotal role
of the H3K27M mutation in its pathogenesis (9). This mutation replaces lysine 27 of
histone H3 with methionine, substantially reducing H3K27
trimethylation (H3K27me3) and causing widespread gene expression
reprogramming. It disrupts epigenetic regulation, promoting tumor
proliferation, invasion and treatment resistance (10). Additionally, the H3K27M mutation
dysregulates multiple key signaling pathways, including MAPK and
PI3K/AKT, driving rapid growth and creating a tumor-supportive
micro-environment (11). These
findings establish H3K27M mutation as a central driver of DMG and
underscore the urgent need for targeted therapeutic strategies.
Despite notable advances in understanding the
molecular basis of DMG, the therapeutic landscape for this disease
remains beset with formidable challenges. Surgery, radiotherapy and
chemotherapy provide minimal improvement in overall survival
currently, as epigenetic alterations driven by the H3K27M mutation
render these tumors highly resistant to conventional treatments.
However, emerging targeted therapies and immunotherapies, including
CAR-T cell therapy, have garnered considerable research interest
(12) though they require further
clinical validation.
Against this backdrop, the present review
systematically summarized the molecular characteristics, genetic
signatures and latest progress in DMG treatment, emphasizing the
effect of histone alterations and innovative therapeutic strategies
to deepen the understanding of DMG and provide theoretical support
and guidance for future research.
Modifications to the definition of DMG
The definition of DMG has evolved markedly with the
2016 and 2021 World Health Organization (WHO) CNS tumor
classifications, shifting towards molecular pathology-based
diagnosis.
Classification and definition in WHO
before 2016
DMG, termed diffuse intrinsic pontine glioma (DIPG)
before 2016, was first defined by Wilfred Harris in 1926 for its
pontine location and histological features (13,14),
predominantly affecting children aged 5–10 with a dismal prognosis
(median OS less than a year and five-year survival rate <1%)
(15). Traditional classification
could not distinguish subtypes or elucidate molecular mechanisms
(16).
The 2016 WHO classification introduced molecular
diagnostics, defining DMG by the H3K27M histone mutation,
specifically the K27M mutation in the H3F3A, HIST1H3B and HIST1H3C
genes (17,18). This mutation disrupts lysine 27
methylation of histone H3, altering gene regulation and driving
tumorigenesis (19), thereby
enhancing diagnostic precision and differentiation.
Updates in the 2021 WHO
classification
The 2021 WHO classification renamed ‘Diffuse Midline
Glioma, H3K27M-mutant’ to ‘Diffuse Midline Glioma, H3 K27-altered’
to encompass broader molecular changes (20), including EZHIP overexpression
(21,22), which reduces H3K27me3 levels,
promoting invasiveness and therapy resistance (23). These updates improved diagnostic
accuracy, highlighted tumor heterogeneity and facilitated tailored,
patient-specific treatment strategies to enhance outcomes (24).
Significance of histones in cancer
Definition and functions of histone
and oncohistones
Histones are the primary structural proteins of
chromatin, packaging DNA into nucleosomes comprising two copies
each of H2A, H2B, H3 and H4. Beyond DNA condensation, histones
regulate gene expression through post-translational modifications
such as methylation, acetylation, phosphorylation and
ubiquitination.
Occurring on lysine residues such as H3K4, H3K27 and
H4K20, methylation can activate or repress transcription based on
its site and extent (mono-, di-, or trimethylation) (25). This process is dynamically regulated
by histone methyltransferases (such as SETD1A, which activates
transcription via H3K4 methylation) and histone demethylases (such
as KDM1A, which represses transcription by demethylating H3K4me2/1)
(26). Catalyzed by histone
acetyltransferases (HATs) such as p300/CBP, acetylation neutralizes
the positive charge of lysine, reducing the electrostatic
interaction of histones with the negatively charged DNA (27), loosening chromatin and facilitating
transcription. Phosphorylation is closely associated with cell
cycle regulation, DNA damage response and transcriptional
activation (28,29). Modifications, such as H2AX serine
139 phosphorylation, signal DNA damage, recruit repair proteins and
coordinate cell cycle responses (30). Ubiquitination, primarily on H2A and
H2B, affects chromatin structure and gene regulation, depending on
the site and mono- or poly-ubiquitination status (31).
Oncohistones, a subset of histones predominantly
observed in H3 (32,33), drive tumorigenesis and cancer
progression by disrupting chromatin structure (34). Mutations alter key modifications
such as H3K27 acetylation, which activates oncogenes such as MYC
and epidermal growth factor receptor (EGFR) (35) and H3K27me, which silences tumor
suppressors by compacting chromatin and blocking transcription
factor access. Elevated H3K27me levels are linked to silenced tumor
suppressors in breast, liver and lung cancers (36–38).
These changes promote tumor proliferation, invasiveness and
resistance, making oncohistones pivotal therapeutic targets.
Systemic effects of histone
modifications
Histone modifications orchestrate key pathways such
as PI3K/AKT/mammalian target of rapamycin (mTOR) and MAPK/ERK
(39,40), influencing tumor proliferation,
apoptosis and drug resistance (41). Aberrant modifications disrupt gene
networks, suppress immune responses and drive metastasis, as seen
with elevated HIST1H2BJ activating MMP9 and VEGF in breast cancer
brain metastases (42,43). These changes highlight the critical
role of histone modifications in cancer progression and
metastasis.
H3K27M mutation in the development of
DMG
Diffuse midline glioma is defined by the H3K27M
oncohistone, which disrupts methylation at histone H3 lysine 27
(H3K27). This missense substitution (lysine-to-methionine) occurs
in H3.1 or H3.3 variants and is associated with a profound
reduction of H3K27me3, a key epigenetic mark of transcriptional
repression. The resulting epigenetic reprogramming reshapes
chromatin accessibility and transcriptional programs, thereby
promoting aggressive phenotypes, including uncontrolled
proliferation, invasion and treatment resistance (44) (Fig.
1).
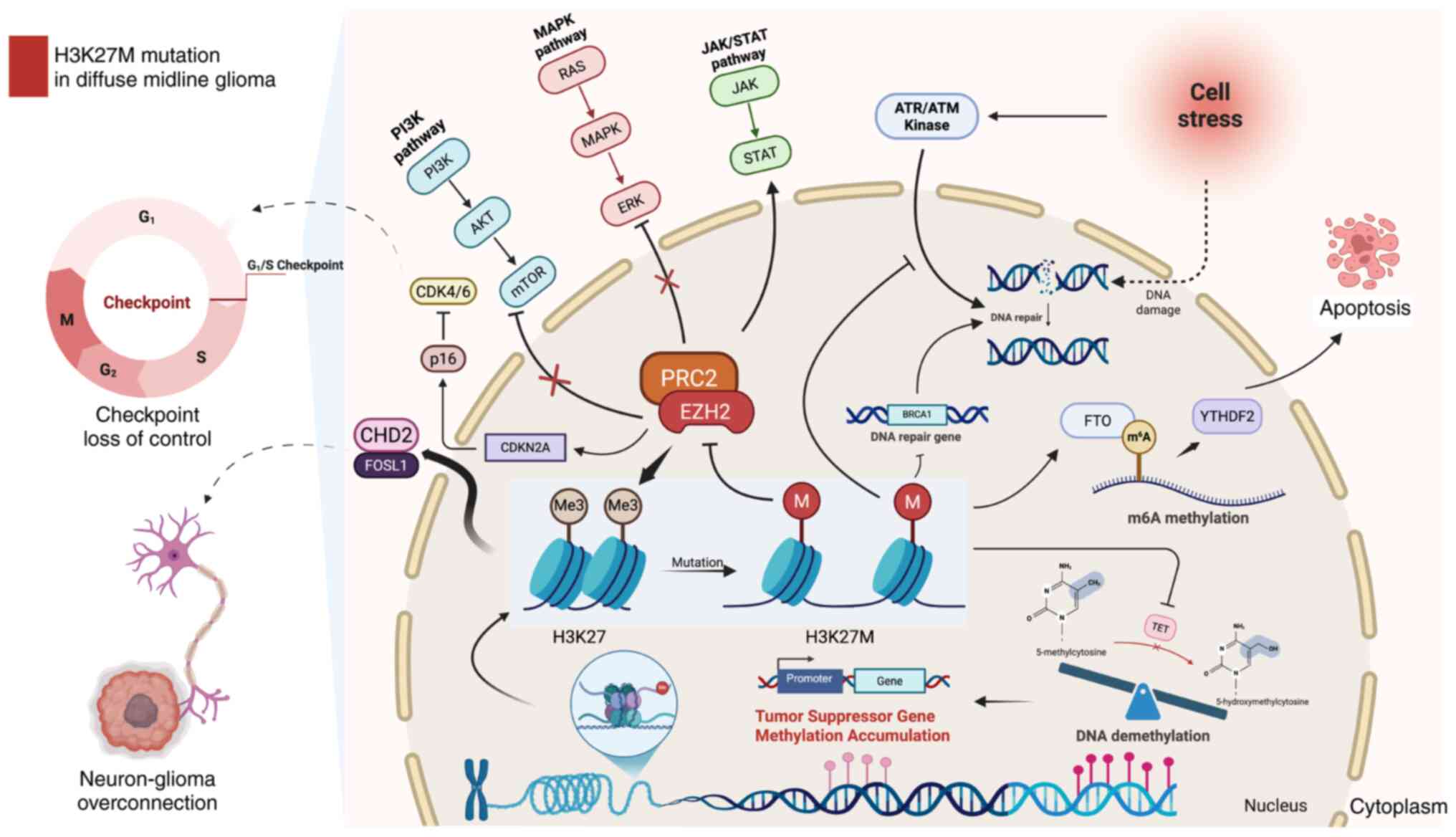 | Figure 1.H3K27M mutation in diffuse midline
glioma. The H3K27M mutation markedly reduces H3K27me3 levels by
inhibiting the EZH2 methyltransferase activity of the PRC2 complex,
leading to disrupted methylation regulation of tumor suppressor
genes and inducing widespread transcriptional abnormalities. This
mutation promotes uncontrolled cell cycle progression, exemplified
by dysregulation of the CDK4/6-p16 pathway and enhances tumor cell
proliferation and invasion through the activation of key signaling
pathways, including MAPK/ERK, PI3K/AKT/mTOR and JAK/STAT. The
H3K27M mutation also induces DNA repair deficiencies by impairing
the homologous recombination repair and base excision repair
pathways. It also affects the cellular stress response by altering
the ATR/ATM kinase pathway, compromising the cell's ability to
manage DNA damage. These defects exacerbate genomic instability and
transcriptional dysregulation, further contributing to tumor
progression. The H3K27M mutation alters m6A methylation and FTO
activity, making DMG cells sensitive to FTO inhibition, which
disrupts cell cycle gene expression and induces apoptosis.
Furthermore, the CHD2/FOSL1 axis plays a crucial role in abnormal
synaptic interactions between neurons and tumor cells, enhancing
tumor growth and invasion by promoting excessive neuronal-tumor
connectivity. Figure created using BioRender.com. H3K27, histone H3
lysine 27, H3K27me3, H3K27 trimethylation; PRC2, polycomb
repressive complex 2; mTOR, mammalian target of rapamycin; FTO,
fat-mass- and obesity-associated protein; DMG, diffuse midline
gliomas. |
Inhibition of H3K27me3 by H3K27M
H3K27 trimethylation is primarily catalyzed by the
EZH2 SET domain within the Polycomb Repressive Complex 2 (PRC2),
which maintains Polycomb-mediated transcriptional repression
programs involved in lineage control and proliferation. Structural
and biochemical studies (45) show
that the H3K27M oncohistone engages the PRC2 catalytic groove: the
substituted methionine inserts into the lysine-access channel of
the EZH2 SET domain, thereby competitively inhibiting PRC2
methyltransferase activity and exhibiting markedly higher affinity
than the corresponding wild-type H3K27 peptide [equilibrium
dissociation constant (Kd) ~3.3±0.4 µM for H3K27M
compared with ~52±12 µM for H3K27]. This inhibitory mode provides a
mechanistic basis for the global reduction and redistribution of
H3K27me3/PRC2 and the ensuing transcriptional reprogramming
observed in pediatric H3K27-altered high-grade gliomas (44).
The suppression of H3K27me3 by H3K27M alters gene
expression and activates multiple signaling pathways essential for
cell proliferation, survival and invasion. In the PI3K/AKT/mTOR
axis, growth-factor/RTK inputs activate PI3K and generate PIP3,
recruiting and activating AKT, which phosphorylates downstream
effectors to promote cell-cycle progression and survival while
suppressing apoptosis (39); mTOR
further sustains biosynthetic and metabolic programs that
facilitate rapid tumor expansion (46).
In parallel, aberrant activation of the JAK/STAT
pathway can reinforce proliferative transcriptional programs and
contribute to immune-evasive phenotypes within the tumor
microenvironment, as JAK-dependent STAT phosphorylation promotes
nuclear transcription of genes linked to growth and
immunoregulation (44,47).
Moreover, MAPK/ERK signaling, often downstream of
RTKs, can be potentiated, enabling RAS-RAF-MEK-ERK propagation and
ERK-driven nuclear transcriptional outputs that promote
proliferation and therapy resistance (48).
Extensive changes in DNA
methylation
The H3K27M mutation disrupts histone methylation and
induces widespread changes in DNA methylation. Research by Vanan
et al (49) and Graham et
al (50) revealed that reduced
H3K27me3 in H3K27M mutant cells extends across the genome, altering
DNA methylation beyond the sites of mutant histone localization.
This indicates that the mutation indirectly affects the regulation
of numerous genes.
Further studies by Paine et al (51) demonstrated significant DNA
methylation changes, particularly in the promoter regions of tumor
suppressor genes such as TP53 and RB1, leading to their silencing
and facilitating the malignant progression of tumors.
Typically, the TET family of enzymes (TET1, TET2 and
TET3) oxidizes 5-methylcytosine (5mC) to 5-hydroxymethylcytosine
(5-hmC) in a process known as DNA demethylation (52). However, the H3K27M mutation inhibits
TET enzyme activity by disrupting chromatin-associated factors,
reducing TET recruitment and markedly decreasing 5-hmC levels. This
results in the accumulation of DNA methylation, particularly in the
promoter regions of tumor suppressor genes (53,54),
creating an epigenetic landscape that promotes DMG invasiveness and
proliferation.
Regulation of m6A RNA methylation
Recent research (55) reveals that H3K27M-mutant DMG
exhibits unique m6A RNA methylation patterns, with m6A marks
enriched on key cell cycle genes. The m6A demethylase fat-mass- and
obesity-associated protein (FTO) is critical for DMG cell
proliferation; inhibition of FTO (such as by FB23-2) increases m6A
levels, disrupts cell cycle progression, reduces tumor cell
survival and induces apoptosis. These effects are partly mediated
by the m6A reader YTHDF2, which is highly expressed in DMG and
promotes degradation of cell cycle transcripts. Targeting
FTO-driven m6A dynamics thus represents a promising therapeutic
strategy for DMG.
Cell cycle regulation by H3K27M
mutation
The H3K27M mutation disrupts cell cycle regulation
by inhibiting the activity of the PRC2 complex and silencing tumor
suppressor genes such as RB1 and CDKN2A. This leads to the loss of
G1/S checkpoint control, allowing cells to bypass
critical regulatory mechanisms and proliferate uncontrollably
(56) (Fig. 1).
Williams et al (57) highlighted the pivotal role of the
p16 protein, encoded by CDKN2A, in maintaining cell cycle control
by inhibiting CDK4/6 activity and preventing the transition from
the G1 to S phase. In H3K27M mutant tumor cells, the
loss of p16 function results in cell cycle imbalance and
accelerated tumor growth.
H3K27M mutation inhibits DNA
repair
The H3K27M mutation additionally impairs DNA repair
processes, exacerbating genomic instability. In normal cells, DNA
damage is repaired to preserve genomic integrity. However, in
H3K27M mutant cells, the capacity for DNA repair, particularly in
the homologous recombination repair and base excision repair
pathways, is markedly compromised (Fig.
1). The expression of essential DNA repair genes, including
BRCA1, is notably diminished, rendering them prone to accumulating
genetic mutations and facilitating tumor progression (58). Furthermore, H3K27M rewires the DNA
damage response (DDR) and checkpoint circuitry (including
ATM/ATR-linked pathways), which can contribute to treatment
resistance but also creates actionable vulnerabilities for
DDR-targeted radio sensitization strategies (59).
Neuron-tumor cell interactions
Beyond epigenetic changes, the H3K27M mutation
promotes tumor expansion by regulating neuron-tumor interactions
(Fig. 1).
Chromodomain-Helicase-DNA-binding protein 2 (CHD2) plays a crucial
role in chromatin remodeling within the neuronal-tumor
microenvironment, regulating genes essential for synaptic
formation, neuronal excitability and nerve transmission, such as
SYP and PSD95, which maintain synaptic integrity (60).
In H3K27M mutant cells, CHD2 remodels chromatin
accessibility and drives an axon-guidance/synaptic transcriptional
program; mechanistically, a FOSL1-CHD2 regulatory axis promotes
neuron-glioma interactions and neuron-induced tumor proliferation
(60). Upregulation of
axon-guidance and synaptic gene programs strengthens neuron-glioma
interactions and neuron-induced proliferation in H3K27-altered
DMG.
Other histone modifications in DMG
Beyond the dysregulation of H3K27 trimethylation by
the H3K27M mutation, other histone modifications markedly
contribute to DMG progression. These modifications regulate
chromatin structure and gene expression, driving tumor cell
proliferation, differentiation and invasion (61). Critical histone modifications and
their roles in DMG are outlined below.
H3K27ac
H3K27ac, the acetylation of lysine 27 on histone H3,
is associated with active chromatin states, promoting gene
transcription by maintaining chromatin openness, predominantly at
promoters and enhancers.
In DMG, the H3K27M mutation indirectly increases
H3K27ac by inhibiting H3K27me3, activating oncogenes such as MYC
and PDGFRA, which drive tumor growth. Additionally, this
upregulation keeps chromatin at promoter and enhancer regions open,
facilitating transcription factors such as SP1 and AP-1 binding
(62). Menez et al (56) demonstrated that HATs such as p300
and CBP reinforce this open state, creating a positive feedback
loop that amplifies the expression of genes promoting
proliferation, survival and invasion.
Mota et al (63) found that the SWI/SNF chromatin
remodeling complex, particularly the BAF subtype, contributed to
H3K27ac accumulation. The antagonistic interaction between BAF and
PRC2 enhances the effects of H3K27ac. Targeting this pathway with
AU-15330, which is a PROTAC compound that degrades SMARCA4, SMARCA2
and PBRM1, effectively reduces H3K27ac levels, impairing chromatin
openness and inhibiting tumor proliferation.
H3K9me3
H3K9me3 is a repressive histone mark enriched in
heterochromatin regions, suppressing gene transcription through
chromatin compaction. In DMG, altered H3K9me3 levels are associated
with silencing tumor suppressor genes.
Xin et al (64) revealed that the histone
methyltransferase SUV39H1 generates H3K9me3 marks, recruiting
protein complexes to compact chromatin and repress tumor suppressor
genes such as CDKN1A, CDKN2B and CDKN2D, thereby promoting tumor
cell proliferation. The fungal metabolite Chaetocin, a SUV39H1
inhibitor, reduces H3K9me3 levels, which restores tumor suppressor
gene expression and induces apoptosis in DMG cells.
Notably, when H3K9me3 levels are inhibited, DMG
cells activate compensatory pathways such as the dopamine receptor
D2 (DRD2) pathway to sustain tumor growth. A combination therapy of
Chaetocin and DRD2 antagonists (such as ONC201) has been shown to
enhance anti-tumor effects and reduce treatment resistance
(64).
H3K36me3
H3K36me3 is an activating histone mark involved in
transcriptional elongation and DNA repair. The H3.3G34R/V mutation
reduces H3K36me3 levels (51),
disrupting transcriptional elongation and mismatch repair,
contributing to a mild ‘hyper-mutator’ phenotype by impairing DNA
repair mechanisms (65).
The loss of H3K36me3 compromises genomic stability,
allowing the accumulation of mutations that drive tumorigenesis.
These findings highlight the essential role of H3K36me3 in
maintaining cellular function and its dysregulation as a key factor
in cancer progression.
Given the pivotal role of these epigenetic
modifications in DMG pathogenesis, targeting these epigenetic
alterations presents a promising avenue for developing novel
therapeutic strategies to combat DMG.
Current treatment regimens
Local treatments, primarily encompassing surgery and
radiotherapy, are central to managing DMG. However, the invasive
nature of DMG and its midline location limit their effectiveness,
underscoring the need for more effective therapeutic options.
Surgical treatments
Surgery for DMG aims to obtain tissue for diagnosis
and molecular analysis, as its midline location makes complete
resection highly risky. Biopsies, particularly stereotactic ones,
highlighted in CPN Paris 2011, are vital for identifying mutations
such as H3K27M, enabling targeted therapies (66). Advances in minimally invasive
techniques and molecular profiling have made biopsy a key clinical
tool, offering critical insights with minimized risks (67).
A multicenter study revealed no survival benefit
from resection over biopsy in H3K27M-mutant DMG patients, with OS
at 12 months for resection and 11 months for biopsy (68). However, resection was associated
with longer ICU stays and higher neurological deficits. Moreover,
the surgical intervention did not improve the Karnofsky Performance
Status (68,69). Similarly, a systematic review
reported a 13.02% complication rate for stereotactic biopsy, with
no biopsy-related deaths and a diagnostic success rate of 97.4%
(69). While biopsies do not
markedly extend survival, they provide invaluable molecular data
for personalized therapies.
Radiotherapies
External beam radiotherapy (EBRT)
EBRT remains the standard treatment for DMG,
delivering 54–59.4 Gy over 30–33 sessions (70). While it temporarily alleviates
symptoms and delays progression for 6–12 months, recurrence is
almost inevitable within a year, with a median OS of 8–12 months
(71).
Hyper-fractionated radiotherapy and
hypo-fractionated radiotherapy
Alternative approaches have been tested, such as
hyper-fractionated (e.g., 70.2 Gy in smaller fractions) and
hypo-fractionated (e.g., 39 Gy in larger fractions) radiotherapy,
but have shown no significant survival benefits. Median survival
remains 7.8–8.5 months (71,72),
with logistical benefits being the primary advantage.
Proton radiotherapy
Proton therapy minimizes damage to the healthy
brain, particularly in pediatric patients, but does not markedly
improve long-term survival. Re-radiation after recurrence offers
limited benefits and poses a higher risk of radiation-induced
damage (73), such as neurological
and cognitive impairments (74,75).
Limitations of radiotherapy
Despite temporary relief, tumor progression is
inevitable, with progression-free survival (PFS) of only 6–8 months
and a 5-year survival rate <1% (76,77).
Combining radiotherapy with agents such as Bromodomain and
Extra-Terminal motif (BET) inhibitors or epigenetic drugs (such as
chitosan) shows potential in augmenting its tumor-killing effects
by disrupting DNA repair (66,78).
Chemotherapies
Chemotherapy serves as a complementary treatment for
DMG, largely as an adjunct to radiotherapy when resection is not
feasible. However, across prospective and registry-era experiences,
conventional cytotoxic chemotherapy has not produced a consistent
survival benefit beyond radiotherapy alone and its use is
increasingly framed within clinical trials or rational combinations
rather than as stand-alone escalation (Table I).
 | Table I.Summary of clinical trials on
chemotherapy, targeted therapy and immunotherapy in the treatment
of DMG. |
Table I.
Summary of clinical trials on
chemotherapy, targeted therapy and immunotherapy in the treatment
of DMG.
| First author/s,
year | Treatment type | Drug | Targets | Patients (n) | PFS (months) | mOS (months) | Other outcomes | (Refs.) |
|---|
| Bailey et
al, 2013 | Chemotherapy | Temozolomide | / | 43 | / | 9.5 | 1-year OS ~35%;
2-year OS ~17% | (79) |
| Chassot et
al, 2013 |
| Temozolomide |
| 22 | 7.5 | 11.7 |
| (80) |
| Massimino et
al, 2008 |
| Cisplatin +
Etoposide |
| 62 | 7 | / | 1-year OS: 45% | (87) |
| Ono et al,
2021 |
|
Cisplatin/Etoposide |
| 1 | / | / | OS >6 years | (89) |
| Veldhuijzen van
Zanten et al, 2017 |
| Gemcitabine |
| 9 | 4.8 | 8.7 |
| (90) |
| Kilburn et
al, 2018 |
| Capecitabine |
| 44 | / | / | 1-year PFS rate:
7.2% | (91) |
| Krystal et
al, 2024 |
| Irinotecan +
Bevacizumab + Mebendazole |
| 10 | 4.7 | 11.4 |
| (93) |
| Lin et al,
2024 |
| Cyclophosphamide +
GD2.CART |
| 11 | / | / | 7/11 neurological
improvement | (95) |
| Venneti et
al, 2023 | Targeted
Therapies | ONC-201 | ClpP | 30 | 3.4 | 9.3 |
| (100) |
| Arrillaga-Romany
et al, 2024 |
| ONC-201 | ClpP | 477 | / | / |
| (101) |
| Su et al,
2022 |
| Vorinostat | HDAC | 79 | / | / | 1-year PFS rate:
5.85%; 1-year OS rate:39.2% | (105) |
| Mueller et
al, 2023 |
| Panobinostat | HDAC | 7 | 7 | 26.1 |
| (104) |
| Neth et al,
2022 |
| Panobinostat | HDAC | 10 | 19 | 42 |
| (106) |
| DeWire et
al, 2020 |
| Ribociclib | CDK4/6 | 10 | / | 16.1 |
| (111) |
| DeWire et
al, 2022 |
| Ribociclib +
Everolimus | CDK4/6 + mTOR | 19 | / | 13.9 |
| (112) |
| Fleischhack et
al, 2019 |
| Nimotuzumab | EGFR | 42 | 5.8 | 9.4 |
| (114) |
| Liu et al,
2025 |
| Nimotuzumab | EGFR | 48 | 7.8 | 10.5 |
| (118) |
| Majzner et
al, 2022 | Immunotherapy | GD2-CAR T | / | 4 | / | / | 3/4 Tumor volume
reduction or stabilization | (120) |
| Lin et al,
2024 |
| C7R-GD2.CART |
| 11 | / | / | Feasibility;
on-target neuroinflammation/TIAN and CRS | (95) |
| Vitanza et
al, 2025 |
| B7-H3 CAR-T |
| 21 | / | / | Repetitive ICV
dosing feasible; DLT reported (intratumoral hemorrhage) | (122) |
| Pérez-Larraya et
al, 2022 |
| DNX-2401 |
| 12 | / | 17.8 |
| (124) |
| Grassl et
al, 2023 |
| H3K27M-vac |
| 8 | 6.2 | 12.8 |
| (125) |
| Mueller et
al, 2020 |
| H3.3K27M peptide
vaccine |
| 29 (A=19;
B=10) | / | 16.1 vs. 9.8 |
| (128) |
Temozolomide
TMZ is an oral alkylating agent whose key cytotoxic
lesion is O6-methylguanine, which can trigger
replication-associated mismatch repair-dependent DNA damage and
apoptosis. In a UK phase II study evaluating dose-dense TMZ with
standard radiotherapy for classical DIPG/DMG, median OS was 9.5
months, with 1-year OS of 35% and the regimen did not demonstrate a
survival benefit over radiotherapy alone; hematologic toxicity led
to treatment withdrawal and frequent dose reductions (79) (Table
I). Similarly, radiotherapy with concurrent and adjuvant TMZ
yielded median OS of 11.7 months and 1-year OS of 50%, but no
significant improvement in outcome and higher hematologic toxicity
compared with radiotherapy alone (80). A major biological constraint is
O6-methylguanine-DNA methyltransferase (MGMT)-mediated repair of
TMZ-induced O6 lesions: H3K27M-altered DMG frequently shows lack of
MGMT promoter methylation and increased MGMT expression, which is
associated with TMZ resistance in DMG models (81). Accordingly, pharmacologic MGMT
inhibition, such as O6-benzylguanine, has been limited by increased
hematologic toxicity and often necessitates substantial TMZ dose
reduction, constraining clinical utility (82,83).
Studies (84–86) demonstrate that BET inhibitors, as
well as other epigenetic modulators, including histone deacetylase
(HDAC) and EZH2 inhibitors, can sensitize DMG cells to TMZ, with
preclinical evidence supporting synergistic antitumor effects when
combined.
Cisplatin and etoposide
Cisplatin primarily induces cytotoxicity by forming
DNA cross-links, whereas etoposide inhibits topoisomerase II and
triggers DNA double-strand breaks; together they provide a
rationale for combination regimens that converge on
DNA-damage-driven apoptosis.
In a mono-institutional 20-year experience of
pediatric diffuse pontine gliomas treated with multi-agent therapy
incorporating cisplatin/etoposide (plus isotretinoin and others),
the reported median survival was 7 months and the 1-year survival
rate was 45% (87). Clinically,
limited benefit is assumed to be related in part to inadequate CNS
drug exposure under an intact/tight blood-brain barrier (BBB) in
DMG (88), although rare long
survivors exist. For example, a pineal-region DMG case with
>6-year survival after multimodal management in which initial
carboplatin-etoposide was ineffective but extensive resection,
high-dose radiotherapy and bevacizumab-based maintenance was
associated with durable control (89).
Gemcitabine
Gemcitabine is a cytidine analog that inhibits DNA
synthesis via DNA polymerase and ribonucleotide reductase and has
been explored as a radiosensitizer in DMG (9,90).
In a phase I/II trial of weekly gemcitabine with
radiotherapy, treatment was safe and well tolerated, with PFS 4.8
months and median OS 8.7 months (90); Quality of life tended to improve,
but survival was not superior to historical controls, supporting
mainly palliative value.
Capecitabine
Capecitabine is an oral prodrug of 5-fluorouracil;
because radiation can induce thymidine phosphorylase, capecitabine
has been tested as a potential RT-sensitizing strategy in DMG
(91). In the PBTC phase Il study
(91), capecitabine + RT was
tolerable, but outcomes were not improved: 1-year PFS 7.21 vs.
15.59% in the historical control (P=0.007) and no OS difference
(P=0.30), leading to the conclusion that capecitabine did not
improve prognosis in newly diagnosed DMG.
Irinotecan
Irinotecan is a topoisomerase I inhibitor that
stabilizes the cleavage complex and induces lethal DNA strand
breaks, thereby suppressing tumor proliferation (92). In a phase I trial, Krystal et
al (93) evaluated irinotecan
plus bevacizumab and mebendazole in 10 pediatric/AYA HGG patients,
including 7 DMG, reporting no dose limiting toxicities and a PFS
and OS of 4.7 and 11.4 months from treatment initiation; the
overall response rate was 33% (two PRs and one CR sustained for 10
months).
Cyclophosphamide
Cyclophosphamide is an alkylating pro-drug that
generates phosphoramide mustard, producing DNA cross-links and
triggering apoptosis (94).
In a phase I trial of intravenous GD2. CAR T cells,
cyclophosphamide (with fludarabine) was used for standard
lymphodepletion in 11 patients and therapy was delivered without
dose-limiting toxicity. The C7R-GD2.CAR T cohort developed grade 1
TIAN in 7/8 and achieved transient neurologic improvement (2 to
>12 months), with partial responses in 2/7 DMG patients by iRANO
(95).
Overall, conventional chemotherapies have shown
limited and largely non-practice-changing benefit in DMG. Future
efforts should prioritize rational combinations (such as chemo with
targeted/epigenetic or immunotherapies) and trial designs that
emphasize clinically meaningful endpoints, including neurologic
function and quality of life.
Targeted therapies
Targeted therapy for diffuse midline glioma aims to
exploit actionable biological dependencies that sustain tumor
growth. Current strategies include mitochondrial stress-based
approaches centered on caseinolytic protease P (ClpP)/imipridones
such as ONC201, epigenetic modulation such as HDAC inhibitors,
panobinostat for epigenetic regulation and combination therapies
targeting CDK4/6 and mTOR and receptor-directed interventions
focusing on EGFR inhibition. These modalities are being evaluated
to counteract core metabolic/epigenetic vulnerabilities and
aberrant signaling programs in DMG (Fig. 2; Table
I).
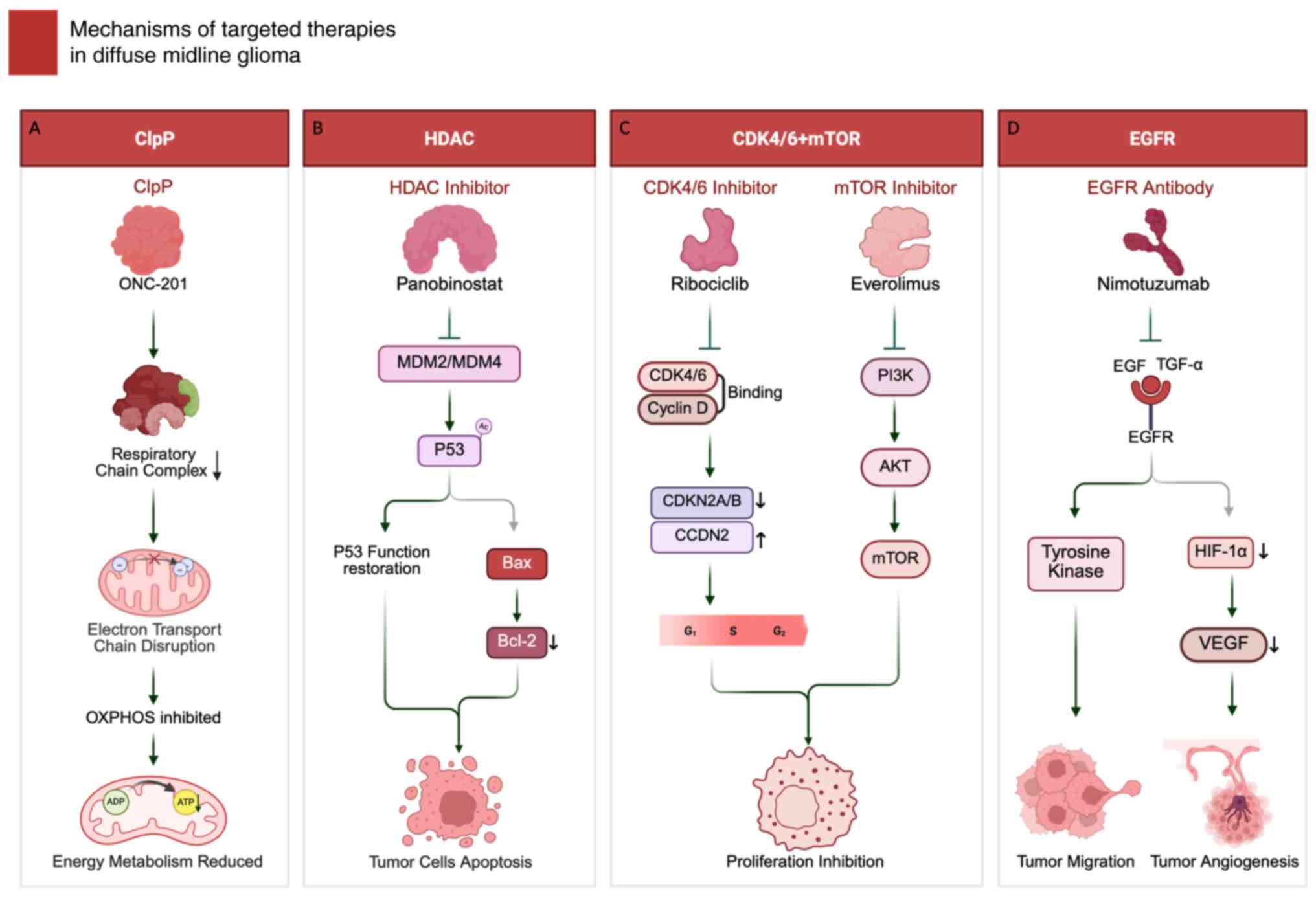 | Figure 2.Mechanisms of targeted therapy in
diffuse midline glioma. (A) ClpP agonist ONC-201 hyperactivates
mitochondrial protease ClpP, leading to respiratory chain protein
degradation, electron transport chain disruption, OXPHOS inhibition
and reduced ATP production. (B) HDAC inhibitor panobinostat
restores p53 function through MDM2/MDM4 modulation and enhanced p53
acetylation, upregulating pro-apoptotic Bax while downregulating
anti-apoptotic Bcl-2 to induce tumor cell apoptosis. (C) CDK4/6
inhibitor ribociclib blocks CDK4/6-cyclin D interaction, inducing
G1 arrest; mTOR inhibitor everolimus suppresses
PI3K/AKT/mTOR signaling to inhibit proliferation and metabolism.
(D) Anti-EGFR antibody nimotuzumab blocks EGF/TGF-α binding,
inhibiting downstream tyrosine kinase signaling and
HIF-1α/VEGF-mediated angiogenesis. Figure created using
BioRender.com. ClpP, caseinolytic protease P; HDAC, histone
deacetylase; mTOR, mammalian target of rapamycin; EGFR, epidermal
growth factor receptor; HIF-1α, hypoxia-inducible factor
1-alpha. |
ClpP
ClpP, a mitochondrial serine protease, forms the
ClpXP complex to maintain mitochondrial proteostasis. Pharmacologic
hyperactivation of ClpP can drive excessive proteolysis of
respiratory-chain proteins, leading to electron transport chain
disruption, OXPHOS inhibition, ATP depletion and apoptotic cell
death (96).
ONC201, an imipridone, acts as an allosteric ClpP
agonist and antagonizes DRD2. In H3K27M-altered DMG models,
ONC201-ClpP engagement promotes degradation of key respiratory
chain components, collapses mitochondrial bioenergetics
(ETC/OXPHOS) and suppresses tumor energy metabolism-features that
match the present study's mechanistic schematic (96,97)
(Fig. 2A). Downstream, ONC201
induces an ATF4/CHOP-linked integrated stress response and can
promote DR5/TRAIL-mediated apoptosis (98), with reported modulation of Akt/ERK
signaling consistent with a pro-apoptotic shift (99).
Clinically, integrated analysis of ONC201-treated
H3K27M-mutant DMG reported longer survival when initiated after
radiotherapy but before recurrence (median OS 21.7 months; PFS 7.3
months) compared with initiation after recurrence (median OS 9.3
months; PFS 3.4 months), with molecular correlates including
increased 2-hydroxyglutarate and partial restoration of H3K27me3
(100). A randomized, double-blind
phase 3 trial (101) is ongoing to
evaluate ONC201 as post-radiotherapy maintenance therapy in newly
diagnosed H3 K27M-mutant diffuse glioma, with OS and PFS as primary
endpoints.
HDAC
HDAC inhibitors reprogram chromatin by increasing
histone acetylation and can reactivate the MDM2/MDM4-p53 axis, in
part by reducing MDM2/MDM4 repression and enhancing p53
acetylation/transcriptional activity, thereby promoting apoptosis
(102). Downstream, they
upregulate pro-apoptotic genes such as Bax while downregulate
anti-apoptotic genes such as Bcl-2, activating the intrinsic
apoptotic pathway (103) (Fig. 2B).
Panobinostat is a multi-HDAC inhibitor with strong
preclinical rationale in DMG, but systemic delivery is constrained
by exposure/toxicity and uncertain BBB penetration, motivating
local delivery approaches (104).
In a phase I/II trial by Su et al (105), panobinostat combined with
radiotherapy and voriostat, followed by maintenance, was
well-tolerated but showed limited survival benefits, with a 1-year
PFS of 5.85% and OS of 39.2%. By contrast, PNOC015 using repeat CED
of MTX110 reported median OS of 26.1 months and median PFS 7
months, supporting intra-tumoral HDAC inhibition with reduced
systemic exposure (104). In
adults, compassionate-use panobinostat showed good tolerability
with median OS 42 months and median PFS 19 months in a small
series, suggesting potential context-dependence that requires
prospective validation (106).
Cyclin-dependent kinases 4 and 6
(CDK4/6) + mTOR
CDK4/6 regulate the G1-to-S phase
transition by phosphorylating retinoblastoma (RB) protein via
cyclin D, activating E2F transcription factors and driving cell
cycle progression (107,108). The mTOR in the PI3K/Akt pathway
governs metabolism, protein synthesis and proliferation, with
aberrations frequently observed in DMG, contributing to unchecked
tumor proliferation and metabolic dependency (107).
Ribociclib, a CDK4/6 inhibitor, halts cell cycle
progression at the G1 phase by disrupting CDK4/6-cyclin
D interaction (109) (Fig. 2C), targeting CDKN2A/B deletions and
CCND2 amplifications seen in DMG. Meanwhile, everolimus, which is a
mTORC1 inhibitor, suppresses tumor proliferation, glycolysis and
angiogenesis (110), particularly
in tumors with H3.1 mutations linked to PIK3CA or PIK3R1
alterations (40). The combination
of ribociclib and everolimus provides dual inhibition of cell cycle
and metabolic pathways, enhancing antitumor effects with reduced
overlapping toxicity.
A phase I/II study of post-radiotherapy single-agent
ribociclib reported feasibility with myelosuppression as the main
severe toxicity and a median OS of 16.1 months in DIPG/DMG,
reinforcing ongoing interest in rational combinations and
biomarker-guided selection (111).
As additional supportive evidence for CDK4/6 targeting in this
setting, A phase I trial assessed this combination in 19 pediatric
DMG patients following radiotherapy, establishing phase II doses of
170 mg/m2 for ribociclib and 1.5 mg/m2 for
everolimus (112). The median OS
was 13.9 months, with 12-, 24- and 36-month survival rates of 53.3,
38.9 and 38.9%, respectively. These promising results warrant
further investigation of this approach.
EGFR
EGFR is a receptor tyrosine kinase that propagates
mitogenic and pro-survival signals mainly through the
RAS-RAF-MEK-ERK and PI3K-AKT cascades, thereby promoting
proliferation, invasion and treatment resistance (113). Additionally, EGFR pathway activity
intersects with hypoxia/angiogenic programs, in part via
PI3K/AKT-hypoxia-inducible factor 1-alpha (HIF-1α)-VEGF, providing
a rationale for EGFR-directed strategies to suppress both tumor
growth and angiogenesis (114).
Nimotuzumab, an anti-EGFR monoclonal antibody,
blocks EGF/TGF-α-EGFR engagement and thereby attenuates downstream
oncogenic signaling; clinically, however, its benefit has been
inconsistent (115). It induces
G1 phase arrest, preventing tumor cell progression into
the S phase (116). Additionally,
it destabilizes HIF-1α, suppresses VEGF transcription and reduces
angiogenesis, collectively curbing tumor growth and metastasis
(117) (Fig. 2D).
A phase III trial in 42 DMG patients evaluated
nimotuzumab with radiotherapy, reporting a median PFS of 5.8 months
and median OS of 9.4 months (114). One- and two-year survival rates
were 33.3 and 4.8%, respectively, with rare serious events
including intratumoral bleeding. More recently, adding nimotuzumab
to chemoradiation was feasible, with median OS 10.5 months and PFS
7.8 months, but did not meet a prespecified ORR-improvement
threshold vs. historical data, supporting continued
biomarker-guided optimization rather than routine adoption
(118).
Immunotherapies
Diffuse midline glioma is characterized as an
immunologically ‘cold tumor’, with low lymphocytic infiltration and
a microenvironment dominated by myeloid-lineage populations, which
collectively constrains adaptive anti-tumor immunity and limits the
efficacy of conventional immunotherapy approaches (119). These features highlight why DMG
immunotherapy remains early-stage and why locoregional,
antigen-directed strategies have become a central development focus
(Table I).
Chimeric antigen receptor (CAR)-T
therapy
CAR-T-cell therapy aims to bypass limited endogenous
T-cell priming by engineering T cells to recognize DMG-associated
surface antigens, most prominently GD2 and B7-H3 (120). Upon antigen engagement, CAR
signaling through CD3Z activates canonical T-cell effector
programs, such as cytokine production and cytotoxicity, enabling
direct killing of antigen-expressing tumor cells.
For GD2 targeting, first-in-human clinical
experience demonstrated feasibility and early signals of activity,
while highlighting neuroinflammation as an on-target risk that
requires proactive monitoring and management (120). More recently, a phase I study of
constitutively active IL-7 receptor-engineered GD2 CAR T cells
(C7R-GD2.CART), further supported feasibility in H3K27-altered DMG,
with tumor-inflammation-associated neurotoxicity and
cytokine-release syndrome as key safety considerations and
preliminary objective responses observed in a subset (95).
In parallel, B7-H3-directed CAR T cells have
advanced through BrainChild-03 using repeated
intra-cerebroventricular dosing; early DIPG evaluable cases showed
no dose-limiting toxicities and evidence of local immune activation
with a durable clinical/radiographic improvement in one patient
(121). Updated Arm C results in
DIPG expanded these observations by establishing feasibility of
repetitive ICV dosing at higher planned dose regimens and providing
benchmark survival and neurotoxicity profiles for future multi-site
evaluation (122).
Oncolytic virus DNX-2401
DNX-2401 is a conditionally replicating adenovirus
designed to preferentially replicate in tumors with RB pathway
dysregulation via a Δ24 deletion in E1A and its RGD modification
can facilitate integrin-mediated entry (123).
In a phase I study of 12 newly diagnosed DMG/DIPG
patients, a single stereotactic intra-tumoral infusion of DNX-2401
(1×1010 or 5×1010 viral particles) followed
by radiotherapy achieved 92% disease control and a median OS of
17.8 months; 12- and 18-month OS were 75 and 50%, respectively
(124). Correlative analysis
supported treatment-associated immune activation, including
increased T-cell-related activity, aligning with an oncolytic
‘viro-immunotherapy’ rationale in DMG.
Cancer vaccine
Cancer vaccines aims to induce tumor
antigen-specific adaptive immunity. In DMG, the shared clonal
neo-antigen H3K27M has enabled peptide-vaccine strategies that
predominantly prime mutation-specific CD4+ T-cell responses, with
potential downstream support for cytotoxic effector programs and
can increase immune infiltration in selected patients (125). Mechanistically, deeper immune
profiling in a long-term recovered responder following H3K27M
vaccination, identified broad HLA background coverage with both B-
and T-cell responses, supporting the concept that vaccine-elicited
anti-H3K27M immunity can be durable in selected cases (126,127).
The first-in-human H3K27M peptide vaccine
(H3K27M-vac) study enrolled eight adults with H3K27M-altered DMG
and showed favorable tolerability with mutation-specific immune
responses dominated by CD4+ T cells; reported outcomes included
median PFS 6.2 months and median OS 12.8 months (125). In younger patients, PNOC007
evaluated an HLA-restricted H3.3K27M peptide vaccine with adjuvant
immunostimulation after radiotherapy and demonstrated
immunogenicity with acceptable safety, reporting ~40% (DIPG) and
~39% (non-pontine DMG) 12-month OS benchmarks (128). INTERCEPT H3 is currently
evaluating the use of H3K27M-vac concurrently with standard
radiotherapy and in combination with the PD-L1 antibody
atezolizumab for maintenance after radiotherapy to clarify its
safety and immunogenicity (129).
Conclusion
Diffuse midline glioma, particularly the
H3K27M-mutated subtype, remains a formidable CNS malignancy due to
its unique molecular features and invasiveness. The present review
emphasized the pivotal role of histone alterations, especially
H3K27M, in reshaping chromatin states and gene expression programs
that drive malignant proliferation, invasion and therapeutic
resistance. Although surgery and radiotherapy remain foundational,
their effect is constrained by anatomical inoperability and diffuse
infiltration. Chemotherapy provides only a modest benefit and is
frequently limited by resistance. By contrast, emerging targeted
approaches, including ONC-201, panobinostat-based strategies and
CDK4/6 inhibition combined with mTOR blockade, directly address
molecular vulnerabilities, while early immunotherapy studies,
including locoregional CAR-T platforms, DNX-2401 virotherapy and
H3K27M-directed vaccination, illustrate feasibility and biological
activity in selected settings.
Limitations of the current evidence base include
small patient numbers, heterogeneous cohorts (age, tumor location,
co-alterations and prior therapy) and variable drug-delivery
strategies, which collectively limit cross-trial comparability and
may inflate apparent signals in single-arm studies. Future progress
will require biomarker-guided stratification, rational combination
regimens aligned to DMG biology (including radiotherapy-integrated
approaches) and delivery-optimized platforms that improve
intra-tumoral exposure while maintaining neurological safety.
Equally important, standardized response assessment and integrated
correlative monitoring (molecular and immune profiling,
longitudinal sampling, where feasible) should be embedded into
multicenter trials to distinguish durable benefit from transient
inflammatory effects and to enable definitive efficacy testing.
Acknowledgements
Not applicable.
Funding
The authors gratefully acknowledge the funding support provided
by the National High-Level Hospital Clinical Research Funding
(grant no. 2022-PUMCH-B-113), the CAMS Innovation Fund for Medical
Sciences (grant no. 2021-12M-1-014) and the National Natural
Science Foundation of China (grant no. 82151302), which markedly
contributed to the advance of this research.
Availability of data and materials
Not applicable.
Authors' contributions
WBW and WLC performed the literature search,
extracted and synthesized data on the epigenetic mechanisms and
therapeutic strategies of diffuse midline glioma and drafted the
original manuscript. WBW prepared and revised the figures and
schematic illustrations and WLC organized the tables. WBM and YW
conceived and supervised the overall topic and structure of the
present review, provided critical revision of the manuscript for
important intellectual content and oversaw the final interpretation
of the literature. YW and WBM are the corresponding authors and
take primary responsibility for the integrity of the work as a
whole. WBW and WLC contributed equally to this work. All authors
read and approved the final manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ostrom QT, Price M, Neff C, Cioffi G,
Waite KA, Kruchko C and Barnholtz-Sloan JS: CBTRUS statistical
report: Primary brain and other central nervous system tumors
diagnosed in the United States in 2016–2020. Neuro Oncol. 25 (12
Suppl 2):iv1–iv99. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fonseca A, Afzal S, Bowes L, Crooks B,
Larouche V, Jabado N, Perreault S, Johnston DL, Zelcer S, Fleming
A, et al: Pontine gliomas a 10-year population-based study: A
report from The Canadian Paediatric Brain Tumour Consortium
(CPBTC). J Neurooncol. 149:45–54. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Patil N, Kelly ME, Yeboa DN, Buerki RA,
Cioffi G, Balaji S, Ostrom QT, Kruchko C and Barnholtz-Sloan JS:
Epidemiology of brainstem high-grade gliomas in children and
adolescents in the United States, 2000–2017. Neuro Oncol.
23:990–998. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Erker C, Lane A, Chaney B, Leary S,
Minturn JE, Bartels U, Packer RJ, Dorris K, Gottardo NG, Warren KE,
et al: Characteristics of patients ≥10 years of age with diffuse
intrinsic pontine glioma: A report from the International DIPG/DMG
registry. Neuro Oncol. 24:141–152. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hoffman LM, Veldhuijzen van Zanten SEM,
Colditz N, Baugh J, Chaney B, Hoffmann M, Lane A, Fuller C, Miles
L, Hawkins C, et al: Clinical, radiologic, pathologic, and
molecular characteristics of long-term survivors of diffuse
intrinsic pontine glioma (DIPG): A collaborative report from the
international and European society for pediatric oncology DIPG
registries. J Clin Oncol. 36:1963–1972. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ying Y, Liu X, Li X, Mei N, Ruan Z, Lu Y
and Yin B: Distinct MRI characteristics of spinal cord diffuse
midline glioma, H3 K27-altered in comparison to spinal cord glioma
without H3 K27-alteration and demyelination disorder. Acta Radiol.
65:284–293. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nafe R, Porto L, Samp PF, You SJ and
Hattingen E: Adult-type and Pediatric-type diffuse gliomas what the
neuroradiologist should know. Clin Neuroradiol. 33:611–624. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Coleman C, Chen K, Lu A, Seashore E,
Stoller S, Davis T, Braunstein S, Gupta N and Mueller S:
Interdisciplinary care of children with diffuse midline glioma.
Neoplasia. 35:1008512023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Saratsis AM, Knowles T, Petrovic A and
Nazarian J: H3K27M mutant glioma: Disease definition and biological
underpinnings. Neuro Oncol. 26 (Suppl 2):S92–S100. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tarapore RS, Arain S, Blaine E, Hsiung A,
Melemed AS and Allen JE: Immunohistochemistry detection of Histone
H3 K27M mutation in human glioma tissue. Appl Immunohistochem Mol
Morphol. 32:96–101. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Duchatel RJ, Jackson ER, Alvaro F, Nixon
B, Hondermarck H and Dun MD: Signal transduction in diffuse
intrinsic pontine glioma. Proteomics. 19:e18004792019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Testa U, Castelli G and Pelosi E: CAR-T
cells in the treatment of nervous system tumors. Cancers (Basel).
16:29132024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Harris W: A case of pontine glioma, with
special reference to the paths of gustatory sensation. Proc R Soc
Med. 19:1–5. 1926.PubMed/NCBI
|
|
14
|
Janssens GO, Kramm CM and von Bueren AO:
Diffuse intrinsic pontine gliomas (DIPG) at recurrence: Is there a
window to test new therapies in some patients? J Neurooncol.
139:5012018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ward E, DeSantis C, Robbins A, Kohler B
and Jemal A: Childhood and adolescent cancer statistics, 2014. CA
Cancer J Clin. 64:83–103. 2014.PubMed/NCBI
|
|
16
|
Gwak HS and Park HJ: Developing
chemotherapy for diffuse pontine intrinsic gliomas (DIPG). Crit Rev
Oncol Hematol. 120:111–119. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 World Health Organization
classification of tumors of the central nervous system: A summary.
Acta Neuropathol. 131:803–820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang TY, Piunti A, Lulla RR, Qi J,
Horbinski CM, Tomita T, James CD, Shilatifard A and Saratsis AM:
Detection of Histone H3 mutations in cerebrospinal fluid-derived
tumor DNA from children with diffuse midline glioma. Acta
Neuropathol Commun. 5:282017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Q, Niu W and Pan H: Targeted therapy
with anlotinib for a H3K27M mutation diffuse midline glioma patient
with PDGFR-alpha mutation: A case report. Acta Neurochir (Wien).
164:2063–2066. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
McNamara C, Mankad K, Thust S, Dixon L,
Limback-Stanic C, D'Arco F, Jacques TS and Löbel U: 2021 WHO
classification of tumours of the central nervous system: A review
for the neuroradiologist. Neuroradiology. 64:1919–1950. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cooney TM, Lubanszky E, Prasad R, Hawkins
C and Mueller S: Diffuse midline glioma: review of epigenetics. J
Neurooncol. 150:27–34. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Park YW, Vollmuth P, Foltyn-Dumitru M,
Sahm F, Ahn SS, Chang JH and Kim SH: The 2021 WHO Classification
for Gliomas and implications on imaging diagnosis: Part 2-summary
of imaging findings on pediatric-type diffuse high-grade gliomas,
pediatric-type diffuse low-grade gliomas, and circumscribed
astrocytic gliomas. J Magn Reson Imaging. 58:690–708. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cassim A, Dun MD, Gallego-Ortega D and
Valdes-Mora F: EZHIP's role in diffuse midline glioma: echoes of
oncohistones? Trends Cancer. 10:1095–1105. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hoffman LM, DeWire M, Ryall S, Buczkowicz
P, Leach J, Miles L, Ramani A, Brudno M, Kumar SS, Drissi R, et al:
Spatial genomic heterogeneity in diffuse intrinsic pontine and
midline high-grade glioma: Implications for diagnostic biopsy and
targeted therapeutics. Acta Neuropathol Commun. 4:12016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Greer EL and Shi Y: Histone methylation: A
dynamic mark in health, disease and inheritance. Nat Rev Genet.
13:343–357. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hyun K, Jeon J, Park K and Kim J: Writing,
erasing and reading histone lysine methylations. Exp Mol Med.
49:e3242017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shvedunova M and Akhtar A: Modulation of
cellular processes by histone and non-histone protein acetylation.
Nat Rev Mol Cell Biol. 23:329–349. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
An T, Liu Y, Gourguechon S, Wang CC and Li
Z: CDK phosphorylation of translation initiation factors couples
protein translation with cell-cycle transition. Cell Rep.
25:3204–3214.e5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Justice JL, Reed TJ, Phelan B, Greco TM,
Hutton JE and Cristea IM: DNA-PK and ATM drive phosphorylation
signatures that antagonistically regulate cytokine responses to
herpesvirus infection or DNA damage. Cell Syst. 15:339–361.e8.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen Q, Bian C, Wang X, Liu X, Ahmad
Kassab M, Yu Y and Yu X: ADP-ribosylation of histone variant H2AX
promotes base excision repair. EMBO J. 40:e1045422021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mattiroli F and Penengo L: Histone
Ubiquitination: An integrative signaling platform in genome
stability. Trends Genet. 37:566–581. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang X and Zhang Z: Oncohistone mutations
in diffuse intrinsic pontine glioma. Trends Cancer. 5:799–808.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schwartzentruber J, Korshunov A, Liu XY,
Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA,
Tönjes M, et al: Driver mutations in histone H3.3 and chromatin
remodelling genes in paediatric glioblastoma. Nature. 482:226–231.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qin J, Wen B, Liang Y, Yu W and Li H:
Histone modifications and their role in colorectal cancer (review).
Pathol Oncol Res. 26:2023–2033. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Caslini C, Hong S, Ban YJ, Chen XS and
Ince TA: HDAC7 regulates histone 3 lysine 27 acetylation and
transcriptional activity at super-enhancer-associated genes in
breast cancer stem cells. Oncogene. 38:6599–6614. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Deng L, Meng T, Chen L, Wei W and Wang P:
The role of ubiquitination in tumorigenesis and targeted drug
discovery. Signal Transduct Target Ther. 5:112020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cole AJ, Clifton-Bligh R and Marsh DJ:
Histone H2B monoubiquitination: Roles to play in human malignancy.
Endocr Relat Cancer. 22:T19–T33. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen X, Song N, Matsumoto K, Nanashima A,
Nagayasu T, Hayashi T, Ying M, Endo D, Wu Z and Koji T: High
expression of trimethylated histone H3 at lysine 27 predicts better
prognosis in non-small cell lung cancer. Int J Oncol. 43:1467–1480.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Stitzlein LM, Adams JT, Stitzlein EN,
Dudley RW and Chandra J: Current and future therapeutic strategies
for high-grade gliomas leveraging the interplay between epigenetic
regulators and kinase signaling networks. J Exp Clin Cancer Res.
43:122024. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pearson AD, DuBois SG, Macy ME, de Rojas
T, Donoghue M, Weiner S, Knoderer H, Bernardi R, Buenger V, Canaud
G, et al: Paediatric strategy forum for medicinal product
development of PI3-K, mTOR, AKT and GSK3beta inhibitors in children
and adolescents with cancer. Eur J Cancer. 207:1141452024.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yan Y and Zhang J: Mechanisms of tamoxifen
resistance: Insight from long non-coding RNAs. Front Oncol.
14:14585882024. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang Y, Tapinos N, Lulla R and El-Deiry
WS: Dopamine pre-treatment impairs the anti-cancer effect of
integrated stress response- and TRAIL pathway-inducing ONC201,
ONC206 and ONC212 imipridones in pancreatic, colorectal cancer but
not DMG cells. Am J Cancer Res. 14:2453–2464. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nandy D, Rajam SM and Dutta D: A three
layered histone epigenetics in breast cancer metastasis. Cell
Biosci. 10:522020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Blasco-Santana L and Colmenero I:
Molecular and pathological features of paediatric high-grade
gliomas. Int J Mol Sci. 25:84982024. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Justin N, Zhang Y, Tarricone C, Martin SR,
Chen S, Underwood E, De Marco V, Haire LF, Walker PA, Reinberg D,
et al: Structural basis of oncogenic histone H3K27M inhibition of
human polycomb repressive complex 2. Nat Commun. 7:113162016.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Arakaki AKS, Szulzewsky F, Gilbert MR,
Gujral TS and Holland EC: Utilizing preclinical models to develop
targeted therapies for rare central nervous system cancers. Neuro
Oncol. 23 (23 Suppl 5):S4–S15. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pal S, Kaplan JP, Nguyen H, Stopka SA,
Savani MR, Regan MS, Nguyen QD, Jones KL, Moreau LA, Peng J, et al:
A druggable addiction to de novo pyrimidine biosynthesis in diffuse
midline glioma. Cancer Cell. 40:957–972.e10. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tsai JW, Cejas P, Coppola M, Wang DK,
Patel S, Wu DW, Arounleut P, Wei X, Zhou N, Syamala S, et al:
Abstract 3562: Dissecting mechanisms underlying FOXR2-mediated
gliomagenesis in diffuse midline gliomas. Cancer Res. 83:35622023.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Vanan MI, Underhill DA and Eisenstat DD:
Targeting epigenetic pathways in the treatment of pediatric diffuse
(High Grade) gliomas. Neurotherapeutics. 14:274–283. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Graham MS and Mellinghoff IK:
Histone-Mutant glioma: Molecular mechanisms, preclinical models,
and implications for therapy. Int J Mol Sci. 21:71932020.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Paine DZ: Molecular Determinants of
Diffuse Midline Glioma of the Pons Vulnerability to the Histone
Deacetylase Inhibitor, Quisinostat. Doctoral dissertation.
University of Arizona; Tucson, USA: 2023
|
|
52
|
Xu Y, Wu F, Tan L, Kong L, Xiong L, Deng
J, Barbera AJ, Zheng L, Zhang H, Huang S, et al: Genome-wide
Regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in
mouse embryonic stem cells. Mol Cell. 42:451–464. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hashizume R: Epigenetic targeted therapy
for diffuse intrinsic pontine glioma. Neurol Med Chir (Tokyo).
57:331–342. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Markouli M, Strepkos D, Papavassiliou KA,
Papavassiliou AG and Piperi C: Crosstalk of epigenetic and
metabolic signaling underpinning glioblastoma pathogenesis. Cancers
(Basel). 14:26552022. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ross SE, Holliday H, Tsoli M, Ziegler DS
and Dinger ME: Abstract B015: RNA N6-methyladenosine (m6A) as a
therapeutic target in Diffuse Midline Glioma (DMG). Cancer Res.
84:B015. 2024. View Article : Google Scholar
|
|
56
|
Menez V, Kergrohen T, Shasha T,
Silva-Evangelista C, Le Dret L, Auffret L, Subecz C, Lancien M,
Ajlil Y, Vilchis IS, et al: VRK3 depletion induces cell cycle
arrest and metabolic reprogramming of pontine diffuse midline
glioma-H3K27 altered cells. Front Oncol. 13:12293122023. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Williams EA, Brastianos PK, Wakimoto H,
Zolal A, Filbin MG, Cahill DP, Santagata S and Juratli TA: A
comprehensive genomic study of 390 H3F3A-mutant pediatric and adult
diffuse high-grade gliomas, CNS WHO grade 4. Acta Neuropathol.
146:515–525. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yan Y: Abstract 1719: Targeting H3.3
serine 28 phosphorylation as a novel therapeutic strategy for
diffuse midline glioma. Cancer Res. 84:1719. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mangoli A, Valentine V, Maingi SM, Wu SR,
Liu HQ, Aksu M, Jain V, Foreman BE, Regal JA, Weidenhammer LB, et
al: Disruption of ataxia telangiectasia-mutated kinase enhances
radiation therapy efficacy in spatially directed diffuse midline
glioma models. J Clin Invest. 135:e1793952025. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhang X, Duan S, Apostolou PE, Wu X,
Watanabe J, Gallitto M, Barron T, Taylor KR, Woo PJ, Hua X, et al:
CHD2 Regulates Neuron-glioma interactions in pediatric glioma.
Cancer Discov. 14:1732–1754. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Diaz AK and Baker SJ: The genetic
signatures of pediatric high-grade glioma: No longer a one-act
play. Semin Radiat Oncol. 24:240–247. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sharma M, Barravecchia I, Magnuson B,
Ferris SF, Apfelbaum A, Mbah NE, Cruz J, Krishnamoorthy V, Teis R,
Kauss M, et al: Histone H3 K27M-mediated regulation of cancer cell
stemness and differentiation in diffuse midline glioma. Neoplasia.
44:1009312023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Mota M, Sweha SR, Pun M, Natarajan SK,
Ding Y, Chung C, Hawes D, Yang F, Judkins AR, Samajdar S, et al:
Targeting SWI/SNF ATPases in H3.3K27M diffuse intrinsic pontine
gliomas. Proc Natl Acad Sci USA. 120:e22211751202023. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Xin DE, Liao Y, Rao R, Ogurek S, Sengupta
S, Xin M, Bayat AE, Seibel WL, Graham RT, Koschmann C and Lu QR:
Chaetocin-mediated SUV39H1 inhibition targets stemness and
oncogenic networks of diffuse midline gliomas and synergizes with
ONC201. Neuro Oncol. 26:735–748. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Fang J, Huang Y, Mao G, Yang S, Rennert G,
Gu L, Li H and Li GM: Cancer-driving H3G34V/R/D mutations block
H3K36 methylation and H3K36me3-MutSα interaction. Proc Natl Acad
Sci USA. 115:9598–9603. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Sheikh SR, Patel NJ and Recinos VMR:
Safety and technical efficacy of pediatric brainstem biopsies: An
updated meta-analysis of 1000+ children. World Neurosurg.
189:428–438.e2. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Walker DA, Liu J, Kieran M, Jabado N,
Picton S, Packer R and St Rose C; CPN Paris 2011 Conference
Consensus Group, : A multi-disciplinary consensus statement
concerning surgical approaches to Low-grade, High-grade
astrocytomas and diffuse intrinsic pontine gliomas in childhood
(CPN Paris 2011) using the Delphi method. Neuro Oncol. 15:462–468.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ryba A, Özdemir Z, Nissimov N, Hönikl L,
Neidert N, Jakobs M, Kalasauskas D, Krigers A, Thomé C, Freyschlag
CF, et al: Insights from a multicenter study on adult H3
K27M-mutated glioma: Surgical resection's limited influence on
overall survival, ATRX as molecular prognosticator. Neuro Oncol.
26:1479–1493. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Dalmage M, LoPresti MA, Sarkar P,
Ranganathan S, Abdelmageed S, Pagadala M, Shlobin NA, Lam S and
DeCuypere M: Survival and neurological outcomes after stereotactic
biopsy of diffuse intrinsic pontine glioma: A systematic review. J
Neurosurg Pediatr. 32:665–672. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Gajjar A, Mahajan A, Abdelbaki M, Anderson
C, Antony R, Bale T, Bindra R, Bowers DC, Cohen K, Cole B, et al:
Pediatric central nervous system cancers, version 2.2023, NCCN
clinical practice guidelines in oncology. J Natl Compr Canc Netw.
20:1339–1362. 2022.PubMed/NCBI
|
|
71
|
Kim HJ and Suh CO: Radiotherapy for
diffuse intrinsic pontine glioma: Insufficient but indispensable.
Brain Tumor Res Treat. 11:79–85. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Cohen KJ, Jabado N and Grill J: Diffuse
intrinsic pontine gliomas-current management and new biologic
insights. Is there a glimmer of hope? Neuro Oncol. 19:1025–1034.
2017.PubMed/NCBI
|
|
73
|
Wawrzuta D, Chojnacka M, Drogosiewicz M,
Pedziwiatr K and Dembowska-Baginska B: Reirradiation for diffuse
intrinsic pontine glioma: Prognostic radiomic factors at
progression. Strahlenther Onkol. 200:797–804. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Esparragosa Vazquez I and Ducray F: The
role of radiotherapy, chemotherapy, and targeted therapies in adult
intramedullary spinal cord tumors. Cancers (Basel). 16:27812024.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yang Z, Sun L, Chen H, Sun C and Xia L:
New progress in the treatment of diffuse midline glioma with H3K27M
alteration. Heliyon. 10:e248772024. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Argersinger DP, Rivas SR, Shah AH, Jackson
S and Heiss JD: New developments in the pathogenesis, therapeutic
targeting, and treatment of H3K27M-mutant diffuse midline glioma.
Cancers (Basel). 13:52802021. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Mandorino M, Maitra A, Armenise D,
Baldelli OM, Miciaccia M, Ferorelli S, Perrone MG and Scilimati A:
Pediatric diffuse midline glioma H3K27-altered: From developmental
origins to therapeutic challenges. Cancers (Basel). 16:18142024.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Watanabe J, Clutter MR, Gullette MJ,
Sasaki T, Uchida E, Kaur S, Mo Y, Abe K, Ishi Y, Takata N, et al:
BET bromodomain inhibition potentiates radiosensitivity in models
of H3K27-altered diffuse midline glioma. J Clin Invest.
134:e1747942024. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Bailey S, Howman A, Wheatley K, Wherton D,
Boota N, Pizer B, Fisher D, Kearns P, Picton S, Saran F, et al:
Diffuse intrinsic pontine glioma treated with prolonged
temozolomide and radiotherapy-results of a United Kingdom phase II
trial (CNS 2007 04). Eur J Cancer. 49:3856–3862. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Chassot A, Canale S, Varlet P, Puget S,
Roujeau T, Negretti L, Dhermain F, Rialland X, Raquin MA, Grill J,
et al: Radiotherapy with concurrent and adjuvant temozolomide in
children with newly diagnosed diffuse intrinsic pontine glioma. J
Neurooncol. 106:399–407. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Abe H, Natsumeda M, Kanemaru Y, Watanabe
J, Tsukamoto Y, Okada M, Yoshimura J, Oishi M and Fujii Y: MGMT
expression contributes to temozolomide resistance in H3K27M-mutant
diffuse midline gliomas and MGMT silencing to temozolomide
sensitivity in IDH-mutant gliomas. Neurol Med Chir (Tokyo).
58:290–295. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Brandes AA, Tosoni A, Cavallo G, Reni M,
Franceschi E, Bonaldi L, Bertorelle R, Gardiman M, Ghimenton C,
Iuzzolino P, et al: Correlations between 06-methylguanine DNA
methyltransferase promoter methylation status, 1p and 19q
deletions, and response to temozolomide in anaplastic and recurrent
oligodendroglioma: A prospective GICNO study. J Clin Oncol.
24:4746–4753. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Warren KE, Aikin AA, Libucha M, Widemann
BC, Fox E, Packer RJ and Balis FM: Phase I study of
O6-benzylguanine and temozolomide administered daily for 5 days to
pediatric patients with solid tumors. J Clin Oncol. 23:7646–7653.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Zhang Y, Dong W, Zhu J, Wang L, Wu X and
Shan H: Combination of EZH2 inhibitor and BET inhibitor for
treatment of diffuse intrinsic pontine glioma. Cell Biosci.
7:562017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Sasaki T, Katagi H, Goldman S, Becher OJ
and Hashizume R: Convection-enhanced delivery of enhancer of zeste
homolog-2 (EZH2) inhibitor for the treatment of diffuse intrinsic
pontine glioma. Neurosurgery. 87:E680–E688. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Singleton WGB, Bienemann AS, Woolley M,
Johnson D, Lewis O, Wyatt MJ, Damment SJP, Boulter LJ, Killick-Cole
CL, Asby DJ and Gill SS: The distribution, clearance, and brainstem
toxicity of panobinostat administered by convection-enhanced
delivery. J Neurosurg Pediatr. 22:288–296. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Massimino M, Spreafico F, Biassoni V,
Simonetti F, Riva D, Trecate G, Giombini S, Poggi G, Pecori E,
Pignoli E, et al: Diffuse pontine gliomas in children: Changing
strategies, changing results? A mono-institutional 20-year
experience. J Neurooncol. 87:355–361. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Tsoli M, Ung C, Upton DH, Venkat P,
Salerno A, Pandher R, Mayoh C, Vittorio O and Ziegler DS: Dipg-35.
Overcoming the blood-brain barrier challenge in diffuse midline
glioma. Neuro Oncol. 26 (Suppl 4):02024. View Article : Google Scholar
|
|
89
|
Ono T, Kuwashige H, Adachi JI, Takahashi
M, Oda M, Kumabe T and Shimizu H: Long-term survival of a patient
with diffuse midline glioma in the pineal region: A case report and
literature review. Surg Neurol Int. 12:6122021. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Veldhuijzen van Zanten SEM, El-Khouly FE,
Jansen MHA, Bakker DP, Sanchez Aliaga E, Haasbeek CJA, Wolf NI,
Zwaan CM, Vandertop WP, van Vuurden DG and Kaspers GJL: A phase
I/II study of gemcitabine during radiotherapy in children with
newly diagnosed diffuse intrinsic pontine glioma. J Neurooncol.
135:307–315. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Kilburn LB, Kocak M, Baxter P, Poussaint
TY, Paulino AC, McIntyre C, Lemenuel-Diot A, Lopez-Diaz C, Kun L,
Chintagumpala M, et al: A pediatric brain tumor consortium phase II
trial of capecitabine rapidly disintegrating tablets with
concomitant radiation therapy in children with newly diagnosed
diffuse intrinsic pontine gliomas. Pediatr Blood Cancer.
65:10.1002/pbc.26832. 2018. View Article : Google Scholar
|
|
92
|
Zhang S, Yang X, Tan Q, Sun H, Chen D,
Chen Y, Zhang H, Yang Y, Gong Q and Yue Q: Cortical myelin and
thickness mapping provide insights into whole-brain tumor burden in
diffuse midline glioma. Cereb Cortex. 34:bhad4912024. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Krystal J, Hanson D, Donnelly D and Atlas
M: A phase 1 study of mebendazole with bevacizumab and irinotecan
in high-grade gliomas. Pediatr Blood Cancer. 71:e308742024.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Voelcker G: The mechanism of action of
cyclophosphamide and its consequences for the development of a new
generation of oxazaphosphorine cytostatics. Sci Pharm. 88:422020.
View Article : Google Scholar
|
|
95
|
Lin FY, Stuckert A, Tat C, White M,
Ruggieri L, Zhang H, Mehta B, Lapteva N, Mei Z, Major A, et al:
Phase I trial of GD2.CART cells augmented with constitutive
interleukin-7 receptor for treatment of high-grade pediatric CNS
tumors. J Clin Oncol. 42:2769–2779. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Ishizawa J, Zarabi SF, Davis RE, Halgas O,
Nii T, Jitkova Y, Zhao R, St-Germain J, Heese LE, Egan G, et al:
Mitochondrial ClpP-mediated proteolysis induces selective cancer
cell lethality. Cancer Cell. 35:721–737.e9. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Jacques S, van der Sloot AM, C Huard C,
Coulombe-Huntington J, Tsao S, Tollis S, Bertomeu T, Culp EJ,
Pallant D, Cook MA, et al: Imipridone anticancer compounds
ectopically activate the ClpP protease and represent a new scaffold
for antibiotic development. Genetics. 214:1103–1120. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Kline CL, Van den Heuvel AP, Allen JE,
Prabhu VV, Dicker DT and El-Deiry WS: ONC201 kills solid tumor
cells by triggering an integrated stress response dependent on ATF4
activation by specific eIF2alpha kinases. Sci Signal. 9:ra182016.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Prabhu VV, Morrow S, Rahman Kawakibi A,
Zhou L, Ralff M, Ray J, Jhaveri A, Ferrarini I, Lee Y, Parker C, et
al: ONC201 and imipridones: Anti-cancer compounds with clinical
efficacy. Neoplasia. 22:725–744. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Venneti S, Kawakibi AR, Ji S, Waszak SM,
Sweha SR, Mota M, Pun M, Deogharkar A, Chung C, Tarapore RS, et al:
Clinical efficacy of ONC201 in H3K27M-mutant diffuse midline
gliomas is driven by disruption of integrated metabolic and
epigenetic pathways. Cancer Discov. 13:2370–2393. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Arrillaga-Romany I, Lassman A, McGovern
SL, Mueller S, Nabors B, van den Bent M, Vogelbaum MA, Allen JE,
Melemed AS, Tarapore RS, et al: ACTION: A randomized phase 3 study
of ONC201 (dordaviprone) in patients with newly diagnosed H3
K27M-mutant diffuse glioma. Neuro Oncol. 26 (Suppl_2):S173–S181.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Jenke R, Ressing N, Hansen FK, Aigner A
and Buch T: Anticancer therapy with HDAC inhibitors:
Mechanism-based combination strategies and future perspectives.
Cancers (Basel). 13:6342021. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Eckschlager T, Plch J, Stiborova M and
Hrabeta J: Histone deacetylase inhibitors as anticancer drugs. Int
J Mol Sci. 18:14142017. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Mueller S, Kline C, Stoller S, Lundy S,
Christopher L, Reddy AT, Banerjee A, Cooney TM, Raber S, Hoffman C,
et al: PNOC015: Repeated convection-enhanced delivery of MTX110
(aqueous panobinostat) in children with newly diagnosed diffuse
intrinsic pontine glioma. Neuro Oncol. 25:2074–2086. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Su JM, Kilburn LB, Mansur DB, Krailo M,
Buxton A, Adekunle A, Gajjar A, Adamson PC, Weigel B, Fox E, et al:
Phase I/II trial of vorinostat and radiation and maintenance
vorinostat in children with diffuse intrinsic pontine glioma: A
Children's Oncology Group report. Neuro Oncol. 24:655–664. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Neth BJ, Balakrishnan SN, Carabenciov ID,
Uhm JH, Daniels DJ, Kizilbash SH and Ruff MW: Panobinostat in
adults with H3 K27M-mutant diffuse midline glioma: A single-center
experience. J Neurooncol. 157:91–100. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Ameratunga M, Kipps E, Okines AFC and
Lopez JS: To cycle or Fight-CDK4/6 inhibitors at the crossroads of
anticancer immunity. Clin Cancer Res. 25:21–28. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Zhang J, Xu D, Zhou Y, Zhu Z and Yang X:
Mechanisms and implications of CDK4/6 inhibitors for the treatment
of NSCLC. Front Oncol. 11:6760412021. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
The JLF and Aplin AE: Arrested
developments: CDK4/6 inhibitor resistance and alterations in the
tumor immune microenvironment. Clin Cancer Res. 25:921–927. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Previtali R, Prontera G, Alfei E, Nespoli
L, Masnada S, Veggiotti P and Mannarino S: Paradigm shift in the
treatment of tuberous sclerosis: Effectiveness of everolimus.
Pharmacol Res. 195:1068842023. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
DeWire M, Fuller C, Hummel TR, Chow LML,
Salloum R, de Blank P, Pater L, Lawson S, Zhu X, Dexheimer P, et
al: A phase I/II study of ribociclib following radiation therapy in
children with newly diagnosed diffuse intrinsic pontine glioma
(DIPG). J Neurooncol. 149:511–522. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
DeWire M, Lazow M, Campagne O, Leach J,
Fuller C, Senthil Kumar S, Stanek J, de Blank P, Hummel TR,
Pillay-Smiley N, et al: Phase I study of ribociclib and everolimus
in children with newly diagnosed DIPG and high-grade glioma: A
CONNECT pediatric neuro-oncology consortium report. Neurooncol Adv.
4:vdac0552022.PubMed/NCBI
|
|
113
|
Greenall SA, McKenzie M, Seminova E,
Dolezal O, Pearce L, Bentley J, Kuchibhotla M, Chen SC, McDonald
KL, Kornblum HI, et al: Most clinical anti-EGFR antibodies do not
neutralize both wtEGFR and EGFRvIII activation in glioma. Neuro
Oncol. 21:1016–1027. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Fleischhack G, Massimino M, Warmuth-Metz
M, Khuhlaeva E, Janssen G, Graf N, Rutkowski S, Beilken A, Schmid
I, Biassoni V, et al: Nimotuzumab and radiotherapy for treatment of
newly diagnosed diffuse intrinsic pontine glioma (DIPG): A phase
III clinical study. J Neurooncol. 143:107–113. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Li S, Wang C, Chen J, Lan Y, Zhang W, Kang
Z, Zheng Y, Zhang R, Yu J and Li W: Signaling pathways in brain
tumors and therapeutic interventions. Signal Transduct Target Ther.
8:82023. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Meel MH, Kaspers GJL and Hulleman E:
Preclinical therapeutic targets in diffuse midline glioma. Drug
Resist Updat. 44:15–25. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Yang K, Wu Z, Zhang H, Zhang N, Wu W, Wang
Z, Dai Z, Zhang X, Zhang L, Peng Y, et al: Glioma targeted therapy:
Insight into future of molecular approaches. Mol Cancer. 21:392022.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Liu Y, Liu C, Wang G, Tao R, Xu J, Shen L,
Wang H, Zhu C, Zhang H, Zeng G, et al: Nimotuzumab combined with
chemoradiation therapy in newly diagnosed pediatric diffuse
intrinsic pontine glioma. Int J Radiat Oncol Biol Phys.
123:839–847. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Pachocki CJ and Hol EM: Current
perspectives on diffuse midline glioma and a different role for the
immune microenvironment compared to glioblastoma. J
Neuroinflammation. 19:2762022. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Majzner RG, Ramakrishna S, Yeom KW, Patel
S, Chinnasamy H, Schultz LM, Richards RM, Jiang L, Barsan V,
Mancusi R, et al: GD2-CART cell therapy for H3K27M-mutated diffuse
midline gliomas. Nature. 603:934–941. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Vitanza NA, Wilson AL, Huang W, Seidel K,
Brown C, Gustafson JA, Yokoyama JK, Johnson AJ, Baxter BA, Koning
RW, et al: Intraventricular B7-H3 CAR T cells for diffuse intrinsic
pontine glioma: Preliminary First-in-Human bioactivity and safety.
Cancer Discov. 13:114–131. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Vitanza NA, Ronsley R, Choe M, Seidel K,
Huang W, Rawlings-Rhea SD, Beam M, Steinmetzer L, Wilson AL, Brown
C, et al: Intracerebroventricular B7-H3-targeting CAR T cells for
diffuse intrinsic pontine glioma: A phase 1 trial. Nat Med.
31:861–868. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Dai B, Roife D, Kang Y, Gumin J, Rios
Perez MV, Li X, Pratt M, Brekken RA, Fueyo-Margareto J, Lang FF, et
al: Preclinical evaluation of sequential combination of oncolytic
adenovirus delta-24-RGD and Phosphatidylserine-targeting antibody
in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 16:662–670.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Gállego Pérez-Larraya J, Garcia-Moure M,
Labiano S, Patiño-García A, Dobbs J, Gonzalez-Huarriz M, Zalacain
M, Marrodan L, Martinez-Velez N, Puigdelloses M, et al: Oncolytic
DNX-2401 virus for pediatric diffuse intrinsic pontine glioma. N
Engl J Med. 386:2471–2481. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Grassl N, Poschke I, Lindner K, Bunse L,
Mildenberger I, Boschert T, Jähne K, Green EW, Hülsmeyer I, Jünger
S, et al: A H3K27M-targeted vaccine in adults with diffuse midline
glioma. Nat Med. 29:2586–2592. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Boschert T, Kromer K, Lerner T, Lindner K,
Haltenhof G, Tan CL, Jähne K, Poschke I, Bunse L, Eisele P, et al:
H3K27M neoepitope vaccination in diffuse midline glioma induces B
and T cell responses across diverse HLA loci of a recovered
patient. Sci Adv. 10:eadi90912024. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Vincent CA and Remeseiro S: The immune
response behind peptide vaccination in diffuse midline glioma. Mol
Oncol. 18:1849–1852. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Mueller S, Taitt JM, Villanueva-Meyer JE,
Bonner ER, Nejo T, Lulla RR, Goldman S, Banerjee A, Chi SN, Whipple
NS, et al: Mass cytometry detects H3.3K27M-specific vaccine
responses in diffuse midline glioma. J Clin Investigation.
130:6325–6337. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Grassl N, Sahm K, Süße H, Poschke I, Bunse
L, Bunse T, Boschert T, Mildenberger I, Rupp AK, Ewinger MP, et al:
INTERCEPT H3: A multicenter phase I peptide vaccine trial for the
treatment of H3-mutated diffuse midline gliomas. Neurol Res Pract.
5:552023. View Article : Google Scholar : PubMed/NCBI
|














