Mitochondria, as highly dynamic organelles with
various biological functions within cells (1,2), serve
a crucial regulatory role in metabolic regulation and the
activation of immune cells (3). As
the center of cellular energy metabolism and biosynthesis,
mitochondria produce ATP through oxidative phosphorylation (OXPHOS)
to maintain cellular energy homeostasis (4). The regulation of mitochondrial
dynamics is notably associated with tumor cell proliferation and
migration, as well as treatment outcomes (5). Mitochondrial release of mitochondrial
DNA (mtDNA), mitochondrial reactive oxygen species (mtROS) and
metabolites is involved in the regulation of various cellular
biological processes, including energy metabolism and immune
responses (6). Dynamic changes in
mitochondrial fusion and fission affect their metabolic function,
and, in turn, the metabolic state regulates the dynamic equilibrium
of mitochondria. This bidirectional interaction influences the
metabolism and effector functions of T cells (7). mtDNA mutations or damage can affect
mitochondrial function, including metabolic efficiency and dynamic
behavior, thereby directly impacting T-cell capability (8). In addition, the metabolic state of
mitochondria can regulate mitochondrial dynamics and mtDNA through
pathways such as mTOR and AMPK (9).
Increasing evidence has indicated that mitochondrial dysfunction is
associated with the occurrence of various diseases, including
autoimmune diseases and tumors (10–12).
To ensure normal mitochondrial structure, function
and quantity, cells have evolved a sophisticated mitochondrial
quality control system that can repair mildly damaged mitochondria
or isolate severely damaged ones through mitochondrial dynamics.
This system can actuate mitochondrial degradation and renewal
through mitophagy and biogenesis, ultimately maintaining
mitochondrial homeostasis to meet cellular metabolic needs
(13).
In the tumor microenvironment (TME), tumor cells
drive nutrient depletion and excessive production of metabolic
byproducts, modulating the metabolic reprogramming of infiltrating
immune cells and the activation of related signaling pathways, such
as the AMPK signaling axis and the mTOR signaling pathway. These
pathways control the polarization of different types of immune
cells, inducing metabolic dysregulation that leads to the loss of
antitumor immune responses (14,15).
Hypoxia and continuous antigen stimulation are common forms of
metabolic stress manifested in the TME. Prolonged exposure to
low-oxygen conditions induces mitochondrial stress leading to
elevated ROS levels (16), which
rapidly causes the exhaustion of tumor-infiltrating CD8+
T cells and mitochondrial damage or depolarization (17). This results in the metabolic
exhaustion and functional decline of CD8+
tumor-infiltrating lymphocytes (TILs), facilitating tumor immune
evasion (18).
The present review explores the relationship between
mitochondria and T-cell antitumor immune responses, investigating
the key regulatory roles of mitochondrial dynamics, energy
metabolism and mtDNA in tumor occurrence, development and immune
evasion. The current review aims to provide a deeper understanding
of the regulatory mechanisms by which mitochondria influence T
cells during tumor development. Additionally, antitumor
interventions targeting mitochondria are discussed, providing a
basis for developing mitochondrial-targeted antitumor immunotherapy
strategies.
Mitochondria within T cells have complex structures,
including an outer membrane, inner membrane, intermembrane space
and matrix (19). The outer
membrane contains selective ion channels and membrane proteins that
regulate intercellular communication (20). The intermembrane space is involved
in protein and lipid biosynthesis, and redox regulation. The inner
membrane folds to form cristae housing the electron transport chain
and respiratory enzyme complexes (21). The matrix is the site of the
tricarboxylic acid (TCA) cycle, providing substrates for OXPHOS and
regulating various metabolites. In addition to energy supply,
mitochondria influence CD8+ T-cell activation, signal
transduction and cytotoxicity through regulation of ROS production
and calcium ion (Ca2+) balance (22).
The TME refers to the dynamic environment
surrounding the tumor, serving as the ‘soil’ for tumor cell
proliferation. Tumor cells can control tumor-associated antigen
(TAA) expression, enhance the expression of immune checkpoint
molecules, secrete immunosuppressive cytokines and accumulate
lactic acid metabolites, altering the metabolic status, TAA
recognition ability and tumor-killing potency of infiltrating
immune cells (23). This creates an
immunosuppressive microenvironment favorable for tumor growth and
spread.
As the tumor progresses, factors such as reduced
oxygen content, increased ROS production, depolarized membrane
potential and nutrient deprivation in the TME disrupt T-cell
mitochondrial metabolism and impair their function (24–26),
manifesting as poor mitochondrial adaptability, reduced abundance,
decreased or lost membrane potential, dynamic imbalance, reduced
cristae number, and disordered shape and arrangement. These factors
can lead to dysfunction of the TCA cycle and OXPHOS, electron
transport chain dysfunction decreased ATP synthesis efficiency and
excessive ROS production (27).
However, mitophagy, as a core mechanism of mitochondrial quality
control, promptly removes dysfunctional mitochondria, preventing
excessive ROS accumulation, mtDNA leakage and T-cell apoptosis,
while maintaining OXPHOS efficiency through mitochondrial renewal,
thereby providing ATP for T-cell proliferation and effector
functions (28,29).
As the energy and metabolic centers of the cell,
mitochondria provide energy through regulating metabolic pathways
such as the TCA cycle, OXPHOS and fatty acid oxidation (FAO).
Mitochondrial metabolism supplies sufficient ATP to meet the energy
demands of cell division and growth. Furthermore, biosynthetic
precursor substances produced by glycolysis and fatty acid
synthesis (FAS), such as glucose-6-phosphate and pyruvate, are used
for the synthesis of nucleic acids, proteins and lipids (30).
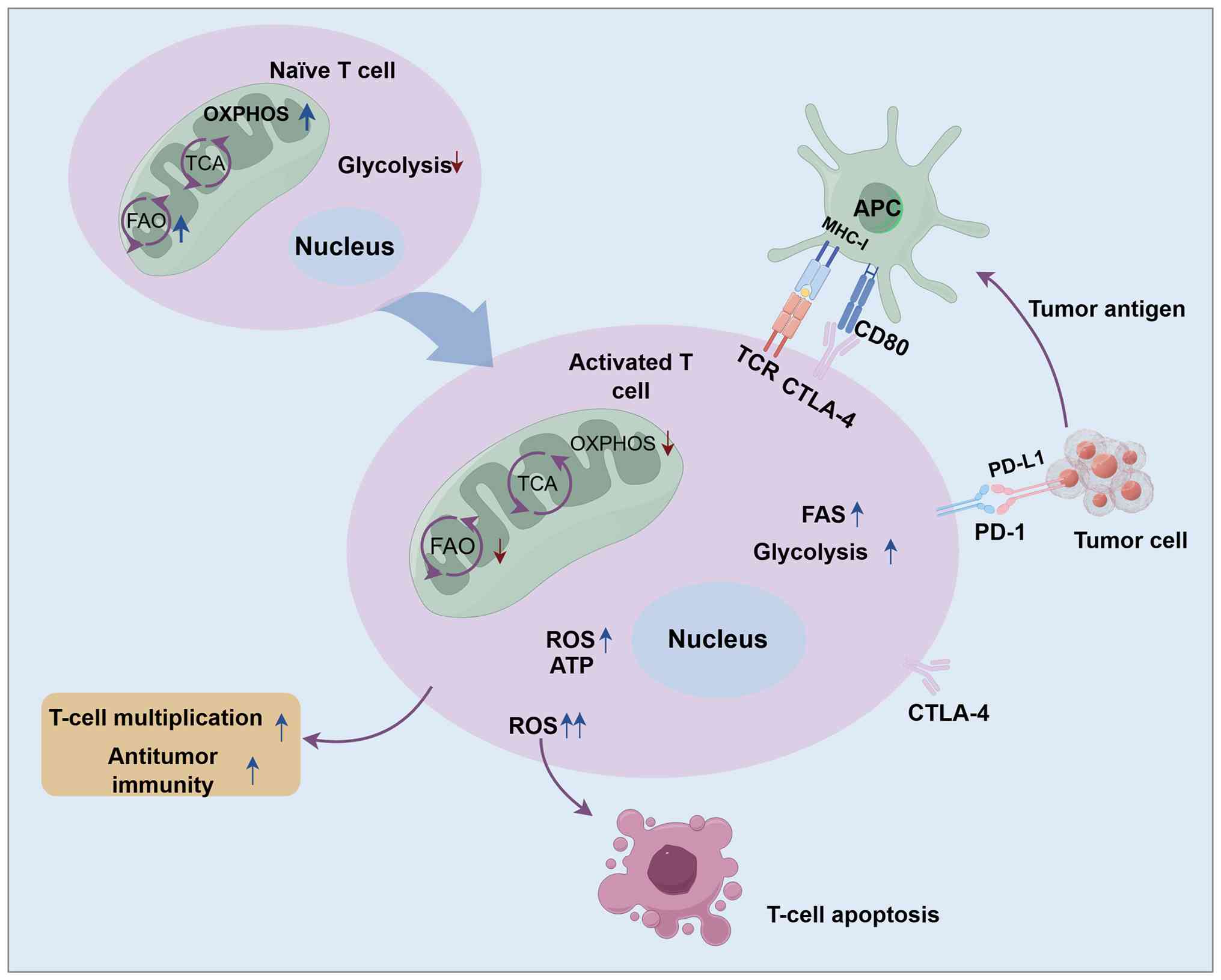
During T-cell activation and proliferation,
mitochondrial metabolic patterns change. The mitochondria of naïve
T cells are often fragmented, relying predominantly on
mitochondrial OXPHOS and FAO to maintain energy requirements in a
resting state (31). Through
regulating the surface expression of GLUT1, which is required for
basal glucose uptake, metabolic homeostasis is maintained, thereby
promoting OXPHOS (32).
Mitochondrial pyruvate carrier 1 (MPC1) promotes mitochondrial
uptake and oxidation of pyruvate, which is crucial for the
development and maturation of T cells in the thymus (33). Within the first 24 h of T-cell
activation, the mitochondrial proteome undergoes notable
restructuring, enhancing mitochondrial one-carbon metabolism to
support rapid T-cell activation (34). Stomatin like protein-2forms
microdomains in mitochondria and cell membranes, promoting membrane
regionalization and assembly of the T-cell receptor (TCR) signaling
complex, thereby enhancing T-cell activation (35).
Upon T-cell activation, mitochondria undergo
morphological changes, transforming from a fragmented state into
longer, more fused forms. Activated T cells exhibit marked
increases in OXPHOS, the TCA cycle and FAS to maintain their
effector functions (36). When T
cells recognize antigens, they transition from a resting state to
an activated state, characterized predominantly by proliferation,
survival and differentiation, which drive the initiation of the
cell cycle, cell growth, IL-2 signaling and enhanced metabolic flux
to sustain anabolic metabolism (37). TCR signals, co-stimulatory signals
and cytokines are necessary conditions for full activation of T
cells, with activation levels assessed by proliferation capacity,
cytokine secretion, effector performance and differentiation state,
all requiring enhanced anabolic metabolism to provide the necessary
energy and material base (38,39).
When TCR and co-receptors recognize the antigen peptide-major
histocompatibility complex (MHC) (signal 1) and receive
co-stimulation (signal 2), it triggers phosphorylation signals such
as PI3K/AKT, activating mTOR complex 1 (mTORC1) and transcription
factors, such as NFAT and MYC in activated T cells, thereby
regulating the expression of key enzymes in glycolysis and
mitochondrial anabolic metabolism (40,41).
In early T-cell activation before cell division, enhanced
glycolytic flux and mitochondrial respiration occur. This process
takes place in mature immune synapses (ISs) lasting several hours,
providing a platform for repeated transmission of T-cell signals
(42,43).
Tumor immune niches, tertiary lymphoid structures
(TLSs) and tumor-draining lymph nodes (TDLNs) are key sites of
intercellular communication (46,47);
notably, interest has been garnered in spatial determinants that
regulate CD8+ T-cell function in these locations. TDLNs
naturally serve as reservoirs for tumor-specific CD8+
TILs. Extensive research has provided evidence of CD8+ T
cells migrating from TDLNs to the tumor, demonstrating that
tumor-specific CD8+ T cells activated in TDLNs generally
exist in a minimally exhausted precursor-exhausted T cell (Tpex) or
Tpex-like state, whereas their clone-related TILs predominantly
exhibit an exhausted terminally differentiated T-cell phenotype
(48–51).
Within the TME, the localization, distribution
patterns and anatomical positions of CD8+ T cells
markedly affect their functional state, differentiation fate and
antitumor efficacy (52). Studies
have demonstrated that the spatial positioning of CD8+ T
cells determines functional subtypes, and the Immunoscore system
can be used to quantify CD8+ T-cell density in the tumor
core and invasive margin region, in order to predict prognosis
(53–55). Unlike traditional memory-like
differentiation, CD8+ T-cell subtypes in the TME show
that Tpex cells share certain stem-like characteristics with memory
T cells (such as TCF1 expression); however, their terminal
exhaustion state is difficult to reverse due to epigenetic
reprogramming and persistent antigen exposure (56).
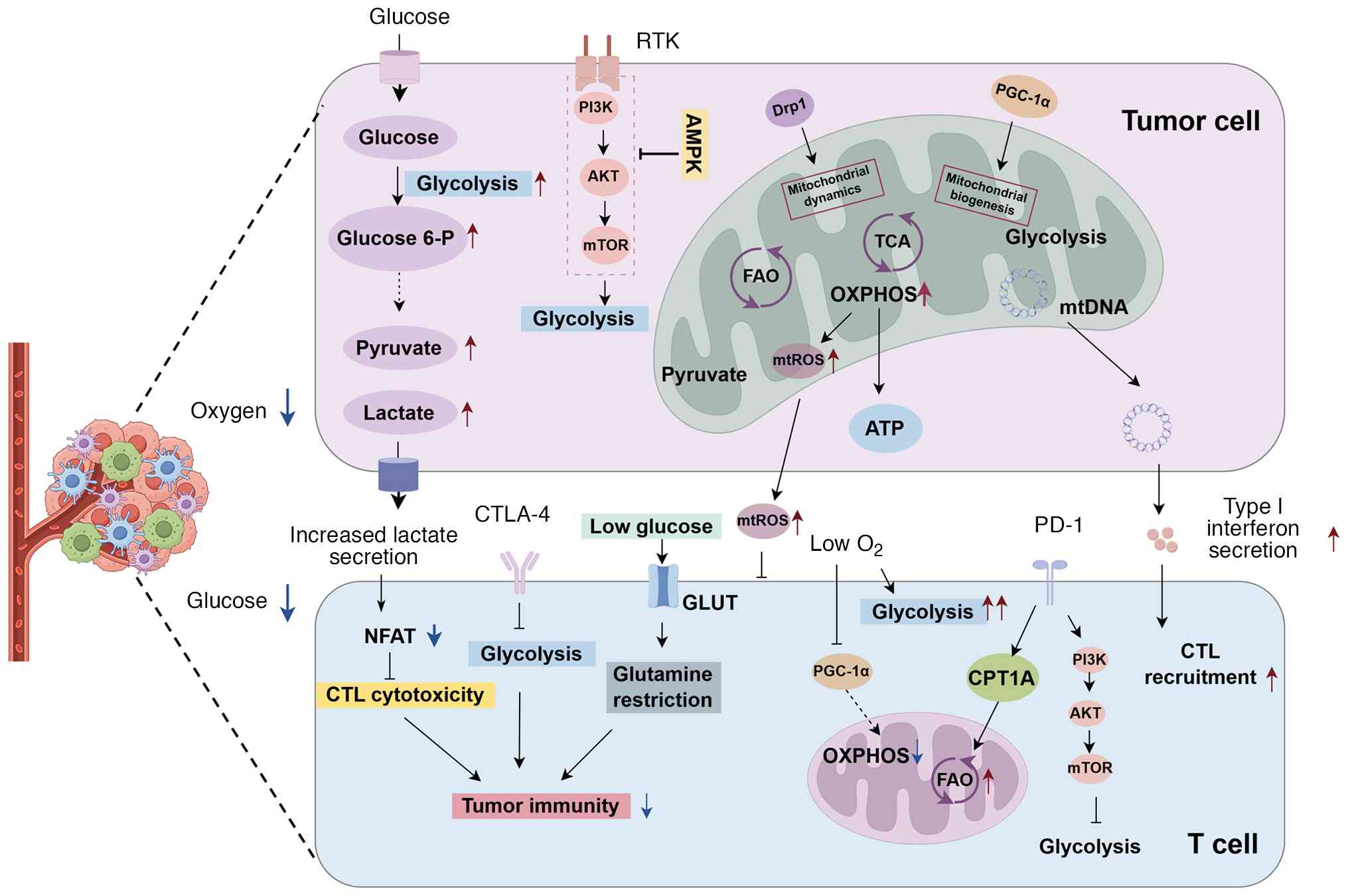
Metabolic heterogeneity appears to contribute to the
heterogeneity of the tumor immune microenvironment. Hypoxic zones
at the tumor core inhibit the mitochondrial respiratory chain
through hypoxia-inducible factor (HIF)-1α, leading to TIL metabolic
reprogramming towards inefficient glycolysis, inhibiting
mitochondrial biogenesis and resulting in functional exhaustion
(57). Conversely, in oxygen-rich
perivascular regions, TILs retain OXPHOS capabilities, maintaining
stem-like characteristics. Malignant cells with high glycolytic
activity can transform their metabolic pathways into anabolic
reactions (58), producing large
amounts of immunosuppressive mediators, such as lactate and
adenosine, inhibiting mitochondrial function, and weakening TIL
survival and cytotoxic functions (59,60).
Mitochondrial functions and metabolic pathways
undergo metabolic reprogramming to adapt to cellular environments
or states (35). Mitochondrial
metabolic reprogramming can enhance the antitumor activity and
immune response of T cells, serving a critical role in the
antitumor activity of T cells infiltrating the TME (65).
Continuous stimulation of T cells in hypoxic
environments can lead to Blimp-1-mediated peroxisome
proliferator-activated receptor-γ coactivator 1α (PGC-1α)-dependent
mitochondrial reprogramming inhibition, resulting in persistent
loss of mitochondrial function and quality (66). Notably, upregulation of PGC-1α
enhances mitochondrial biogenesis and metabolic abilities, boosting
the antitumor effects of CD8+ T cells (67). In glucose-deficient TMEs, activation
of the endoplasmic reticulum (ER) to nucleus signaling 1α-X-box
binding protein 1 signaling axis inhibits the abundance of
glutamine transporters in T cells, limiting the availability of
glutamine crucial for maintaining mitochondrial respiration and
leading to reduced T-cell antitumor function (68). Additionally, under
glucose-restricted conditions, T cells suppress signaling pathways
via the mTORC1 pathway, reducing the expression of antigen-induced
genes, and weakening CD8+ T-cell adhesion, proliferation
capacity and their ability to secrete cytokines, such as IFN-γ,
granulocyte-macrophage colony-stimulating factor and granzyme B
(36,69).
Immune checkpoints are a class of immunosuppressive
molecules that regulate the degree of immune activation to prevent
overly activated T cells from damaging normal tissues. Programmed
death-1 (PD-)1 and cytotoxic T-lymphocyte-associated protein 4
(CTLA-4) are common immune checkpoints, which have notable
immunoregulatory roles under normal circumstances. When programmed
death-ligand 1 (PD-L1) is highly expressed on tumor cells and binds
with PD-1 receptors on T cells, it transmits negative regulatory
signals to inhibit TCR signaling and CD28-mediated co-stimulatory
signals, leading to T-cell exhaustion and loss of immune response,
promoting tumor immune escape (73). The costimulatory molecule CTLA-4 on
T cells binds with B7 molecules on antigen-presenting cells (APCs),
suppressing T-cell proliferation and activation, as well as
antitumor cell capabilities, promoting tumor immune escape
(74).
The expression level of immune checkpoint molecules
can influence T-cell differentiation and function by affecting
mitochondrial structure, function and metabolism. Mitochondrial
structural changes occur in response to immune checkpoint
molecules, with PD-1 stimulation leading to a reduction in the
number and length of mitochondrial cristae (75). When PD-1 binds to PD-L1, it
phosphorylates the immunoreceptor tyrosine-based switch motif and
immunoreceptor tyrosine-based inhibitory motif on the PD-1 tail,
recruiting the SHP2 phosphatase, which inhibits the Ras/MAPK and
PI3K/AKT/mTOR glycolytic regulatory pathways critical for T-cell
activation. This reduces the glycolytic capacity of T cells,
weakening their proliferation and activation functions (76,77).
As well as inhibiting T-cell glycolysis, the PD-1/PD-L1 pathway can
upregulate the expression of adipose triglyceride lipase and
carnitine palmitoyltransferase 1A (CPT1A), promoting endogenous
lipid FAO. This shift enables T cells to utilize fatty acid
metabolism for sustained survival, thereby delaying T-cell
exhaustion (75). Furthermore, PD-1
signaling phosphorylates STAT3, promoting the expression of CPT1B,
a key rate-limiting enzyme in FAO, which accelerates fatty acid
catabolism, and inhibits the glycolytic process and functions of
CD8+ T effector cells.
When tumor cell-secreted PD-L1 exosomes bind to
T-cell PD-1, they activate the CREB and STAT signaling pathways,
upregulating cholesterol and phospholipid synthases, promoting
lipid metabolism, and accelerating T-cell aging (78,79).
In lung cancer, PD-1 on CD8+ T cells inhibits the AKT
signaling pathway, promoting the nuclear translocation of the
transcription factor GATA1, which inhibits the transcription of
phospholipid phosphatase 1. This leads to abnormal phospholipid
metabolism, converting unsaturated fatty acids in the TME to
unsaturated phospholipids, inducing ferroptosis in CD8+
T cells, reducing cytokine secretion and weakening the antitumor
immune response (80).
Mitophagy is the selective degradation of damaged or
dysfunctional mitochondria by cells to maintain homeostasis. There
are two main mechanisms of mitophagy: One is the ubiquitin pathway
mediated by the Parkin-PINK1 signaling cascade, and the other is
the receptor-dependent pathway mediated by BNIP3 and NIX. The
Parkin-PINK1 pathway facilitates degradation by adding ubiquitin
chains on the surface of damaged mitochondria (81), whereas NIX or BNIP3 are expressed on
the mitochondrial surface, and directly recruit and assemble
autophagosomes upon mitochondrial damage (82). In healthy mitochondria, PINK1 is
constantly cleaved and degraded (83); when mitochondria are depolarized or
damaged, PINK1 stabilizes on the outer mitochondrial membrane (OMM)
and phosphorylates, recruiting the E3 ubiquitin ligase Parkin.
Parkin catalyzes the formation of multiple types of ubiquitin
chains on OMM proteins, such as mitofusin (MFN)1/2 and
voltage-dependent anion channel (VDAC) (84). By clearing damaged mitochondria,
mitophagy suppresses the production of IFN-1 and the activation of
inflammasomes, thereby reducing the secretion of IL-1β and IL-18,
and preventing the accumulation of mitochondria-derived
damage-associated molecular patterns such as ROS and mtDNA
(85). BNIP3 can interact with
PINK1 to facilitate its stabilization and regulate the recruitment
of Parkin. NIX can act as a Parkin substrate, enhancing the
recruitment of autophagic adapter proteins via Parkin-mediated
ubiquitination, thereby accelerating clearance.
Mitochondria are the primary organelles for
intracellular production of ROS through the electron transport
chain and OXPHOS process in aerobic respiration (86). The level and sustained generation
capacity of ROS serve a crucial role in the antitumor immunity of T
cells (87,88). In tumor immunity, activated T cells
achieve tumor cell killing by increasing ROS production, recruiting
neutrophils and macrophages (89).
Extracellular ROS can also affect T-cell activation by altering the
immunogenicity of antigen peptides in APCs (90).
Excessive ROS can cause damage to nuclear and mtDNA,
and oxidative damage to proteins and lipids, leading to cellular
damage (91). It promotes T-cell
apoptosis by upregulating pro-apoptotic factors such as Fas and
downregulating the expression of the anti-apoptotic protein Bcl-2
(92). Elevated ROS can further
promote the transformation of immunosuppressive cells, such as
myeloid-derived suppressor cells, tumor-activated macrophages and
regulatory T cells, through various pathways, including HIF-1α
(93), supporting tumor cell
proliferation (91,94). Targeted removal of tumor
extracellular ROS can increase T-lymphocyte tumor infiltration,
restoring T-cell antitumor immunity induced by immunogenic cell
death (95). Additionally, the
deficiency of reduced glutathione in regulatory T cells can lead to
serine metabolism abnormalities and downregulation of the
transcriptional regulator Foxp3 expression, ultimately weakening
the immunosuppressive function of regulatory T cells (96).
Mitochondrial dynamics refer to the continuous
processes of fission and fusion, transfer and positioning within
cells under physiological or stimulated states, regulating the
dynamic equilibrium of mitochondrial distribution, quantity, size
and shape within cells (97). The
synergistic interactions and balance between mitochondrial dynamics
are important for maintaining mitochondrial adaptation to varying
metabolic demands and stress environments.
Mitochondrial fusion is the process whereby two
mitochondria merge into a single organelle, undergoing
morphological changes in response to cellular energy demands and
the need for material exchange between mitochondria. Proteins
involved in mitochondrial fusion in mammalian cells include MFN1,
MFN2 and optic atrophy 1 protein (OPA1). MFN1 and MFN2 primarily
mediate the fusion of the mitochondrial outer membrane, whereas
OPA1 is involved in the inner membrane fusion process.
Additionally, MFN2 is located on the ER membrane, mediating
physical contact between the ER and mitochondria, and
Ca2+ homeostasis (98).
During the process of T-cell expansion,
mitochondrial fusion prompts a metabolic shift in T cells from
glycolysis to FAO and OXPHOS, facilitating the differentiation of
naïve T cells into memory phenotypes and enhancing antitumor
persistence (99). By regulating
Ca2+ homeostasis and ROS levels, mitochondrial fusion
maintains TCR signaling and transcription factor activation such as
NFAT, promoting the expression of effector molecules including
IL-2. Mitochondria that are partially damaged can receive NADH,
FADH2 and TCA intermediates from healthy mitochondria
through fusion, sustaining their ATP production capabilities.
Mitochondrial fusion allows adjacent mitochondria to share mtDNA,
metabolites and enzymes, maintaining OXPHOS activity, which is
particularly important in the oxidative stress of TME. Moreover,
MFN1 is involved in the clearance of damaged mitochondria through
the ER-associated degradation pathway mediated by the
polyubiquitination action of the E3 ubiquitin ligase Parkin
(100).
Mitochondrial fission refers to the process where a
mitochondrion divides to produce smaller and more dispersed
mitochondria, assisting in the movement of mitochondria to regions
requiring energy. Mitochondrial fission is primarily mediated by
dynamin-related protein 1 (Drp1), which is recruited to the
mitochondrial outer membrane, inducing mitochondrial constriction
and fragmentation. Moderate fission benefits cell division and
autophagy, but excessive fission can lead to mitochondrial
fragmentation, loss of membrane potential, decreased ATP production
and subsequently, cell apoptosis (104).
During T-cell activation, mitochondrial fission
accelerators are required to facilitate mitochondrial division and
upregulate calcineurin activity to activate Drp1 (105,106), inducing mitochondrial fission and
inner membrane cristae remodeling, which ultimately leads to
mitochondrial aggregation below the TCR clusters (43). An increase in mitochondrial numbers
and inner membrane cristae remodeling aids in controlling
glycolysis-associated signaling, serving an essential regulatory
role in effector T-cell activation. T cells recognize antigens
through forming an IS with APCs. During the activation process
induced by TCR and CD28 co-stimulation, mitochondria rapidly
produce and release ATP at the IS, increasing intracellular
Ca2+ concentration, promoting T-cell activation and
proliferation (107,108). T-cell activation requires
mitochondrial translocation along the microtubule network to the
IS, supporting sustained activation through maintenance of
Ca2+ signals (109). At
the IS, Drp1-mediated mitochondrial fission helps maintain
intracellular Ca2+ levels, activating mTOR and cMyc
signaling pathways, thereby promoting T-cell proliferation.
Suppression of Drp1 expression hampers CD8+ T-cell
migration and effector functions (110). Mitochondrial fission, driven by
GTPase Drp1 oligomerization, is vital for CD8+ T-cell
chemotaxis and infiltration, and controls mitochondrial
distribution during cell proliferation. Changes in mitochondrial
dynamics transform mitochondria from isolated ‘energy factories’
into functionally integrated metabolic communities, markedly
enhancing the ability of the cell to adapt to environmental stress.
This is particularly critical for maintaining T-cell function
within the TME (70).
Mitochondrial transfer is part of mitochondrial
dynamics, involving various mechanisms such as gap junctions,
nanotubes and extracellular vesicles (111). Tunnel nanotubes (TNTs) are one of
the primary routes for mitochondrial transfer. Transport through
nanotubes is a GTP-dependent process, with GTP primarily generated
in the mitochondrial TCA cycle providing energy for mitochondrial
dynamic changes and transport (112). Tumor cells can scavenge
mitochondria from immune cells via nanotubes, causing
unidirectional mitochondrial transfer from immune cells to tumor
cells, enhancing mitochondrial respiration function, proliferation
capacity and accelerating tumor progression (113). Tumor cells can transfer
mitochondria to T cells through TNTs or extracellular vesicles,
replacing existing mitochondria in T cells with mutated tumor cell
mitochondria, thereby inhibiting the antitumor immune response of T
cells (8). TNTs can effectively
transfer mitochondria from bone marrow stromal cells to T cells,
enhancing their metabolic abilities. When ‘reengineered’ T cells
are transferred into tumor-bearing hosts, they exhibit vigorous
proliferation, higher infiltration efficiency into tumors, reduced
apoptosis rate and stronger antitumor capabilities (114).
Mitochondria and the nucleus maintain cellular
homeostasis and mtDNA integrity through bidirectional signaling,
coordinating stress responses, metabolic adaptation and cell
survival (115,116). Anterograde signaling from the
nucleus involves nuclear-encoded proteins such as TFAM, PGC-1α and
NRF1/2 to regulate mitochondrial replication and repair in response
to cellular needs (117,118). Conversely, mitochondria utilize
retrograde signaling to relay metabolic stress signals such as
changes in Ca2+, ROS or NAD+/NADH back to the
nucleus, reprogramming gene expression to restore function
(119,120). The integration of anterograde and
retrograde signals constitutes mitonuclear communication, which is
crucial for maintaining mtDNA integrity, particularly under stress
conditions such as radiation. mtDNA is the genetic material within
mitochondria, which has a key role in OXPHOS and energy metabolism
in cells. Mutations in the mitochondrial genome are an important
component of cancer mutant genomes (121). As a common intracellular
damage-associated molecular pattern, mtDNA breakdown and release
are crucial factors in mitochondrial dysfunction-mediated
inflammation (122,123). Abnormal mitochondrial gene copy
number, gene expression alterations and mtDNA epigenetic
modifications often influence cancer occurrence and malignant
transformation by regulating cellular metabolism, ROS production
and intercellular interactions (124).
Excessive production of ROS causes oxidative stress,
a major source of DNA damage (125). When cells experience oxidative
stress, the integrity of the mitochondrial membrane is compromised,
promoting the release of oxidized mtDNA into the cytoplasm,
triggering IFN and pro-inflammatory responses (126,127). In the TME, the extracellular
leakage of mtDNA is mainly mediated by extracellular vesicles
composed of exosomes, microvesicles and apoptotic bodies. mtDNA can
enter extracellular vesicles through direct contact or
mitochondrial-derived vesicles, and then be transported into the
extracellular space where it can be passively released during cell
death through mechanical damage-mediated rupture of the cell
membrane (128). Once released
into the cytoplasm, mtDNA can be recognized by pattern-recognition
receptors such as cyclic GMP-AMP synthase (cGAS), Toll-like
receptor (TLR)9, and NOD-, LRR-, and pyrin domain-containing
protein 3 (NLRP3), activating downstream inflammatory signaling
pathways, including the cGAS-stimulator of interferon genes (STING)
pathway, TLR9-myeloid differentiation primary response 88-NF-κB
pathway and NLRP3 inflammasome pathway. mtDNA stress elevates
immunoproteasomes and MHC class I antigen presentation pathways
through the cGAS/STING/type I IFN signaling pathway, leading to
autonomous activation and proliferation of CD8+ T cells
(129).
Direct or indirect cell contact facilitates the
transfer of mtDNA-mutated mitochondria from cancer cells to TILs,
which exhibit metabolic abnormalities, cell senescence, functional
defects, impaired memory formation and effector function (8). In melanoma models, mtDNA is
kinetically released in an ATP-dependent manner into the cytosol,
inducing PD-L1 expression through the STING-IFN pathway, promoting
immune escape (130). mtDNA
mutations render tumors more sensitive to immune checkpoint
blockade (ICB) therapies; alterations in redox balance caused by
mtDNA mutations increase tumor sensitivity to ICB (131). How mitochondria regulate T-cell
tumor immunity pathways is illustrated in Fig. 2.
Mitochondrial dysfunction within the TME is a
notable reason for the impaired antitumor immune response and
antitumor activity in response to diverse immunotherapies.
Targeting mitochondrial energy metabolism, mitochondrial dynamics
and other mitochondrial physiological processes to rebuild T-cell
antitumor function has proven effective in some cancer models
(70,132,133). Therefore, regulating mitochondrial
metabolism, dynamics or other physiological processes, and
increasing mitochondrial quality to restore T-cell antitumor
activity may be a new strategy for cancer treatment in the
future.
Research on melanoma models has revealed that the
use of PPARα agonists to promote FAO can enhance the ability of
CD8+ TILs to slow tumor progression (139). In addition, TILs with high
expression of PGC-1α exhibit stronger mitochondrial adaptability
and longer survival times within the tumor (66). These two pathways help recover
energy and overcome exhaustion in tumor-infiltrating chimeric
antigen receptor (CAR)-T cells. Mitochondrial dysfunction and
HIF-1α-mediated glycolytic reprogramming contribute to T-cell
exhaustion; using 2-deoxy-D-glucose pharmacological inhibitors to
suppress glycolytic reprogramming is a viable metabolic
intervention strategy to maintain CAR-T cell stemness, longevity
and function during tumor immunotherapy (29). Targeting Regnase-1 enhances
mitochondrial adaptability through basic leucine zipper ATF-like
transcription factor, promoting effector T cells for tumor therapy
and boosting tumor adoptive cell therapy (140). VDAC2, a protein located on the
mitochondrial outer membrane, increases CD8+ T-cell
proportion in tumors lacking VDAC2, exhibiting stronger effector
functions; targeting mitochondrial VDAC2 directly kills tumor cells
and recruits more CD8+ T cells to reshape the immune
microenvironment (141).
Mitochondrial fusion is a key regulatory hub that
determines the metabolic mode selection and functional vitality of
T cells. In exhausted T cells, restricted mitochondrial fusion
leads to abnormal mitochondrial morphology, impacting quality
control and biosynthesis, inducing mitochondrial dysfunction and
metabolic reprogramming (142).
Targeting mitochondrial fusion to restore T-cell vitality and
reverse exhaustion may become a novel strategy in tumor
immunotherapy (143).
PGC-1α regulates mitochondrial function and quality
by modulating fusion and fission processes. In the Lewis lung
carcinoma mouse model, the PGC-1α agonist bezafibrate has been
reported to markedly increase ROS in tumor-infiltrating effector
CTLs and the expression of FAO-related genes in tumor tissues. This
promoted an increase in C-X-C motif chemokine ligand (CXCL)9,
CXCL10 and C-X-C motif chemokine receptor 3 receptor expression in
infiltrating CTLs, supporting CD8+ T-cell survival,
infiltration and activation (144). Bezafibrate may further enhance CTL
mitochondrial activation by upregulating OXPHOS and glycolysis,
improving their proliferation and effector functions, increasing
FAO rate and mitochondrial respiratory capacity to meet cellular
energy demands, enhancing antitumor immunity (145).
The process by which tumor cells ‘hijack’ functional
mitochondria from T cells via TNTs may constitute an alternative
mechanism of immune evasion. Selectively inhibiting TNT formation
could be considered a strategic approach to target mitochondrial
involvement in antitumor activity. Current pan-inhibitors (such as
farnesyltransferase inhibitors and cytochalasin) only partially
suppress TNT formation due to a lack of specific markers (113,146). Studies have shown that
mitochondrial transfer is highly related to actin filament
regulators Pim-1 proto-oncogene, serine/threonine kinase, myosin
IB, profilin-1, and Abl-interactor 1, which may serve as targets to
disrupt TNTs (147,148). The cell adhesion molecule
poliovirus receptor-related 2 (encoding Nectin-2) is also one of
the top predictive markers for mitochondrial transfer (149). Additionally, mitochondrial
transfer can improve CD8+ T-cell mitochondrial quality
and metabolic adaptability, proving useful in adoptive
immunotherapy (114).
Another direction in mitochondrial gene therapy is
mtDNA genomic editing, a field where traditional tools such as
CRISPR-Cas9 have excelled in nuclear DNA editing but face
challenges, such as delivering Cas9 to mitochondria and off-target
effects. The innovative transcription activator-like
effector-linked deaminase (TALED) technology offers precise
single-base conversion without DNA double-strand breaks, enabling
base editing. TALEDs facilitate adenine to guanine conversions,
crucial for repairing mutations associated with mitochondrial
diseases (152). An advanced study
into TALED-induced mitochondria-driven precise base editing has
elucidated reliance on the base excision repair pathway of cells,
leading to enhanced TALEDs, which enable efficient mtDNA adenine
base editing (153). Introducing
or correcting mutations in cell and animal models allows more
accurate exploration of the role of mtDNA in cancer metabolism
(153). Future aims may include
in vitro mtDNA editing to promote mitochondrial biogenesis
and OXPHOS to address progressive mitochondrial quality loss in
unmodified TILs.
Abnormal mitochondrial function in tumor cells has
positioned compound-based mitochondrial targeting as a promising
strategy to eradicate chemoresistant cancer cells. Studies on small
molecules/nano-inducers have focused primarily on mitophagy
regulation and mitochondrial-targeted damage strategies (154,155). Development of mitochondrial
nano-inducers targets selective mitophagy within autophagosomes to
degrade mitochondria, markedly enhancing CD8+ T-cell
immune attacks by increasing MHC-I molecule expression, reducing
the anti-apoptotic protein BCL-XL and releasing signaling molecules
that activate T cells, thus improving antitumor immune responses
(156). KCKT is an in situ
transformable nanoparticle, and KCKT platform nanoparticles
facilitate lysosomal escape and direct mitochondrial damage while
blocking protective autophagy, notably enhancing drug delivery and
mitochondrial targeting efficiency, providing a novel option for
therapy (157).
Azocarbonyl-propargyl-modified supramolecular albumin nanoparticles
achieve targeted drug release in hypoxic tumor zones. For example,
SHC4H nanoparticles co-release hydroxychloroquine and a
mitochondrial-targeted photosensitizer to treat hypoxic tumors by
modulating mitosis-induced oxidative stress cascades (158). TAEN is a developed modular
acid/enzyme dual-gated nanotechnology platform, designed for
efficient cascade drug delivery from lysosomes to mitochondria.
TAEN uses hierarchical targeting to stabilize drug loading during
circulation, selectively accumulate at tumor sites, and
cascade-deliver drugs to mitochondria starting from lysosomes at
the subcellular level. This nanotechnology effectively induces
mtROS stress to cleave gasdermin-E, provoking tumor cell
pyroptosis, enhancing tumor-killing efficiency by 30–50 times and
activating antitumor immune responses (159).
Mitochondria are crucial organelles in tumor cell
metabolic reprogramming. In addition to providing energy, they are
critical in tumor cell survival, immune evasion, tumor progression
and the immunosuppressive environment of hypoxic TMEs.
Mitochondrial metabolic reprogramming provides energy and
biosynthetic precursors, forming the basis for T-cell activation,
proliferation and differentiation. Dynamic remodeling regulates the
functional state of T cells, and maintaining mitochondrial quality
is a key target to prevent T-cell exhaustion in the tumor TME and
to enhance the effectiveness of immunotherapy. The present review
discusses mitochondrial metabolism and biogenesis, dynamics, mtDNA
interactions, mtROS involvement and the interplay with T cells in
the TME, and antitumor immunity. The study aims to provide insights
for developing innovative antitumor immunotherapies from the
mitochondrial-tumor immunity perspective.
However, the discussion on mitochondrial targeted
therapy in the present review lacks validation through large
clinical cohorts, and prospective studies on mtDNA mutations and
the efficacy of immunotherapy are still limited. More clinical
research targeting mitochondria is needed in the future to
facilitate the translation of theories into treatments.
Additionally, although the study explores T-cell spatial
heterogeneity, it lacks a systematic analysis of the patterns of
mitochondrial defects and their immune impacts across different
cancer types. Cross-cancer comparative studies are urgently needed
to guide the development of precise immunotherapy strategies.
Current mitochondria-targeting immunotherapies
incorporate mitochondrial-targeting drugs and immune checkpoint
inhibitors to improve T-cell metabolic adaptability, reduce ROS
damage and limit tumor energy supply, enhancing T-cell
cytotoxicity. Targeting Drp1/MFN2 can restore T-cell mitochondrial
morphology and function, reversing the exhausted state. Specific
targeting and elimination of mutated DNA in the mitochondrial
genome enables precise delivery of therapeutic genes. Small
molecule/nano-inducers targeting mitochondria increase targeting
precision and reduce off-target toxicity. However, mitochondrial
targeting strategies, despite their theoretical promise, face
multiple challenges in clinical translation. Firstly, there is a
technical lack of efficient and specific mitochondrial targeting
delivery systems. The dual-layer structure of the mitochondrial
membrane and low lysosomal escape efficiency are major reasons for
delivery failure (160,161). This requires a mechanistic
analysis of the interaction between carriers and mitochondrial
membranes, and the development of non-membrane potential-dependent
targeting strategies. Secondly, viral vectors may induce immune
responses, and the accumulation of nanomaterials in organs requires
optimization through renal clearance engineering, with toxicity
risks urgently needing evaluation (162,163). Lastly, regarding the demand for
personalized treatment, mtDNA heterogeneity necessitates the
development of patient-specific editing strategies, and clinical
translation requires integration with patient-specific barrier
profiles. With advancements in single-cell sequencing technology
enhancing the resolution of mtDNA heterogeneity analysis, combined
with artificial intelligence-driven predictive models, the future
of mitochondrial medicine will focus on transforming mitochondrial
disease treatment models from ‘one-way intervention’ to ‘dynamic
precision regulation’, accelerating the clinical implementation of
precision treatment technologies.
Not applicable.
This work was supported by the Joint Innovation Fund of Health
Commission of Chengdu and Chengdu University of Traditional Chinese
Medicine (grant no. WXLH202405010).
Not applicable.
MZ, ZL, YH and YY wrote the original draft. YX and
KC restructured the manuscript's organization, edited the
manuscript and created the figures. ZZ and DC reviewed and edited
the manuscript. CZ acquired funding reviewed and edited the
manuscript. Data authentication is not applicable. All authors read
and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Bereiter-Hahn J: Behavior of mitochondria
in the living cell. Int Rev Cytol. 122:1–63. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chan DC: Mitochondrial dynamics and its
involvement in disease. Annu Rev Pathol. 15:235–259. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mills EL, Kelly B and O'Neill LAJ:
Mitochondria are the powerhouses of immunity. Nat Immunol.
18:488–498. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chandel NS: Mitochondria. Cold Spring
Harbor Perspect Biol. 13:a0405432021. View Article : Google Scholar
|
|
5
|
Seo JH, Agarwal E, Bryant KG, Caino MC,
Kim ET, Kossenkov AV, Tang HY, Languino LR, Gabrilovich DI, Cohen
AR, et al: Syntaphilin ubiquitination regulates mitochondrial
dynamics and tumor cell movements. Cancer Res. 78:4215–4228. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wellen KE and Thompson CB: A two-way
street: Reciprocal regulation of metabolism and signalling. Nat Rev
Mol Cell Biol. 13:270–276. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chan DC: Mitochondrial fusion and fission
in mammals. Annu Rev Cell Dev Biol. 22:79–99. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ikeda H, Kawase K, Nishi T, Watanabe T,
Takenaga K, Inozume T, Ishino T, Aki S, Lin J, Kawashima S, et al:
Immune evasion through mitochondrial transfer in the tumour
microenvironment. Nature. 638:225–236. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Suzuki J, Yamada T, Inoue K, Nabe S,
Kuwahara M, Takemori N, Takemori A, Matsuda S, Kanoh M, Imai Y, et
al: The tumor suppressor menin prevents effector CD8 T-cell
dysfunction by targeting mTORC1-dependent metabolic activation. Nat
Commun. 9:32962018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Harrington JS, Ryter SW, Plataki M, Price
DR and Choi AMK: Mitochondria in health, disease, and aging.
Physiol Rev. 103:2349–2422. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
El Hiani Y, Ahidouch A, Lehen'kyi V, Hague
F, Gouilleux F, Mentaverri R, Kamel S, Lassoued K, Brûlé G and
Ouadid-Ahidouch H: Extracellular signal-regulated kinases 1 and 2
and TRPC1 channels are required for calcium-sensing
receptor-stimulated MCF-7 breast cancer cell proliferation. Cell
Physiol Biochem. 23:335–346. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
DeBerardinis RJ and Chandel NS:
Fundamentals of cancer metabolism. Sci Adv. 2:e16002002016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu BH, Xu CZ, Liu Y, Lu ZL, Fu TL, Li GR,
Deng Y, Luo GQ, Ding S, Li N and Geng Q: Mitochondrial quality
control in human health and disease. Mil Med Res.
11:322024.PubMed/NCBI
|
|
14
|
Vodnala SK, Eil R, Kishton RJ, Sukumar M,
Yamamoto TN, Ha NH, Lee PH, Shin M, Patel SJ, Yu Z, et al: T cell
stemness and dysfunction in tumors are triggered by a common
mechanism. Science. 363:eaau01352019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lyssiotis CA and Kimmelman AC: Metabolic
interactions in the tumor microenvironment. Trends Cell Biol.
27:863–875. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Scharping NE, Rivadeneira DB, Menk AV,
Vignali PDA, Ford BR, Rittenhouse NL, Peralta R, Wang Y, Wang Y,
DePeaux K, et al: Mitochondrial stress induced by continuous
stimulation under hypoxia rapidly drives T cell exhaustion. Nat
Immunol. 22:205–215. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu YR, Imrichova H, Wang H, Chao T, Xiao
Z, Gao M, Rincon-Restrepo M, Franco F, Genolet R, Cheng WC, et al:
Disturbed mitochondrial dynamics in CD8+ TILs reinforce
T cell exhaustion. Nat Immunol. 21:1540–1551. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vardhana SA, Hwee MA, Berisa M, Wells DK,
Yost KE, King B, Smith M, Herrera PS, Chang HY, Satpathy AT, et al:
Impaired mitochondrial oxidative phosphorylation limits the
self-renewal of T cells exposed to persistent antigen. Nat Immunol.
21:1022–1033. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang MQ, Zhang SL, Sun L, Huang LT, Yu J,
Zhang JH, Tian Y, Han CB and Ma JT: Targeting mitochondria:
Restoring the antitumor efficacy of exhausted T cells. Mol Cancer.
23:2602024. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
König T, Nolte H, Aaltonen MJ, Tatsuta T,
Krols M, Stroh T, Langer T and McBride HM: MIROs and DRP1 drive
mitochondrial-derived vesicle biogenesis and promote quality
control. Nat Cell Biol. 23:1271–1286. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mangalhara KC, Varanasi SK, Johnson MA,
Burns MJ, Rojas GR, Esparza Moltó PB, Sainz AG, Tadepalle N, Abbott
KL, Mendiratta G, et al: Manipulating mitochondrial electron flow
enhances tumor immunogenicity. Science. 381:1316–1323. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sullivan LB, Gui DY and Vander Heiden MG:
Altered metabolite levels in cancer: Implications for tumour
biology and cancer therapy. Nat Rev Cancer. 16:680–693. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
De Martino M, Rathmell JC, Galluzzi L and
Vanpouille-Box C: Cancer cell metabolism and antitumour immunity.
Nat Rev Immunol. 24:654–669. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tian W, Liu Y, Cao C, Zeng Y, Pan Y, Liu
X, Peng Y and Wu F: Chronic stress: Impacts on tumor
microenvironment and implications for anti-cancer treatments. Front
Cell Dev Biol. 9:7770182021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lei Y, Liao F, Tian Y, Wang Y, Xia F and
Wang J: Investigating the crosstalk between chronic stress and
immune cells: Implications for enhanced cancer therapy. Front
Neurosci. 17:13211762023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu Y, Tian S, Ning B, Huang T, Li Y and
Wei Y: Stress and cancer: The mechanisms of immune dysregulation
and management. Front Immunol. 13:10322942022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kawano I, Bazila B, Ježek P and Dlasková
A: Mitochondrial dynamics and cristae shape changes during
metabolic reprogramming. Antioxid Redox Signaling. 39:684–707.
2023. View Article : Google Scholar
|
|
28
|
Xu X, Araki K, Li S, Han JH, Ye L, Tan WG,
Konieczny BT, Bruinsma MW, Martinez J, Pearce EL, et al: Autophagy
is essential for effector CD8(+) T cell survival and memory
formation. Nat Immunol. 15:1152–1161. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu H, Zhao X, Hochrein SM, Eckstein M,
Gubert GF, Knöpper K, Mansilla AM, Öner A, Doucet-Ladevèze R,
Schmitz W, et al: Mitochondrial dysfunction promotes the transition
of precursor to terminally exhausted T cells through
HIF-1α-mediated glycolytic reprogramming. Nat Commun. 14:68582023.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chang CH, Curtis JD, Maggi LB Jr, Faubert
B, Villarino AV, O'Sullivan D, Huang SC, van der Windt GJ, Blagih
J, Qiu J, et al: Posttranscriptional control of T cell effector
function by aerobic glycolysis. Cell. 153:1239–1251. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ron-Harel N, Santos D, Ghergurovich JM,
Sage PT, Reddy A, Lovitch SB, Dephoure N, Satterstrom FK, Sheffer
M, Spinelli JB, et al: Mitochondrial biogenesis and proteome
remodeling promote one-carbon metabolism for T cell activation.
Cell Metab. 24:104–117. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guerrero JA, Klysz DD, Chen Y,
Malipatlolla M, Lone J, Fowler C, Stuani L, May A, Bashti M, Xu P,
et al: GLUT1 overexpression in CAR-T cells induces metabolic
reprogramming and enhances potency. Nat Commun. 15:86582024.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ramstead AG, Wallace JA, Lee SH, Bauer KM,
Tang WW, Ekiz HA, Lane TE, Cluntun AA, Bettini ML, Round JL, et al:
Mitochondrial pyruvate carrier 1 promotes peripheral T cell
homeostasis through metabolic regulation of thymic development.
Cell Rep. 30:2889–2899.e6. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Christie DA, Kirchhof MG, Vardhana S,
Dustin ML and Madrenas J: Mitochondrial and plasma membrane pools
of stomatin-like protein 2 coalesce at the immunological synapse
during T cell activation. PLoS One. 7:e371442012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fan X, Yang M, Lang Y, Lu S, Kong Z, Gao
Y, Shen N, Zhang D and Lv Z: Mitochondrial metabolic reprogramming
in diabetic kidney disease. Cell Death Dis. 15:4422024. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
MacIver NJ, Michalek RD and Rathmell JC:
Metabolic regulation of T lymphocytes. Annu Rev Immunol.
31:259–283. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chapman NM and Chi H: Hallmarks of T-cell
exit from quiescence. Cancer Immunol Res. 6:502–508. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mescher MF, Curtsinger JM, Agarwal P,
Casey KA, Gerner M, Hammerbeck CD, Popescu F and Xiao Z: Signals
required for programming effector and memory development by CD8+ T
cells. Immunol Rev. 211:81–92. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Makowski L, Chaib M and Rathmell JC:
Immunometabolism: From basic mechanisms to translation. Immunol
Rev. 295:5–14. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang R, Dillon CP, Shi LZ, Milasta S,
Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger
J and Green DR: The transcription factor Myc controls metabolic
reprogramming upon T lymphocyte activation. Immunity. 35:871–882.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Vaeth M, Maus M, Klein-Hessling S,
Freinkman E, Yang J, Eckstein M, Cameron S, Turvey SE, Serfling E,
Berberich-Siebelt F, et al: Store-operated Ca2+ entry
controls clonal expansion of T cells through metabolic
reprogramming. Immunity. 47:664–679.e6. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Stirnweiss A, Hartig R, Gieseler S,
Lindquist JA, Reichardt P, Philipsen L, Simeoni L, Poltorak M,
Merten C, Zuschratter W, et al: T cell activation results in
conformational changes in the Src family kinase Lck to induce its
activation. Sci Signaling. 6:ra132013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Schwindling C, Quintana A, Krause E and
Hoth M: Mitochondria positioning controls local calcium influx in T
cells. J Immunol. 184:184–190. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Velnati S, Massarotti A, Antona A, Talmon
M, Fresu LG, Galetto AS, Capello D, Bertoni A, Mercalli V, Graziani
A, et al: Structure activity relationship studies on Amb639752:
toward the identification of a common pharmacophoric structure for
DGKα inhibitors. J Enzyme Inhib Med Chem. 35:96–108. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Araki K, Turner AP, Shaffer VO, Gangappa
S, Keller SA, Bachmann MF, Larsen CP and Ahmed R: mTOR regulates
memory CD8 T-cell differentiation. Nature. 460:108–112. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Klemm F, Maas RR, Bowman RL, Kornete M,
Soukup K, Nassiri S, Brouland JP, Iacobuzio-Donahue CA, Brennan C,
Tabar V, et al: Interrogation of the microenvironmental landscape
in brain tumors reveals disease-specific alterations of immune
cells. Cell. 181:1643–1660.e17. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cabrita R, Lauss M, Sanna A, Donia M,
Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K,
Vallon-Christersson J, et al: Tertiary lymphoid structures improve
immunotherapy and survival in melanoma. Nature. 577:561–565. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Connolly KA, Kuchroo M, Venkat A, Khatun
A, Wang J, William I, Hornick NI, Fitzgerald BL, Damo M, Kasmani
MY, et al: A reservoir of stem-like CD8+ T cells in the
tumor-draining lymph node preserves the ongoing antitumor immune
response. Sci Immunol. 6:eabg78362021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Prokhnevska N, Cardenas MA, Valanparambil
RM, Sobierajska E, Barwick BG, Jansen C, Reyes Moon A, Gregorova P,
delBalzo L, Greenwald R, et al: CD8+ T cell activation
in cancer comprises an initial activation phase in lymph nodes
followed by effector differentiation within the tumor. Immunity.
56:107–124.e5. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nagasaki J, Inozume T, Sax N, Ariyasu R,
Ishikawa M, Yamashita K, Kawazu M, Ueno T, Irie T, Tanji E, et al:
PD-1 blockade therapy promotes infiltration of tumor-attacking
exhausted T cell clonotypes. Cell Rep. 38:1103312022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pai JA, Hellmann MD, Sauter JL, Mattar M,
Rizvi H, Woo HJ, Shah N, Nguyen EM, Uddin FZ, Quintanal-Villalonga
A, et al: Lineage tracing reveals clonal progenitors and long-term
persistence of tumor-specific T cells during immune checkpoint
blockade. Cancer Cell. 41:776–790.e7. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liang H, Huang J, Li H, He W, Ao X, Xie Z,
Chen Y, Lv Z, Zhang L, Zhong Y, et al: Spatial proximity of
CD8+ T cells to tumor cells predicts neoadjuvant therapy
efficacy in breast cancer. NPJ Breast Cancer. 11:132025. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Galon J, Costes A, Sanchez-Cabo F,
Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M,
Berger A, Wind P, et al: Type, density, and location of immune
cells within human colorectal tumors predict clinical outcome.
Science. 313:1960–1964. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Pagès F, Mlecnik B, Marliot F, Bindea G,
Ou FS, Bifulco C, Lugli A, Zlobec I, Rau TT, Berger MD, et al:
International validation of the consensus Immunoscore for the
classification of colon cancer: A prognostic and accuracy study.
Lancet. 391:2128–2139. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Angell HK, Bruni D, Barrett JC, Herbst R
and Galon J: The immunoscore: Colon cancer and beyond. Clin Cancer
Res. 26:332–339. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sen DR, Kaminski J, Barnitz RA, Kurachi M,
Gerdemann U, Yates KB, Tsao HW, Godec J, LaFleur MW, Brown FD, et
al: The epigenetic landscape of T cell exhaustion. Science.
354:1165–1169. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Reina-Campos M, Scharping NE and Goldrath
AW: CD8+ T cell metabolism in infection and cancer. Nat
Rev Immunol. 21:718–738. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhu J and Thompson CB: Metabolic
regulation of cell growth and proliferation. Nat Rev Mol Cell Biol.
20:436–450. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Brand A, Singer K, Koehl GE, Kolitzus M,
Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, et
al: LDHA-associated lactic acid production blunts tumor
immunosurveillance by T and NK cells. Cell Metab. 24:657–671. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Vigano S, Alatzoglou D, Irving M,
Ménétrier-Caux C, Caux C, Romero P and Coukos G: Targeting
adenosine in cancer immunotherapy to enhance T-cell function. Front
Immunol. 10:9252019. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Qin M, Hamanishi J, Ukita M, Yamanoi K,
Takamatsu S, Abiko K, Murakami R, Miyamoto T, Suzuki H, Ueda A, et
al: Tertiary lymphoid structures are associated with favorable
survival outcomes in patients with endometrial cancer. Cancer
Immunol Immunother. 71:1431–1442. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhang MJ, Wen Y and Sun ZJ: The impact of
metabolic reprogramming on tertiary lymphoid structure formation:
Enhancing cancer immunotherapy. BMC Med. 23:2172025. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Jia D, Wang Q, Qi Y, Jiang Y, He J, Lin Y,
Sun Y, Xu J, Chen W, Fan L, et al: Microbial metabolite enhances
immunotherapy efficacy by modulating T cell stemness in pan-cancer.
Cell. 187:1651–1665.e21. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Guo Y, Xie YQ, Gao M, Zhao Y, Franco F,
Wenes M, Siddiqui I, Bevilacqua A, Wang H, Yang H, et al: Metabolic
reprogramming of terminally exhausted CD8+ T cells by
IL-10 enhances anti-tumor immunity. Nat Immunol. 22:746–756. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sukumar M, Kishton RJ and Restifo NP:
Metabolic repro-graming of anti-tumor immunity. Curr Opin Immunol.
46:14–22. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Scharping NE, Menk AV, Moreci RS,
Whetstone RD, Dadey RE, Watkins SC, Ferris RL and Delgoffe GM: The
tumor microenvironment represses T cell mitochondrial biogenesis to
drive intratumoral T cell metabolic insufficiency and dysfunction.
Immunity. 45:374–388. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Dumauthioz N, Tschumi B, Wenes M, Marti B,
Wang H, Franco F, Li W, Lopez-Mejia IC, Fajas L, Ho PC, et al:
Enforced PGC-1α expression promotes CD8 T cell fitness, memory
formation and antitumor immunity. Cell Mol Immunol. 18:1761–1771.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Song M, Sandoval TA, Chae CS, Chopra S,
Tan C, Rutkowski MR, Raundhal M, Chaurio RA, Payne KK, Konrad C, et
al: IRE1α-XBP1 controls T cell function in ovarian cancer by
regulating mitochondrial activity. Nature. 562:423–428. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Cham CM, Driessens G, O'Keefe JP and
Gajewski TF: Glucose deprivation inhibits multiple key gene
expression events and effector functions in CD8+ T cells. Eur J
Immunol. 38:2438–2450. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Wenes M, Jaccard A, Wyss T,
Maldonado-Pérez N, Teoh ST, Lepez A, Renaud F, Franco F, Waridel P,
Yacoub Maroun C, et al: The mitochondrial pyruvate carrier
regulates memory T cell differentiation and antitumor function.
Cell Metab. 34:731–746.e9. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Nava Lauson CB, Tiberti S, Corsetto PA,
Conte F, Tyagi P, Machwirth M, Ebert S, Loffreda A, Scheller L,
Sheta D, et al: Linoleic acid potentiates CD8+ T cell
metabolic fitness and antitumor immunity. Cell Metab.
35:633–650.e9. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Tang Y, Chen Z, Zuo Q and Kang Y:
Regulation of CD8+ T cells by lipid metabolism in cancer
progression. Cell Mol Immunol. 21:1215–1230. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Pauken KE and Wherry EJ: Overcoming T cell
exhaustion in infection and cancer. Trends Immunol. 36:265–276.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Greenwald RJ, Freeman GJ and Sharpe AH:
The B7 family revisited. Annu Rev Immunol. 23:515–548. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Patsoukis N, Bardhan K, Chatterjee P, Sari
D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, et al:
PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis
and promoting lipolysis and fatty acid oxidation. Nat Commun.
6:66922015. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Boussiotis VA and Patsoukis N: Effects of
PD-1 signaling on immunometabolic reprogramming. Immunometabolism.
4:e2200072022. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Baldanzi G: Immune checkpoint receptors
signaling in T cells. Int J Mol Sci. 23:35292022. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Zhao M, Yuan H, Yang G, Wang Y, Bu Y,
Zhang H, Zhao L, Lv P, Yun H, Geng Y, et al: Tumour cell-expressed
PD-L1 reprograms lipid metabolism via EGFR/ITGB4/SREBP1c signalling
in liver cancer. JHEP Rep. 6:1010092024. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Ma F, Liu X, Zhang Y, Tao Y, Zhao L,
Abusalamah H, Huffman C, Harbison RA, Puram SV, Wang Y and Peng G:
Tumor extracellular vesicle-derived PD-L1 promotes T cell
senescence through lipid metabolism reprogramming. Sci Transl Med.
17:eadm72692025. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ping Y, Shan J, Qin H, Li F, Qu J, Guo R,
Han D, Jing W, Liu Y, Liu J, et al: PD-1 signaling limits
expression of phospholipid phosphatase 1 and promotes intratumoral
CD8+ T cell ferroptosis. Immunity. 57:2122–2139.e9.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Alsayyah C, Singh MK, Morcillo-Parra MA,
Cavellini L, Shai N, Schmitt C, Schuldiner M, Zalckvar E, Mallet A,
Belgareh-Touzé N, et al: Mitofusin-mediated contacts between
mitochondria and peroxisomes regulate mitochondrial fusion. PLoS
Biol. 22:e30026022024. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Sandoval H, Thiagarajan P, Dasgupta SK,
Schumacher A, Prchal JT, Chen M and Wang J: Essential role for Nix
in autophagic maturation of erythroid cells. Nature. 454:232–235.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Lu Y, Li Z, Zhang S, Zhang T, Liu Y and
Zhang L: Cellular mitophagy: Mechanism, roles in diseases and small
molecule pharmacological regulation. Theranostics. 13:736–766.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Wang S, Long H, Hou L, Feng B, Ma Z, Wu Y,
Zeng Y, Cai J, Zhang DW and Zhao G: The mitophagy pathway and its
implications in human diseases. Signal Transduction Targeted Ther.
8:3042023. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
White E, Lattime EC and Guo JY: Autophagy
regulates stress responses, metabolism, and anticancer immunity.
Trends Cancer. 7:778–789. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Silwal P, Kim JK, Kim YJ and Jo EK:
Mitochondrial reactive oxygen species: Double-edged weapon in host
defense and pathological inflammation during infection. Front
Immunol. 11:16492020. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Mougiakakos D, Johansson CC, Jitschin R,
Böttcher M and Kiessling R: Increased thioredoxin-1 production in
human naturally occurring regulatory T cells confers enhanced
tolerance to oxidative stress. Blood. 117:857–861. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Mougiakakos D, Johansson CC and Kiessling
R: Naturally occurring regulatory T cells show reduced sensitivity
toward oxidative stress-induced cell death. Blood. 113:3542–3545.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Seledtsov VI, Goncharov AG and Seledtsova
GV: Clinically feasible approaches to potentiating cancer
cell-based immunotherapies. Hum Vaccin Immunother. 11:851–869.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Weiskopf D, Schwanninger A, Weinberger B,
Almanzar G, Parson W, Buus S, Lindner H and Grubeck-Loebenstein B:
Oxidative stress can alter the antigenicity of immunodominant
peptides. J Leukocyte Biol. 87:165–172. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Cheung EC and Vousden KH: The role of ROS
in tumour development and progression. Nat Rev Cancer. 22:280–297.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Hildeman DA, Mitchell T, Teague TK, Henson
P, Day BJ, Kappler J and Marrack PC: Reactive oxygen species
regulate activation-induced T cell apoptosis. Immunity. 10:735–744.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Bailly C: Regulation of PD-L1 expression
on cancer cells with ROS-modulating drugs. Life Sci.
246:1174032020. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Kuo CL, Ponneri Babuharisankar A, Lin YC,
Lien HW, Lo YK, Chou HY, Tangeda V, Cheng LC, Cheng AN and Lee AY:
Mitochondrial oxidative stress in the tumor microenvironment and
cancer immunoescape: Foe or friend? J Biomed Sci. 29:742022.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Deng H, Yang W, Zhou Z, Tian R, Lin L, Ma
Y, Song J and Chen X: Targeted scavenging of extracellular ROS
relieves suppressive immunogenic cell death. Nat Commun.
11:49512020. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Kurniawan H, Franchina DG, Guerra L,
Bonetti L, -Baguet LS, Grusdat M, Schlicker L, Hunewald O, Dostert
C, Merz MP, et al: Glutathione restricts serine metabolism to
preserve regulatory T cell function. Cell Metab. 31:920–936.e7.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Kyriakoudi S, Drousiotou A and Petrou PP:
When the balance tips: Dysregulation of mitochondrial dynamics as a
culprit in disease. Int J Mol Sci. 22:46172021. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
de Brito OM and Scorrano L: Mitofusin 2
tethers endoplasmic reticulum to mitochondria. Nature. 456:605–610.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Buck MD, O'Sullivan D, Klein Geltink RI,
Curtis JD, Chang CH, Sanin DE, Qiu J, Kretz O, Braas D, van der
Windt GJ, et al: Mitochondrial dynamics controls T cell fate
through metabolic programming. Cell. 166:63–76. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Picca A, Mankowski RT, Burman JL, Donisi
L, Kim JS, Marzetti E and Leeuwenburgh C: Mitochondrial quality
control mechanisms as molecular targets in cardiac ageing. Nat Rev
Cardiol. 15:543–554. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Yang JF, Xing X, Luo L, Zhou XW, Feng JX,
Huang KB, Liu H, Jin S, Liu YN, Zhang SH, et al: Mitochondria-ER
contact mediated by MFN2-SERCA2 interaction supports
CD8+ T cell metabolic fitness and function in tumors.
Sci Immunol. 8:eabq24242023. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Tilokani L, Nagashima S, Paupe V and
Prudent J: Mitochondrial dynamics: Overview of molecular
mechanisms. Essays Biochem. 62:341–360. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Menk AV, Scharping NE, Rivadeneira DB,
Calderon MJ, Watson MJ, Dunstane D, Watkins SC and Delgoffe GM:
4-1BB costimulation induces T cell mitochondrial function and
biogenesis enabling cancer immunotherapeutic responses. J Exp Med.
215:1091–1100. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Hu C, Huang Y and Li L: Drp1-dependent
mitochondrial fission plays critical roles in physiological and
pathological progresses in mammals. Int J Mol Sci. 18:1442017.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Cereghetti GM, Stangherlin A, Martins De
Brito O, Chang CR, Blackstone C, Bernardi P and Scorrano L:
Dephosphorylation by calcineurin regulates translocation of Drp1 to
mitochondria. Proc Natl Acad Sci USA. 105:15803–15808. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Smith-Garvin JE, Koretzky GA and Jordan
MS: T cell activation. Annu Rev Immunol. 27:591–619. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Woehrle T, Ledderose C, Rink J, Slubowski
C and Junger WG: Autocrine stimulation of P2Y1 receptors is part of
the purinergic signaling mechanism that regulates T cell
activation. Purinergic Signalling. 15:127–137. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Ledderose C, Bromberger S, Slubowski CJ,
Sueyoshi K and Junger WG: Frontline science: P2Y11 receptors
support T cell activation by directing mitochondrial trafficking to
the immune synapse. J Leukocyte Biol. 109:497–508. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Quintana A, Schwindling C, Wenning AS,
Becherer U, Rettig J, Schwarz EC and Hoth M: T cell activation
requires mitochondrial translocation to the immunological synapse.
Proc Natl Acad Sci USA. 104:14418–14423. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Simula L, Pacella I, Colamatteo A,
Procaccini C, Cancila V, Bordi M, Tregnago C, Corrado M, Pigazzi M,
Barnaba V, et al: Drp1 controls effective T cell
immune-surveillance by regulating T cell migration, proliferation,
and cMyc-dependent metabolic reprogramming. Cell Rep.
25:3059–3073.e10. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Mohammadalipour A, Dumbali SP and Wenzel
PL: Mitochondrial transfer and regulators of mesenchymal stromal
cell function and therapeutic efficacy. Front Cell Dev Biol.
8:6032922020. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Lambeth DO: What is the function of GTP
produced in the Krebs citric acid cycle? IUBMB Life. 54:143–144.
2002.PubMed/NCBI
|
|
113
|
Saha T, Dash C, Jayabalan R, Khiste S,
Kulkarni A, Kurmi K, Mondal J, Majumder PK, Bardia A, Jang HL and
Sengupta S: Intercellular nanotubes mediate mitochondrial
trafficking between cancer and immune cells. Nat Nanotechnol.
17:98–106. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Baldwin JG, Heuser-Loy C, Saha T, Schelker
RC, Slavkovic-Lukic D, Strieder N, Hernandez-Lopez I, Rana N,
Barden M, Mastrogiovanni F, et al: Intercellular nanotube-mediated
mitochondrial transfer enhances T cell metabolic fitness and
antitumor efficacy. Cell. 187:6614–6630.e21. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Monaghan RM and Whitmarsh AJ:
Mitochondrial proteins moonlighting in the nucleus. Trends Biochem
Sci. 40:728–735. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
English J, Son JM, Cardamone MD, Lee C and
Perissi V: Decoding the rosetta stone of mitonuclear communication.
Pharmacol Res. 161:1051612020. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Quirós PM, Mottis A and Auwerx J:
Mitonuclear communication in homeostasis and stress. Nat Rev Mol
Cell Biol. 17:213–226. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Whelan SP and Zuckerbraun BS:
Mitochondrial signaling: Forwards, backwards, and in between. Oxid
Med Cell Longev. 2013:3516132013. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Jazwinski SM: The retrograde response: A
conserved compensatory reaction to damage from within and from
without. Prog Mol Biol Transl Sci. 127:133–154. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Walker BR and Moraes CT:
Nuclear-mitochondrial interactions. Biomolecules. 12:4272022.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Ju YS, Alexandrov LB, Gerstung M,
Martincorena I, Nik-Zainal S, Ramakrishna M, Davies HR,
Papaemmanuil E, Gundem G, Shlien A, et al: Origins and functional
consequences of somatic mitochondrial DNA mutations in human
cancer. Elife. 3:e029352014. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Chen S, Liao Z and Xu P: Mitochondrial
control of innate immune responses. Front Immunol. 14:11662142023.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Riley JS and Tait SW: Mitochondrial DNA in
inflammation and immunity. EMBO Rep. 21:e497992020. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Lin Y, Yang B, Huang Y, Zhang Y, Jiang Y,
Ma L and Shen YQ: Mitochondrial DNA-targeted therapy: A novel
approach to combat cancer. Cell Insight. 2:1001132023. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
West AP, Shadel GS and Ghosh S:
Mitochondria in innate immune responses. Nat Rev Immunol.
11:389–402. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
West AP, Khoury-Hanold W, Staron M, Tal
MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff
DA, et al: Mitochondrial DNA stress primes the antiviral innate
immune response. Nature. 520:553–557. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Shimada K, Crother TR, Karlin J, Dagvadorj
J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, et
al: Oxidized mitochondrial DNA activates the NLRP3 inflammasome
during apoptosis. Immunity. 36:401–414. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
De Gaetano A, Solodka K, Zanini G, Selleri
V, Mattioli AV, Nasi M and Pinti M: Molecular mechanisms of
mtDNA-mediated inflammation. Cells. 10:28982021. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Wang X, Zhang H, Wang Y, Bramasole L, Guo
K, Mourtada F, Meul T, Hu Q, Viteri V, Kammerl I, et al: DNA
sensing via the cGAS/STING pathway activates the immunoproteasome
and adaptive T-cell immunity. EMBO J. 42:e1105972023. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Cheng AN, Cheng LC, Kuo CL, Lo YK, Chou
HY, Chen CH, Wang YH, Chuang TH, Cheng SJ and Lee AY: Mitochondrial
lon-induced mtDNA leakage contributes to PD-L1-mediated
immunoescape via STING-IFN signaling and extracellular vesicles. J
Immunother Cancer. 8:e0013722020. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Mahmood M, Liu EM, Shergold AL, Tolla E,
Tait-Mulder J, Huerta-Uribe A, Shokry E, Young AL, Lilla S, Kim M,
et al: Mitochondrial DNA mutations drive aerobic glycolysis to
enhance checkpoint blockade response in melanoma. Nat Cancer.
5:659–672. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Corrado M, Samardžić D, Giacomello M, Rana
N, Pearce EL and Scorrano L: Deletion of the mitochondria-shaping
protein Opa1 during early thymocyte maturation impacts mature
memory T cell metabolism. Cell Death Differ. 28:2194–2206. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Peng J, Hu X, Fan S, Zhou J, Ren S, Sun R
and Chen Y, Shen X and Chen Y: Inhibition of mitochondrial
biosynthesis using a ‘right-side-out’ membrane-camouflaged micelle
to facilitate the therapeutic effects of shikonin on
triple-negative breast cancer. Adv Healthc Mater. 11:e22007422022.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Gemta LF, Siska PJ, Nelson ME, Gao X, Liu
X, Locasale JW, Yagita H, Slingluff CL Jr, Hoehn KL, Rathmell JC
and Bullock TNJ: Impaired enolase 1 glycolytic activity restrains
effector functions of tumor-infiltrating CD8+ T cells.
Sci Immunol. 4:eaap95202019. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Al-Habsi M, Chamoto K, Matsumoto K, Nomura
N, Zhang B, Sugiura Y, Sonomura K, Maharani A, Nakajima Y, Wu Y, et
al: Spermidine activates mitochondrial trifunctional protein and
improves antitumor immunity in mice. Science. 378:eabj35102022.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Cronin SJF, Seehus C, Weidinger A, Talbot
S, Reissig S, Seifert M, Pierson Y, McNeill E, Longhi MS, Turnes
BL, et al: The metabolite BH4 controls T cell proliferation in
autoimmunity and cancer. Nature. 563:564–568. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Malinee M, Pandian GN and Sugiyama H:
Targeted epigenetic induction of mitochondrial biogenesis enhances
antitumor immunity in mouse model. Cell Chem Biol. 29:463–475.e6.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Chamoto K, Chowdhury PS, Kumar A, Sonomura
K, Matsuda F, Fagarasan S and Honjo T: Mitochondrial activation
chemicals synergize with surface receptor PD-1 blockade for T
cell-dependent antitumor activity. Proc Natl Acad Sci USA.
114:E761–E770. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang
G, Hudaihed A, Filisio F, Giles-Davis W, Xu X, Karakousis GC, et
al: Enhancing CD8+ T cell fatty acid catabolism within a
metabolically challenging tumor microenvironment increases the
efficacy of melanoma immunotherapy. Cancer Cell. 32:377–391.e9.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
He J, Shangguan X, Zhou W, Cao Y, Zheng Q,
Tu J, Hu G, Liang Z, Jiang C, Deng L, et al: Glucose limitation
activates AMPK coupled SENP1-Sirt3 signalling in mitochondria for T
cell memory development. Nat Commun. 12:43712021. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Yuan S, Sun R, Shi H, Chapman NM, Hu H,
Guy C, Rankin S, Kc A, Palacios G, Meng X, et al: VDAC2 loss
elicits tumour destruction and inflammation for cancer therapy.
Nature. 640:1062–1071. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Liu YN, Yang JF, Huang DJ, Ni HH, Zhang
CX, Zhang L, He J, Gu JM, Chen HX, Mai HQ, et al: Hypoxia induces
mitochondrial defect that promotes T cell exhaustion in tumor
microenvironment through MYC-regulated pathways. Front Immunol.
11:19062020. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Teijeira A, Labiano S, Garasa S,
Etxeberria I, Santamaría E, Rouzaut A, Enamorado M, Azpilikueta A,
Inoges S, Bolaños E, et al: Mitochondrial morphological and
functional reprogramming following CD137 (4-1BB) costimulation.
Cancer Immunol Res. 6:798–811. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Wan H, Xu B, Zhu N and Ren B: PGC-1α
activator-induced fatty acid oxidation in tumor-infiltrating CTLs
enhances effects of PD-1 blockade therapy in lung cancer. Tumori.
106:55–63. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Chowdhury PS, Chamoto K, Kumar A and Honjo
T: PPAR-induced fatty acid oxidation in T cells increases the
number of tumor-reactive CD8+ T cells and facilitates
anti-PD-1 therapy. Cancer Immunol Res. 6:1375–1387. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Bukoreshtliev NV, Wang X, Hodneland E,
Gurke S, Barroso JFV and Gerdes HH: Selective block of tunneling
nanotube (TNT) formation inhibits intercellular organelle transfer
between PC12 cells. FEBS Lett. 583:1481–1488. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Liu K, Ji K, Guo L, Wu W, Lu H, Shan P and
Yan C: Mesenchymal stem cells rescue injured endothelial cells in
an in vitro ischemia-reperfusion model via tunneling nanotube like
structure-mediated mitochondrial transfer. Microvasc Res. 92:10–18.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Onfelt B, Nedvetzki S, Benninger RKP,
Purbhoo MA, Sowinski S, Hume AN, Seabra MC, Neil MA, French PM and
Davis DM: Structurally distinct membrane nanotubes between human
macrophages support long-distance vesicular traffic or surfing of
bacteria. J Immunol. 177:8476–8483. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Zhang H, Yu X, Ye J, Li H, Hu J, Tan Y,
Fang Y, Akbay E, Yu F, Weng C, et al: Systematic investigation of
mitochondrial transfer between cancer cells and T cells at
single-cell resolution. Cancer Cell. 41:1788–1802.e10. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Pierini S, Fang C, Rafail S, Facciponte
JG, Huang J, De Sanctis F, Morgan MA, Uribe-Herranz M, Tanyi JL and
Facciabene A: A tumor mitochondria vaccine protects against
experimental renal cell carcinoma. J Immunol. 195:4020–4027. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Shang L, Jiang X, Zhao X, Huang X, Wang X,
Jiang X, Kong X, Yao M, Jiang S and Wong PP: Mitochondrial
DNA-boosted dendritic cell-based nanovaccination triggers antitumor
immunity in lung and pancreatic cancers. Cell Rep Med.
5:1016482024. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Mok BY, de Moraes MH, Zeng J, Bosch DE,
Kotrys AV, Raguram A, Hsu F, Radey MC, Peterson SB, Mootha VK, et
al: A bacterial cytidine deaminase toxin enables CRISPR-free
mitochondrial base editing. Nature. 583:631–637. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Fan Y, Xu W, Gao BQ, Qin H, Wu X, Wei J,
Ni Q, Zhou L, Xiang J, Wu J, et al: Leveraging base excision repair
for efficient adenine base editing of mitochondrial DNA. Nat
Biotechnol. Mar 25–2025.(Epub ahead of print). View Article : Google Scholar
|
|
154
|
Guo Y, Zhang H, Yan C, Shen B, Zhang Y,
Guo X, Sun S, Yu F, Yan J, Liu R, et al: Small molecule agonist of
mitochondrial fusion repairs mitochondrial dysfunction. Nat Chem
Biol. 19:468–477. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Chen L, Fan Y, Jiang N, Huang X, Yu M,
Zhang H, Xu Z, He D, Wang Y, Ding C, et al: An energy
metabolism-engaged nanomedicine maintains mitochondrial homeostasis
to alleviate cellular ageing. Nat Nanotechnol. 20:1332–1344. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Pan X, Wang Z, Tan M, Fu Z, Nie G and Wang
H: Nanoinducer-mediated mitochondria-selective degradation enhances
T cell immunotherapy against multiple cancers. Nat Nanotechnol.
20:947–958. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Lv Y, Song B, Yang G, Wang Y, Wu Z, Si M,
Yang Z, Chen H, Liu C, Li M, et al: In situ transformable
nanoparticle effectively suppresses bladder cancer by damaging
mitochondria and blocking mitochondrial autophagy flux. Adv Sci
(Weinh). 12:e24094252025. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Wang W, Yao SY, Luo J, Ding C, Huang Q,
Yang Y, Shi Z, Lin J, Pan YC, Zeng X, et al: Engineered
hypoxia-responsive albumin nanoparticles mediating mitophagy
regulation for cancer therapy. Nat Commun. 16:5962025. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Liu J, Yan Y, Zhang Y, Pan X, Xia H, Zhou
J, Wan F, Huang X, Zhang W, Zhang Q, et al: Lysosome-mitochondria
cascade targeting nanoparticle drives robust pyroptosis for cancer
immunotherapy. J Am Chem Soc. 146:34568–34582. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Pei D and Buyanova M: Overcoming endosomal
entrapment in drug delivery. Bioconjug Chem. 30:273–283. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Goswami R, Jeon T, Nagaraj H, Zhai S and
Rotello VM: Accessing intracellular targets through
nanocarrier-mediated cytosolic protein delivery. Trends Pharmacol
Sci. 41:743–754. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Kang H, Han M, Xue J, Baek Y, Chang J, Hu
S, Nam H, Jo MJ, El Fakhri G, Hutchens MP, et al: Renal clearable
nanochelators for iron overload therapy. Nat Commun. 10:51342019.
View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Feng K, Xu Z, Wang Y, Wu X, Xiong F, Ruan
Y, Wu X, Ye L, Su D, Yu J and Sun X: Renal-clearable porous hollow
copper iron oxide nanoparticles for trimodal
chemodynamic-photothermal-chemo anti-tumor therapy. Nanoscale.
15:3188–3198. 2023. View Article : Google Scholar : PubMed/NCBI
|