Introduction
The occurrence and development of tumors are complex
processes driven by multiple factors, including accumulation of
gene mutations (1), imbalance in
epigenetic regulation (2), and
abnormalities in key signaling pathways (3). Although high-throughput sequencing
technology has systematically depicted the mutation landscapes of
different tumor types (4), gene
mutations alone are insufficient to fully explain the high degree
of plasticity and adaptability exhibited by tumor cells under
different microenvironments and treatment pressures. By contrast,
epigenetic regulation is dynamic and reversible, enabling tumor
cells to rapidly reshape their transcriptional processes without
altering the DNA sequence, which allows them to adapt to
environmental changes and gain survival advantages (5). Therefore, chromatin-level regulation
is an important mechanistic basis for understanding tumor
progression and differences in treatment responses.
Of numerous chromatin-remodeling complexes, the
nucleosome-remodeling and deacetylase (NuRD) complex holds a unique
position due to its simultaneous adenosine triphosphate
(ATP)-dependent chromatin-remodeling activity and histone
deacetylase (HDAC) activity (6).
Structural studies have revealed that the NuRD complex has a highly
modular organizational form. The HDAC-metastasis-associated protein
(MTA)-retinoblastoma-binding protein subunits form a relatively
stable deacetylase core, while chromodomain helicase DNA-binding
protein 3 or 4 (CHD3, CHD4) constitutes the ATP-dependent
chromatin-remodeling module. The selective assembly of different
subunits determines the functional characteristics of different
NuRD subtypes (7) (Fig. 1).
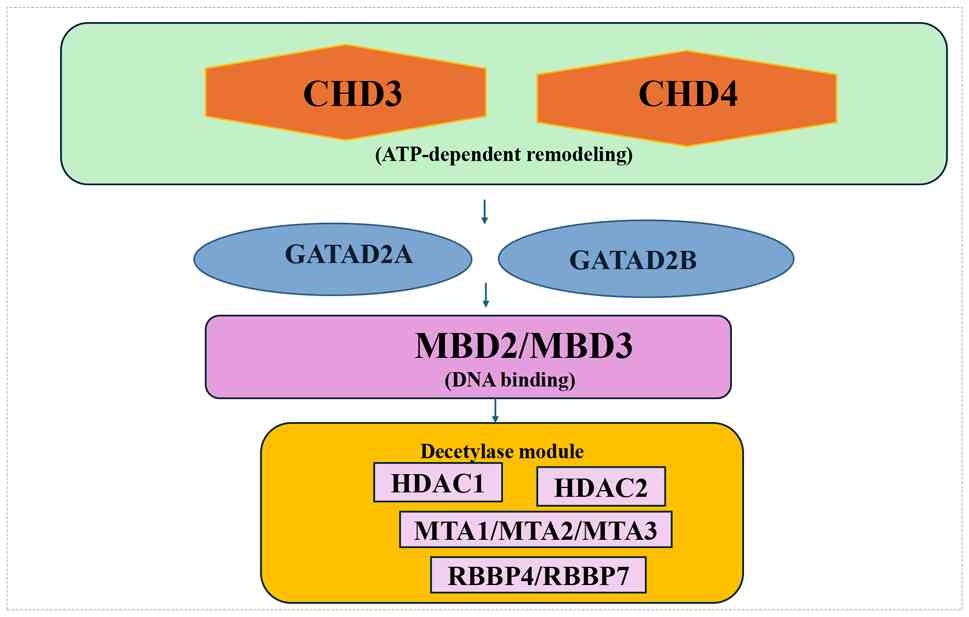 | Figure 1.Modular organization of the NuRD
Complex. CHD3 and CHD4 constitute the ATP-dependent
chromatin-remodeling module, which interacts with GATAD2A/B and
MBD2/3. The deacetylase module contains HDAC1/2, MTA1/2/3 and
RBBP4/7. NuRD, nucleosome remodeling and deacetylase; CHD3,
chromodomain helicase DNA-binding protein 3; CHD4, chromodomain
helicase DNA-binding protein 4; GATAD2A, GATA zinc finger domain
containing 2A; GATAD2B, GATA zinc finger domain containing 2B;
MBD2, methyl-CpG-binding domain protein 2; MBD3, methyl-CpG-binding
domain protein 3; HDAC1, histone deacetylase 1; HDAC2, histone
deacetylase 2; MTA1/2/3, metastasis-associated proteins 1/2/3;
RBBP4, retinoblastoma-binding protein 4; RBBP7,
retinoblastoma-binding protein 7. |
CHD4 belongs to the CHD family of
chromatin-remodeling proteins and contains multiple conserved
functional domains. Its N-terminus has two tandem plant homeodomain
(PHD) finger domains that can recognize histone H3 tail
modifications, thereby promoting localization of the NuRD complex
to chromatin regions associated with gene silencing (8). Following the PHD finger domains, CHD4
has two tandem chromodomains that can interact with nucleosomal DNA
and participate in regulation of ATPase activity. Its central
region encompasses a highly conserved sucrose non-fermentable 2
(SNF2) family ATPase/helicase domain, which provides energy via ATP
hydrolysis to drive nucleosome sliding (9). In addition to the ATPase core domain,
multiple auxiliary regions of CHD4 are also involved in its
functional regulation. For example, the N-terminal disordered
region can enhance chromatin-remodeling activity, while the
C-terminal region has a certain self-inhibitory effect, thereby
limiting the catalytic ability of the ATPase. When the SWItch
3-adenosine deaminase 2-nuclear receptor corepressor
1-transcription factor IIIB/SANT-like Imitation SWItch domain
(SANT/SLIDE) DNA-binding domain binds to DNA, this self-inhibitory
state can be relieved, thereby activating the nucleosome-remodeling
function of CHD4 (10). Through the
synergistic action of these domains, CHD4 plays important roles in
gene expression regulation (11),
DNA damage response (DDR) (12),
and chromatin structure regulation (13,14).
Therefore, abnormal expression or gene mutations of Chd4 are
closely associated with various human diseases, including
congenital developmental defects and neurological disorders
(6). Meanwhile, an increasing
number of studies have shown that CHD4 also plays a key role in
tumor initiation, tumor evolution, and the development of treatment
resistance (15).
Context-dependent roles of CHD4 in tumor
progression
CHD4 is a typical context-dependent epigenetic
regulator. In different cell types, chromatin environments, and
signaling pathways, CHD4 can either promote tumorigenesis or
exhibit tumor-suppressive functions (16).
Molecular mechanisms by which CHD4
promotes tumor progression
In various solid tumors, high expression of CHD4 is
often associated with increased tumor aggressiveness, enhanced
metastatic potential, and poor prognosis, suggesting that its role
in tumor progression is crucial (17). Recent studies have shown that CHD4
does not function through a single molecular pathway but is more
likely to influence tumor cell behavior by regulating multiple
interconnected transcriptional processes (18–20).
Of these transcriptional regulatory networks (TRNs),
oncogenic signaling pathways form an important basis for CHD4′s
involvement in tumor migration and invasion. Wingless/Integrated
(Wnt)/β-catenin is a typical signaling pathway through which CHD4
regulates tumor metastasis. In gastric cancer (GC), CHD4 interacts
with myosin heavy chain 9, thereby inhibiting the activity of
glycogen synthase kinase-3β and stabilizing β-catenin, which
activates the Wnt/epithelial-mesenchymal transition (EMT) signaling
pathway, thereby promoting tumor invasion and metastasis (18). Similar regulatory patterns can also
be observed in ovarian cancer, where CHD4 enhances nuclear
accumulation and transcriptional activity of β-catenin through the
enhancer of zeste homolog 2 (EZH2)/β-catenin signaling axis
(19). These studies suggest that
CHD4 may repeatedly participate in the regulation of
β-catenin-related transcriptional processes in different tumor
types, thereby promoting EMT and tumor metastasis. In addition to
the Wnt/β-catenin pathway, CHD4′s influence on tumor cell migration
is also closely related to cytoskeleton-related signaling pathways.
In non-small cell lung cancer, CHD4 activates the Ras
homolog gene family, member A (RhoA)/ρ-associated protein kinase
signaling pathway by regulating PHD finger protein 5A, thereby
enhancing the proliferation and migratory ability of tumor cells
(20). This mechanism suggests that
CHD4 might help regulate tumor cell movement by affecting
cytoskeletal dynamics and act as a bridge between different
signaling networks.
The regulatory role of CHD4 is not limited to cell
migration but also extends to regulation of tumor cell
proliferation. For example, in breast cancer (BC), CHD4 upregulates
the transcriptional levels of estrogen receptor-α (ERα) and
inhibits its ubiquitination degradation, thereby continuously
enhancing the transcriptional activity of the ERα signaling pathway
(21). This phenomenon indicates
that CHD4 can not only regulate classical signaling pathways but
might also further amplify oncogenic signals by stabilizing key
transcription factors (TFs).
A hypoxic state in the tumor microenvironment (TME)
further expands the functions of CHD4, which can co-activate
hypoxia-inducible factors 1α and 2α (HIF-1α, HIF-2α),
synergistically promoting the transcription of hypoxia-responsive
genes with the HIF complex. Under normoxic conditions, CHD4 is
already enriched in the promoter regions of HIF target genes and
promotes the loading of RNA polymerase II via p300, while HIF
activation under hypoxic conditions further enhances the
recruitment of CHD4 to chromatin, forming a positive-feedback
regulatory loop and amplifying the hypoxia response signal
(22). This process suggests that
the chromatin-binding and transcriptional regulatory activity of
CHD4 may be further enhanced under hypoxic conditions. Such changes
may further affect the downstream TRN of CHD4. For example, CHD4
can promote citrullination of the key glycolytic enzyme pyruvate
kinase muscle isozyme 2 (PKM2) by regulating the expression of
peptidyl arginine deiminases 1 and 3 (PADI1, PADI3), thereby
altering PKM2′s metabolic regulatory mode and enhancing glycolytic
activity (23). Given that enhanced
glycolysis is an important means by which tumor cells adapt to
hypoxic environments, this CHD4-PADI1/3-PKM2 regulatory axis may be
a mechanism by which CHD4 participates in tumor metabolic
adaptation under hypoxic conditions.
In addition to signaling and metabolic regulation,
CHD4 also participates in tumorigenesis by maintaining the
silencing of tumor suppressor genes. In colorectal cancer (CRC),
CHD4 is recruited to DNA damage sites and further recruits DNA
methyltransferases (DNMTs) to establish abnormal DNA methylation
and maintain suppressive chromatin structures at the promoter
regions of multiple tumor suppressor genes (24). Moreover, CHD4 can synergize with
DNMT1 and DNMT3B to jointly maintain the stable silencing of tumor
suppressor genes through multiple epigenetic mechanisms, including
DNA methylation, histone deacetylation, and nucleosome remodeling
(25).
It is worth noting that in certain tumor types, CHD4
also helps maintain chromatin structural homeostasis. For example,
in Ewing sarcoma, CHD4 deletion leads to chromatin structural
disorders and induces spontaneous DNA damage accumulation, thereby
significantly inhibiting tumor cell proliferation (13). This phenomenon suggests that the
role of CHD4 may be highly dependent on the context of the specific
tumor environment.
Taken together, these studies paint an increasingly
clearer picture: CHD4 reshapes tumor cell behavior through multiple
mechanisms, including regulation of signaling pathways,
microenvironment adaptation, metabolic reprogramming, and
epigenetic regulation, thereby promoting tumor progression.
Tumor-suppressive role of CHD4 in
specific transcriptional environments
Although numerous studies have shown that CHD4 has
oncogenic effects in various tumors, under specific transcriptional
regulatory conditions, it may also exhibit tumor-suppressive
functions. For example, in luminal-type BC, the TF transcriptional
repressor GATA binding 1 (TRPS1) can recruit the CHD4/NuRD (MTA2)
complex to the promoter region of tp63, inhibiting tp63 expression
via chromatin remodeling and thereby limiting tumor cell migration
and invasion (26). Further
research has found that TRPS1 can also recruit CHD4 to the
regulatory region of sex-determining region Y (SRY)-related
high-mobility group (HMG) box 2 (SOX2), inhibiting SOX2
transcription by altering local chromatin structure (27). Since SOX2 is an important factor for
maintaining cancer stem cell characteristics, the TRPS1-CHD4-SOX2
regulatory axis can exert tumor-suppressive effects by limiting
tumor stemness. In addition, CHD4 can help inhibit tumor
progression by regulating enhancer activity. Studies have shown
that AT-rich interactive domain-containing protein 1A (ARID1A)
promotes the binding of the CHD4-zinc finger myeloid, Nervy, and
deformed epidermal autoregulatory factor 1 homolog, or MYND, type
containing 8 (ZMYND8) complex to super-enhancers by maintaining the
distribution of the histone variant H3.3 on chromatin, thereby
exerting transcriptional inhibitory effects on genes related to EMT
and cell migration. When ARID1A function is lost, the
transcriptional inhibition mediated by CHD4-ZMYND8 is disrupted,
leading to excessive activation of super-enhancers and promoting
tumor cell migration and invasion (16).
The results of the aforementioned studies indicate
that when CHD4 synergizes with specific TFs or chromatin
regulators, it can limit the activation of abnormal transcriptional
processes by maintaining suppressive chromatin structures, thereby
exerting tumor-suppressive effects under specific contexts.
Effect of Chd4 mutations on tumor
progression
In addition to changes in expression levels,
mutations to the Chd4 gene can also reshape the TRN related
to tumor progression. It has been shown that Chd4 mutations
do not necessarily lead to complete loss of its function but may
reshape the cell's transcriptional process by altering its
chromatin-remodeling activity or target gene selectivity. This view
was first supported by functional studies. Researchers used the
Drosophila homolog dMi-2 as a model to systematically
analyze various missense mutations of Chd4 obtained from
patients with tumors. The results revealed that these mutations
could alter the ATPase activity, nucleosome-binding ability, and
nucleosome-sliding efficiency of Chd4 in a mutation-specific
manner. Some mutations reduce ATP hydrolysis ability or weaken
chromatin-remodeling efficiency, while others may enhance related
activities (28). Similar mutation
effects have also been observed in human tumors. For example, in
endometrial cancer, the common mutations R975H and R1162W can
reduce CHD4 protein stability and weaken its function, thereby
activating the transforming growth factor-β signaling pathway and
enhancing cancer cell stemness characteristics (29). Another study also found that the
R975H mutation could activate multiple oncogenic signaling
pathways, such as tumor necrosis factor-α/nuclear factor
κ-light-chain-enhancer of activated B cells, Kirsten Ras
oncogene homolog, mammalian target of rapamycin, and
myelocytomatosis oncogene; and promote polarization of
tumor-associated macrophages toward an immunosuppressive M2
phenotype, thereby enhancing tumor immune escape ability (15). In addition to missense mutations,
some truncating mutations located in the SNF2 domain (such as
p.Trp736Ter) may also disrupt the integrity of the NuRD complex and
lead to abnormal chromatin remodeling, thereby promoting
tumorigenesis and metastasis (30).
In conclusion, the effects of Chd4 mutations on tumor
progression show significant heterogeneity.
CHD4 in DDR and tumor treatment
resistance
Regulatory role of CHD4 in DDR
DDR, the core defense system allowing cells to
maintain genomic integrity, encompasses multiple stages such as
damage recognition, signal transduction, chromatin remodeling and
DNA repair (31). It has been shown
that dynamic changes in chromatin structure are an indispensable
regulatory link in the DDR chain. As an important
chromatin-remodeling factor, CHD4 plays a crucial role in this
process (32).
After the occurrence of DNA double-strand breaks
(DSBs), cells first rapidly recognize the damaged sites through
poly [adenosine diphosphate (ADP)-ribose] polymerase 1/2
(PARP1/2)-mediated poly-ADP-ribosylation reactions. Poly [adenosine
diphosphate (ADP)-ribose] (PAR) chains not only serve as a scaffold
for recruiting DNA repair factors but they can also induce local
chromatin relaxation, thereby promoting recruitment of repair
factors (33). Studies have
demonstrated that at this stage, CHD4 can be rapidly recruited to
DNA damage sites in a PAR-dependent manner and participate in early
chromatin remodeling (32). In the
early stages of DDR, CHD4 usually works in concert with the
acetyltransferase p300. P300 reduces chromatin compaction through
histone acetylation, while CHD4 relies on its ATPase activity to
regulate nucleosome positions. This maintains a relatively open and
dynamic chromatin state in the damaged region, thereby providing
optimal conditions for the entry of DNA repair factors (34).
As DDR signals are further amplified, ataxia
telangiectasia mutated protein (ATM) kinase is activated and
initiates a series of signal cascades. Really Interesting New Gene
finger protein 8 (RNF8)-mediated histone ubiquitination is
considered an important step in signal amplification. RNF8 can
catalyze the ubiquitination modification of H2A and H2A histone
family member X, providing a molecular platform for the stable
binding of DNA repair complexes such as breast cancer type 1
(BRCA1) (35). During this
catalytic process, CHD4 enhances the spatial accessibility between
RNF8 and its substrates by regulating the chromatin structure
around the damaged sites, thereby promoting formation of ubiquitin
chains. Meanwhile, RNF8 can also promote recruitment of additional
CHD4 to the damaged region via a non-catalytic mechanism, forming a
positive-feedback regulatory loop at damaged sites and further
stabilizing the assembly of DNA repair complexes (36).
Regulation of DDR also relies on the synergistic
action of multiple chromatin regulatory factors. For example, the
silent mating type information regulation 2 homolog 6 (SIRT6)/CHD4
pathway is considered an important regulatory branch for DSB
repair. SIRT6 can act as a damage sensor and promote early signal
amplification by activating PARP1 (37); under ATM-dependent conditions,
interaction between SIRT6 and CHD4 is significantly enhanced, and
they jointly promote chromatin remodeling at the damaged sites and
homologous recombination repair (HRR). When ATM activity is
inhibited or absent, this synergistic effect is significantly
weakened, leading to a decrease in HRR efficiency (38). In addition, it has been demonstrated
that CHD4-mediated chromatin remodeling may also affect the choice
of DNA repair pathways via regulation of R-loop-related mechanisms.
When DSBs occur in transcriptionally active chromatin regions, the
bromodomain and extra-terminal domain family protein
bromodomain-containing protein 3 can recruit CHD4 and work in
concert with the Tat interacting protein, 60 kDa complex to
initiate chromatin remodeling, promoting histone H4-lysine 16
acetylation and expelling heterochromatin protein 1. This inhibits
the binding of p53 binding protein 1 at damaged sites and promotes
the recruitment of BRCA1 and R-loop-related repair factors
(39), a process that ultimately
promotes R-loop-mediated HRR. Taken together, these findings
suggest that CHD4 can affect DNA repair pathway selection by
regulating R-loop-related chromatin remodeling, thereby maintaining
genomic stability.
As DNA damage is gradually repaired, cells must
terminate DDR signals in a timely manner and restore normal cell
cycle progression. The ATM-checkpoint kinase 2-p53 signaling axis
plays a central role in this process: p53 can induce p21 expression
to mediate G1/S checkpoint arrest, thereby providing a window of
time for DNA repair (40). It has
been revealed that as a core subunit of the NuRD complex, CHD4 can
mediate p53 deacetylation and inhibit its transcriptional activity
(41). This suggests that CHD4 may
be involved in terminating DDR signals and restoring cell cycle
after DNA repair is completed.
In the more complex chromatin regulatory network
(CRN), the overall synergistic action of the NuRD complex is also
crucial for DDR. For example, the R-loop structure formed on DNA
damage can promote the establishment of chromatin boundaries via
the GATA zinc finger domain containing 2B/NuRD complex in the
damaged region, thereby limiting excessive DNA end resection and
maintaining the stability of the repair process (42). In addition, in the PARP-dependent
repair pathway, chromatin regulatory factors such as lysine
(K)-specific demethylase 5A and ZMYND8 can further stabilize the
enrichment of CHD4 at damaged sites by interacting with NuRD
complex subunits (43). The
aforementioned CRNs jointly participate in the dynamic remodeling
of chromatin structure in the damaged region, thereby affecting the
localization of DNA repair factors and choice of repair pathways
(Fig. 2).
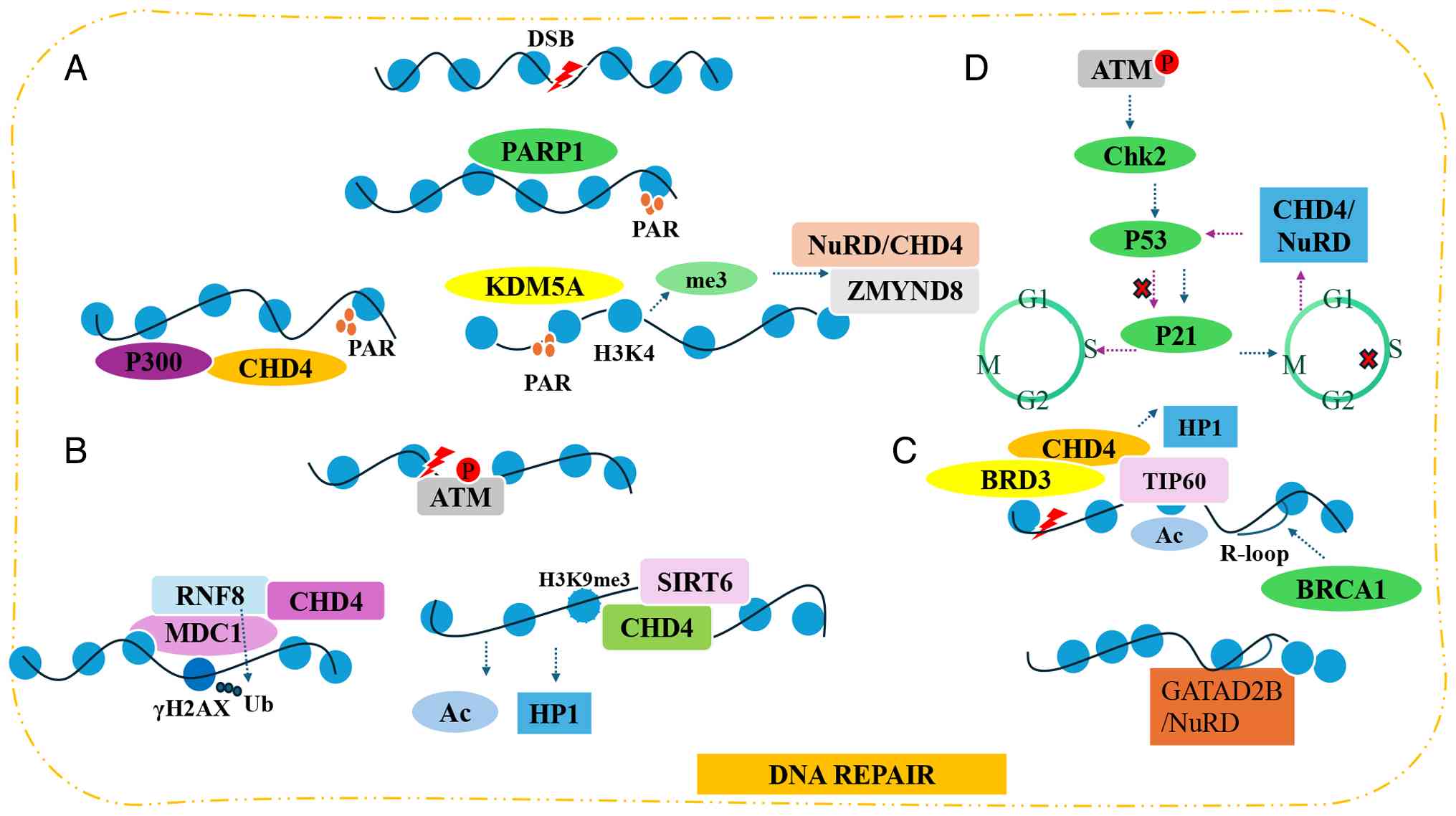 | Figure 2.CHD4′s involvement in DDR. (A) After
DNA damage occurs, PARP is activated and recruits the CHD4/p300
complex or the ZMYND8/NuRD complex, thereby promoting DNA repair.
(B) ATM drives chromatin remodeling mediated by RNF8-CHD4 or
SIRT6-CHD4. (C) CHD4 is involved in R-loop-mediated DNA repair. (D)
After DNA repair is completed, CHD4/NuRD inhibits p53 activity via
deacetylation, thereby terminating DDR signals. DSB, DNA
double-strand break; PARP1, poly(ADP-ribose) polymerase 1; PAR,
poly(ADP-ribose); P300, E1A binding protein p300; CHD4,
chromodomain helicase DNA-binding protein 4; KDM5A, lysine-specific
demethylase 5A; H3K4, histone H3 lysine 4; me3, trimethylation;
NuRD, nucleosome remodeling and deacetylase complex; ZMYND8, zinc
finger MYND-type containing 8; ATM, ataxia telangiectasia mutated;
RNF8, RING finger protein 8; MDC1, mediator of DNA damage
checkpoint 1; γH2AX, phosphorylated H2A histone family member X;
Ub, ubiquitin; SIRT6, sirtuin 6; HP1, heterochromatin protein 1;
Ac, acetylation; BRD3, bromodomain-containing protein 3; TIP60, Tat
interacting protein, 60 kDa; R-loop, RNA-DNA hybrid structure;
BRCA1, breast cancer type 1 susceptibility protein; GATAD2B, GATA
zinc finger domain containing 2B; Chk2, checkpoint kinase 2; p53,
tumor protein p53; p21, cyclin-dependent kinase inhibitor 1A. |
CHD4 and tumor treatment
resistance
Abnormal CHD4 expression is closely related to
insensitivity of tumors to radiotherapy (44) and chemotherapy (CT) (45), suggesting that it plays an important
role in the development of tumor treatment resistance. In the
context of DNA-damaging treatment, CHD4 can regulate chromatin
structure to keep DDR-related genes ready to be rapidly activated,
thereby enhancing tumor cells' ability to repair treatment-induced
DNA damage. This effect is relatively typical in glioblastoma,
where CHD4 maintains the expression of key HRR factors such as
RAD51, thereby increasing tumor cell resistance to radiotherapy and
DNA-damaging CT drugs (46). This
phenomenon suggests that the treatment resistance mediated by CHD4
is not merely due to changes in a single repair factor but rather
reflects CHD4′s regulation of the DNA repair transcriptional
process's overall accessibility.
In addition to enhanced DNA repair ability, CHD4 can
also participate in CT resistance by regulating pro-survival
signaling pathways and drug efflux mechanisms. In GC, high CHD4
expression is closely related to tumor progression and CT
resistance. CHD4 promotes the interaction between mitogen-activated
protein kinase (MEK1/2) and extracellular signal-regulated kinase
1/2 (ERK1/2) and activates the MEK/ERK signaling pathway, thereby
maintaining continuous phosphorylation of ERK and enhancing the
proliferation and survival ability of tumor cells. Meanwhile, this
pathway can also upregulate major vault protein expression, promote
drug efflux, and reduce the intracellular concentration of CT drugs
(for example, cisplatin), ultimately leading to CT resistance in GC
cells (47). Similar drug efflux
mechanisms can also be observed in ovarian cancer, where high CHD4
expression can upregulate that of multidrug resistance mutation 1
and enhance drug efflux ability, thereby reducing the accumulation
of cisplatin in cells and leading to platinum-based CT resistance
(48). These findings collectively
indicate that CHD4 not only affects the DNA repair ability of tumor
cells but may also expand drug resistance by remodeling the
intracellular-drug disposition and survival signaling networks.
The effect of CHD4 on treatment response also
extends to the level of cell cycle regulation. In BC, CHD4 forms
the NuRD complex with HDAC1 and acts on the p21 promoter region,
inhibiting the transcription and expression of p21 via chromatin
remodeling and histone deacetylation (49). Since p21 is an important cell cycle
inhibitor, such a decrease in its expression can promote cell cycle
progression, making tumor cells more likely to bypass cell cycle
checkpoints after DNA damage and thereby enhancing their resistance
to CT drugs. This shows that CHD4-mediated treatment resistance not
only depends on ‘repairing more damage’ but also involves the
reshaping of cell cycle timing, enabling tumor cells to maintain a
proliferative advantage under treatment pressure.
In addition to the intrinsic mechanisms of tumor
cells, CHD4 can also affect the response to immunotherapy by
regulating the tumor immune microenvironment. In
microsatellite-stable CRC, CHD4 can recruit the histone
methyltransferase euchromatic histone lysine methyltransferase 2
(EHMT2) to form a co-transcriptional inhibitory complex, thereby
inhibiting the expression of galectin-7 (GAL7) and maintaining an
immunosuppressive ‘cold-tumor’ microenvironment. Inhibition of
EHMT2 can restore GAL7 expression and enhance Cluster of
Differentiation 8+ (CD8+) T-cell-mediated
antitumor immune responses, thereby increasing sensitivity to
programmed cell death protein 1 inhibitor therapy (50). Pan-cancer data analysis further
supports the important role of CHD4 in tumor treatment resistance.
CHD4 promotes the development of genomic-instability
characteristics via epigenetic regulation while shaping an
immunosuppressive TME characterized by reduced CD8+
T-cell infiltration and increased immune escape (51). This suggests that the role of CHD4
extends beyond tumor cells themselves to the tumor immune system
interactions on which treatment responses depend.
Notably, CHD4′s effect on treatment resistance does
not always manifest in the same way but is shaped by specific
genetic backgrounds. In BRCA1/2-deficient tumors, CHD4 deletion
does not restore HRR ability but instead enhances replication fork
stability by inhibiting meiotic recombination 11 homolog 1-mediated
replication fork degradation, thereby increasing the resistance of
tumor cells to cisplatin and PARP inhibitors (52). This phenomenon shows that the role
of CHD4 cannot be simply summarized as ‘promoting repair’ or
‘inhibiting repair’; its deeper function may lie in regulating how
tumor cells respond to replication and treatment stresses.
Therefore, its role in tumor treatment resistance is obviously
context dependent, and its biological consequences depend on the
specifics of the genetic background, repair pathway state, and TME
characteristics (Fig. 3).
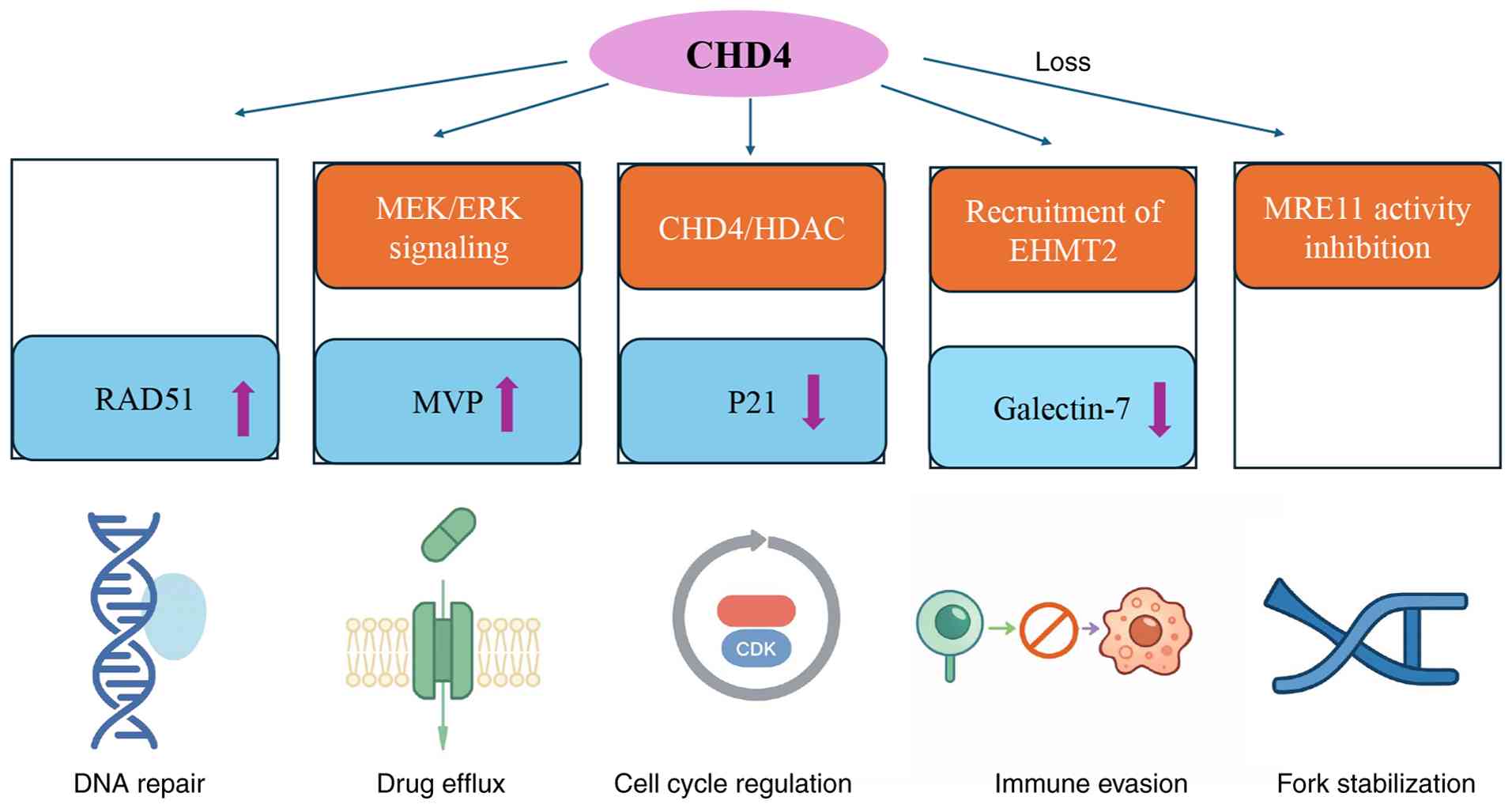 | Figure 3.Role of CHD4 in tumor therapy
resistance. CHD4 mediates tumor therapy resistance via regulation
of DNA repair, drug efflux, cell cycle progression, immune evasion
and replication fork stability. CHD4, chromodomain helicase
DNA-binding protein 4; RAD51, RAD51 recombinase; MEK,
mitogen-activated protein kinase; ERK, extracellular
signal-regulated kinase; MVP, major vault protein; HDAC, histone
deacetylase; EHMT2, euchromatic histone lysine methyltransferase 2;
MRE11, MRE11 homolog double-strand break repair nuclease; p21,
cyclin-dependent kinase inhibitor 1A. |
Non-coding RNA (ncRNA)-mediated regulatory
network of CHD4
Although CHD4 plays a crucial role in chromatin
remodeling, DDR, and tumor therapy resistance, its function is not
solely determined by the protein complex itself. In recent years,
ncRNAs have increasingly been recognized as significant regulators
of CHD4, a key epigenetic hub (53). In transcriptionally active regions,
RNA molecules can directly bind to CHD4, thereby influencing its
chromatin-binding capacity and nucleosome-remodeling activity.
Studies have shown that CHD4 exhibits a high affinity for RNA
molecules rich in guanine (G) and that RNA binding inhibits CHD4′s
interaction with chromatin and weakens its nucleosome-remodeling
ability. Mechanistically, RNA and CHD4 have a competitive binding
relationship, which hinders CHD4 from establishing repressive
chromatin structures in transcriptional regions and maintains a
relatively open chromatin state (54). Under DNA damage conditions, long
ncRNAs can also participate in functional regulation of CHD4. For
example, nuclear paraspeckle assembly transcript 1 (Neat1)
undergoes spatial relocation via N6-methyladenosine modification
after DNA damage and acts as a ‘molecular scaffold’ at the damage
site, thereby redistributing the subnuclear localization of CHD4.
This process not only helps amplify the damage signal but also
promotes assembly of DNA repair complexes (53). Another class of ncRNAs with active
regulatory roles is developmental pluripotency associated 2
upstream binding RNA (Dubr). Research has found that
Dubr can directly interact with the NuRD complex and inhibit
the expression of genes related to cell differentiation and
morphogenesis by regulating chromatin accessibility at activator
protein 1 enhancer regions (55).
In addition to directly regulating CHD4 activity,
various microRNAs (miRNAs or miRs) and circular RNAs (circRNAs) can
modulate CHD4 expression levels by affecting its transcription,
mRNA stability, or protein degradation. In GC, circFBXL4
acts as a competing endogenous RNA for miR-146a-5p, relieving its
inhibition of signal transducer and activator of transcription 1
and thereby indirectly promoting CHD4 transcription (56). Meanwhile, hsa-circ-0007396 can
‘adsorb’ miR-767-3p, weakening its targeting effect on CHD4 mRNA
and thus upregulating CHD4 expression (57). In oral squamous-cell carcinoma,
hsa-miR-194-5p can directly target CHD4 and regulate the
phosphoinositide 3-kinase/protein kinase B signaling pathway,
thereby enhancing cell anti-apoptotic ability and promoting drug
resistance (58). Furthermore, some
circRNAs can participate in tumor progression by regulating CHD4
protein stability. For example, in CRC, circWBSCR22 can
inhibit up-frameshift mutation 1 homolog (UPF1)-mediated CHD4
ubiquitination and degradation by binding to UPF1, thereby
stabilizing CHD4 protein and promoting EMT, cell invasion and tumor
metastasis (59) (Fig. 4).
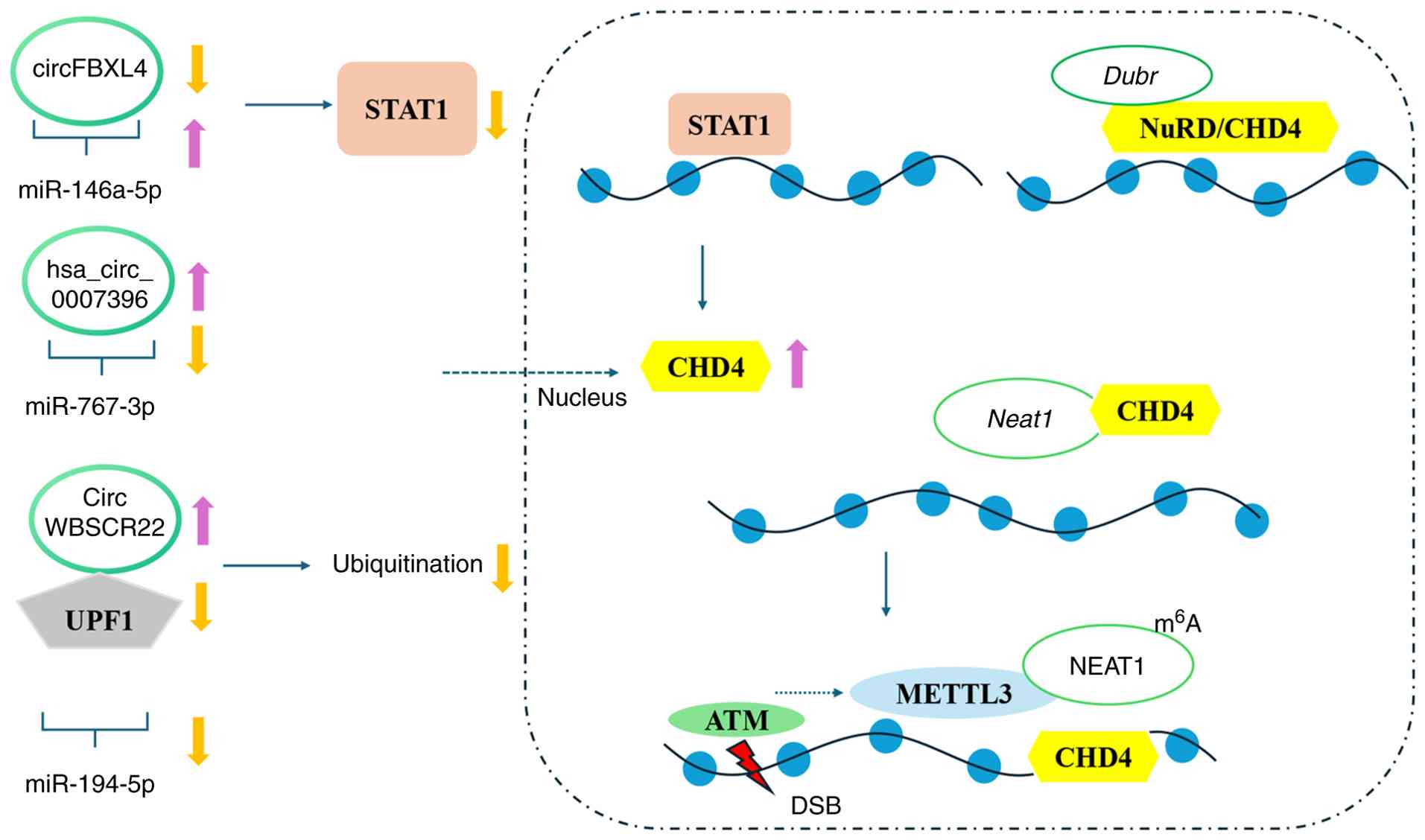 | Figure 4.Non-coding RNA regulation of CHD4.
lncRNAs (such as Neat1 and Dubr) can influence
chromatin remodeling or DNA repair signals by regulating CHD4
localization or NuRD complex activity. Meanwhile, miRNAs and
circRNAs mainly regulate CHD4 expression levels via competing
endogenous RNA mechanisms, transcriptional regulation, or protein
stability regulation. circFBXL4, circular RNA FBXL4; miRNA
or miR, microRNA; hsa_circ_0007396, human circular RNA 0007396;
circWBSCR22, circular RNA WBSCR22; UPF1, up-frameshift
mutation 1 homolog; STAT1, signal transducer and activator of
transcription 1; CHD4, chromodomain helicase DNA-binding protein 4;
NuRD, nucleosome remodeling and deacetylase complex; Neat1,
nuclear paraspeckle assembly transcript 1; Dubr,
developmental pluripotency associated 2 upstream binding RNA;
METTL3, methyltransferase-like 3; ATM, ataxia telangiectasia
mutated; DSB, DNA double-strand break; m6A, N6-methyladenosine. |
In summary, these ncRNAs construct a multi-layered
regulatory network by modulating CHD4 localization, activity,
transcriptional level and protein stability. This network plays a
crucial role in shaping the context-dependent functions of CHD4 and
provides new insights into understanding the high plasticity
exhibited by tumor cells under therapeutic pressure. Moreover, the
interaction between RNA and CHD4 may also serve as a potential
therapeutic-intervention target, particularly in tumor therapy
resistance and DDR regulation.
Discussion
Recent studies have revealed the high functional
plasticity of CHD4 in different biological contexts, a
characteristic that has been demonstrated with relative clarity in
developmental systems. For example, during cardiac development,
CHD4 can form specific regulatory complexes with different TFs,
resulting in different transcriptional regulatory outcomes. Studies
have shown that the TF T-box transcription factor 5 can recruit
CHD4 to the regulatory regions of atrial-related genes, promoting
atrial-specific gene expression and maintaining cardiac-rhythm
homeostasis in a specific chromatin environment (60). In other developmental contexts, CHD4
can synergize with TFs such as SET and MYND domain containing 1
(61) or GATA4/natural killer 2
homeobox 5 (62) to inhibit
non-myocardial-gene expression through the NuRD complex, thereby
maintaining the specific transcriptional process of myocardial
cells. These studies suggest that the biological function of CHD4
largely depends on its binding TF partners and the local chromatin
environment.
Similar context-dependent regulation is also greatly
significant in tumors. Current research indicates that CHD4
function shows obvious context dependence by tumor type, which is
mainly reflected in tumor progression and therapy resistance.
During tumor progression, CHD4-mediated chromatin remodeling
(11) and transcriptional
regulation (13) promote tumor cell
proliferation, migration and invasion. However, in specific TRNs,
CHD4 may also limit tumor progression by inhibiting abnormal
transcriptional processes. In terms of therapy resistance, high
expression of CHD4 is usually associated with enhanced DDR capacity
(46) and CT resistance (47), while against certain genetic
backgrounds, CHD4 functional loss may generate alternative drug
resistance mechanisms by altering DDR modes or replication fork
stability. Therefore, the role of CHD4 in tumors is not
unidirectional but is instead influenced by multiple factors such
as genetic background, TRNs and chromatin environment.
From the perspective of epigenetic regulation, CHD4
can integrate multiple chromatin modification mechanisms to shape
transcriptional states. For example, CHD4 can synergize with
HDAC1/2 to regulate chromatin compaction, thereby inhibiting gene
transcription. Meanwhile, it can also work with histone
methyltransferases [for example, SET domain bifurcated histone
lysine methyltransferase 1 (SETDB1) (63) or EHMT2] to further reinforce gene
silencing by maintaining repressive chromatin marks like histone
H3-lysine 9 (H3K9) methylation. In addition, in some tumors, CHD4
can synergize with DNA methyltransferases DNMT1 and DNMT3B to
maintain abnormal DNA methylation states at the promoter regions of
tumor suppressor genes, thereby strengthening transcriptional
repression. These different levels of epigenetic modifications
jointly determine the transcriptional regulatory effects of
CHD4.
In addition to the aforementioned epigenetic
modifications, the local chromatin environment may also affect
recruitment and function of CHD4. For example, some
cancer-associated histone mutations (such as H3.3 G34R) can alter
the conformation and dynamic behavior of the histone tail, thereby
affecting the interactions between chromatin regulatory proteins
such as CHD4 and nucleosomes (64).
Furthermore, studies on SelectID technology have found that the
CHD4/NuRD complex can be significantly enriched in methylated DNA
regions (65), suggesting that DNA
methylation may help regulate gene expression by recruiting
chromatin-remodeling complexes.
At the therapeutic level, the CRN mediated by CHD4
may provide new interventional ideas for epigenetic targeted
therapy. For example, the HDAC subunits in the NuRD complex are
already important targets of various antitumor drugs, indicating
that interfering with CHD4/NuRD complex function may affect
tumor-related transcriptional processes. In addition, it has been
identified that synthetic-lethality relationships may exist between
CHD4 and other epigenetic regulatory factors. For example, dual
inactivation of CHD4 and the H3K9 methyltransferase SETDB1 can
significantly reduce tumor cell viability (63). These findings suggest that
exploiting the dependencies within the epigenetic regulatory
network may provide an important theoretical basis for new
combination therapy strategies. The abnormalities of Chd4 in
different tumor types and their corresponding mechanisms are
summarized in Table I.
 | Table I.CHD4 abnormalities and mechanisms in
different tumor types. |
Table I.
CHD4 abnormalities and mechanisms in
different tumor types.
| First author/s,
year | Tumor type | Expression/mutation
status | Outcome | Main mechanism | (Refs.) |
|---|
| Wu et al,
2023 | Gastric cancer | High
expression | Oncogenic | Enhances the
interaction between MEK1/2 and ERK1/2, leading to higher ERK
phosphorylation levels and sustained pathway activation | (47) |
| Shi et al,
2025 |
| High
expression | Oncogenic | Binds MYH9 via the
ATPase domain and promotes its nuclear export; cytoplasmic MYH9
then inhibits GSK3β, resulting in β-catenin stabilization | (18) |
| Kim et al,
2011 |
| Gene mutation; loss
of function | Oncogenic | Alters gene
expression by affecting chromatin structure | (66) |
| Li et al,
2018 | Endometrial
cancer | R975H and R1162W
mutations | Oncogenic | Activates
transforming growth factor beta signaling | (29) |
| Zhang et al,
2022 | Breast cancer | Not specified | Tumor
suppressor | Is recruited to the
SOX2 promoter region by TRPS1, repressing transcription | (27) |
| Wang et al,
2020 |
| High
expression | Oncogenic | Under hypoxic
conditions, its interaction with HIF is enhanced, promoting the
transcription of HIF target genes | (22) |
| Sattout et
al, 2024 |
| High
expression | Oncogenic | Enhances ERα
transcriptional activity | (21) |
| Luo et al,
2018 |
| High
expression | Oncogenic | Binds to the
promoter region of the E-cadherin gene and represses its
transcription | (67) |
| Ou-Yang et
al, 2019 |
| High
expression | Oncogenic | Promotes the
transcription of β1-integrin | (68) |
| Xia et al,
2017 | Colorectal
cancer | High
expression | Oncogenic | After its
localization to oxidative-damage sites via OGG1, it recruits DNA
methyltransferase and suppresses multiple tumor suppressor
genes | (24) |
| Kim et al,
2011 |
| Gene mutation; loss
of function | Oncogenic | Alters gene
expression by affecting chromatin structure | (66) |
| Sun et al,
2024 |
| Not specified | Oncogenic | Recruits EHMT2 to
form a co-transcriptional silencing complex, repressing GAL7
expression | (50) |
| Pratheeshkumar
et al, 2021 | Thyroid cancer | High
expression | Oncogenic | Regulates the
expression of EMT-related genes | (69) |
| Wang et al,
2023 | Ovarian cancer | High
expression | Oncogenic | Interacts with
EZH2, promoting nuclear accumulation of β-catenin | (19) |
| Nio et al,
2015 | Liver cancer | High
expression | Oncogenic | Regulates the
epigenetic state of cells through the NuRD complex | (70) |
| Xu et al,
2020 | non-small cell lung
cancer | High
expression | Oncogenic | Interacts with
PHF5A and activates the RhoA/ROCK pathway | (20) |
In conclusion, the research value of CHD4 lies not
only in whether it can be directly targeted but also in how it, as
an integration node of the CRN, reshapes the stress response
trajectory of tumor cells in different contexts. Future research
should analyze the context-dependent regulatory mechanisms of CHD4
at the system level by, for example, systematically analyzing its
chromatin-binding profile, NuRD complex assembly mode, and DNA
repair pathway selection and combining emerging technologies such
as single-cell multi-omics and spatial transcriptomics to provide
new theoretical bases for CRN-based intervention strategies in
precision tumor therapy.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 81971087).
Availability of data and materials
Not applicable.
Authors' contributions
SL contributed to the conception and design of the
review, performed comprehensive literature analysis and
interpretation, and drafted the manuscript. QM contributed to
literature acquisition, analysis and interpretation, and
participated in manuscript drafting and revision. KL contributed to
literature analysis and organization, figure and table preparation,
and manuscript revision. ZJ contributed to critical analysis and
interpretation of the literature and revised the manuscript
critically for important intellectual content. YM conceived and
supervised the study, contributed to interpretation of the
literature, critically revised the manuscript, and gave final
approval of the version to be published. All authors read and
approved the final version of the manuscript and agree to be
accountable for all aspects of the work. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hanahan D: Hallmarks of cancer: New
dimensions. Cancer Discov. 12:31–46. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Esteller M, Dawson MA, Kadoch C, Rassool
FV, Jones PA and Baylin SB: The epigenetic hallmarks of cancer.
Cancer Discov. 14:1783–1809. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sanchez-Vega F, Mina M, Armenia J, Chatila
WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia
S, et al: Oncogenic signaling pathways in the cancer genome atlas.
Cell. 173:321–337.e10. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
ICGC/TCGA Pan-Cancer Analysis of Whole
Genomes Consortium: Pan-cancer analysis of whole genomes. Nature.
578:82–93. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xu X, Peng Q, Jiang X, Tan S, Yang Y, Yang
W, Han Y, Chen Y, Oyang L, Lin J, et al: Metabolic reprogramming
and epigenetic modifications in cancer: From the impacts and
mechanisms to the treatment potential. Exp Mol Med. 55:1357–1370.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Boulasiki P, Tan XW, Spinelli M and Riccio
A: The NuRD complex in neurodevelopment and disease: A case of
sliding doors. Cells. 12:11792023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Low JKK, Silva APG, Sharifi Tabar M,
Torrado M, Webb SR, Parker BL, Sana M, Smits C, Schmidberger JW,
Brillault L, et al: The nucleosome remodeling and deacetylase
complex has an asymmetric, dynamic, and modular architecture. Cell
Rep. 33:1084502020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Musselman CA, Ramírez J, Sims JK,
Mansfield RE, Oliver SS, Denu JM, Mackay JP, Wade PA, Hagman J and
Kutateladze TG: Bivalent recognition of nucleosomes by the tandem
PHD fingers of the CHD4 ATPase is required for CHD4-mediated
repression. Proc Natl Acad Sci USA. 109:787–792. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Farnung L, Ochmann M and Cramer P:
Nucleosome-CHD4 chromatin remodeler structure maps human disease
mutations. eLife. 9:e561782020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhong Y, Moghaddas Sani H, Paudel BP, Low
JKK, Silva APG, Mueller S, Deshpande C, Panjikar S, Reid XJ,
Bedward MJ, et al: The role of auxiliary domains in modulating CHD4
activity suggests mechanistic commonality between enzyme families.
Nat Commun. 13:75242022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Saotome M, Poduval DB, Grimm SA, Nagornyuk
A, Gunarathna S, Shimbo T, Wade PA and Takaku M: Genomic
transcription factor binding site selection is edited by the
chromatin remodeling factor CHD4. Nucleic Acids Res. 52:3607–3622.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pan MR, Hsieh HJ, Dai H, Hung WC, Li K,
Peng G and Lin SY: Chromodomain helicase DNA-binding protein 4
(CHD4) regulates homologous recombination DNA repair, and its
deficiency sensitizes cells to poly(ADP-ribose) polymerase (PARP)
inhibitor treatment. J Biol Chem. 287:6764–6772. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Graca Marques J, Pavlovic B, Ngo QA, Pedot
G, Roemmele M, Volken L, Kisele S, Perbet R, Wachtel M and Schäfer
BW: The chromatin remodeler CHD4 sustains ewing sarcoma cell
survival by controlling global chromatin architecture. Cancer Res.
84:241–257. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sperlazza J, Rahmani M, Beckta J, Aust M,
Hawkins E, Wang SZ, Zu Zhu S, Podder S, Dumur C, Archer K, et al:
Depletion of the chromatin remodeler CHD4 sensitizes AML blasts to
genotoxic agents and reduces tumor formation. Blood. 126:1462–1472.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Q, Zhu F, Tong Y, Shi D and Zhang J:
CHD4 R975H mutant activates tumorigenic pathways and promotes
stemness and M2-like macrophage polarization in endometrial cancer.
Sci Rep. 14:186172024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reske JJ, Wilson MR, Armistead B, Harkins
S, Perez C, Hrit J, Adams M, Rothbart SB, Missmer SA, Fazleabas AT
and Chandler RL: ARID1A-dependent maintenance of H3.3 is required
for repressive CHD4-ZMYND8 chromatin interactions at
super-enhancers. BMC Biol. 20:2092022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Goswami K, Venkatachalam K, Singh SP, Rao
CV and Madka V: Chromatin Remodulator CHD4: A Potential Target for
Cancer Interception. Genes (Basel). 16:2252025. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shi Y, Zhao Z, Zhou S, Zhou Z, Huang Z,
Zhou Z and Zhang C: CHD4 drives gastric cancer metastasis via
MYH9/GSK3β/β-catenin axis and WNT/EMT pathway activation. Cancer
Lett. 628:2178132025. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang J, Zhong F, Li J, Yue H, Li W and Lu
X: The epigenetic factor CHD4 contributes to metastasis by
regulating the EZH2/β-catenin axis and acts as a therapeutic target
in ovarian cancer. J Transl Med. 21:382023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu N, Liu F, Wu S, Ye M, Ge H, Zhang M,
Song Y, Tong L, Zhou J and Bai C: CHD4 mediates proliferation and
migration of non-small cell lung cancer via the RhoA/ROCK pathway
by regulating PHF5A. BMC Cancer. 20:2622020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sattout A, Yu X, Sun Z, Li Y, Li Y, Li S,
Huo W and Wu H: CHD4-induced up-regulation of ERα activity
contributes to breast cancer progression. Genes Dis. 11:1011082024.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang Y, Chen Y, Bao L, Zhang B, Wang JE,
Kumar A, Xing C, Wang Y and Luo W: CHD4 promotes breast cancer
progression as a coactivator of Hypoxia-inducible factors. Cancer
Res. 80:3880–3891. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Coassolo S, Davidson G, Negroni L, Gambi
G, Daujat S, Romier C and Davidson I: Citrullination of pyruvate
kinase M2 by PADI1 and PADI3 regulates glycolysis and cancer cell
proliferation. Nat Commun. 12:17182021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xia L, Huang W, Bellani M, Seidman MM, Wu
K, Fan D, Nie Y, Cai Y, Zhang YW, Yu LR, et al: CHD4 has oncogenic
functions in initiating and maintaining epigenetic suppression of
multiple tumor suppressor genes. Cancer Cell. 31:653–668.e7. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cai Y, Geutjes EJ, de Lint K, Roepman P,
Bruurs L, Yu LR, Wang W, van Blijswijk J, Mohammad H, de Rink I, et
al: The NuRD complex cooperates with DNMTs to maintain silencing of
key colorectal tumor suppressor genes. Oncogene. 33:2157–2168.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Y, Lin X, Gong X, Wu L, Zhang J, Liu
W, Li J and Chen L: Atypical GATA transcription factor TRPS1
represses gene expression by recruiting CHD4/NuRD(MTA2) and
suppresses cell migration and invasion by repressing TP63
expression. Oncogenesis. 7:962018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang J, Lv X, Wei B, Gong X and Chen L:
CHD4 mediates SOX2 transcription through TRPS1 in luminal breast
cancer. Cell Signal. 100:1104642022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kovač K, Sauer A, Mačinković I, Awe S,
Finkernagel F, Hoffmeister H, Fuchs A, Müller R, Rathke C, Längst G
and Brehm A: Tumour-associated missense mutations in the dMi-2
ATPase alters nucleosome remodelling properties in a
mutation-specific manner. Nat Commun. 9:21122018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li Y, Liu Q, McGrail DJ, Dai H, Li K and
Lin SY: CHD4 mutations promote endometrial cancer stemness by
activating TGF-beta signaling. Am J Cancer Res. 8:903–914.
2018.PubMed/NCBI
|
|
30
|
Dong H, Zhao J, Zhang B, Wu X and Liu G:
Case report: Novel mutation in CHD4 triggers occult breast cancer
with bone metastases. Front Oncol. 15:16827942025. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gong P, Guo Z, Wang S, Gao S and Cao Q:
Histone phosphorylation in DNA damage response. Int J Mol Sci.
26:24052025. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Smith R, Sellou H, Chapuis C, Huet S and
Timinszky G: CHD3 and CHD4 recruitment and chromatin remodeling
activity at DNA breaks is promoted by early
poly(ADP-ribose)-dependent chromatin relaxation. Nucleic Acids Res.
46:6087–6098. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang H and Zha S: The dynamics and
regulation of PARP1 and PARP2 in response to DNA damage and during
replication. DNA Repair (Amst). 140:1036902024. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qi W, Chen H, Xiao T, Wang R, Li T, Han L
and Zeng X: Acetyltransferase p300 collaborates with chromodomain
helicase DNA-binding protein 4 (CHD4) to facilitate DNA
double-strand break repair. Mutagenesis. 31:193–203. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tan Q, Niu K, Zhu Y, Chen Z, Li Y, Li M,
Wei D, Balajee AS, Fang H and Zhao Y: RNF8 ubiquitinates RecQL4 and
promotes its dissociation from DNA double strand breaks.
Oncogenesis. 10:242021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Luijsterburg MS, Acs K, Ackermann L,
Wiegant WW, Bekker-Jensen S, Larsen DH, Khanna KK, van Attikum H,
Mailand N and Dantuma NP: A new non-catalytic role for ubiquitin
ligase RNF8 in unfolding higher-order chromatin structure. EMBO J.
31:2511–2527. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Onn L, Portillo M, Ilic S, Cleitman G,
Stein D, Kaluski S, Shirat I, Slobodnik Z, Einav M, Erdel F, et al:
SIRT6 is a DNA double-strand break sensor. ELife. 9:e516362020.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hou T, Cao Z, Zhang J, Tang M, Tian Y, Li
Y, Lu X, Chen Y, Wang H, Wei FZ, et al: SIRT6 coordinates with CHD4
to promote chromatin relaxation and DNA repair. Nucleic Acids Res.
48:2982–3000. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Qian J, Watanabe T, Watanabe R, Kanno SI,
Takahashi A, Kohsaka S, Yoshino Y, Chiba N, Tanaka K, Kohno T, et
al: BET family BRD3 initiates DSB-induced chromatin remodeling with
TIP60 to promote R-loop-mediated HR. Cell Reps. 44:1164612025.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Neizer-Ashun F and Bhattacharya R: Reality
CHEK: Understanding the biology and clinical potential of CHK1.
Cancer Lett. 497:202–211. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Geyer F, Geyer M, Reuning U, Klapproth S,
Wolff KD and Nieberler M: CHD4 acts as a prognostic factor and
drives radioresistance in HPV negative HNSCC. Sci Rep. 14:82862024.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu Z, Ajit K, Wu Y, Zhu WG and Gullerova
M: The GATAD2B-NuRD complex drives DNA:RNA hybrid-dependent
chromatin boundary formation upon DNA damage. EMBO J. 43:2453–2485.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gong F, Clouaire T, Aguirrebengoa M,
Legube G and Miller KM: Histone demethylase KDM5A regulates the
ZMYND8-NuRD chromatin remodeler to promote DNA repair. J Cell Biol.
216:1959–1974. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang HC, Chou CL, Yang CC, Huang WL, Hsu
YC, Luo CW, Chen TJ, Li CF and Pan MR: Over-Expression of CHD4 is
an independent biomarker of poor prognosis in patients with rectal
cancers receiving concurrent chemoradiotherapy. Int J Mol Sci.
20:40872019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chang CL, Huang CR, Chang SJ, Wu CC, Chen
HH, Luo CW and Yip HK: CHD4 as an important mediator in regulating
the malignant behaviors of colorectal cancer. Int J Biol Sci.
17:1660–1670. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
McKenzie LD, LeClair JW, Miller KN, Strong
AD, Chan HL, Oates EL, Ligon KL, Brennan CW and Chheda MG: CHD4
regulates the DNA damage response and RAD51 expression in
glioblastoma. Sci Rep. 9:44442019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wu J, Zhou Z, Li J, Liu H, Zhang H, Zhang
J, Huang W, He Y, Zhu S, Huo M, et al: CHD4 promotes acquired
chemoresistance and tumor progression by activating the MEK/ERK
axis. Drug Resist Updat. 66:1009132023. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Oyama Y, Shigeta S, Tokunaga H, Tsuji K,
Ishibashi M, Shibuya Y, Shimada M, Yasuda J and Yaegashi N: CHD4
regulates platinum sensitivity through MDR1 expression in ovarian
cancer: A potential role of CHD4 inhibition as a combination
therapy with platinum agents. PLoS One. 16:e02510792021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hou MF, Luo CW, Chang TM, Hung WC, Chen
TY, Tsai YL, Chai CY and Pan MR: The NuRD complex-mediated p21
suppression facilitates chemoresistance in BRCA-proficient breast
cancer. Exp Cell Res. 359:458–465. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sun L, Liu R, Wu ZJ, Liu ZY, Wan AH, Yan
S, Liu C, Liang H, Xiao M, You N, et al: Galectin-7 Induction by
EHMT2 inhibition enhances immunity in microsatellite stability
colorectal cancer. Gastroenterology. 166:466–482. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Fu G, Tao Y, Feng K, Chen Y, Zhang W,
Zhang Z, Hu G and Ou Y: CHD4 epigenetically coordinates genomic
instability and immunosuppression to drive pan-cancer progression
and confer HDAC inhibitor sensitivity. Clin Exp Med. 26:1502026.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ray Chaudhuri A, Callen E, Ding X, Gogola
E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et
al: Replication fork stability confers chemoresistance in
BRCA-deficient cells. Nature. 535:382–387. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mamontova V, Trifault B, Gribling-Burrer
AS, Bohn P, Boten L, Preckwinkel P, Gallant P, Solvie D, Ade CP,
Papadopoulos D, et al: NEAT1 promotes genome stability via m6A
methylation-dependent regulation of CHD4. Genes Dev. 38:915–930.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ullah I, Thölken C, Zhong Y, John M,
Rossbach O, Lenz J, Gößringer M, Nist A, Albert L, Stiewe T, et al:
RNA inhibits dMi-2/CHD4 chromatin binding and nucleosome
remodeling. Cell Rep. 39:1108952022. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hall SD, Tran K, Zhu J, Su T and McHugh
CA: DUBR non-coding RNA regulates gene expression by affecting AP-1
enhancer accessibility. Funct Integr Genomics. 25:682025.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhang Y, Feng Z, Xu Y, Jiang S, Zhang Q,
Zhang Z, Wang K, Li X, Xu L, Yuan M, et al: Novel roles of LSECtin
in gastric cancer cell adhesion, migration, invasion, and lymphatic
metastasis. Cell Death Dis. 13:5932022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang Y, Huang X, Wang P, Zeng Y and Zhou
G: The hsa_circ_0007396-miR-767-3p-CHD4 axis is involved in the
progression and carcinogenesis of gastric cancer. J Gastrointest
Oncol. 13:2885–2902. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Li Q, Wang K, Shen Y, Lin C, Miao J and Hu
X: Bioinformatics based exploration of hsa-miR-194-5p regulation of
CHD4 through PI3K/AKT signal pathway to enhance tumor resistance to
apoptosis due to loss of nests and participate in poor prognosis of
oral squamous cell carcinoma. Ann Transl Med. 11:1072023.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Luo J, Xu S, Wang J, He L and Li Z:
Circular RNA circWBSCR22 facilitates colorectal cancer metastasis
by enhancing CHD4′s protein stability. Int J Biol Macromol.
282:1371352024. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sweat ME, Shi W, Sweat YY, Li J, Li J,
Keating EM, Ponek A, Ma Q, Park C, Trembley MA, et al: TBX5 and
CHD4 coordinately activate atrial cardiomyocyte genes to maintain
cardiac rhythm homeostasis. Circulation. 152:784–801. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Shi W, Wasson LK, Dorr KM, Robbe ZL,
Wilczewski CM, Hepperla AJ, Davis IJ, Seidman CE, Seidman JG and
Conlon FL: CHD4 and SMYD1 repress common transcriptional programs
in the developing heart. Development. 151:dev2025052024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Robbe ZL, Shi W, Wasson LK, Scialdone AP,
Wilczewski CM, Sheng X, Hepperla AJ, Akerberg BN, Pu WT, Cristea
IM, et al: CHD4 is recruited by GATA4 and NKX2-5 to repress
noncardiac gene programs in the developing heart. Genes Dev.
36:468–482. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Moraes-Almeida MS, Sogayar MC and Demasi
MAA: Evidence of a synthetic lethality interaction between SETDB1
histone methyltransferase and CHD4 chromatin remodeling protein in
a triple negative breast cancer cell line. Braz J Med Biol Res.
56:e128542023. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Fuchs HA, Peng Y, Ayyapan S, Rosas R,
Zhang H, Panchenko AR and Musselman CA: G34R cancer mutation alters
the conformational ensemble and dynamics of the histone H3.3 tails.
Nucleic Acids Res. 54:gkaf13812026. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Qian W, Jiang P, Niu M, Fu Y, Huang D,
Zhang D, Liang Y, Wang Q, Han Y, Zeng X, et al: Selective
identification of epigenetic regulators at methylated genomic sites
by SelectID. Nat Commun. 16:37092025. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kim MS, Chung NG, Kang MR, Yoo NJ and Lee
SH: Genetic and expressional alterations of CHD genes in gastric
and colorectal cancers. Histopathology. 58:660–668. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Luo CW, Wu CC, Chang SJ, Chang TM, Chen
TY, Chai CY, Chang CL, Hou MF and Pan MR: CHD4-mediated loss of
E-cadherin determines metastatic ability in triple-negative breast
cancer cells. Exp Cell Res. 363:65–72. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ou-Yang F, Pan MR, Chang SJ, Wu CC, Fang
SY, Li CL, Hou MF and Luo CW: Identification of CHD4-β1 integrin
axis as a prognostic marker in triple-negative breast cancer using
next-generation sequencing and bioinformatics. Life Sci.
238:1169632019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Pratheeshkumar P, Siraj AK, Divya SP,
Parvathareddy SK, Alobaisi K, Al-Sobhi SS, Al-Dayel F and Al-Kuraya
KS: CHD4 predicts aggressiveness in PTC patients and promotes
cancer stemness and EMT in PTC cells. Int J Mol Sci. 22:5042021.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Nio K, Yamashita T, Okada H, Kondo M,
Hayashi T, Hara Y, Nomura Y, Zeng SS, Yoshida M, Hayashi T, et al:
Defeating EpCAM(+) liver cancer stem cells by targeting chromatin
remodeling enzyme CHD4 in human hepatocellular carcinoma. J
Hepatol. 63:1164–1172. 2015. View Article : Google Scholar : PubMed/NCBI
|














