Introduction
Colorectal cancer (CRC) is a leading cause of
mortality from cancer worldwide and is among the most frequent
malignancies of the digestive tract. CRC has been calculated to be
the third cancer type globally with regard to cancer incidence and
second in relation to cancer mortality, with this imposing large
economic and psychosocial burdens on patients, families and
healthcare systems (1). The
pathogenesis of CRC is multifactorial in origin, exhibiting a
complex causative association with obesity, dietary patterns,
physical inactivity, inherited susceptibility and chronic
intestinal inflammation, including inflammatory bowel disease (IBD)
(2). Standard management techniques
rely on surgical resection, complemented by chemotherapy,
molecularly targeted agents and immunotherapy (3). Despite this, high rates of
postoperative relapse, a limited response to systemic therapy and a
restricted set of actionable targets contribute to adverse
outcomes; the 5-year survival estimate for patients with
unresectable disease or distant metastases remains ~15% (4). These challenges emphasize the pressing
need to formulate novel therapeutic strategies to better manage CRC
and enhance patient survival.
Ferroptosis is a form of programmed cell death
driven by iron-dependent lipid peroxidation (LPO), first proposed
and explained by Dixon et al (5). Ferroptosis differs from apoptosis,
autophagy and necroptosis in both morphological and biochemical
characteristics. Ferroptosis is defined by the presence of shrunken
mitochondria with increased membrane density, reduced mitochondrial
volume and diminished or absent cristae (6,7). In
the typical ferroptotic event, classical apoptotic hallmarks such
as chromatin condensation or apoptotic body formation are absent.
The initiation and execution of this pathway are orchestrated
across multiple cellular organelles, namely the mitochondria,
endoplasmic reticulum and Golgi apparatus, which collectively
integrate the lipid metabolism, iron handling and antioxidant
defenses of the cell. This integration defines the cellular
vulnerability to ferroptosis (8).
In the context of tumors, ferroptosis has been revealed as a
crucial modulator of malignant phenotypes, exerting a notable
influence on processes such as proliferation and migration, while
concomitantly shaping therapeutic resistance (9).
Accumulating evidence indicates that pharmacological
induction of ferroptosis can significantly boost the therapeutic
efficacy of conventional treatment regimens for CRC (10–13).
However, ferroptosis inhibition has been reported to limit
intestinal epithelial cells (IECs) injury and dampen inflammatory
signalling, thus ameliorating IBD phenotypes, and potentially
influencing CRC prevention and therapeutic outcomes (14,15).
The present review aims to delineate the core molecular circuitry
and principal signalling networks that govern ferroptosis. In
addition, it highlights key recent advances and discusses emerging
opportunities for CRC intervention through modulation of LPO, iron
metabolism and antioxidant defense systems. Furthermore, it
evaluates the translational potential of ferroptosis-oriented
therapies, provides an overview of the current limitations in the
extant evidence base and establishes a conceptual framework for the
development of microbiota-informed and nanotechnology-enabled
approaches.
Molecular mechanisms of ferroptosis
Enhanced iron accumulation and exacerbation of LPO
are the core triggers of ferroptosis. To counter this, cells deploy
antioxidant programs. These can be dependent on glutathione (GSH)
peroxidase 4 (GPX4) or independent of it. The purpose of these
programs is to detoxify lipid hydroperoxides and limit oxidative
damage. When the ferroptotic drive overcomes these defenses,
peroxidized phospholipids build up in cellular membranes. This
accumulation breaches membrane integrity and leads to cell death
(Fig. 1). Ferroptosis is thus a
terminal outcome of compromised redox homeostasis.
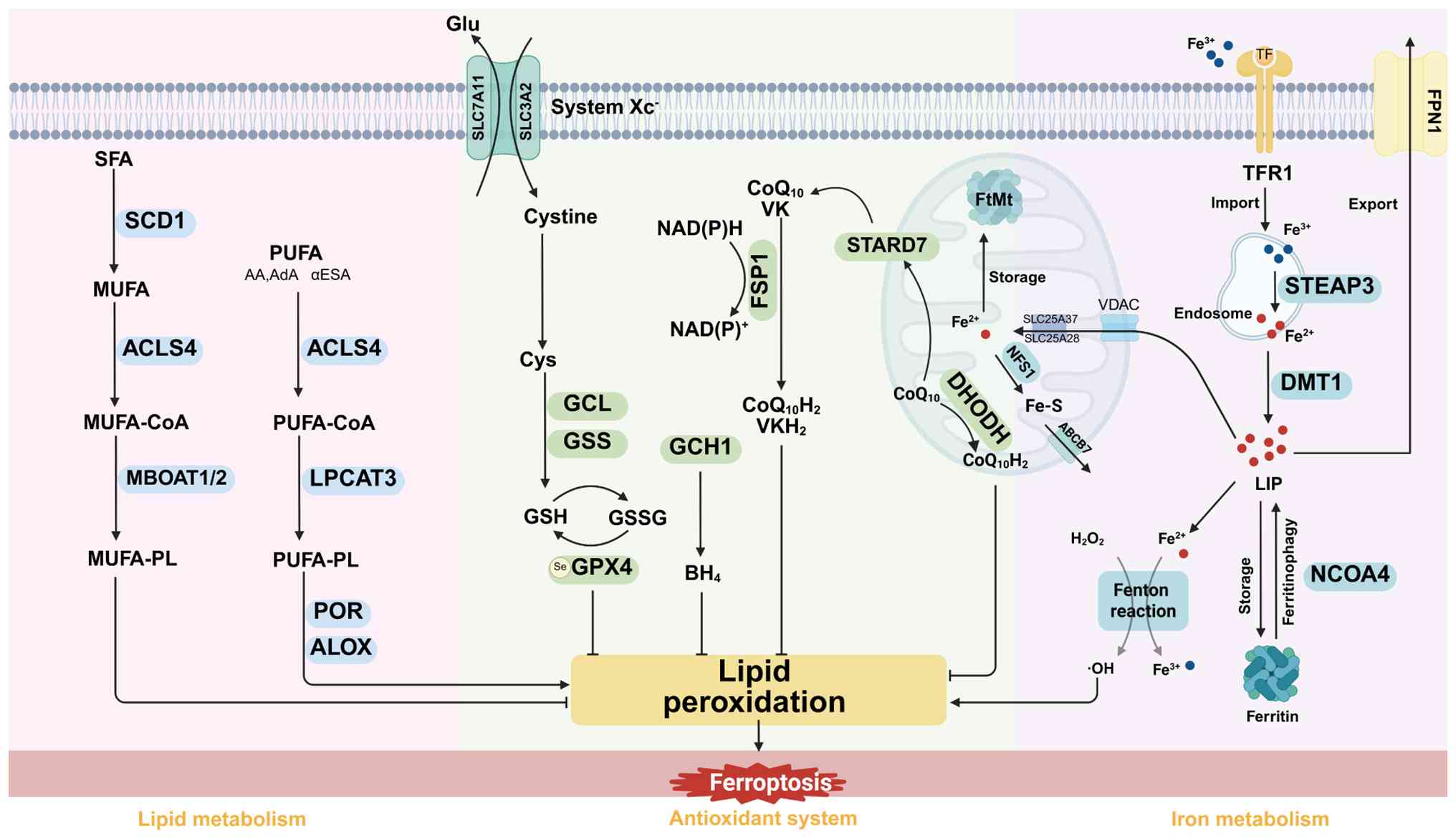 | Figure 1.Molecular mechanisms of ferroptosis.
Several key pathways are involved in regulating ferroptosis,
interconnected through iron metabolism, lipid metabolism and
antioxidant systems. Iron metabolism plays a dual regulatory role
in ferroptosis, both promoting and inhibiting the process.
Circulating Fe3+ ions bind to TF and TFR1 and are
internalized. They then act through two mechanisms: i) Promoting
the formation of the LIP, which activates the Fenton reaction,
triggering ferroptosis; and ii) being stored in ferritin to limit
free iron accumulation and reduce its redox activity, thereby
inhibiting ferroptosis. In lipid metabolism, PUFAs are esterified
by ACSL4 and incorporated into the cell membrane via LPCAT3. LOX
catalyzes the oxidation of PUFA-PL into peroxide derivatives,
leading to membrane instability. SFAs and MUFAs protect against
ferroptosis by antagonizing LPO. In the antioxidant system, GPX4
effectively inhibits LPO by converting GSH to GSSG and reducing
toxic lipid hydroperoxides to non-toxic phosphatidyl alcohols.
Additionally, the FSP1/CoQH2, DHODH/CoQH2 and
GCH1/BH4 systems contribute to mitigating LPO in a
GPX4-independent manner. TF, transferrin; TFR1, TF receptor 1;
PUFAs, polyunsaturated fatty acids; ACSL4, acyl-CoA synthetase
long-chain family member 4; LPCAT3, lysophosphatidylcholine
acyltransferase 3; PUFA-PLs, PUFA-containing phospholipids; SFAs,
saturated fatty acids; MUFAs, monounsaturated fatty acids; LPO,
lipid peroxidation; GPX4, glutathione peroxidase 4; GSH,
glutathione; GSSG, GSH disulfide; FSP1-CoQ10,
ferroptosis suppressor protein 1-coenzyme Q10;
DHODH-CoQ10, dihydroorotate
dehydrogenase-CoQ10; GCH1-BH4, guanosine
triphosphate cyclohydrolase 1-tetrahydrobiopterin; SCD1,
stearoyl-CoA desaturase 1; MBOAT1/2, membrane-bound
O-acyltransferase domain-containing 1 and 2; POR, cytochrome P450
reductase; Glu, glutamate; Cys, cysteine; GSH, glutathione; GPX4,
glutathione peroxidase 4; GCL, glutamate-cysteine ligase; GSS,
glutathione synthetase; NAD(P)H, nicotinamide adenine dinucleotide
phosphate; ALOXs, Arachidonate lipoxygenases; AA, arachidonic acid;
AdA, adrenic acid; αESA, α-eleostearic acid; STARD7, StAR-related
lipid transfer domain containing 7; FtMt, ferritin mitochondrial;
SLC25A37, solute carrier family 25 member 37; CoQ10H2,
reduced coenzyme Q10; ABCB7, ATP binding cassette subfamily B
member 7; NFS1, cysteine desulfurase 1; STEAP3, six transmembrane
epithelial antigen of the prostate 3; DMT1, divalent metal
transporter 1; LIP, labile iron pool; FPN1, ferroportin 1; NCOA4,
nuclear receptor coactivator 4; VDAC, voltage-dependent anion
channel. |
Lipid metabolism
Lipid remodeling has been defined as a key feature
of ferroptosis. In addition, peroxidation of polyunsaturated fatty
acids (PUFAs) within membrane phospholipids is widely acknowledged
as the proximal driver of ferroptotic death, whereas
monounsaturated fatty acids (MUFAs) have been found to counteract
ferroptosis initiation by diminishing the availability of
oxidizable substrates (16). PUFA
oxidation is a process that can take place through both enzymatic
and non-enzymatic routes. In the non-enzymatic axis, acyl-CoA
synthetase long-chain family member 4 (ACSL4) has been observed to
display a marked preference for the stimulation of PUFAs, with a
particular affinity for arachidonic acid (AA) and adrenic acid
(AdA), which results in the formation of the corresponding acyl-CoA
thioesters (17). These activated
species are then esterified into phosphatidylethanolamine by
lysophosphatidylcholine acyltransferase 3 (LPCAT3), producing
PUFA-containing phospholipids that are vulnerable to peroxidative
damage (18). On the phospholipid
molecules, AA and AdA participate in LPO by forming acyl chains,
becoming key substrates that trigger ferroptosis. In the enzymatic
pathway, lipoxygenase and cytochrome P450 reductase (POR) are key
enzymes that initiate LPO. Arachidonate lipoxygenases (ALOXs)
participate in the catalysis of PUFAs through various mechanisms,
including the generation of reactive oxygen species (ROS), lipid
signalling, modification of the structure and function of complex
lipid-protein complexes and regulation of cellular redox states
(19). POR provides electrons to
cytochrome P450 enzymes, activates molecular oxygen and inserts it
into the PUFA chain, catalyzing the LPO reaction, which ultimately
produces phospholipid hydroperoxides (20).
Conversely, saturated fatty acids can be desaturated
by stearoyl-CoA desaturase 1, thereby generating MUFAs. Subsequent
to activation by ACSL3, MUFAs are incorporated into membrane
phospholipids by membrane-bound O-acyltransferase domain-containing
1/2, thus generating MUFA-containing phospholipids (21–23).
MUFA-enriched lipids are relatively resistant to oxidation, a
process that contributes to the conservation of the membrane
architecture and the mitigation of oxidative injury. This results
in the attenuation of ferroptosis through competitive dilution of
peroxidation-prone substrates. Of note, the sensitivity of tumor
cells to ferroptosis appears to be limited less by the total levels
of intracellular PUFAs than by the efficiency with which highly
unsaturated PUFAs are directed into vulnerable phospholipid pools
(24). Accordingly, the equilibrium
between PUFA- and MUFA-containing phospholipids within cellular
membranes is a fundamental determinant of ferroptotic
susceptibility.
Iron metabolism
Intracellular iron metabolism is a dynamic process
involving the uptake, transport, storage and utilization of iron,
and is tightly regulated by multiple signaling molecules at various
levels. The core mechanism of cellular iron uptake relies on
transferrin (TF) receptor 1 (TFR1), which mediates the
transmembrane transport of TF-ferric iron complexes via endocytosis
(25). TF ensures the stability of
the Fe3+ oxidation state during transport into the cell,
preventing ion displacement. Following endocytic internalization of
the TF-receptor complex, the process of endosomal acidification
promotes the dissociation of Fe3+, which is subsequently
reduced to Fe2+ by six-transmembrane epithelial antigen
of prostate 3, which then channels the Fe2+ into the
labile iron pool (LIP), thus facilitating the support of cellular
metabolism. Cytosolic Fe2+ trafficking and buffering are
complex processes involving several routes, including export from
endosomes via divalent metal transporter 1 (DMT1), sequestration
within ferritin and intercellular redistribution via multivesicular
bodies and exosomes (26). DMT1
delivers Fe2+ into the cytoplasm, directly expanding the
LIP and contributing to intracellular iron homeostasis.
Pharmacological DMT1 inhibition or disruption of multivesicular
body-exosome biogenesis can lead to iron accumulation by
restricting iron efflux (27,28).
Ferroportin 1 (FPN1) is the sole recognized iron exporter in
mammalian cells and is subject to tight control by hepcidin. Under
iron-replete conditions, hepatocyte-derived hepcidin binds FPN1 and
prompts its internalization and degradation, blocking iron release
to preserve systemic and cellular iron balance (29). Iron storage is regulated by
ferritin, a heteropolymer composed of ferritin heavy (FTH1) and
light chains. Nuclear receptor coactivator 4 (NCOA4) functions as a
selective cargo receptor for ferritinophagy, targeting ferritin to
autophagosomes for subsequent lysosomal degradation, resulting in
the liberation of redox-active iron (30). As indicated by Liu et al
(31), activation of the NCOA4-FTH1
axis instigates ferritinophagy and promotes ferroptosis in CRC.
Fe2+ in the LIP catalyzes the generation of hydroxyl
radicals and other ROS via the Fenton reaction. Excessive
accumulation triggers a LPO chain reaction, ultimately leading to
cellular damage or ferroptosis. Therefore, regulating the
transmembrane transport of iron ions is a core strategy for
maintaining LIP homeostasis.
Beyond the cytosol, mitochondria constitute an
important source of ROS and represent a central node for iron
utilization. The handling of iron is linked to electron transport
chain activity and redox homeostasis in ferroptosis. Mitochondrial
iron metabolism relies on a highly regulated transfer of iron
across both the outer and inner mitochondrial membranes. On the
outer membrane, the voltage-dependent anion channel enables the
interchange of iron and other metabolites between the cytoplasm and
the intermembrane space. The process of import across the inner
membrane is predominantly mediated by solute carrier family 25
members 37 and 28, which function as dedicated mitochondrial iron
importers to regulate matrix iron availability (32,33).
Following delivery to the matrix, iron is predominantly utilized as
a substrate for the biosynthesis of iron-sulfur (Fe-S) clusters.
This process relies on the mitochondrial iron-sulfur cluster (ISC)
system, which serves as the central hub for Fe-S cluster synthesis,
coordinating both cluster assembly and transmembrane transport.
Cysteine desulfurase 1 (NFS1), an Fe-S cluster biosynthesis enzyme,
regulates ISC assembly. Downregulation of NFS1 impedes ISC
assembly, leading to an iron starvation response, intracellular
iron overload, ROS accumulation and ultimately ferroptosis
(34). Mitochondrial ferritin, as
an iron storage protein in mitochondria, converts free
Fe2+ to a stable form through its ferrous oxidase
activity, preventing iron-induced oxidative stress as well as
damage to mitochondria and cells (35). Additionally, mitochondrial iron
transporter ATP binding cassette subfamily B member 7 assists in
transporting Fe-S clusters from mitochondria to the cytoplasm,
thereby contributing to the regulation of iron transfer and its
utilization within the mitochondria (36). Therefore, mitochondria play a
central hub role in cellular iron homeostasis. Targeted disruption
of mitochondrial iron metabolism can selectively induce cellular
ferroptosis, providing novel intervention strategies for disease
treatment.
Antioxidant system in ferroptosis
GPX4-dependent pathway
To mitigate the oxidative stress that drives
ferroptosis, organisms employ antioxidant defense strategies. GPX4
is a crucial suppressor of ferroptosis across diverse cell types
and in various tissues. GSH has been identified as a core redox
metabolite within this network. In the ferroptosis context, GPX4
uses GSH as a substrate for the reduction of phospholipid and
cholesterol hydroperoxides, with the subsequent conversion of the
hydroperoxides to their corresponding alcohols. Simultaneously, GSH
is oxidized to glutathione disulfide (GSSG). By limiting the
accumulation of peroxidized PUFA-containing lipids, this
biochemical reaction preserves membrane bilayer integrity and halts
the self-propagating LPO cascade, thereby preventing ferroptotic
death (37). In accordance with
this mechanism, RAS-selective lethal 3 (RSL3), an archetypal
ferroptosis inducer, covalently modifies the catalytic
selenocysteine residue (Sec46) of GPX4, disabling its peroxidase
activity and precipitating ferroptosis (38).
The Xc−/GSH/GPX4 axis forms a core
antioxidant defense pathway that effectively blocks LPO-driven
ferroptosis by maintaining redox homeostasis. The system
Xc− consists of solute carrier family 7 member 11
(SLC7A11) and SLC3A2, also known as the light chain subunit xCT and
the heavy chain partner protein CD98. System Xc− is a
sodium-independent antiporter that catalyzes the 1:1 equilibrium
exchange of extracellular cystine for intracellular glutamate
(Glu). Following importation, cystine is rapidly reduced to
cysteine (Cys), which provides the rate-limiting substrate for GSH
biosynthesis (39). Inhibition of
system Xc− depletes intracellular GSH reserves, reduces
the antioxidant capacity of the cell and ultimately triggers
membrane rupture and ferroptosis. Erastin, a reductive inducer of
ferroptosis, promotes lipid ROS accumulation and triggers oxidative
damage and ferroptosis by binding to SLC7A11, inhibiting Cys2
uptake, disrupting GSH synthesis and suppressing GPX4 enzyme
activity (40). Notably, erastin
inhibits system Xc−, progressively depleting GSH
reserves and promoting the accumulation of lipid ROS and
mitochondrial ROS, ultimately leading to ferroptosis. By contrast,
RSL3 inhibits the GPX4 active site, rapidly suppressing its ability
to reduce phospholipid hydroperoxides, leading to a rapid
accumulation of mitochondrial ROS and ultimately resulting in
ferroptosis (41). Thus, both
inducers directly or indirectly inactivate GPX4 function,
highlighting its regulatory role in the ferroptosis pathway as a
classic ferroptosis inducer. Additionally, GPX4 is a selenoprotein
with a selenocysteine-containing catalytic center, and its activity
depends on the biological availability of selenium. The addition of
selenium to cells or its administration to animals can inhibit
ferroptosis (42). Therefore,
selenium may regulate GPX4 expression levels to dynamically control
cellular sensitivity to ferroptosis. In addition to GPX4, other
selenoproteins (such as selenophosphate synthetase 2 and
selenophosphate) play roles in ferroptosis by scavenging free
radicals or reducing mitochondrial oxidative stress (42). In summary, targeting the
Xc−-GSH-GPX4 axis can induce ferroptosis by inhibiting
either system Xc− or GPX4. This dual strategy provides a
new approach for treating ferroptosis-related diseases.
GPX4-independent pathway
GPX4 is a pivotal regulator of ferroptosis,
safeguarding membrane integrity by reducing phospholipid
hydroperoxides and consequently limiting lipid peroxide
accumulation. In the same way, various GPX4-independent defense
modules also restrain ferroptotic signalling, which includes the
ferroptosis suppressor protein 1 (FSP1)-coenzyme Q10
(CoQ10; ubiquinone) axis, the dihydroorotate
dehydrogenase (DHODH)-CoQ10 pathway and the guanosine
triphosphate cyclohydrolase 1 (GCH1) - tetrahydrobiopterin
(BH4) system. In essence, these programs act to impede
the propagation of lipid radicals and the progression of the LPO
cascade, consequently delaying or preventing ferroptotic collapse.
FSP1, which resides within lipid droplets (LDs) and the plasma
membrane, functions as an NAD(P)H-dependent oxidoreductase that
catalyzes the reduction of CoQ10 to its oxidized form,
CoQ10H2, also known as ubiquinol (43), which functions as a lipophilic
radical-trapping antioxidant, exerting a direct quenching impact on
lipid radicals and effectively terminating chain-propagating
peroxidation processes.
Additionally, it enhances antioxidant function by
reducing α-tocopherol, indirectly preventing ferroptosis. At the
cell membrane, aldehyde dehydrogenase 7A1 generates NADH, which
supports the antioxidant activity of FSP1. It also reduces membrane
damage by consuming harmful aldehydes such as 4-hydroxynonenal
(4-HNE) and malondialdehyde (44).
Furthermore, FSP1 is involved in the reduction of vitamin K and
mediates endosomal sorting complex required for
transport-III-dependent membrane repair to inhibit ferroptosis
(45). Similar to FSP1′s strategy
of scavenging lipid-free radicals, DHODH in mitochondria also
reduces CoQ10 to ubiquinol, inhibiting ferroptosis
(46). StAR-related lipid transfer
domain protein 7 facilitates a mechanistic connection between the
FSP1-CoQ10 and DHODH-CoQ10 antioxidant
programs by acting as a shuttle for CoQ10 between the
mitochondria and the plasma membrane, extending ubiquinone-based
protection beyond the organelle (47). In the same manner, guanosine
triphosphate GCH1 catalyzes the pivotal step in BH4 biosynthesis.
BH4 functions as a radical-trapping antioxidant that suppresses
lipid peroxyl radicals and LPO. It also plays an essential role as
a cofactor for nitric oxide synthases, forming a GPX4-independent
ferroptosis defense module (48).
Through its actions in ROS control and membrane protection against
autoxidation, the GCH1-BH4 axis offers resistance to ferroptotic
stress; however, genetic or pharmacological blockade of GCH1
depletes BH4, increases peroxide accumulation and precipitates
ferroptosis (49). Taken together,
the human body harbors multiple antioxidant defense networks that
attenuate iron-dependent oxidative stress via synergistic
mechanisms, thereby sustaining cellular homeostasis.
Ferroptosis in tumor development and
regulation
A broad range of tumor-associated stimuli and
signaling pathways contribute to tumor development and cancer cell
proliferation by regulating ferroptosis, either through its
activation or suppression. These insights position ferroptosis at
the forefront of cancer pathophysiology. The principal regulatory
factors and signalling pathways that govern ferroptosis in cancer
are described below.
p53
p53 is a pivotal tumor suppressor that orchestrates
cell-cycle arrest, DNA repair and metabolic homeostasis,
constituting a paramount intrinsic barrier to malignant
transformation. The available evidence also points to p53 as a
context-dependent regulator of ferroptosis, with both pro- and
anti-ferroptotic functions (50).
From a mechanistic perspective, p53 is able to repress the
transcription of SLC7A11, limiting cystine uptake and consequently
constraining GSH biosynthesis and redox buffering. p53 can promote
ferroptotic vulnerability by enabling ALOX12 activity, thereby
driving ALOX12-dependent LPO and increasing the sensitivity of
tumor cells to ferroptosis (51,52).
In support of this axis, the DNA-replication factor GINS4 has been
observed to antagonize p53 acetylation, leading to a reduction in
p53 stability and an increase in SLC7A11 expression (53). In turn, this has been shown to
diminish ferroptosis in cancer cells. Beyond its regulation of
system Xc−, p53 also induces the expression of
spermidine/spermine N1-acetyltransferase 1 (SAT1), a rate-limiting
enzyme in polyamine catabolism, further supporting a link between
p53 signalling and metabolic pathways involved in ferroptosis. In
various tumors, p53 promotes ferroptosis, and ALOX15 inhibitors can
block SAT1-mediated ferroptosis (54). Notably, p53 also indirectly
influences ferroptosis by regulating metabolic target genes such as
glutaminase 2, prostaglandin-endoperoxide synthase 2 and ferredoxin
reductase (55). However, under
certain stress conditions, p53 exhibits an inhibitory effect on
ferroptosis.
For illustrative purposes, p53 has been found to
transcriptionally induce cyclin-dependent kinase inhibitor 1A, thus
delaying GSH deficiency under cystine limitation and concomitantly
diminishing the ferroptosis sensitivity of lung cancer cells
(56). In CRC, p53 has furthermore
been demonstrated to impede ferroptosis by attenuating dipeptidyl
peptidase-4 (DPP4) activity and diminishing the DPP4-NADPH oxidase
1 interaction (57). In sum, these
findings accentuate the context-dependent nature of p53 control
over ferroptosis and highlight the necessity for meticulous,
tumor-specific modification of p53-ferroptosis signalling in the
development of ferroptosis-based anticancer therapies.
Nuclear factor E2-related factor 2
(Nrf2)
Nrf2 was initially recognized as a crucial component
in the process of cellular antioxidant responses. Further research
has since revealed the vital function of Nrf2 in the regulation of
iron metabolism and the antioxidant defense system, consequently
counteracting ferroptosis (58).
Nrf2 regulates iron metabolism by activating downstream target
genes involved in iron metabolism, such as ferritin, FPN1 and
ferrochelatase, which subsequently reduces the free iron levels in
the LIP and inhibits the occurrence of ferroptosis (59). Heme oxygenase 1 (HO-1), an
Nrf2-dependent inducible enzyme, protects tumor cells from
ferroptosis by alleviating oxidative stress. However, the free iron
produced by HO-1 can increase the cell's sensitivity to
ferroptosis, with this dual effect depending on the dynamic balance
between oxidative stress and ferroptosis (60). In the antioxidant system, activation
of Nrf2 triggers the transcription of a series of downstream target
genes. These genes encode enzymes such as SLC7A11, the catalytic
subunit of glutamate-cysteine ligase and its regulatory subunit
GCLM, which together regulate GSH synthesis and metabolism. These
proteins facilitate the entry of Cys into the cell, and the
resulting GSH maintains cellular redox homeostasis, enabling the
cell to sustain GPX4 activity and inhibit ferroptosis (61).
The activity of Nrf2 is tightly regulated by
Kelch-like ECH-associated protein 1 (KEAP1). Under normal
conditions, KEAP1 binds to NRF2, inhibiting its nuclear
translocation and promoting its ubiquitination and degradation,
thereby limiting Nrf2 activity (62). The binding and dissociation of these
two proteins regulate Nrf2 stability, thereby influencing the
cellular redox balance. Notably, transmembrane protein 160, ring
finger protein 217 and cathepsin S interact with KEAP1, promoting
its ubiquitination and proteasomal degradation (63–65).
On the other hand, DPP9 competes with NRF2 and can
non-enzymatically bind to KEAP1 (66). These distinct interference
mechanisms disrupt the normal binding of the KEAP1-NRF2 complex,
maintaining Nrf2 stability and activating its downstream
antioxidant responses, thereby inhibiting ferroptosis in tumor
cells. Furthermore, protein arginine methyltransferase 5-mediated
KEAP1 methylation enhances its stability, leading to negative
regulation of Nrf2 activity (67).
Therefore, exploring how Nrf2 selectively regulates specific target
genes to prevent ferroptosis is crucial for developing novel cancer
therapies and prevention strategies based on ferroptosis
mechanisms.
Autophagy
Autophagy acts as a mediator of ferroptosis and
interacts with various forms of autophagy, including
ferritinophagy, lipophagy, mitophagy, clock autophagy and
chaperone-mediated autophagy (CMA). These types of autophagy
influence cellular iron metabolism, lipid metabolism and the redox
system. Ferritinophagy is a selective autophagy process mediated by
NCOA4. After forming a complex with ferritin, NCOA4 is transported
to the lysosome for degradation, which increases the level of free
iron within the cell (68).
Previous studies have shown that ataxia telangiectasia mutated
kinase phosphorylates NCOA4 and decreases HO-1 expression. Both
mechanisms enhance the interaction between NCOA4 and ferritin,
thereby inducing ferroptosis in tumor cells (69,70).
On the other hand, the E3 ubiquitin ligases Deltex 2 and S-phase
kinase-associated protein 2 promote the degradation of NCOA4
through ubiquitination, thereby inhibiting ferritinophagy and its
associated ferroptosis (71,72).
Lipophagy reduces intracellular lipid reserves by
degrading LDs, thereby increasing the sensitivity of cells to
ferroptosis. Ras related protein Rab 7a, a member of the RAS
oncogene family, is a key factor linking lipophagy to ferroptosis.
It specifically recognizes and degrades LDs, promoting ferroptosis
in tumor cells (73). By contrast,
the p53 target gene phospholipid transfer protein promotes LD
formation and inhibits lipophagy-dependent ferroptosis (74). Of note, clock autophagy further
promotes ferroptosis by inhibiting lipid storage. This process is
mediated by the autophagic cargo receptor sequestosome-1, which
degrades brain and muscle ARNT-like 1 (BMAL1) (75). DDB1- and CUL4-associated factor 7
stabilizes BMAL1 through deubiquitination, preventing its
degradation and thus inhibiting clock autophagy (76).
Mitophagy inhibits ferroptosis by removing damaged
or excess mitochondria, thereby reducing ROS production. This
process also helps maintain mitochondrial network stability
(77). LPO-induced endoplasmic
reticulum stress activates ATF4, which upregulates the E3 ubiquitin
ligase Parkin, initiating the transport of damaged mitochondria to
autophagosomes, thereby effectively inhibiting
mitochondria-associated ferroptosis (78). GPX4 interacts with chaperone heat
shock cognate protein 70 (HSC70) and lysosomal membrane protein
type 2A (LAMP2A), promoting GPX4 degradation in the lysosome via
the CMA pathway, ultimately inducing ferroptosis (79). Notably, HSP90 has been found to
enhance LAMP2A stability, thereby promoting GPX4 degradation and
ferroptosis (80). On the other
hand, prostaglandin E synthase 3 (also known as p23) competes with
HSC70 for binding, while creatine kinase B phosphorylates GPX4,
thus preventing its interaction with HSC70. Both mechanisms
stabilize GPX4 protein levels, thereby inhibiting ferroptosis
(81,82). Therefore, further investigation into
the complex interplay between ferroptosis and autophagy will
provide new intervention strategies for cancer treatment (Fig. 2).
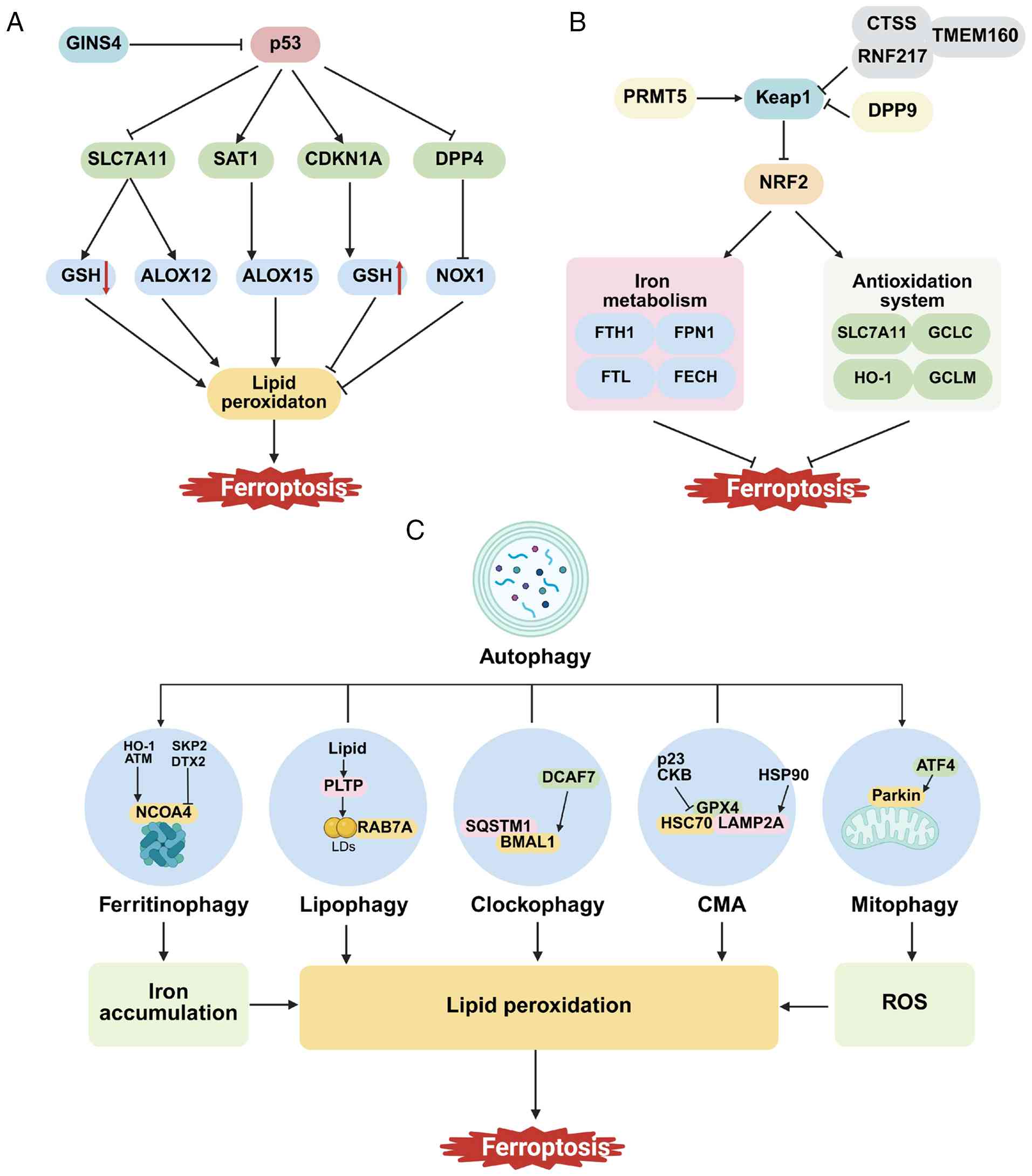 | Figure 2.Cancer-related signaling pathways
regulating ferroptosis. (A) GINS4 inhibits the stability of p53,
affecting its function. p53 promotes ferroptosis by downregulating
SLC7A11 and upregulating SAT1. Additionally, p53 inhibits
ferroptosis by promoting CDKN1A or suppressing DPP4. (B) NRF2
inhibits ferroptosis by regulating components related to the
antioxidant system and iron metabolism. During ferroptosis, NRF2
activity is regulated by target genes such as PRMT5, DPP9 and CTSS.
(C) Interaction between autophagy and ferroptosis. Ferritinophagy,
lipophagy, mitophagy, clock autophagy and CMA regulate ferroptosis
by modulating iron accumulation, ROS levels and LPO. SLC7A11,
solute carrier family 7 member 11; SAT1, spermidine/spermine
N1-acetyltransferase 1; CDKN1A, cyclin-dependent kinase inhibitor
1A; DPP4, dipeptidyl peptidase-4; PRMT5, protein arginine
methyltransferase 5; CTSS, cathepsin S; CMA, chaperone-mediated
autophagy; GINS4, GINS complex subunit 4; GSH, glutathione; ALOXs,
Arachidonate lipoxygenases; NOX1, NADPH oxidase 1; Keap1,
Kelch-1ike ECH- associated protein l; RNF217, ring finger protein
217; TMEM160, transmembrane protein 160; FTH1, ferritin heavy chain
1; FPN1, ferroportin 1; FTL, ferritin light chain; FECH,
ferrochelatase; GCLC, glutamate-cysteine ligase catalytic subunit;
GCLM, glutamate-cysteine ligase modifier subunit; HO-1, heme
oxygenase 1; ATM, ataxia-telangiectasia mutated; SKP2, S-phase
kinase-associated protein 2; DTX2, E3 ubiquitin ligase deltex 2;
PLTP, phospholipid transfer protein; RAB7A, Ras related protein Rab
7a; SQSTM1, sequestosome 1; BMAL1, brain and muscle ARNT-like 1;
DCAF7, DDB1- and CUL4-associated factor 7; CKB, Creatine kinase B;
HSC70, heat shock cognate protein 70; HSP90, heat shock protein 90;
LAMP2A, lysosomal membrane protein type 2A; ATF4, activation
transcription factor 4; ROS, reactive oxygen species; LDs, lipid
droplets. |
Role of ferroptosis in the TME
The TME comprises neoplastic cells in addition to
stromal and immune compartmental elements, whose dynamic
interactions shape tumor growth, metastatic dissemination and
therapeutic sensitivity in a coordinated manner. Within this
biological system, reciprocal intercellular signalling is an
important factor in the evolution of tumors, and it is
progressively evident that ferroptosis is firmly incorporated into
these bidirectional circuits. Non-malignant cells, particularly
immune populations, can influence the susceptibility of cancer
cells to ferroptosis via the secretion of cytokines, metabolites
and lipid mediators. Conversely, ferroptotic cancer cells secrete
signals that can reprogram neighbouring stromal and immune cells,
thereby augmenting or diminishing antitumor immunity (83). Notably, TME-associated stresses and
risk factors have the potential to induce ferroptosis within immune
cells themselves, thereby compromising immunoregulatory capacity
and fostering tumor progression. It is imperative to analyze these
ferroptosis-centered crosstalk networks between tumor and non-tumor
cells to achieve mechanistic insight and to develop rational
therapies that exploit ferroptosis in cancer.
CD8+ T cells
CD8+ T cells primarily rely on secreting
interferons (IFNs) and cytotoxic granules to eliminate infected or
tumorigenic cells. IFN-γ secretion was found to promote ferroptosis
through at least two complementary mechanisms: First, IFN-γ exerts
a suppressive effect on the expression of the system Xc−
subunits SLC3A2 and SLC7A11, thereby constraining cystine import
and depleting intracellular GSH. These effects consequently
sensitize tumor cells to LPO-driven cell death. Second, in the
occurrence of AA and related fatty acids, IFN-γ upregulates ACSL4,
reshaping the tumor-cell lipid composition by means of facilitating
AA incorporation into phospholipids bearing C16 or C18 acyl chains,
which in turn increases the pool of peroxidation-prone substrates
and ultimately precipitates ferroptosis (84,85).
Recent studies indicate that, under the combined treatment of IFN-γ
and AA, a small subset of resistant tumor cells use
VPS33B-interacting protein as a regulatory factor to export ACSL4
via exosomes, thereby evading ferroptosis (86). Furthermore, the synergistic effect
of AA and IFN-κ activates the transcription factor STAT1 signaling
pathway, thus upregulating ACSL4 expression in tumor cells and
ultimately increasing their susceptibility to ferroptosis (87). CD8+ T cells not only
initiate ferroptosis in tumor cells, but are also modulated by
ferroptotic programs within the tumor, thereby establishing a
bidirectional regulatory circuit. As tumor cells are subject to
ferroptosis, they release damage-associated molecular patterns
(DAMPs) that serve to promote the maturation and activation of
dendritic cells (DCs). Activated DCs enhance the function of
CD8+ T cells, thereby reinforcing tumor cell killing and
establishing a positive-feedback cycle that amplifies antitumor
immunity (88).
Ferroptosis exhibits cell type dependence during
tumor development. Inducing ferroptosis in tumor cells effectively
inhibits tumor growth; however, ferroptosis in CD8+ T
cells significantly reduces their cytotoxic activity, potentially
indirectly promoting tumor progression. Lipid accumulation is a
common metabolic alteration in the TME, closely linked to immune
suppression (89). Within the TME,
elevated cholesterol can lead to CD36 upregulation in
CD8+ T cells, resulting in enhanced fatty-acid uptake
and a greater susceptibility to ferroptosis. This lipid-driven
susceptibility has been observed to attenuate the production of
cytotoxic cytokines and compromise antitumor activity (90). Sickle cell disease has been observed
to induce changes in the three-dimensional chromatin architecture
of CD8+ T cells, effectively repressing the
ferroptosis-associated gene SLC7A11. Reductions in hydrogen sulfide
levels concomitantly impede SLC7A11 recovery, thereby potentiating
the risk of ferroptosis (91). Li
et al (92) further
elucidated that the loss of the epilepsy-susceptibility gene DEP
domain-containing protein (DEPDC)5 increased mechanistic target of
rapamycin signalling, elevated intracellular ROS and led to the
sensitization of CD8+ T cells to ferroptotic death.
Notably, vitamin E supplementation or an iron-restricted diet
restored peripheral CD8+ T-cell abundance in DEPDC5-deficient mice,
improving cellular homeostasis and strengthening antitumor
immunity. Collectively, these findings underscore a pivotal
therapeutic challenge, namely, the need for selectively inducing
ferroptosis in tumor cells while preserving the effector function
of CD8+ T cells, a prerequisite for precision
immunotherapy.
Tumor-associated macrophages
(TAMs)
TAMs exist in two phenotypes: M1 and
M2. M1 macrophages primarily secrete
pro-inflammatory factors with antitumor immune effects and
molecules that inhibit angiogenesis, while M2
macrophages produce factors that promote tissue remodeling and
angiogenesis, facilitating tumor initiation and progression
(93). In triple-negative breast
cancer, the cytokine TGF-β1 secreted by TAMs stimulates
the synthesis of GSH in malignant cells, consequently increasing
their tolerance to ferroptosis and inducing tumor progression
(94). Furthermore, targeting
ferroptosis not only directly affects tumor cells but also
modulates the functional state of TAMs, influencing the overall
antitumor immune response. Ferroptosis inducers can impair the
ability of TAMs to clear ferroptotic tumor cells by inducing
phospholipid peroxidation in TAMs, thereby promoting tumor
resistance to ferroptosis-based therapy. Upregulating the
expression of Toll-like receptor 2 in TAMs can restore their
phagocytic function, representing a synergistic stratagem to
enhance the efficacy of ferroptosis-inducing therapy (95).
In multiple settings, M2-like macrophages
are more predisposed to ferroptosis than their M1-like
counterparts, a discrepancy that can influence macrophage
polarization and assist in the perpetuation of pro-inflammatory
programs (96). Significantly,
despite the generally comparable levels of LPO along with the
expression of core ferroptosis regulators in M1 and
M2 macrophages, M1 cells typically exhibit
high inducible nitric oxide synthase (iNOS) activity. The resulting
NO· radicals can quench lipid-derived radicals and attenuate
LPO-associated injury, whereas the relatively low or negligible
iNOS expression in M2 macrophages leaves them less
protected and more susceptible to oxidative membrane damage
(97). The inhibition of
apolipoprotein C1 has been reported to perturb iron and lipid
metabolic pathways, elevate ROS, engage ferroptotic signalling and
drive the repolarization of macrophages from an M2- to
an M1-like state. This reshapes the immune
microenvironment in hepatocellular carcinoma and enhances the
effectiveness of immunotherapy (98). Additionally, changes in TME
homeostasis influence the behavior of macrophages. For instance,
short-term acidosis (24–72 h) upregulates zinc finger AN1
domain-containing protein 5, which regulates SLC3A2 protein via
ubiquitination, promoting the polarization of TAMs towards the
M1 phenotype. This enhances their phagocytic ability and
ferroptosis-inducing effects on breast cancer cells (99). Accordingly, therapeutic strategies
that target ferroptosis in M2-like macrophages, thereby
facilitating their repolarization towards an M1 state,
have the potential to enhance macrophage phagocytic capacity,
mitigate immunosuppressive constraints within the TME and improve
antitumor efficacy.
T regulatory cells (Tregs)
Tregs are a CD4+ T-cell subset with
immunosuppressive properties, often referred to as the ‘brakes’ of
the immune system. In the TME, Tregs are often excessively
accumulated and hyperactive, inhibiting the activation and
proliferation of effector T cells, weakening the body's antitumor
immunity and creating an immunosuppressive state (100). The survival and function of Tregs
depend on iron homeostasis, with FTH being a key regulator of iron
homeostasis and a suppressor of ferroptosis in Tregs. FTH is
involved in iron metabolism and influences the intracellular redox
state, thereby maintaining the activity of ten-eleven translocation
(TET) enzymes, which require iron ions as essential cofactors
(101). The transcription factor
forkhead box protein P3 (Foxp3) is a key regulator of Treg cell
function, controlling the expression of specific genes that define
their suppressive program. TET enzymes regulate the methylation and
transcription of the Foxp3 gene, influencing Treg transcriptional
activity and function, and ultimately affecting autoimmune and
antitumor responses (102).
Notably, Tregs from the TME exhibit higher basal levels of LPO,
indicating that the antioxidant enzyme GPX4 is crucial for
maintaining the lipid redox balance, preventing ferroptosis and
preserving their suppressive activity (103). When GPX4 is specifically deleted
in Tregs, LPO accumulates excessively, inducing ferroptosis,
particularly when T-cell receptor and co-stimulatory signals are
activated. These cells also release pro-inflammatory factors such
as interleukin-1β, thus promoting type 17 T-helper cell-mediated
inflammatory responses and disrupting immune tolerance (104). Therefore, selectively inducing
ferroptosis in Tregs within the TME could specifically weaken their
immunosuppressive function, thereby releasing cytotoxic T cells to
target the tumor.
DCs
DCs are defined as designated antigen-presenting
cells that interface innate and adaptive immunity (105). By capturing tumor-derived antigens
and consequent presentation to T cells, DCs orchestrate T
cell-mediated anti-tumor responses. Emerging evidence signifies
that lipid metabolic rewiring during ferroptosis has the ability to
compromise DC function. The LPO-derived aldehyde 4-HNE activates
the endoplasmic reticulum stress sensor X-box binding protein 1,
resulting in aberrant lipid accumulation and disruption of DC lipid
homeostasis. Functionally, this DC impairment suppresses local
T-cell responses in the TME (106). Peroxisome proliferator-activated
receptor γ (PPARγ), a crucial nuclear receptor in lipid metabolism,
can be activated by the GPX4 inhibitor RSL3 to induce ferroptosis
in DCs. Specific knockdown of PPARγ using genetic methods
effectively reverses the ferroptosis induced by RSL3, restoring DC
maturation (107,108). Recent research has shown that
programmed death ligand-1 (PD-L1) binds to SLC7A11 mRNA to prevent
its degradation, thereby maintaining the ferroptosis resistance of
DCs and mitigating the damage caused by ferroptosis inducers
(109). Additionally, in
inflammatory diseases, Sestrin2 protects DCs from ferroptosis
triggered by lipopolysaccharide. However, its protective mechanisms
in the tumor context still require further investigation (110). Notably, ferroptosis in tumor cells
can also regulate DC function. Li et al (111) demonstrated that inducing
ferroptosis in head and neck squamous cell carcinoma cells
triggered an endogenous double-stranded DNA (dsDNA) cascade, which
significantly promotes DCs infiltration and maturation, thereby
enhancing the suppression of tumor growth. Therefore, ferroptosis
can both weaken the antitumor activity of DCs and influence immune
responses through the tumor cell-DCs interaction network.
Understanding the molecular mechanisms involved and restoring DC
functionality may provide new strategies to enhance tumor
immunotherapy.
Natural killer (NK) cells
NK cells play a role in tumor immune surveillance by
releasing cytolytic granules containing perforin and granzyme B,
which can differentiate and eliminate target cells that are
transformed, infected or under stress (112). NK cells in the TME undergo
ferroptosis, characterized by morphological changes, increased
expression of LPO and oxidative stress-related proteins, which
impair NK cell function (113).
Previous research has shown that NK cells activate an integrated
stress response centered around activating transcription factor 3
(ATF3), which induces ferroptosis through NCOA4-mediated iron
overload by inhibiting Nrf2, reducing NK cell survival and
tumor-killing efficacy in the TME (114). Of note, tumor cells and their
associated components play a crucial role in regulating NK cell
ferroptosis. In gastric cancer, cancer-associated fibroblasts
(CAFs) not only promote iron transfer into the TME, expanding the
unstable iron pool within NK cells, but also promote NCOA4-mediated
ferritinophagy via follostatin-like-protein 1 derived from CAFs,
ultimately inducing ferroptosis (115). However, the microbiota in
hepatocellular carcinoma (such as B. parabrevis) enhances
lipolysis, generating higher levels of acetyl-CoA and increasing
RAR-related orphan receptor C acetylation to upregulate NEDD4L
expression, which promotes the ubiquitination of iron transporters
and inhibits NK cell ferroptosis (116). Tumors often exhibit antigen
heterogeneity and immune evasion, enabling them to escape the
cytotoxic effects of immune cells. Chimeric antigen receptor
(CAR)-NK cell therapy has garnered increasing attention in recent
years due to its ability to target both antigen-specific and
non-specific cancer cells through CAR-dependent and independent
pathways (117). The combination
of this therapy with ferroptosis not only induces ferroptosis by
releasing IFN-γ to downregulate the expression of system
Xc− subunits SLC7A11/SLC3A2, leading to GSH depletion in
tumor cells, but also achieves targeted delivery of ferroptosis
inducers via exosomes derived from CAR-NK cells, showing potential
in eliminating malignant tumors (118,119). Therefore, protecting NK cells in
the TME from ferroptosis and optimizing CAR-NK therapy can more
effectively induce ferroptosis in tumor cells, providing an
important approach for expanding clinical applications.
CAFs
CAFs, as an essential component of the TME, regulate
tumor progression through multiple mechanisms. CAFs influence the
remodelling of local immunity, thus facilitating immune evasion and
diminishing antitumor surveillance (120). Recent research has positioned CAFs
as key mediators of ferroptotic vulnerability within tumors. For
illustrative purposes, consider the findings of Zhang et al
(121), which revealed that
CAF-derived lactate suppresses ferroptosis in triple-negative
breast cancer cells, contributing to resistance to the
anthracycline doxorubicin. By contrast, the ferroptotic status of
malignant cells has been observed to have the capacity to reprogram
the behaviour of CAFs. In cases of gastrointestinal tumors, the
inhibition of ferroptosis subsequent to anoctamin 1 has been
observed to be associated with increased transforming growth
factor-β release. Such release has been hypothesized to drive CAF
accumulation and activation, in turn limiting CD8+ T cell
infiltration and weakening anti-tumor immunity, ultimately
promoting resistance to immunotherapy (122). Exosome-mediated intercellular
communication provides an additional tier of regulatory control:
CAF-derived exosomes enriched in microRNA-432-5p repress CHAC1,
decrease LPO in prostate cancer cells and reduce ferroptosis,
thereby rendering resistance to docetaxel. Furthermore, engineering
CAF-derived exosomes can enhance targeting efficiency,
significantly promoting ferroptosis and improving chemotherapy
efficacy (123,124). Therefore, a deeper exploration of
the ferroptosis regulatory network between CAFs and tumor cells,
and targeting this mechanism, may provide a new path for overcoming
tumor resistance (Fig. 3 and
Table I).
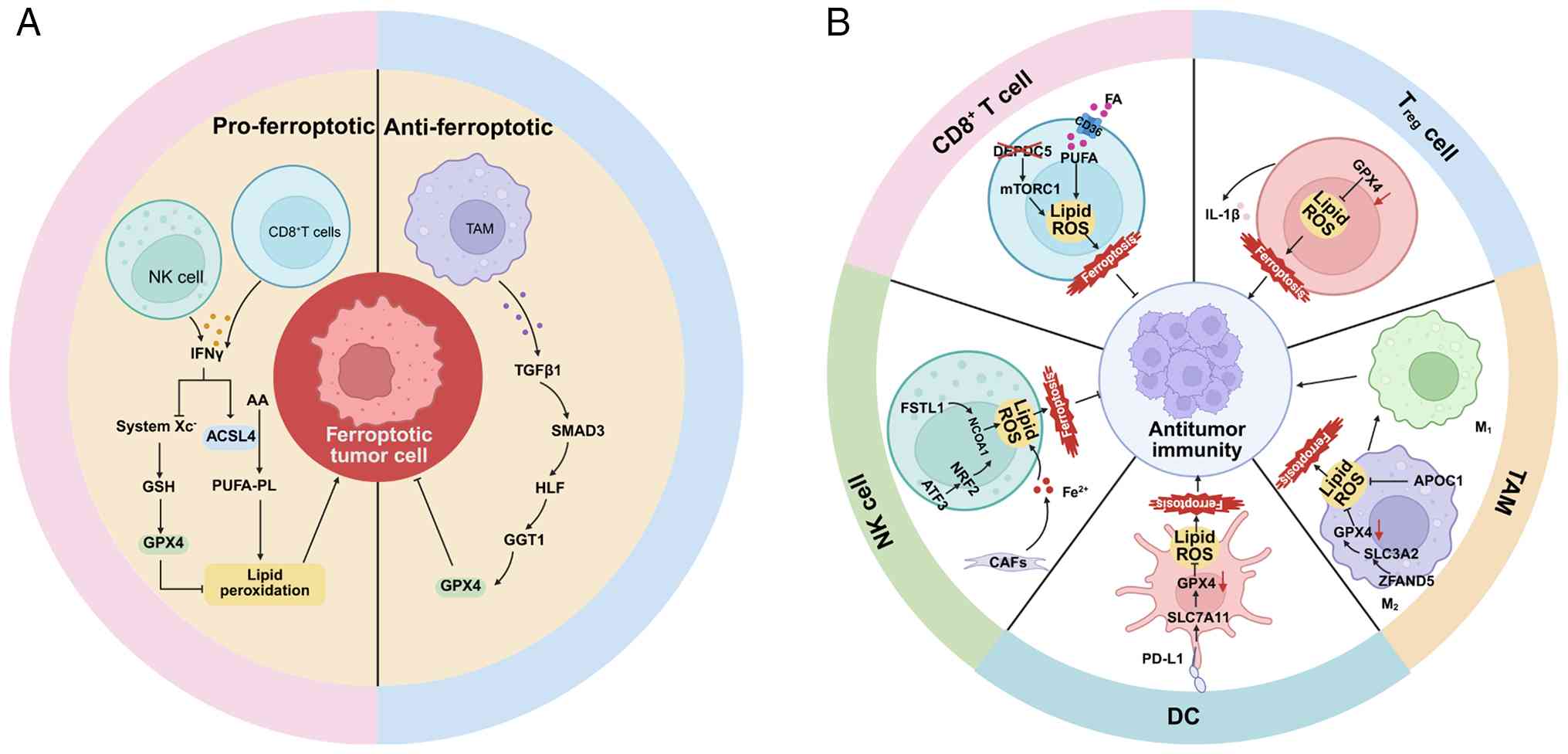 | Figure 3.Ferroptosis-mediated crosstalk in the
TME. (A) Ferroptosis of tumor-associated immune cells can either
promote or suppress antitumor immunity. (B) Immune cells act on
tumor cells by regulating ferroptosis in the TME. TME, tumor
microenvironment; IFNγ, interferon γ; ACSL4, acyl-CoA synthetase
long-chain family member 4; AA, arachidonic acid; GSH, glutathione;
GPX4, glutathione peroxidase 4; PUFA-PLs, PUFA-containing
phospholipids; NK cell, natural killer cell; TAMs, tumor-associated
macrophages; DC, dendritic cell; Tregs, regulatory T cell; TGF-β,
transforming growth factor β; SMAD3, SMAD family member 3; HLF,
hepatic leukemia factor; GGT1, γ-glutamyltransferase 1; DEPDC5, DEP
domain-containing protein 5; PUFA, polyunsaturated fatty acid;
MTORC1, mechanistic target of rapamycin complex 1; FA, fatty acid;
IL-1β, interleukin 1β; APOC1, apolipoprotein C1; SLC3A2, solute
carrier family 3 member 2; ZFAND5, zinc finger AN1
domain-containing protein 5; PD-L1, programmed death ligand-1;
SLC7A11, solute carrier family 7 members 11; CAF, cancer-associated
fibroblast; Nrf2, nuclear factor E2-related factor 2; ATF3,
activation transcription factor 3; FSTL1, follistatin-like protein
1. |
 | Table I.Roles and regulatory mechanisms of
ferroptosis in the tumor microenvironment. |
Table I.
Roles and regulatory mechanisms of
ferroptosis in the tumor microenvironment.
| Cell type |
Regulator/target | Regulatory
mechanism | Effect on
ferroptosis | (Refs.) |
|---|
| CD8 + T
cells | IFN-γ↑ | Downregulates
system Xc− and upregulates ACSL4 in the presence of
AA | Promotes tumor cell
ferroptosis | (84,85) |
|
| CD36↑ | Enhances fatty acid
uptake | Promotes
ferroptosis in CD8+ T cells | (90) |
|
| SCD | Downregulates
SLC7A11 | Promotes
ferroptosis in CD8+ T cells | (91) |
|
| DEPDC5↓ | Increases lipid ROS
accumulation | Promotes
ferroptosis in CD8+ T cells | (92) |
| TAMs | TGF-β1↑ | Increases GSH
levels | Inhibits tumor cell
ferroptosis | (94) |
|
| iNOS↑ | Attenuates
LPO-induced cellular injury | Ferroptosis in
M2 macrophages | (97) |
|
| APOC1↓ | Increases lipid ROS
accumulation | Facilitates
M2-to-M1 repolarization through
ferroptosis | (98) |
|
| ZFAND5↑ | Downregulates
GSH | Promotes tumor cell
ferroptosis | (99) |
| Tregs | FTH↑ | Maintain iron
homeostasis | Inhibits
ferroptosis in Treg cells | (101,102) |
|
| GPX4↓ | Promotes lipid
peroxide accumulation | Promotes
ferroptosis in Treg cells | (104) |
| DCs | RSL3 | Activates the PPARγ
pathway | Promotes
ferroptosis in DCs | (107,108) |
|
| PD-L1 | Protects SLC7A11
mRNA from degradation | Inhibits
ferroptosis in DCs | (109) |
| NK cells | ATF3↑ | Induces
NCOA4-mediated iron overload | Promotes
ferroptosis in NK cells | (114) |
|
| FSTL1↑ | Induces
NCOA4-mediated ferritinophagy | Promotes
ferroptosis in NK cells | (115) |
|
| NEDD4L↑ | Promotes
ferroportin ubiquitination | Inhibits
ferroptosis in NK cells | (116) |
|
| IFN-γ↑ | Downregulates
system Xc− | Promotes tumor cell
ferroptosis | (118) |
Ferroptosis inducers in CRC combination
therapy
Patients with CRC face remarkable clinical
challenges, including high mortality rates, widespread drug
resistance and limited effective treatment options. There is a
correlation between cancer-related genes and ferroptosis-associated
pathways, making tumor cells (including CRC cells) more prone to
ferroptosis compared to normal cells. Accordingly, integrating
ferroptosis-inducing agents with established therapeutic modalities
has the potential to offer a compelling strategy for significantly
enhancing therapeutic efficacy in CRC.
Chemotherapy
Despite notable progress in CRC treatment,
chemotherapy remains a key approach for treating patients with
unresectable metastatic tumors. Oxaliplatin, a first-line
chemotherapy drug for CRC, forms a 1,2-diaminocyclohexane-platinum
complex that inserts into dsDNA, inhibiting DNA repair enzyme
activity and enhancing its cytotoxic effects (125). Although oxaliplatin-based
chemotherapy regimens have improved response rates in patients with
CRC, chemotherapy resistance remains an important challenge
(126). Since the majority of
tumor cells are iron-dependent and prone to ferroptosis induction,
triggering ferroptosis can overcome resistance and is considered an
effective anticancer strategy.
The transcription factor forkhead box A (FOXA)
inhibits ferroptosis by activating the Nrf2/GPX4 pathway, thereby
increasing oxaliplatin resistance in CRC cells. Conversely, the E3
ubiquitin ligase tripartite motif containing 36 (TRIM36) mediates
FOXA2 degradation, acting as a key factor in restoring ferroptosis
sensitivity and overcoming CRC resistance (127). Mitochondrial carrier homolog 2
(MTCH2) blocks its ubiquitin-proteasomal degradation pathway,
stabilizing E2F4 protein, inhibiting TFRC transcription and
reducing iron uptake, thereby blocking ferroptosis. Targeting MTCH2
and combining it with the ferroptosis inducer sorafenib effectively
inhibits CRC cell proliferation and metastasis (128). In another study, E3
ubiquitin-protein ligase UBR5 maintains signaling pathway stability
through Lys-11-linked polyubiquitination, inhibiting ferroptosis
and mediating chemotherapy resistance in CRC. The combination of
UBR5 inhibitors and ferroptosis inducers enhances the chemotherapy
sensitivity to oxaliplatin (129).
This suggests that dual or triple therapy combining oxaliplatin,
ferroptosis inducers and targeted inhibitors holds great research
potential, particularly for CRC and other drug-resistant tumors.
SLC7A11 is a critical factor for Cys uptake, and its inhibition
reduces GSH synthesis, triggering ferroptosis in cancer cells.
Previous research indicates that the long non-coding RNA (lncRNA)
HMG recruits the E3 ubiquitin ligase MDM2 to trigger p53
ubiquitination and proteasomal degradation, thus derepressing
SLC7A11 expression and inducing resistance to ferroptosis and
chemotherapy in CRC cells (130).
In a previous study, Qiu et al (131) reported that p52-zinc finger
estrogen receptor interaction clone 6 drives DAZAP1 transcription
and, independently of p53, stabilizes SLC7A11 mRNA, ultimately
reinforcing ferroptosis resistance in CRC cells. Therefore,
investigating the regulatory mechanisms of SLC7A11 and developing
targeted inhibitors to reverse SLC7A11-mediated ferroptosis
resistance could effectively suppress CRC growth, metastasis and
resistance.
In summary, ferroptosis has great potential for
overcoming CRC chemotherapy resistance. Developing new inhibitors
and combining them with ferroptosis inducers can better target and
regulate ferroptosis-related genes, providing an effective strategy
to improve CRC chemotherapy resistance.
Immunotherapy
Immunotherapy has emerged as a central therapeutic
modality in CRC, acting principally by inducing coordinated innate
and adaptive immune responses. However, the overall efficacy of
this approach in CRC remains limited due to strong immune
suppressive mechanisms in the TME, including impaired T cell
infiltration, high genomic heterogeneity and low PD-L1 expression
(132). Based on the
aforementioned discussion regarding ferroptosis and immune
regulation, inducing ferroptosis holds promise as a strategy to
enhance CRC response to immunotherapy.
Immune checkpoint inhibitors (ICIs) have
demonstrated antitumor potential. Because ferroptosis exerts
immunomodulatory effects and can contribute to ICI-mediated
antitumor activity, combining ICIs with ferroptosis inducers can
synergistically suppress CRC growth in vitro and in
vivo (133). Specifically,
Icariin induces ferroptosis in CRC cells by triggering
mitochondrial dysfunction. When combined with anti-PD-1 therapy,
Icariin produces a dose-dependent antitumor effect (134). Conversely, overexpression of
apolipoprotein L3 (APOL3) increases CRC cell susceptibility to
ferroptosis and enhances CD8+ T-cell cytotoxicity by downregulating
LDHA. In the setting of combined RSL3 and PD-1 blockade, APOL3
overexpression further amplifies this synergistic antitumor effect
(135). Collectively, the synergy
between inducing tumor-cell ferroptosis and reprogramming immune
effector function may enhance the therapeutic efficacy of ICIs.
Monoclonal antibodies (mAbs) are widely used in
oncology due to their target specificity and high affinity.
Increasing evidence also highlights their potential to trigger
ferroptosis and thereby reverse therapy resistance in tumor cells
(136). A mAb targeting the
extracellular domain of LGR4 reportedly blocked LGR4-Wnt signaling
and downregulated SLC7A11, promoting ferroptosis and increasing the
chemosensitivity of drug-resistant CRC cells (137).
Antibody-drug conjugates (ADCs) achieve targeted
delivery to tumor cells by coupling mAbs to potent cytotoxic
small-molecule payloads. By contrast, bispecific ADCs (BsADCs)
recognize two tumor-associated targets, which can improve tumor
binding and internalization relative to conventional single-target
ADCs (138). Based on the
overexpression of CDH17 and guanylate cyclase 2C in CRC, Zhang
et al (139) designed a
BsADC that recognizes both antigens and is conjugated to the
ferroptosis inducer RSL3. This BsADC markedly increased binding to
and internalization by CRC cells, enabling dual-targeted delivery
and activation of ferroptosis. Its antitumor efficacy and safety
were superior to those of the corresponding single-target ADCs.
Although CAR-T cell therapy is highly promising, its
efficacy in solid tumors such as CRC remains inferior to that
observed in hematological malignancies (140). Li et al (141) showed that combining CAR-T cells
with ferroptosis inducers can trigger ferroptosis via LPO and the
ACSL4 axis, thereby increasing CAR-T responsiveness and improving
outcomes in non-small cell lung cancer. This combination strategy
may be extendable to solid tumors, including CRC, in future
studies.
On the other hand, while ferroptosis can enhance
antitumor immunity, it may also exert detrimental effects on immune
cells themselves. Within the TME, CD8+ T cells are more
susceptible to ferroptosis than CRC cells; therefore, preserving a
stable antioxidant defense system is essential for maintaining
their antitumor function. Combined treatment with an adenosine A2A
receptor inhibitor and liproxstatin-1 could preserve GSH/GPX4
homeostasis in CD8+ T cells, thereby inhibiting
ferroptosis and enhancing the antitumor immune response (142). Therefore, the application of
ferroptosis-based immunotherapy in CRC should carefully balance its
dual effects by enhancing CRC cells' susceptibility to ferroptosis
while minimizing adverse effects on immune cells.
Gut microbiota-based therapy
The gut microbiota comprises a multifaceted
ecosystem within the intestinal lumen that influences the
epithelial and immune compartments by means of microbe-derived
metabolites, proteins and other macromolecules, thereby exerting a
pivotal influence on CRC initiation and progression (143). Increasing evidence continues to
implicate a potentially tractable therapeutic axis involving ‘gut
microbiota-ferroptosis’. A more comprehensive understanding of the
mechanisms by which microbial signals influence ferroptotic
vulnerability in CRC has the potential to reveal novel intervention
opportunities.
For example, the probiotic Lactobacillus
plantarum MM89 secretes γ-linolenic acid, which induces
ferroptosis centered on mitochondrial damage and thereby suppresses
CRC progression (144). By
contrast, Fusobacterium nucleatum, an oncogenic bacterium
enriched in patients with CRC, activates the
E-cadherin/β-catenin/TCF4 axis, upregulates GPX4 and suppresses
ferroptosis, thereby promoting oxaliplatin resistance in CRC cells
(145). Collectively, a strategy
that eliminates pathogenic bacteria using antimicrobials, while
combining ferroptosis inducers with probiotics, may help reverse
chemotherapy resistance, reduce recurrence and improve
prognosis.
Beyond the microbiota itself, gut microbiota-derived
metabolites can also shape CRC outcomes through ferroptosis
(146). An anaerobic
Peptostreptococcus species enriched in CRC produces the
tryptophan metabolite trans-3-indoleacrylic acid, which
specifically activates aryl hydrocarbon receptor and upregulates
aldehyde dehydrogenase 1 family member A3 (ALDH1A3) transcription.
ALDH1A3 promotes NADH production via retinaldehyde metabolism,
thereby suppressing the antioxidant pathway mediated by the
FSP1-CoQ10 system and inhibiting ferroptosis, which in turn
accelerates CRC progression (147).
In addition, fecal microbiota transplantation (FMT),
which can restore gut microbial metabolites involved in redox
homeostasis, has the capacity to modulate ferroptosis (148). In mice, curcumin-conditioned FMT
alters the abundance of microbes such as Lactobacillus and
Akkermansia, reshapes host metabolism, and downregulates
GPX4 and SLC7A11 to induce ferroptosis in CRC. It also enhances
intratumoral infiltration of CD8+ T cells, further
suppressing CRC progression (149). Notably, dietary patterns can
influence the availability of microbial enzymes and
microbiota-derived metabolites, thereby modulating the development
of CRC (150). Recent studies
indicate that rational dietary interventions, such as creatine
supplementation, increasing butyrate levels and adopting a
low-arginine diet, can enhance CRC cell sensitivity to ferroptosis
by modulating ferroptosis-related pathways, highlighting their
potential as adjunct strategies for CRC management (151–153).
Therefore, a systematic investigation into how the
gut microbiota and its metabolites regulate ferroptosis and
intestinal homeostasis, in conjunction with healthy dietary habits
and microbiome-informed precision interventions, may substantially
improve therapeutic outcomes in patients with CRC.
Nanotherapy
Although ferroptosis inducers have attracted
increasing interest in oncology, the majority of candidates remain
limited by poor aqueous solubility and low in vivo
bioavailability, which substantially hampers their potential as
therapeutics. By contrast, nanotechnology can enhance the
druggability of ferroptosis inducers by improving solubility,
prolonging systemic circulation, facilitating efficient drug
loading and reducing toxicity (154). Therefore, nanocarriers engineered
to deliver ferroptosis inducers hold strong promise for the
treatment of CRC.
Zhang et al (155) developed a biomimetic nanocarrier,
RSV-NPs@RBCm, by encapsulating
resveratrol (RSV) in poly(ε-caprolactone)-poly(ethylene glycol)
nanoparticles and subsequently coating the RSV-loaded nanoparticles
with a red blood cell membrane (RBCm). This engineered system
markedly improved RSV solubility, while the RBCm coating conferred
immune-evasive properties and prolonged systemic circulation. When
co-administered with the iRGD peptide, the system further increased
intratumoral accumulation and penetration, ultimately
downregulating SLC7A11 and GPX4 to induce ferroptosis in CRC cells.
However, because ferroptosis is governed by a coupled, multi-target
and multi-pathway regulatory network, interventions that act on a
single pathway often fail to achieve durable and clinically
meaningful efficacy. Accordingly, integrating ferroptosis-based
strategies with other therapeutic modalities on nanoplatforms may
produce synergistic antitumor effects.
Phototherapy is a widely used synergistic approach,
primarily including photodynamic therapy (PDT) and photothermal
therapy (PTT). In PDT, photoactivated sensitizers generate ROS
within tumors, disrupting redox homeostasis and amplifying LPO to
promote ferroptosis (156). Luo
et al (157) reported a
ferroptosis-sensitizing nano-photosensitizer that releases chlorin
e6 and Fe3+ in the acidic TME. This release initiates a
ROS cascade that increases LPO. Co-delivery of evodiamine further
inhibits GPX4, thereby intensifying ferroptosis and enhancing PDT
sensitivity. By contrast, PTT uses photothermal agents to raise
local temperature, disrupt cellular membranes and facilitate iron
influx, which enhances the Fenton reaction and accelerates
ferroptosis (158). For example, a
photothermal nanoplatform assembled from camptothecin (CPT) and
IR820 (L820/CPT-CPT NPs) triggers ferroptosis after cellular
uptake. Cleavage of diselenide bonds continuously depletes
intracellular GSH in CRC cells, leading to GPX4 inactivation.
Meanwhile, linoleic acid and the IR820-mediated photothermal effect
further elevate LPO, producing an amplified ferroptosis-driven
antitumor response (159).
Ferroptosis also has immunostimulatory potential
and can synergize with immunotherapy through a positive-feedback
mechanism (160). Li et al
(161) developed a photothermal
metal-phenolic network platform. This ovalbumin-coated
metal-phenolic network leverages the phenomenon of enhanced
permeability and retention to achieve selective accumulation at
tumor sites. Within this system, Fe3+-gallic acid MPNs
simultaneously produce a photothermal effect and drive an
Fe3+-mediated Fenton reaction, sustaining high ROS
levels. In addition, buthionine sulfoximine suppresses GSH
biosynthesis, weakening GPX4-dependent antioxidant defense and
thereby amplifying ferroptosis. Subsequently, DAMPs released from
ferroptotic cells stimulate antitumor immunity. Activated
CD8+ T cells secrete IFN-γ, which downregulates
antioxidant components (including GPX4) in CRC cells. This
‘closed-loop’ positive-feedback circuit increases CRC sensitivity
to ferroptosis.
Therefore, under a multimodal combination
framework, nanocarriers should be engineered for co-loading of
photo-/thermo-agents, ferroptosis modulators and immunoregulatory
agents within a single platform, with on-demand release triggered
by signals such as pH, ROS, light or heat. Notably, conventional
systemic administration is often associated with off-target damage,
limited targeting specificity and suboptimal delivery efficiency,
whereas local administration can increase drug accumulation at the
lesion site and reduce adverse effects caused by systemic exposure
(162). For example, Ye et
al (163) designed a
liposome-loaded ROS-responsive hydrogel for intratumoral injection
that undergoes network cleavage in response to elevated ROS in the
TME, enabling localized release of the NAMPT inhibitor FK866 and
the STAT3 inhibitor C188-9. FK866 depletes NAD+, thereby
inhibiting STAT3 activation and downregulating its downstream
effector GPX4 to induce ferroptosis in CRC cells. Co-administration
of C188-9 further strengthens ferroptosis and promotes immune
activation (164–167) (Fig.
4 and Table II).
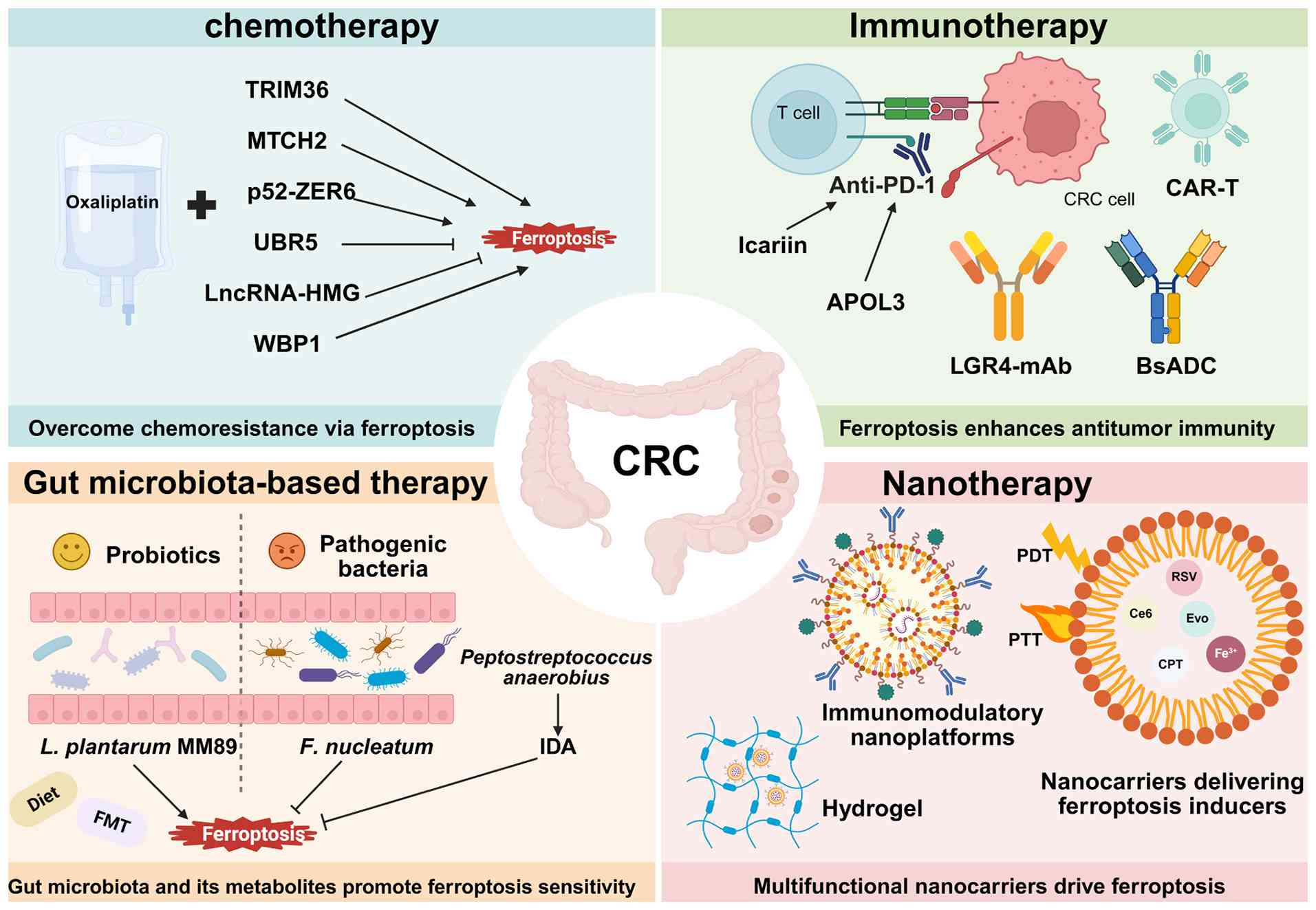 | Figure 4.Integration of ferroptosis inducers
with chemotherapy, immunotherapy, gut microbiota-based therapy and
nanotherapy to suppress CRC progression, overcome therapeutic
resistance and limit immune evasion. BsADCs, bispecific ADCs; FMT,
fecal microbiota transplantation; CAR-T, chimeric antigen receptor
T-cell; PDT, photodynamic therapy; PTT, photothermal therapy; CRC,
colorectal cancer; TRIM36, tripartite motif containing 36; MTCH2,
mitochondrial carrier homolog 2; ZER6, zinc-finger estrogen
receptor interaction clone 6; UBR5, ubiquitin protein ligase E3
component N-recognition protein 5; lncRNAs, long non-coding RNAs;
WBP1, WW domain-binding protein 1; APOL3, apolipoprotein L3; PD-1,
programmed cell death protein 1; mAbs, monoclonal antibodies; IDA,
trans-3-indoleacrylic acid; L. plantarum MM89,
Lactobacillus plantarum MM89; F. nucleatum, Fusobacterium
nucleatum; Ce6, chlorin e6; Evo, evodiamine; CPT, camptothecin;
RSV, resveratrol. |
| Table II.Ferroptosis-modulating therapeutic
strategies in CRC. |
Table II.
Ferroptosis-modulating therapeutic
strategies in CRC.
| Therapeutic
method | Representative
agent/target | CRC cell
models | Main mechanism | (Refs.) |
|---|
| Chemotherapy | TRIM36 | HCT116, LoVo, HT2,
Caco-2, SW480, SW620 | Promotes FOXA2
degradation, inhibits the NRF2 pathway | (127) |
|
| MTCH2 | RKO, SW620, HCT116,
SW480 | MTCH2 loss induces
ferroptosis through the E2F4-TFRC axis | (128) |
|
| UBR5 | SW1116, HCT116,
SW620 | Maintains
Smad3-SLC7A11 signaling | (129) |
|
| LncRNA-HMG | LoVo, HCT116,
SW480 | Promotes p53
degradation and upregulates SLC7A11 and VKORC1L1 | (130) |
|
| p52-ZER6 | HCT116, LoVo,
HT29 | Regulates DAZAP1
transcription to upregulate SLC7A11 expression | (131) |
|
|
Trifluridine/tipiracil | RKO, HT29, HCT116,
DLD-1 | Activates p53,
suppresses SLC7A11 expression | (164) |
|
| WBP1 | HCT116, SW480 | WBP1 inhibition
impairs mitochondrial function | (165) |
|
| SNX1 | HCT116, SW480,
SW948, SK-CO-1, Caco-2, LoVo, HT29 | Modulates the EGFR
downstream PPAR-ACSL1/4 axis | (166) |
| Immunotherapy | Icariin | HCT116, SW620,
MC38, CT26 | Induces
mitochondrial dysfunction | (134) |
|
| APOL3 | RKO, HCT116,
Caco-2 | Promotes LDHA
degradation, and enhances CD8+ T-cell function | (135) |
|
| LGR4-mAb | HCT116, HT-29 | Inhibits LGR4-Wnt
signaling and downregulates SLC7A11 | (137) |
|
| CDH17-GUCY2C | COLO-205, HT-29,
HCT116, T84, | Dual targeting of
CDH17 and | (139) |
|
| BsADC | LS1034, LS-180,
SW620, LoVo, COLO-201, RKO, SW1463, SW403 | GUCY2C enables RSL3
delivery to CRC cells |
|
|
| CYP1B1 | RKO, HCT116, HT29,
MC38 | CYP1B1 inhibition
is reported to promote ACSL4 degradation | (167) |
| Gut
microbiota-based therapy | L. plantarum
MM89 | DLD-1, HT-29 | Secretion of
γ-linolenic acid induces mitochondrial damage | (144) |
|
| F.
nucleatum | HCT116, HT-29 | Upregulates
GPX4 | (145) |
|
| IDA | HT29, MC38 | Enhances
FSP1-mediated antioxidant defense | (147) |
|
| Curcumin | RKO, CT26 | Curcumin-treated
FMT downregulates GPX4 and SLC7A11 expression | (149) |
|
| Arginine | HCT15, LoVo,
SW620 | Activates the
AMPK-p53-p21 pathway | (151) |
|
| Butyrate | HCT116, SW480,
SW620, RKO | Inhibits xCT and
reduces GSH synthesis | (152) |
|
| Creatine | DLD-1, SW480,
MC38 | Induces FSP1
phosphorylation | (153) |
| Nanotherapy | RSV-NPs@RBCm | HT29, HCT116 | Downregulates
SLC7A11 and GPX4 expression | (155) |
|
| FCE NPs | CT26 | Triggers a
Fe3+-mediated Fenton reaction, Ce6-induced ROS
generation, and Evo- mediated GPX4 inhibition | (157) |
|
| L820/CPT-CPT
NPs | DLD-1, HCT116,
SW480, RKO | Combines GPX4
inactivation with linoleic acid oxidation | (159) |
|
| BOFG | CT26 | Impairs
GPX4-mediated antioxidant defense; IFN-γ secreted by immune cells
further downregulates GPX4 | (161) |
|
| Liposome-loaded
hydrogel | MC38, CT26 | Depletes NAD,
activates the STAT3/GPX4 axis | (163) |
In summary, the use of rationally designed
multifunctional nanocarriers that integrate biomimetic engineering,
stimulus-responsive release and localized delivery while enabling
deep synergy between ferroptosis and complementary therapies may
support the development of ferroptosis-driven treatment paradigms
and improve therapeutic outcomes in CRC.
Effects of ferroptosis inhibition on
IBD-to-CRC transition
Chronic inflammation triggered by infectious
agents, dysregulated immune function or unhealthy lifestyle
behaviors is a major high-risk factor that promotes tumorigenesis.
Although only a minority of CRC cases can be unequivocally
attributed to chronic inflammatory conditions, intestinal
inflammation can impair the epithelial barrier through oxidative
stress. This dysfunction increases epithelial susceptibility to
environmental mutagens and thereby elevates the risk of somatic
mutations. Therefore, elucidating the mechanisms by which chronic
inflammation contributes to CRC remains important (14,168).
IBD, primarily encompassing Crohn's disease and ulcerative colitis,
is an immune-mediated chronic intestinal disorder. Its hallmark
features include epithelial barrier disruption, aberrant immune
regulation and structural remodeling of the gut. Under persistent
inflammatory pressure, IBD progression arises from multifactorial
interactions, including genetic and epigenetic alterations,
oxidative stress, immune dysregulation and microbial dysbiosis.
Together, these processes drive lesions along the
‘inflammation-dysplasia-carcinoma’ sequence, making CRC among the
most severely affecting complications of IBD (169–171).
During the active inflammatory phase of IBD,
inhibition of ferroptosis generally exerts a protective effect. The
maintenance of intestinal mucosal barrier integrity is imperative
for the preservation of gut homeostasis. Mucus and digestive
secretions produced by IECs dilute toxins and inhibit or eliminate
bacteria, thereby forming a critical line of defense. However,
ferroptosis in IECs can compromise tissue repair, disrupt the
mucosal barrier and weaken immune function, thereby increasing the
risk of CRC (172,173). Previous evidence indicates that
NEDD4 like E3 ubiquitin protein ligase (NEDD4L) regulates the
stability of SLC3A2 via K63-linked ubiquitination, which enhances
GPX4 activity and suppresses ferroptosis in IECs. Loss of NEDD4L
exacerbates intestinal barrier injury and amplifies inflammatory
responses. Consistently, ferroptosis inhibitors can alleviate
NEDD4L-deficiency-driven colitis and its associated CRC progression
(174).
This antiferroptotic effect also indirectly reduces
the risk of inflammation-driven carcinogenesis. In immune
regulation, pro-inflammatory M1 macrophages play a
pivotal role in maintaining intestinal homeostasis. During host
defense against external insults, M1 macrophages elicit
a controlled pro-inflammatory response that facilitates pathogen
clearance and preserves the intestinal microenvironmental balance.
However, once the threat is resolved, this inflammatory program
must be restrained to prevent excessive tissue damage and reduce
the likelihood of progression from IBD to CRC (175,176). OTSSP167, an inhibitor of maternal
embryonic leucine zipper kinase, not only ameliorates gut dysbiosis
and suppresses ferroptosis in IECs, but also markedly reduces
macrophage infiltration in murine colitis models. It promotes
polarization toward an antitumor-associated M1 phenotype
and decreases the secretion of pro-inflammatory mediators, thereby
exerting anti-inflammatory effects and suppressing the initiation
and progression of colitis-associated CRC (177). In the early stages of
inflammation-driven carcinogenesis, moderate inhibition of
ferroptosis in specific cell populations may help interrupt the
transition from IBD to CRC.
However, once tissues enter the
inflammation-to-cancer transition or tumor cell selection stage,
resistance to ferroptosis is no longer merely an adaptive stress
response, but gradually becomes a critical event that promotes
tumor survival and progression. To adapt to a microenvironment
characterized by high ROS levels, hypoxia and persistent exposure
to inflammatory cytokines, tumor cells often actively establish
anti-ferroptotic programs, thereby sustaining survival,
proliferation and malignant evolution. On the one hand, during
chronic inflammation-driven colorectal carcinogenesis, serine
protease 22 (PRSS22) is progressively upregulated in IECs. Its
overexpression attenuates ferroptosis by suppressing heme oxygenase
1-mediated iron release and ROS accumulation, thereby supporting
the survival of CRC cells. In addition, PRSS22 can proteolytically
cleave osteopontin, generating bioactive fragments that enhance the
migratory and metastatic potential of CRC cells, and further
accelerate tumor progression (178). On the other hand, at the CRC
stage, the RNA-binding protein interleukin enhancer-binding factor
3 (ILF3) reduces CRC cells' sensitivity to ferroptosis by
stabilizing SLC3A2 mRNA, thereby enhancing cystine uptake and GSH
synthesis. Furthermore, TNF-α in the inflammatory microenvironment
can further upregulate ILF3, reinforcing this anti-ferroptotic
state and consequently promoting CRC progression (179). In premalignant and malignant cell
stages, inhibition of ferroptosis essentially confers a survival
advantage that supports continued tumor progression.
Therefore, across the continuous disease course
from IBD to CRC, ferroptosis regulation does not exert a uniformly
protective or pathogenic effect, but instead displays marked stage
dependency and dual biological roles. During the early or active
phase of intestinal inflammation, moderate inhibition of
ferroptosis may help reduce epithelial cell injury, preserve
mucosal barrier integrity and, to a certain extent, limit the
persistent amplification of chronic inflammation, thereby exerting
a protective effect against inflammation-associated tumorigenesis.
However, once the disease progresses to the inflammation-to-cancer
transition stage or an established TME has formed, sustained
enhancement of anti-ferroptotic capacity may impair the elimination
of abnormally proliferating cells and create favorable conditions
for tumor cell survival and malignant progression. Accordingly, the
therapeutic relevance of ferroptosis in the IBD-CRC axis should be
defined as a stage-specific precision regulatory strategy rather
than a simple approach of global activation or complete inhibition.
Future studies should systematically elucidate the specific
functions of distinct regulatory nodes of ferroptosis during the
transition from IBD to CRC, with consideration of disease stage,
key target cell populations and underlying molecular mechanisms,
thereby providing a theoretical basis for precision therapy.
Clinical research progress targeting
ferroptosis
Clinical trials
The translation of ferroptosis research from bench
to bedside has progressed substantially and numerous clinical
studies have explored the therapeutic potential of targeting
ferroptosis-related pathways in CRC. In this context, therapeutic
strategies targeting ferroptosis have attracted considerable
attention from both researchers and clinicians, and an increasing
number of ferroptosis modulators have entered clinical trials
(180–191) (Table
III). Accordingly, a variety of candidate drugs, technical
approaches and therapeutic strategies targeting ferroptosis have
been developed, and related clinical studies are gradually shifting
from early empirical exploration toward mechanism-based design. In
clinical practice, ferroptosis-targeted interventions are more
likely to be used as combination therapies tailored to specific
pathological contexts of CRC, with the potential to overcome
apoptotic resistance in tumor cells, enhance the responsiveness of
CRC to immunotherapy and chemotherapy, and suppress metastatic
growth. Future research should improve the targeting specificity of
these interventions for colorectal tissues and promote the
integration of multi-mechanistic agents to enhance overall efficacy
while reducing drug dosage and minimizing off-target effects as
much as possible.
| Table III.Clinical trials targeting ferroptosis
in colorectal cancer. |
Table III.
Clinical trials targeting ferroptosis
in colorectal cancer.
| Drug | Target | Trial ID | Phase | (Refs.) |
|---|
| Sorafenib | SLC7A11 | NCT07505472 | Marketed | (128,180–182) |
|
|
| NCT01715441 |
|
|
|
|
| NCT01471353 |
|
|
|
|
| NCT01383343 |
|
|
| Sulfasalazine | SLC7A11 | NCT06134388 | Marketed as an
anti-inflammatory agent; phase I in cancer | (164) |
| Neratinib | Iron | NCT03919292 | Marketed | (183) |
| Artesunate | Iron | NCT07095309 | Marketed as an
antimalarial drug, in oncology phase II trials | (184) |
| Cisplatin | GSH | NCT07433673 | Marketed | (185–188) |
|
|
| NCT06704724 |
|
|
|
|
| NCT06621563 |
|
|
|
|
| NCT04457284 |
|
|
| Gemcitabine | GPX4 | NCT05733000 | Marketed | (189) |
| Carbon
nanoparticle-loaded iron | Iron
metabolism | NCT06048367 | Phase I | (190,191) |
|
|
| NCT07433283 |
|
|
Potential ferroptosis biomarkers
The pathophysiological role of ferroptosis in CRC
has attracted increasing attention and the associated molecular
alterations provide an important basis for the development of
highly sensitive biomarkers. To date, research on ferroptosis
biomarkers has mainly focused on molecular events that reflect
dysregulated iron metabolism, LPO accumulation and abnormalities in
antioxidant systems. Because these changes often precede the
appearance of canonical ferroptotic phenotypes, they may serve as
pharmacodynamic biomarkers for assessing ferroptosis-inducing
effects (192). For example,
malondialdehyde, 4-HNE, ACSL4, TFR1 and GPX4 have been proposed as
candidate biomarkers of ferroptosis susceptibility. However, most
of these indicators are indirect measures, with limited ability to
distinguish ferroptosis from nonspecific oxidative damage, and they
are readily influenced by multiple factors, including
pre-analytical sample stability, dietary factors and comorbidities
(193). Although imaging
approaches based on ROS-sensitive probes can provide a certain
degree of spatial information, their application remains limited by
insufficient probe specificity and poor tissue penetration
(194). In addition to molecular
and imaging biomarkers, ferroptosis-related lncRNAs are also
considered promising biomarkers in CRC. Accumulating evidence
indicates that these molecules are involved not only in the
regulation of CRC cell proliferation, metastasis and drug
resistance, but are also closely associated with patient prognosis,
the risk of recurrence and metastasis, and treatment response
(195). For instance, a prognostic
model comprising 15 ferroptosis-related lncRNAs constructed by Ge
et al (196) could
independently predict survival outcomes in patients with CRC and
potentially help evaluate treatment sensitivity. However, these
biomarkers remain largely at an early stage of research, and their
stability, consistency and reproducibility still require further
validation. Notably, CRC exhibits substantial heterogeneity in
genetic drivers, metabolic reprogramming and the TME, suggesting
that ferroptosis-directed therapy will more likely depend on
biomarker-guided precision patient stratification rather than
uniform application.
A recent large-scale TME atlas constructed from
single-cell CRC data further classified patients into six subtypes
with distinct immune evasion mechanisms, indicating that CRC
stratification should not be limited to a simple ‘immune-hot’ vs.
‘immune-cold’ classification (197). On this basis, multimodal
machine-learning strategies integrating lipidomic features,
oxidized phospholipid burden, pathological parameters and radiomic
data may help identify those patients who are most likely to
benefit from ferroptosis-related interventions, and will be
critical for translating ferroptosis-targeted therapy into clinical
practice in CRC.
Discussion
As an iron-dependent and non-reversible mode of
cell death, ferroptosis has emerged as a promising target for CRC
therapy. However, despite the exploration of various
ferroptosis-centered therapeutic strategies, important challenges
remain to be overcome before these strategies can be broadly
translated into clinical applications.
The regulatory network governing ferroptosis is
highly complex, involving multiple interconnected signaling
pathways and metabolic axes. However, the key molecular events and
hierarchical regulatory logic remain incompletely understood,
particularly their association with other cell death programs such
as autophagy, which remains controversial. Autophagy may suppress
ferroptosis in certain contexts, while in others,
autophagy-dependent ferroptosis can be initiated. Conversely,
ferroptosis can also occur independently of autophagy. These
context-dependent interactions require further mechanistic
clarification (198). Current
evidence indicates that, among the GPX4-independent ferroptosis
defense pathways, the GCH1-BH4 axis is the most promising
regulatory target for therapeutic translation in CRC. Inhibition of
this pathway enhances erastin-induced ferroptosis by activating
ferritinophagy and produces synergistic antitumor effects. By
contrast, the biological roles and therapeutic potential of other
GPX4-independent pathways in CRC remain to be systematically
evaluated (199). Notably,
canonical ferroptosis inducers such as erastin and RSL3 primarily
act by disrupting iron homeostasis and redox balance. However,
their limited target selectivity can cause off-target toxicity, and
high doses may induce additional injury in normal tissues,
including the liver and kidney (200). Therefore, future studies should
aim to increase the mechanistic understanding of
ferroptosis-associated networks and exploit CRC-specific metabolic
vulnerabilities, such as dysregulated iron metabolism and impaired
redox homeostasis, to guide precision drug design. This approach
may enable the development of novel ferroptosis modulators with
higher selectivity and fewer off-target effects, maximizing
antitumor efficacy while maintaining an acceptable safety
profile.
Ferroptosis and antitumor immune responses exhibit
a complex bidirectional association. Combining ferroptosis inducers
with immunotherapy can improve efficacy in certain settings.
Ferroptotic tumor cells have also been observed to remodel the TME
towards immunosuppression, dampening T-cell function and thereby
enabling immune escape. This highlights the necessity for
strategies that selectively trigger ferroptosis in tumor cells
while preserving immune effector cells from ferroptotic
destruction. One strategy that has been suggested is the
disturbance of inhibitory feedback circuits in the TME that
propagate danger- or signal-induced secondary ferroptosis to immune
cells in the subsequent sequence of tumor-cell ferroptosis. Another
approach is to develop antigen-guided drug-delivery systems that
release ferroptosis inducers specifically within tumor cells,
thereby minimizing off-target toxicity to immune cells. Notably,
post-translational modifications (PTMs) can markedly alter cellular
sensitivity to ferroptosis by regulating the stability, activity
and intermolecular interactions of key ferroptosis-related proteins
(201). Combining PTM-based
ferroptosis-targeting strategies with ICIs may both reprogram
immune-cell function to strengthen antitumor immunity and offer a
new combinatorial approach to overcome therapeutic resistance in
CRC (202).
With the widespread adoption of single-cell
sequencing, researchers can more precisely characterize how the gut
microbiota and its metabolites regulate immune cell populations.
Emerging evidence suggests that the gut microbiota may shape the
antitumor immune microenvironment by selectively promoting or
inhibiting ferroptosis (146,203). Although the gut microbiome plays
essential roles in numerous physiological processes, its specific
contribution to ferroptosis remains insufficiently and
systematically investigated. This gap may stem from relatively
small fecal sample sizes and the difficulty of definitively
identifying specific microbial taxa or small-molecule metabolites
that modulate ferroptosis. Consequently, current profiles of gut
microbiota alterations in patients with cancer may be incomplete.
Therefore, existing microbiome-related evidence for ferroptosis is
largely indirect and remains limited. Further studies are needed to
elucidate associations between key microbial taxa and
ferroptosis-related genes.
Although nanocarrier delivery systems can improve
drug bioavailability and pharmacokinetics, their clinical
manufacturability and long-term safety require more rigorous
evaluation. Overly complex nanosystems often compromise
reproducibility and process scalability. By contrast, carriers with
well-defined physicochemical properties and simpler architectures
are more likely to achieve batch-to-batch consistency and meet the
clinical trial quality control requirements. Certain nanodrug
delivery systems are poorly biodegradable and may accumulate in
organs such as the liver and kidneys, increasing metabolic burden
and causing organ-specific toxicity (160). Furthermore, numerous current
nano-assemblies are designed around biochemical and immunological
profiles representative of the main population, which may limit
their ability to address the tumor heterogeneity frequently
encountered in clinical practice.
Notably, artificial intelligence (AI) and machine
learning are increasingly used to screen novel compounds, optimize
carrier architectures and enhance drug-loading efficiency. By
modeling in vivo pathophysiological responses, these
approaches also accelerate the development of nanotherapeutic
systems with improved tumor targeting and microenvironmental
adaptability (204). Therefore,
AI-enabled nanomedicine strategies could be leveraged to optimize
the delivery of ferroptosis inducers. Priorities include developing
biodegradable, scalable and multi-stimuli-responsive delivery
platforms, alongside strengthening systematic safety-evaluation
frameworks to reduce systemic toxicity and off-target effects.
The inflammatory consequences of inducing
ferroptosis and the associated potential pro-tumor risks should be
rigorously evaluated. Ferroptosis has substantial cytotoxic
potential against tumor cell populations that are refractory to
conventional therapies. However, accompanying processes, including
inflammatory responses, increased genomic instability, epigenetic
remodeling and activation of cytokine signaling pathways, may under
certain conditions facilitate tumor initiation or progression
toward increased malignancy (205). The goal of anti-inflammatory
therapy is to suppress pro-tumor inflammation while preserving or
even enhancing antitumor immune responses. Given the systemic and
multifactorial nature of inflammation, interventions that target
multiple proteins implicated in inflammatory signaling and
tumorigenesis may be more effective than approaches focused on a
single gene, protein or pathway. Furthermore, the clinical efficacy
of inflammation-targeted therapies remains inconsistent, partly due
to the lack of validated biomarkers for treatment selection and to
interpatient heterogeneity, including age and tumor molecular
profiles (206). Therefore,
integrating multi-omics data with AI-based modeling may enable the
identification of disease-specific inflammatory phenotypes in
patients with intestinal disorders. This framework could guide the
selective inhibition of ferroptosis in IECs, thereby improving the
balance between inflammation control and immune surveillance, and
facilitating the implementation of personalized therapies.
The majority of previous studies rely on simplified
cell lines and conventional animal models; however, neither of them
fully captures the complex physiological and pathological features
of CRC progression. Human three-dimensional in vitro models
derived from stem-cell technologies such as organoids and
organ-on-a-chip platforms can more accurately recapitulate human
tissue architecture and microenvironmental dynamics. These systems
provide a more translational platform for screening personalized
combination therapies. Future research should prioritize clinical
trials to systematically evaluate the efficacy and safety of
ferroptosis-targeted therapies and to accelerate their translation
into clinical practice. Notably, the application of AI may further
accelerate clinical translation (207,208). AI-driven models that evaluate
patient-specific redox trajectories and ferroptosis sensitivity may
help optimize therapeutic benefit and improve clinical
outcomes.
In summary, current evidence supports ferroptosis
as a promising therapeutic target in CRC. Its involvement is
primarily linked to several key biological processes, including
dysregulated iron metabolism, enhanced LPO and impaired antioxidant
defense systems. Furthermore, the induction of ferroptosis shows
considerable potential for increasing the sensitivity of CRC to
chemotherapy, immunotherapy and emerging drug-delivery strategies.
However, these advances remain far from clinical translation.
First, the core regulatory network that determines ferroptosis
sensitivity in CRC has not yet been fully defined, and differences
in vulnerability across distinct biological contexts remain to be
clarified. Second, ferroptosis-based interventions have a dual
nature. A major challenge for optimizing combination immunotherapy
is how to enhance tumor cell killing while preserving the
functional integrity of immune cells, including CD8+ T
cells, NK cells and TAMs, to the greatest extent possible. Third,
current evidence for interactions between the gut microbiota and
ferroptosis is largely correlative, and their causal association
and underlying molecular basis require further investigation.
Finally, biomarkers for patient stratification, treatment-response
prediction and dynamic monitoring, as well as delivery platforms
with high targeting specificity and low toxicity, are not yet
sufficiently mature. Therefore, future research should leverage
single-cell omics, spatial multi-omics and AI-assisted analysis to
focus on identifying responsive patient populations, determining
the optimal timing for intervention and developing precise delivery
strategies. Such efforts will help advance ferroptosis research
from mechanistic exploration toward clinical application in
CRC.
Acknowledgements
The figures were created with BioRender (biorender.com).
Funding
This study was supported by Hubei Provincial Natural Science
Foundation (grant no. JCZRLH20260143).
Availability of data and materials
Not applicable.
Authors' contributions
MW and MZ conceived the study, constructed figures
and edited the manuscript. MW performed the literature review and
wrote the manuscript. MZ provided overall supervision. Data
authentication is not applicable. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263.
2024.PubMed/NCBI
|
|
2
|
Dekker E, Tanis PJ, Vleugels JLA, Kasi PM
and Wallace MB: Colorectal cancer. Lancet. 394:1467–1480. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Morris VK, Kennedy EB, Baxter NN, Benson
AB III, Cercek A, Cho M, Ciombor KK, Cremolini C, Davis A, Deming
DA, et al: Treatment of metastatic colorectal cancer: ASCO
guideline. J Clin Oncol. 41:678–700. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siegel RL, Miller KD, Goding Sauer A,
Fedewa SA, Butterly LF, Anderson JC, Cercek A, Smith RA and Jemal
A: Colorectal cancer statistics, 2020. CA Cancer J Clin.
70:145–164. 2020.PubMed/NCBI
|
|
5
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu A, Ni Y, Zhou Y and Shi J: Emerging
dual role of ferroptosis in lung cancer (Review). Oncol Rep.
54:1412025. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu Y, Lu S, Wu LL, Yang L, Yang L and
Wang J: The diversified role of mitochondria in ferroptosis in
cancer. Cell Death Dis. 14:5192023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dixon SJ and Olzmann JA: The cell biology
of ferroptosis. Nat Rev Mol Cell Bio. 25:424–442. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang S, Guo Q, Zhou L and Xia X:
Ferroptosis: A double-edged sword. Cell Death Discov. 10:2652024.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Masci D, Ling L, Yang L, Puxeddu M, Colla
C, Coluccia A, Santelli M, Sciò P, Cuřínová P, Ansari MSZ, et al:
Ferroptosis induction by a new pyrrole derivative in triple
negative breast cancer and colorectal cancer. J Med Chem.
68:17840–17858. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xia W, Lv Y, Zou Y, Kang Z, Li Z, Tian J,
Zhou H, Su W and Zhong J: The role of ferroptosis in colorectal
cancer and its potential synergy with immunotherapy. Front Immunol.
15:15267492025. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Neagu AI, Bostan M, Ionescu VA, Gheorghe
G, Hotnog CM, Roman V, Mihaila M, Stoica SI, Diaconu CC, Diaconu
CC, et al: The impact of the microbiota on the immune response
modulation in colorectal cancer. Biomolecules. 15:10052025.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu K, Huang L, Qi S, Liu S, Xie W, Du L,
Cui J, Zhang X, Zhang B, Liu L, et al: Ferroptosis: The
Entanglement between Traditional drugs and nanodrugs in tumor
therapy. Adv Healthc Mater. 12:e22030852023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schmitt M and Greten FR: The inflammatory
pathogenesis of colorectal cancer. Nat Rev Immunol. 21:653–667.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xie H, Cao C, Shu D, Liu T and Zhang T:
The important role of ferroptosis in inflammatory bowel disease.
Front Med (Lausanne). 11:14490372024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mouawad N, El Jaafari N, El Sibai M and
Abi-Habib RJ: Harnessing ferroptosis for cancer therapy: Mechanisms
and therapeutic strategies (Review). Oncol Rep.
55:242026.PubMed/NCBI
|
|
17
|
Ding K, Liu C, Li L, Yang M, Jiang N, Luo
S and Sun L: Acyl-CoA synthase ACSL4: An essential target in
ferroptosis and fatty acid metabolism. Chin Med J (Engl).
136:2521–2537. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee JY, Kim WK, Bae KH, Lee SC and Lee EW:
Lipid metabolism and ferroptosis. Biology (Basel).
10:1842021.PubMed/NCBI
|
|
19
|
Gündem E, Stehling S, Borchert A and Kuhn
H: The reaction specificity of mammalian ALOX15B orthologs does not
depend on the evolutionary ranking of the animals. J Lipid Res.
66:1007682025. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dai E, Chen X, Linkermann A, Jiang X, Kang
R, Kagan VE, Bayir H, Yang WS, Garcia-Saez AJ, Ioannou MS, et al: A
guideline on the molecular ecosystem regulating ferroptosis. Nat
Cell Biol. 26:1447–1457. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tesfay L, Paul BT, Konstorum A, Deng Z,
Cox AO, Lee J, Furdui CM, Hegde P, Torti FM and Torti SV:
Stearoyl-CoA Desaturase 1 protects ovarian cancer cells from
ferroptotic cell death. Cancer Res. 79:5355–5366. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rodencal J, Kim N, He A, Li VL, Lange M,
He J, Tarangelo A, Schafer ZT, Olzmann JA, Long JZ, et al:
Sensitization of cancer cells to ferroptosis coincident with cell
cycle arrest. Cell Chem Biol. 31:234–248.e13. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liang D, Feng Y, Zandkarimi F, Wang H,
Zhang Z, Kim J, Cai Y, Gu W, Stockwell BR and Jiang X: Ferroptosis
surveillance independent of GPX4 and differentially regulated by
sex hormones. Cell. 186:2748–2764.e22. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sokol KH, Lee CJ, Rogers TJ, Waldhart A,
Ellis AE, Madireddy S, Daniels SR, House RRJ, Ye X, Olesnavich M,
et al: Lipid availability influences ferroptosis sensitivity in
cancer cells by regulating polyunsaturated fatty acid trafficking.
Cell Chem Biol. 32:408–422.e6. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Feng H, Schorpp K, Jin J, Yozwiak CE,
Hoffstrom BG, Decker AM, Rajbhandari P, Stokes ME, Bender HG, Csuka
JM, et al: Transferrin receptor is a specific ferroptosis marker.
Cell Rep. 30:3411–3423.e7. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu D, Hu Z, Lu J and Yi C:
Redox-regulated iron metabolism and ferroptosis in ovarian cancer:
Molecular insights and therapeutic opportunities. Antioxidants
(Basel). 13:7912024. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shi J, Yang MM, Yang S, Fan F, Zheng G,
Miao Y, Hua Y, Zhang J, Cheng Y, Liu S, et al: MaiJiTong granule
attenuates atherosclerosis by reducing ferroptosis via activating
STAT6-mediated inhibition of DMT1 and SOCS1/p53 pathways in LDLR-/-
mice. Phytomedicine. 128:1554892024. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Brown CW, Amante JJ, Chhoy P, Elaimy AL,
Liu H, Zhu LJ, Baer CE, Dixon SJ and Mercurio AM: Prominin2 drives
ferroptosis resistance by stimulating iron export. Dev Cell.
51:575–586.e4. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Katsarou A and Pantopoulos K: Basics and
principles of cellular and systemic iron homeostasis. Mol Aspects
Med. 75:1008662020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mancias JD, Wang X, Gygi SP, Harper JW and
Kimmelman AC: Quantitative proteomics identifies NCOA4 as the cargo
receptor mediating ferritinophagy. Nature. 509:105–109. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu L, Liu Y, Zhou X, He H, Chen N, Qin Y,
Sun X, Bian Z, Zhang Q, Mao L and Sun S: Sodium butyrate induces
ferroptosis in colorectal cancer cells by promoting
NCOA4-FTH1-mediated ferritinophagy. Int Immunopharmacol.
163:1151882025. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jin B, Zhang Z, Zhang Y, Yang M, Wang C,
Xu J, Zhu Y, Mi Y, Jiang J and Sun Z: Ferroptosis and myocardial
ischemia-reperfusion: Mechanistic insights and new therapeutic
perspectives. Front Pharmacol. 15:14829862024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kunji ERS, King MS, Ruprecht JJ and
Thangaratnarajah C: The SLC25 carrier family: Important transport
proteins in mitochondrial physiology and pathology. Physiology
(Bethesda). 35:302–327. 2020.PubMed/NCBI
|
|
34
|
She G, Hai XX, Jia LY, Zhang YJ, Ren YJ,
Pang ZD, Wu LH, Han MZ, Zhang Y, Li JJ, et al: Hippo pathway
activation mediates cardiomyocyte ferroptosis to promote dilated
cardiomyopathy through downregulating NFS1. Redox Biol.
82:1035972025. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Song Y, Gao M, Wei B, Huang X, Yang Z, Zou
J and Guo Y: Mitochondrial ferritin alleviates ferroptosis in a
kainic acid-induced mouse epilepsy model by regulating iron
homeostasis: Involvement of nuclear factor erythroid 2-related
factor 2. CNS Neurosci Ther. 30:e146632024. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Banerjee R, Bintee B, Hegde M,
BharathwajChetty B, Alqahtani MS, Abbas M, Alqahtani A, Sethi G,
Rangan L, Ma Z and Kunnumakkara AB: Protein iron transporters as
potential therapeutic targets in cancer: A review. Int J Biol
Macromol. 327((Pt 2)): 1466952025. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bai S, Guo Y, Qiang J, Dai Q and Yang Y:
Research progress on the regulation of ferroptosis in NPC (Review).
Oncol Rep. 55:332026.PubMed/NCBI
|
|
38
|
Zhao C, Huang X, Wu Z, Sun W, Pan Y and
Liu Y: pH-Responsive self-assembled upconversion photosensitizer
and RSL3 synergistically induce tumor ferroptosis. J Med Chem.
68:15110–15119. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tang D, Chen X, Kang R and Kroemer G:
Ferroptosis: Molecular mechanisms and health implications. Cell
Res. 31:107–125. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Song X, Hao X and Zhu BT: Role of
mitochondrial reactive oxygen species in chemically-induced
ferroptosis. Free Radical Biol Med. 223:473–492. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lee J and Roh JL: Selenium and
selenoproteins: Key regulators of ferroptosis and therapeutic
targets in cancer. J Mol Med (Berl). 103:899–911. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gao R, Wang J, Huang J, Wang T, Guo L, Liu
W, Guan J, Liang D, Meng Q and Pan H: FSP1-mediated ferroptosis in
cancer: from mechanisms to therapeutic applications. Apoptosis.
29:1019–1037. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang JS, Morris AJ, Kamizaki K, Chen J,
Stark J, Oldham WM, Nakamura T, Mishima E, Loscalzo J, Minami Y, et
al: ALDH7A1 protects against ferroptosis by generating membrane
NADH and regulating FSP1. Cell. 188:2569–2585.e20. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li W, Liang L, Liu S, Yi H and Zhou Y:
FSP1: A key regulator of ferroptosis. Trends Mol Med. 29:753–764.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee
H, Koppula P, Wu S, Zhuang L, Fang B, et al: DHODH-mediated
ferroptosis defence is a targetable vulnerability in cancer.
Nature. 593:586–590. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Deshwal S, Onishi M, Tatsuta T, Bartsch T,
Cors E, Ried K, Lemke K, Nolte H, Giavalisco P and Langer T:
Mitochondria regulate intracellular coenzyme Q transport and
ferroptotic resistance via STARD7. Nat Cell Biol. 25:246–257.
2023.PubMed/NCBI
|
|
48
|
Liu Z, Kang R, Yang N, Pan X, Yang J, Yu
H, Deng W, Jia Z, Zhang J and Shen Q: Tetrahydrobiopterin
inhibitor-based antioxidant metabolic strategy for enhanced cancer
ferroptosis-immunotherapy. J Colloid Interf Sci. 658:100–113. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Du J, Zhu X, Zhang Y, Huang X, Wang X,
Yang F, Xia H and Hou J: CTRP13 attenuates atherosclerosis by
inhibiting endothelial cell ferroptosis via activating GCH1. Int
Immunopharmacol. 143((Pt 3)): 1136172024. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liu Y and Gu W: p53 in ferroptosis
regulation: The new weapon for the old guardian. Cell Death Differ.
29:895–910. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Jiang L, Kon N, Li T, Wang SJ, Su T,
Hibshoosh H, Baer R and Gu W: Ferroptosis as a p53-mediated
activity during tumour suppression. Nature. 520:57–62. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chu B, Kon N, Chen D, Li T, Liu T, Jiang
L, Song S, Tavana O and Gu W: ALOX12 is required for p53-mediated
tumour suppression through a distinct ferroptosis pathway. Nat Cell
Biol. 21:579–591. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chen L, Cai Q, Yang R, Wang H, Ling H, Li
T, Liu N, Wang Z, Sun J, Tao T, et al: GINS4 suppresses ferroptosis
by antagonizing p53 acetylation with Snail. Proc Natl Acad Sci USA.
120:e22195851202023. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang Y, Song XW, Zhang N, Li XH, Wu FC,
Wei YA, Xu DL, Xu LF and Yuan FW: Ezetimibe engineered L14-8
suppresses advanced prostate cancer by activating PLK1/TP53-SAT1
-induced ferroptosis. Adv Sci (Weinh). 12:e041922025. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Shin D, Lee J and Roh J: Pioneering the
future of cancer therapy: Deciphering the p53-ferroptosis nexus for
precision medicine. Cancer Lett. 585:2166452024. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Tarangelo A, Magtanong L, Bieging-Rolett
KT, Li Y, Ye J, Attardi LD and Dixon SJ: p53 suppresses metabolic
stress-induced ferroptosis in cancer cells. Cell Rep. 22:569–575.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J,
Zhong M, Yuan H, Zhang L, Billiar TR, et al: The tumor suppressor
p53 limits ferroptosis by blocking DPP4 activity. Cell Rep.
20:1692–1704. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yang X, Liang B, Zhang L, Zhang M, Ma M,
Qing L, Yang H, Huang G and Zhao J: Ursolic acid inhibits the
proliferation of triple-negative breast cancer stem-like cells
through NRF2-mediated ferroptosis. Oncol Rep. 52:942024. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Jiang X, Yu M, Wang WK, Zhu LY, Wang X,
Jin HC and Feng LF: The regulation and function of Nrf2 signaling
in ferroptosis-activated cancer therapy. Acta Pharmacol Sin.
45:2229–2240. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Chen Y, Jiang Z and Li X: New insights
into crosstalk between Nrf2 pathway and ferroptosis in lung
disease. Cell Death Dis. 15:8412024. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Xiang Y, Song X and Long D: Ferroptosis
regulation through Nrf2 and implications for neurodegenerative
diseases. Arch Toxicol. 98:579–615. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Bollong MJ, Lee G, Coukos JS, Yun H,
Zambaldo C, Chang JW, Chin EN, Ahmad I, Chatterjee AK, Lairson LL,
et al: A metabolite-derived protein modification integrates
glycolysis with KEAP1-NRF2 signalling. Nature. 562:600–604. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Huang C, Zeng Q, Chen J, Wen Q, Jin W, Dai
X, Ruan R, Zhong H, Xia Y, Wu Z, et al: TMEM160 inhibits KEAP1 to
suppress ferroptosis and induce chemoresistance in gastric cancer.
Cell Death Dis. 16:2872025. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wang S, Zhang C, Zhou S, Liu S, Li Q,
Cheng X, Wang R, Chen B, Li Y and Xi M: RNF217-KEAP1-NRF2 feedback
loop confers therapeutic resistance by inhibiting ferroptosis in
esophageal squamous cell carcinoma. Drug Resist Update.
83:1012962025. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Xu RC, Sun JL, Wang F, Liu HH, Qi ZR, Shi
X, Yu XN, Liu TT, Weng SQ, Dong L, et al: Cathepsin S regulates
ferroptosis sensitivity in hepatocellular carcinoma through the
KEAP1-NRF2 signaling pathway. Redox Biol. 86:1038152025. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chang K, Chen Y, Zhang X, Zhang W, Xu N,
Zeng B, Wang Y, Feng T, Dai B, Xu F, et al: DPP9 stabilizes NRF2 to
suppress ferroptosis and induce sorafenib resistance in clear cell
renal cell carcinoma. Cancer Res. 83:3940–3955. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wang Z, Li R, Hou N, Zhang J, Wang T, Fan
P, Ji C, Zhang B, Liu L, Wang Y, et al: PRMT5 reduces immunotherapy
efficacy in triple-negative breast cancer by methylating KEAP1 and
inhibiting ferroptosis. J Immunother Cancer. 11:e0068902023.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Li S, Huang P, Lai F, Zhang T, Guan J, Wan
H and He Y: Mechanisms of ferritinophagy and ferroptosis in
diseases. Mol Neurobiol. 61:1605–1626. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wu H, Liu Q, Shan X, Gao W and Chen Q: ATM
orchestrates ferritinophagy and ferroptosis by phosphorylating
NCOA4. Autophagy. 19:2062–2077. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Wang H, Zhang P, Cheng Q, Chang K, Bao L
and Yi X: Heme oxygenase-1 (HO-1) depletion promotes ferroptosis to
reverse cisplatin-resistance via enhancing NCOA4-mediated
ferritinophagy in ovarian cancer. Int J Biol Macromol.
327:1472572025. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Liu Z, Liu C, Fan C, Li R, Zhang S, Liu J,
Li B, Zhang S, Guo L, Wang X, et al: E3 ubiquitin ligase DTX2
fosters ferroptosis resistance via suppressing NCOA4-mediated
ferritinophagy in non-small cell lung cancer. Drug Resist Update.
77:1011542024. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhang Q, Xu Z, Liu W, Cheng Z, Ding Y, Xie
Y and Yan S: Gastrodin promotes ferroptosis in CRC cells by
inhibiting SKP2 to reduce NCOA4 ubiquitination. Tissue Cell.
95:1027932025. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Shen C, Liu J, Liu H, Li G, Wang H, Tian
H, Mao Y and Hua D: Timosaponin AIII induces lipid peroxidation and
ferroptosis by enhancing Rab7-mediated lipophagy in colorectal
cancer cells. Phytomedicine. 122:1550792024. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Gnanapradeepan K, Indeglia A, Stieg DC,
Clarke N, Shao C, Dougherty JF, Murali N and Murphy ME: PLTP is a
p53 target gene with roles in cancer growth suppression and
ferroptosis. J Biol Chem. 298:1026372022. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yang M, Chen P, Liu J, Zhu S, Kroemer G,
Klionsky DJ, Lotze MT, Zeh HJ, Kang R and Tang D: Clockophagy is a
novel selective autophagy process favoring ferroptosis. Sci Adv.
5:eaaw22382019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Jiang H, Wang X, Zhu Z, Song C, Li D, Yun
Y, Hui L, Bao L, O Connor DP, Ma J and Xu G: DCAF7 recruits USP2 to
facilitate hepatocellular carcinoma progression by suppressing
clockophagy-induced ferroptosis. Cell Death Dis. 16:6542025.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Granata S, Votrico V, Spadaccino F,
Catalano V, Netti GS, Ranieri E, Stallone G and Zaza G: Oxidative
stress and ischemia/reperfusion injury in kidney transplantation:
Focus on ferroptosis, mitophagy and new antioxidants. Antioxidants
(Basel). 11:7692022. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Liu S, Chen JH, Li LC, Ye ZP, Liu JN, Chen
YH, Hu BX, Tang JH, Feng GK, Li ZM, et al: Susceptibility of
mitophagy-deficient tumors to ferroptosis induction by relieving
the suppression of lipid peroxidation. Adv Sci (Weinh).
12:e24125932025. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Yu S, Li Z, Zhang Q, Wang R, Zhao Z, Ding
W, Wang F, Sun C, Tang J, Wang X, et al: GPX4 degradation via
chaperone-mediated autophagy contributes to antimony-triggered
neuronal ferroptosis. Ecotoxicol Environ Saf. 234:1134132022.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M,
Shan B, Pan H and Yuan J: Chaperone-mediated autophagy is involved
in the execution of ferroptosis. Proc Natl Acad Sci USA.
116:2996–3005. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Chen J, Peng Y, Zhou M, Che Y, Zhao S, He
C, Zhang W, Tian X, Zhang W, Liu Z, et al: P23 acts as a negative
regulator of ferroptosis in NSCLC by blocking GPX4 degradation via
chaperone-mediated autophagy. Mol Cancer. 24:2342025. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Wu K, Yan M, Liu T, Wang Z, Duan Y, Xia Y,
Ji G, Shen Y, Wang L, Li L, et al: Creatine kinase B suppresses
ferroptosis by phosphorylating GPX4 through a moonlighting
function. Nat Cell Biol. 25:714–725. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Lei G, Zhuang L and Gan B: The roles of
ferroptosis in cancer: Tumor suppression, tumor microenvironment,
and therapeutic interventions. Cancer Cell. 42:513–534. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Wang W, Green M, Choi JE, Gijón M, Kennedy
PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, et al: CD8+ T
cells regulate tumour ferroptosis during cancer immunotherapy.
Nature. 569:270–274. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Liao P, Wang W, Wang W, Kryczek I, Li X,
Bian Y, Sell A, Wei S, Grove S, Johnson JK, et al: CD8+ T cells and
fatty acids orchestrate tumor ferroptosis and immunity via ACSL4.
Cancer Cell. 40:365–378.e6. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Jiang Y, Li J, Wang T, Gu X, Li X, Liu Z,
Yue W and Li M: VIPAS39 confers ferroptosis resistance in
epithelial ovarian cancer through exporting ACSL4. Ebiomedicine.
114:1056462025. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Gao Y, Liu S, Huang Y, Wang H, Zhao Y, Cui
X, Peng Y, Li F and Zhang Y: CAR T cells engineered to secrete IFNκ
induce tumor ferroptosis via an IFNAR/STAT1/ACSL4 axis. Cancer
Immunol Res. 12:1691–1702. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Liang Y and Zhao Y, Qi Z, Li X and Zhao Y:
Ferroptosis: CD8+T cells' blade to destroy tumor cells or poison
for self-destruction. Cell Death Discov. 11:1282025. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Xu S, Chaudhary O, Rodríguez-Morales P,
Sun X, Chen D, Zappasodi R, Xu Z, Pinto AFM, Williams A, Schulze I,
et al: Uptake of oxidized lipids by the scavenger receptor CD36
promotes lipid peroxidation and dysfunction in CD8+ T cells in
tumors. Immunity. 54:1561–1577.e7. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ma X, Xiao L, Liu L, Ye L, Su P, Bi E,
Wang Q, Yang M, Qian J and Yi Q: CD36-mediated ferroptosis dampens
intratumoral CD8+ T cell effector function and impairs their
antitumor ability. Cell Metab. 33:1001–1012.e5. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Zhao Z, Hu B, Deng Y, Soeung M, Yao J, Bei
L, Zhang Y, Gong P, Huang LA, Jiang Z, et al: Sickle cell disease
induces chromatin introversion and ferroptosis in CD8+ T cells to
suppress anti-tumor immunity. Immunity. 58:1484–1501.e11. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Li S, Ouyang X, Sun H, Jin J, Chen Y, Li
L, Wang Q, He Y, Wang J, Chen T, et al: DEPDC5 protects CD8+ T
cells from ferroptosis by limiting mTORC1-mediated purine
catabolism. Cell Discov. 10:532024. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Wang C, Wang X, Zhang D, Sun X, Wu Y, Wang
J, Li Q and Jiang G: The macrophage polarization by miRNAs and its
potential role in the treatment of tumor and inflammation (Review).
Oncol Rep. 50:1902023. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Li H, Yang P, Wang J, Zhang J, Ma Q, Jiang
Y, Wu Y, Han T and Xiang D: HLF regulates ferroptosis, development
and chemoresistance of triple-negative breast cancer by activating
tumor cell-macrophage crosstalk. J Hematol Oncol. 15:22022.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Luo X, Gong HB, Gao HY, Wu YP, Sun WY, Li
ZQ, Wang G, Liu B, Liang L, Kurihara H, et al: Oxygenated
phosphatidylethanolamine navigates phagocytosis of ferroptotic
cells by interacting with TLR2. Cell Death Differ. 28:1971–1989.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Xiao L, Xian M, Zhang C, Guo Q and Yi Q:
Lipid peroxidation of immune cells in cancer. Front Immunol.
14:13227462024. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Kapralov AA, Yang Q, Dar HH, Tyurina YY,
Anthonymuthu TS, Kim R, St Croix CM, Mikulska-Ruminska K, Liu B,
Shrivastava IH, et al: Redox lipid reprogramming commands
susceptibility of macrophages and microglia to ferroptotic death.
Nat Chem Biol. 16:278–290. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Hao X, Zheng Z, Liu H, Zhang Y, Kang J,
Kong X, Rong D, Sun G, Sun G, Liu L, et al: Inhibition of APOC1
promotes the transformation of M2 into M1 macrophages via the
ferroptosis pathway and enhances anti-PD1 immunotherapy in
hepatocellular carcinoma based on single-cell RNA sequencing. Redox
Biol. 56:1024632022. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Xiong H, Zhai Y, Meng Y, Wu Z, Qiu A, Cai
Y, Wang G and Yang L: Acidosis activates breast cancer ferroptosis
through ZFAND5/SLC3A2 signaling axis and elicits M1 macrophage
polarization. Cancer Lett. 587:2167322024. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Huang WB, Lai HZ, Long J, Dai ZL, Ma Q,
Xiao C and You FM: Biomechanics of the tumor extracellular matrix
and regulatory T cells: Regulatory mechanisms and potential
therapeutic targets. Cell Commun Signal. 23:3752025. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Wu Q, Carlos AR, Braza F, Bergman ML,
Kitoko JZ, Bastos-Amador P, Cuadrado E, Martins R, Oliveira BS,
Martins VC, et al: Ferritin heavy chain supports stability and
function of the regulatory T cell lineage. EMBO J. 43:1445–1483.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Sumida TS, Cheru NT and Hafler DA: The
regulation and differentiation of regulatory T cells and their
dysfunction in autoimmune diseases. Nat Rev Immunol. 24:503–517.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Yan J, Zeng Y, Guan Z, Li Z, Luo S, Niu J,
Zhao J, Gong H, Huang T, Li Z, et al: Inherent preference for
polyunsaturated fatty acids instigates ferroptosis of Treg cells
that aggravates high-fat-diet-related colitis. Cell Rep.
43:1146362024. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Xu C, Sun S, Johnson T, Qi R, Zhang S,
Zhang J and Yang K: The glutathione peroxidase Gpx4 prevents lipid
peroxidation and ferroptosis to sustain Treg cell activation and
suppression of antitumor immunity. Cell Rep. 35:1092352021.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Wculek SK, Cueto FJ, Mujal AM, Melero I,
Krummel MF and Sancho D: Dendritic cells in cancer immunology and
immunotherapy. Nat Rev Immunol. 20:7–24. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Cubillos-Ruiz JR, Silberman PC, Rutkowski
MR, Chopra S, Perales-Puchalt A, Song M, Zhang S, Bettigole SE,
Gupta D, Holcomb K, et al: ER stress sensor XBP1 controls
anti-tumor immunity by disrupting dendritic cell homeostasis. Cell.
161:1527–1538. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Qiu Y, Gan M, Wang X, Liao T, Chen Q, Lei
Y, Chen L, Wang J, Zhao Y, Niu L, et al: The global perspective on
peroxisome proliferator-activated receptor γ (PPARγ) in ectopic fat
deposition: A review. Int J Biol Macromol. 253((Pt 5)): 1270422023.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Han L, Bai L, Qu C, Dai E, Liu J, Kang R,
Zhou D, Tang D and Zhao Y: PPARG-mediated ferroptosis in dendritic
cells limits antitumor immunity. Biochem Biophys Res Commun.
576:33–39. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Xiao K, Zhang S, Peng Q, Du Y, Yao X, Ng
II and Tang H: PD-L1 protects tumor-associated dendritic cells from
ferroptosis during immunogenic chemotherapy. Cell Rep.
43:1148682024. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Li JY, Ren C, Wang LX, Yao RQ, Dong N, Wu
Y, Tian YP and Yao YM: Sestrin2 protects dendrite cells against
ferroptosis induced by sepsis. Cell Death Dis. 12:8342021.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Li M, Jin S, Ma H, Yang X and Zhang Z:
Reciprocal regulation between ferroptosis and STING-type I
interferon pathway suppresses head and neck squamous cell carcinoma
growth through dendritic cell maturation. Oncogene. 44:1922–1935.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Liu Y, Su C, Wei X, Wei N, Qian Q and Xu
Z: Prospects and applications of NK therapy in the treatment of
gliomas (Review). Oncol Rep. 54:882025. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Poznanski SM, Singh K, Ritchie TM, Aguiar
JA, Fan IY, Portillo AL, Rojas EA, Vahedi F, El-Sayes A, Xing S, et
al: Metabolic flexibility determines human NK cell functional fate
in the tumor microenvironment. Cell Metab. 33:1205–1220.e5. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Chen T, Zhou S, Zhang Y, Meng H, Miao H,
Feng M, Jiang Y, Wan Y, Zhang L and Cheng W: Overcoming
ferroptosis-induced exhaustion of NK cells through inhibition of
the ATF3-Mediated integrated stress response in ovarian cancer. Int
J Biol Sci. 21:5531–5546. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Yao L, Hou J, Wu X, Lu Y, Jin Z, Yu Z, Yu
B, Li J, Yang Z, Li C, et al: Cancer-associated fibroblasts impair
the cytotoxic function of NK cells in gastric cancer by inducing
ferroptosis via iron regulation. Redox Biol. 67:1029232023.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Pan B, Zhang X, Ye D, Yao Y, Zhang Z, Luo
Y, Wu H, Wang X and Tang N: Intratumoral Brevibacillus parabrevis
enhances antitumor immunity by inhibiting NK cell ferroptosis in
hepatocellular carcinoma. Cell Death Dis. 16:4072025. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Dagher OK and Posey AD Jr: Forks in the
road for CAR T and CAR NK cell cancer therapies. Nat Immunol.
24:1994–2007. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Wu L, Liu F, Yin L, Wang F, Shi H, Zhao Q,
Yang F, Chen D, Dong X, Gu Y and Xing N: The establishment of
polypeptide PSMA-targeted chimeric antigen receptor-engineered
natural killer cells for castration-resistant prostate cancer and
the induction of ferroptosis-related cell death. Cancer Commun
(Lond). 42:768–783. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Tao B, Du R, Zhang X, Jia B, Gao Y, Zhao Y
and Liu Y: Engineering CAR-NK cell derived exosome disguised
nano-bombs for enhanced HER2 positive breast cancer brain
metastasis therapy. J Control Release. 363:692–706. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Zhang F, Ma Y, Li D, Wei J, Chen K, Zhang
E, Liu G, Chu X, Liu X, Liu W, et al: Cancer associated fibroblasts
and metabolic reprogramming: unraveling the intricate crosstalk in
tumor evolution. J Hematol Oncol. 17:802024. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Zhang K, Guo L, Li X, Hu Y and Luo N:
Cancer-associated fibroblasts promote doxorubicin resistance in
triple-negative breast cancer through enhancing ZFP64 histone
lactylation to regulate ferroptosis. J Transl Med. 23:2472025.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Jiang F, Jia K, Chen Y, Ji C, Chong X, Li
Z, Zhao F, Bai Y, Ge S, Gao J, et al: ANO1-Mediated inhibition of
cancer ferroptosis confers immunotherapeutic resistance through
recruiting cancer-associated fibroblasts. Adv Sci (Weinh).
10:e23008812023. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Zhao J, Shen J, Mao L, Yang T, Liu J and
Hongbin S: Cancer associated fibroblast secreted miR-432-5p targets
CHAC1 to inhibit ferroptosis and promote acquired chemoresistance
in prostate cancer. Oncogene. 43:2104–2114. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Du J, Meng X, Yang M, Chen G, Li J, Zhu Z,
Wu X, Hu W, Tian M, Li T, et al: NGR-Modified CAF-Derived exos
targeting tumor vasculature to induce ferroptosis and overcome
chemoresistance in osteosarcoma. Adv Sci (Weinh). 12:e24109182025.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Rottenberg S, Disler C and Perego P: The
rediscovery of platinum-based cancer therapy. Nat Rev Cancer.
21:37–50. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Koeberle SC, Kipp AP, Stuppner H and
Koeberle A: Ferroptosis-modulating small molecules for targeting
drug-resistant cancer: Challenges and opportunities in manipulating
redox signaling. Med Res Rev. 43:614–682. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Liu X, Yan C, Chang C, Meng F, Shen W,
Wang S and Zhang Y: FOXA2 suppression by TRIM36 exerts anti-tumor
role in colorectal cancer via inducing NRF2/GPX4-Regulated
Ferroptosis. Adv Sci (Weinh). 10:e23045212023. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Xing P, Chen J, Hao H, Qiao X, Yang X,
Weng K, Chen J, Song L, Liu T, Hou Y, et al: MTCH2 deficiency
promotes E2F4/TFRC-Mediated ferroptosis and sensitizes colorectal
cancer liver metastasis to sorafenib. Adv Sci (Weinh).
12:e000192025. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Song M, Huang S, Wu X, Zhao Z, Liu X, Wu
C, Wang M, Gao J, Ke Z, Ma X and He W: UBR5 mediates colorectal
cancer chemoresistance by attenuating ferroptosis via Lys 11
ubiquitin-dependent stabilization of Smad3-SLC7A11 signaling. Redox
Biol. 76:1033492024. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Xin Z, Hu C, Zhang C, Liu M, Li J, Sun X,
Hu Y, Liu X and Wang K: LncRNA-HMG incites colorectal cancer cells
to chemoresistance via repressing p53-mediated ferroptosis. Redox
Biol. 77:1033622024. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Qiu L, Li W, Zhang L, Zhang X, Zhao H,
Miyagishi M, Wu S and Kasim V: p52-ZER6/DAZAP1 axis promotes
ferroptosis resistance and colorectal cancer progression via
regulating SLC7A11 mRNA stabilization. Acta Pharm Sin B.
15:2039–2058. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Huo Z, Liu G and Li J: Recent research
progress and clinical status of immunotherapy for colorectal
cancer. J Adv Res. 82:729–750. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Lu Y, Xie X and Luo L: Ferroptosis
crosstalk in anti-tumor immunotherapy: Molecular mechanisms, tumor
microenvironment, application prospects. Apoptosis. 29:1914–1943.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Haoyue W, Kexiang S, Shan TW, Jiamin G,
Luyun Y, Junkai W and Wanli D: Icariin promoted ferroptosis by
activating mitochondrial dysfunction to inhibit colorectal cancer
and synergistically enhanced the efficacy of PD-1 inhibitors.
Phytomedicine. 136:1562242025. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Lv Y, Tang W, Xu Y, Chang W, Zhang Z, Lin
Q, Ji M, Feng Q, He G and Xu J: Apolipoprotein L3 enhances CD8+ T
cell antitumor immunity of colorectal cancer by promoting
LDHA-mediated ferroptosis. Int J Biol Sci. 19:1284–1298. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Chen P, Li X, Zhang R, Liu S, Xiang Y,
Zhang M, Chen X, Pan T, Yan L, Feng J, et al: Combinative treatment
of β-elemene and cetuximab is sensitive to KRAS mutant colorectal
cancer cells by inducing ferroptosis and inhibiting
epithelial-mesenchymal transformation. Theranostics. 10:5107–5119.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Zheng H, Liu J, Cheng Q, Zhang Q, Zhang Y,
Jiang L, Huang Y, Li W, Zhao Y, Chen G, et al: Targeted activation
of ferroptosis in colorectal cancer via LGR4 targeting overcomes
acquired drug resistance. Nat Cancer. 5:572–589. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Gu Y, Wang Z and Wang Y: Bispecific
antibody drug conjugates: Making 1+1>2. Acta Pharm Sin B.
14:1965–1986. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Zhang Y, Du J, Cui X, Ling Y and Tang C:
Development of a bispecific CDH17-GUCY2C ADC bearing the
ferroptosis inducer RSL3 for the treatment of colorectal cancer.
Cell Death Discov. 11:3472025. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Ouladan S and Orouji E: Chimeric antigen
receptor-T cells in colorectal cancer: Pioneering new avenues in
solid tumor immunotherapy. J Clin Oncol. 43:994–1005. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Li D, Zhang W, Wang R, Xie S, Wang Y, Guo
W, Huang Z, Lu C, Shan L, Liu H, et al: ROR1 CAR-T cells and
ferroptosis inducers orchestrate tumor ferroptosis via PC-PUFA2.
Biomark Res. 13:172025. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Chen S, Fan J, Xie P, Ahn J, Fernandez M,
Billingham LK, Miska J, Wu JD, Wainwright DA, Fang D, et al: CD8+ T
cells sustain antitumor response by mediating crosstalk between
adenosine A2A receptor and glutathione/GPX4. J Clin Invest.
134:e1700712024. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Wong CC and Yu J: Gut microbiota in
colorectal cancer development and therapy. Nat Rev Clin Oncol.
20:429–452. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Chen Y, Zhang Y, Dai M, Qiu C, Sun Q, Fan
T, Guo Y, Zhao L and Jiang Y: γ-Linolenic acid derived from
Lactobacillus plantarum MM89 induces ferroptosis in colorectal
cancer. Food Funct. 16:1760–1771. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Li B, Wei Z, Wang Z, Xu F, Yang J, Lin B,
Chen Y, Wenren H, Wu L, Guo X, et al: Fusobacterium nucleatum
induces oxaliplatin resistance by inhibiting ferroptosis through
E-cadherin/β-catenin/GPX4 axis in colorectal cancer. Free Radical
Bio Med. 220:125–138. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Liu R, Wang J, Liu Y, Gao Y and Yang R:
Regulation of gut microbiota on immune cell ferroptosis: A novel
insight for immunotherapy against tumor. Cancer Lett.
598:2171152024. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Cui W, Guo M, Liu D, Xiao P, Yang C, Huang
H, Liang C, Yang Y, Fu X, Zhang Y, et al: Gut microbial metabolite
facilitates colorectal cancer development via ferroptosis
inhibition. Nat Cell Biol. 26:124–137. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Zhou J, Lu P, He H, Zhang R, Yang D, Liu
Q, Liu Q, Liu M and Zhang G: The metabolites of gut microbiota:
Their role in ferroptosis in inflammatory bowel disease. Eur J Med
Res. 30:2482025. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Zhou H, Zhuang Y, Liang Y, Chen H, Qiu W,
Xu H and Zhou H: Curcumin exerts anti-tumor activity in colorectal
cancer via gut microbiota-mediated CD8+ T Cell tumor infiltration
and ferroptosis. Food Funct. 16:3671–3693. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
O'Keefe SJ: Diet, microorganisms and their
metabolites, and colon cancer. Nat Rev Gastroenterol Hepatol.
13:691–706. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Lin Y, Zhang Y, Huang T, Chen J, Li G,
Zhang B, Xu L, Wang K, He H, Chen H, et al: Arginine deprivation
induces quiescence and confers vulnerability to ferroptosis in
colorectal cancer. Cancer Res. 85:1663–1679. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
He Y, Ling Y, Zhang Z, Mertens RT, Cao Q,
Xu X, Guo K, Shi Q, Zhang X, Huo L, et al: Butyrate reverses
ferroptosis resistance in colorectal cancer by inducing
c-Fos-dependent xCT suppression. Redox Biol. 65:1028222023.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Zhou X, Wang G, Wu Y, Wu M, Zhai X, Tian
C, Prochownik EV, Jiang C and Li Y: SLC6A8-mediated creatine uptake
suppresses ERK2-FSP1 signaling and induces ferroptosis in
colorectal cancer. Cell Rep. 44:1161392025. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Liu Q, Zhao Y, Zhou H and Chen C:
Ferroptosis: Challenges and opportunities for nanomaterials in
cancer therapy. Regen Biomater. 10:rbad0042023. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Zhang Z, Ji Y, Hu N, Yu Q, Zhang X, Li J,
Wu F, Xu H, Tang Q and Li X: Ferroptosis-induced anticancer effect
of resveratrol with a biomimetic nano-delivery system in colorectal
cancer treatment. Asian J Pharm Sci. 17:751–766. 2022.PubMed/NCBI
|
|
156
|
Wang H, Qiao C, Guan Q, Wei M and Li Z:
Nanoparticle-mediated synergistic anticancer effect of ferroptosis
and photodynamic therapy: Novel insights and perspectives. Asian J
Pharm Sci. 18:1008292023.PubMed/NCBI
|
|
157
|
Luo J, Xu L, Feng J, Xu K, Tian P, Bai X,
Xu S, Wen L, Lu C and Song J: Tumor microenvironment-activated and
ROS-Augmented nanoplatform amplified PDT against colorectal cancer
through impairing GPX4 to induce ferroptosis. ACS Appl Mater
Interfaces. 17:41586–41596. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
He M, Du C, Xia J, Zhang ZG and Dong CM:
Multivalent polypeptide and tannic acid cooperatively
iron-coordinated nanohybrids for synergistic cancer photothermal
ferroptosis therapy. Biomacromolecules. 23:2655–2666. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Deng K, Tian H, Zhang T, Gao Y, Nice EC,
Huang C, Xie N, Ye G and Zhou Y: Chemo-photothermal nanoplatform
with diselenide as the key for ferroptosis in colorectal cancer. J
Control Release. 366:684–693. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Yang H, Yao X, Liu Y, Shen X, Li M and Luo
Z: Ferroptosis nanomedicine: Clinical challenges and opportunities
for modulating tumor metabolic and immunological landscape. ACS
Nano. 17:15328–15353. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Li Y, Duan Y, Li Y, Gu Y, Zhou L, Xiao Z,
Yu X, Cai Y, Cheng E, Liu Q, et al: Cascade loop of ferroptosis
induction and immunotherapy based on metal-phenolic networks for
combined therapy of colorectal cancer. Exploration (Beijing).
5:202301172024. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
McCoubrey LE, Favaron A, Awad A, Orlu M,
Gaisford S and Basit AW: Colonic drug delivery: Formulating the
next generation of colon-targeted therapeutics. J Control Release.
353:1107–1126. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Ye C, Mi M, Shi S, Qi L, Weng S, Wang L,
Lu Y, Chen C, Tan Y, Yang M, et al: ROS-Responsive hydrogel for
localized delivery of nampt and Stat3 inhibitors exhibits
synergistic antitumor effects in colorectal cancer through
ferroptosis induction and immune microenvironment remodeling. Adv
Sci (Weinh). 12:e065992025. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Huang M, Wu Y, Wei X, Cheng L, Fu L, Yan
H, Wei W, Li B, Ru H, Mo X, et al: Trifluridine/tipiracil induces
ferroptosis by targeting p53 via the p53-SLC7A11 axis in colorectal
cancer 3D organoids. Cell Death Dis. 16:2552025. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Wang Y, Qi D, Ge G, Cao N, Liu X, Zhu N,
Li F, Huang X, Yu K, Zheng J, et al: WBP1 regulates mitochondrial
function and ferroptosis to modulate chemoresistance in colorectal
cancer. Mol Med. 31:932025. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Qian LH, Wen KL, Guo Y, Liao YN, Li MY, Li
ZQ, Li SX and Nie HZ: Nutrient deficiency-induced downregulation of
SNX1 inhibits ferroptosis through PPARs-ACSL1/4 axis in colorectal
cancer. Apoptosis. 30:1391–1409. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Chen C, Yang Y, Guo Y, He J, Chen Z, Qiu
S, Zhang Y, Ding H, Pan J and Pan Y: CYP1B1 inhibits ferroptosis
and induces anti-PD-1 resistance by degrading ACSL4 in colorectal
cancer. Cell Death Dis. 14:2712023. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Chen S, Ma J, Tang J, Yang Y, Zhou S and
Feng P: Research progress of macrophage ferroptosis in inflammatory
bowel disease and inflammation-cancer transformation. Front
Immunol. 16:16582802025. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Zhang Y, Zhang Y, Yao S, Chen L, Dong Q,
Luo R, Zheng K, Liu J, Liu Y, Chen Y, et al: Inflammatory bowel
disease induces colorectal cancer: Risk factors, triggering
mechanisms, and treatment with phyto-derivatives. Phytother Res.
39:3386–3418. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Iacucci M, Nardone OM, Ditonno I,
Capobianco I, Pugliano CL, Maeda Y, Majumder S, Zammarchi I,
Santacroce G and Ghosh S: Advancing inflammatory bowel
disease-driven colorectal cancer management: Molecular insights and
endoscopic breakthroughs towards precision medicine. Clin
Gastroenterol Hepatol. 23:2361–2373. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Burgos-Molina AM, Téllez Santana T,
Redondo M and Bravo Romero MJ: The Crucial role of inflammation and
the immune system in colorectal cancer carcinogenesis: A
comprehensive perspective. Int J Mol Sci. 25:61882024. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Liu Y, Zhang JT, Sun M, Song J, Sun HM,
Wang MY, Wang CM and Liu W: Targeting ferroptosis in the treatment
of ulcerative colitis by traditional Chinese medicine: A novel
therapeutic strategies. Phytomedicine. 139:1565392025. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Zhang K, Guo J, Yan W and Xu L: Macrophage
polarization in inflammatory bowel disease. Cell Commun Signal.
21:3672023. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Liang J, Wang N, Yao Y, Wang Y, An X, Wang
H, Liu H, Jiang Y, Li H, Cheng X, et al: NEDD4L mediates intestinal
epithelial cell ferroptosis to restrict inflammatory bowel diseases
and colorectal tumorigenesis. J Clin Invest. 135:e1739942024.
View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Na YR, Stakenborg M, Seok SH and Matteoli
G: Macrophages in intestinal inflammation and resolution: A
potential therapeutic target in IBD. Nat Rev Gastroenterol Hepatol.
16:531–543. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Meng EX, Verne GN and Zhou Q: Macrophages
and gut barrier function: Guardians of gastrointestinal health in
post-inflammatory and post-infection responses. Int J Mol Sci.
25:94222024. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Tang B, Zhu J, Fang S, Wang Y, Vinothkumar
R, Li M, Weng Q, Zheng L, Yang Y, Qiu R, et al: Pharmacological
inhibition of MELK restricts ferroptosis and the inflammatory
response in colitis and colitis-propelled carcinogenesis. Free
Radic Biol Med. 172:312–329. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Kuang Z, Su Q, Liang W, Shen K and Xie J:
PRSS22 inhibits HMOX1-mediated ferroptosis and induces osteopontin
cleavage to promote M2 macrophage polarization and
colitis-associated carcinogenesis. Oncogene. 45:1275–1286. 2026.
View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Wang S, Zhu L, Wang Y, Han Y, Wang Q, Yang
W, Zhao L, Zhang Y, Pei D, Huang W, et al: ILF3 promotes colorectal
cancer cell resistance to ferroptosis by enhancing cysteine uptake
and GSH synthesis via stabilizing SLC3A2 mRNA. Cell Death Dis.
16:5492025. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Xiang Y, Zheng J, Zhao X, Zhou L, Yan Q,
Ma Y, Zhou Y, Jiang P, Fang Y, Li W, et al: Sodium butyrate
enhances sorafenib-induced ferroptosis and immunogenic cell death
by modulating IRF2-Oasl2-cGAS pathway in colorectal cancer. Mater
Today Bio. 35:1024982025. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Wang L, Wang J and Chen L: TIMP1 represses
sorafenib-triggered ferroptosis in colorectal cancer cells by
activating the PI3K/Akt signaling pathway. Immunopharmacol
Immunotoxicol. 45:419–425. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Wang R, Xing R, Su Q, Yin H, Di Wu, Lv C
and Yan Z: Knockdown of SFRS9 inhibits progression of colorectal
cancer through triggering ferroptosis mediated by GPX4 Reduction.
Front Oncol. 11:6835892021. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Jacobs SA, Lee JJ, George TJ, Wade JL III,
Stella PJ, Wang D, Sama AR, Piette F, Pogue-Geile KL, Kim RS, et
al: Neratinib-plus-cetuximab in quadruple-WT (KRAS, NRAS, BRAF,
PIK3CA) metastatic colorectal cancer resistant to cetuximab or
panitumumab: NSABP FC-7, A phase Ib study. Clin Cancer Res.
27:1612–1622. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Ren M, Jian Z, Li C, Cao J, Ma X, He L,
Zhang Y, Chen Y and Xu Y: Design, synthesis, and in vitro/in vivo
biological evaluation of Artesunate-Ebselen derivatives:
GPX4-targeted ferroptosis induction and synergistic antitumor
immune activation in colorectal cancer. Bioorg Chem.
174:1097552026. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Chen HC, Chen CY, Wang PY, Su PY, Tsai SP,
Hsu CP, Liu HS, Huang CF, Cheng WH, Lee MF and Su CL: Using
integrated bioinformatics analysis to identify saponin formosanin C
as a ferroptosis inducer in colorectal cancer with p53 and
oncogenic KRAS. Antioxidants (Basel). 14:10272025. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Niu X, Wang M, Wang M, Liu X, Zhang Y,
Zheng P, Zhang S, Liu T, Cao Z and Zhang C: Dracorhodin
perochlorate sensitizes colorectal cancer to ferroptosis by
activating HMOX1 and inhibiting the SLC7A11/GPX4 axis. Int
Immunopharmacol. 158:1148272025. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Zeng L, Zhao W, Han T, Qing F, He Z, Zhao
Q, Luo A, Hu P, Ding X and Zhang Z: Ropivacaine prompts ferroptosis
to enhance the cisplatin-sensitivity of human colorectal cancer
through SIRT1/Nrf2 signaling pathway. Chem Biol Interact.
400:1111632024. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Huang JY, Hsu TW, Chen YR and Kao SH:
Rosmarinic acid potentiates cytotoxicity of cisplatin against
colorectal cancer cells by enhancing apoptotic and ferroptosis.
Life (Basel). 14:10172024.PubMed/NCBI
|
|
189
|
Zeng X, Sun L, Ling X, Jiang Y, Shen J,
Liang L and Zhang X: Comprehensive analysis identifies novel
targets of gemcitabine to improve chemotherapy treatment strategies
for colorectal cancer. Front Endocrinol (Lausanne). 14:11705262023.
View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Zhang H, Song X, Zhao L, Wang C, Chen S,
Wang Y, Zhang L, Xu F, Zhang H, Wang Q and Luo C: Singlet
oxygen-precharged injectable hydrogel triggers ferroptosis and
lactate metabolism reprogramming for enhanced colorectal cancer
immunotherapy. J Nanobiotechnol. 24:1862026. View Article : Google Scholar
|
|
191
|
Xie P, Yang ST, Huang Y, Zeng C, Xin Q,
Zeng G, Yang S, Xia P, Tang X and Tang K: Carbon
nanoparticles-Fe(II) complex for efficient tumor inhibition with
low toxicity by amplifying oxidative stress. ACS Appl Mater
Interfaces. 12:29094–29102. 2020.PubMed/NCBI
|
|
192
|
Rochette L, Dogon G, Rigal E, Zeller M,
Cottin Y and Vergely C: Lipid peroxidation and iron metabolism: Two
corner stones in the homeostasis control of ferroptosis. Int J Mol
Sci. 24:4492022. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Chen X, Comish PB, Tang D and Kang R:
Characteristics and biomarkers of ferroptosis. Front Cell Dev Biol.
9:6371622021. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Matsuoka Y, Katsumata Y, Chu PS, Morikawa
R, Nakamoto N, Iguchi K, Takahashi K, Kou T, Ito R, Taura K, et al:
Monitoring ferroptosis in vivo: Iron-driven volatile oxidized
lipids as breath biomarkers. Redox Biol. 86:1038582025. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Zhao YP, Liu JL, Wang S and Li X: Role of
non-coding RNA-regulated ferroptosis in colorectal cancer. Cell
Death Discov. 11:3152025. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Ge X, Xu J, He J, Wang J and Qian Y:
Identification and functional characterization of prognosis-related
ferroptosis-associated lncRNAs in colorectal cancer. Front Immunol.
16:15612102025. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Chu X, Li X, Zhang Y, Dang G, Miao Y, Xu
W, Wang J, Zhang Z and Cheng S: Integrative single-cell analysis of
human colorectal cancer reveals patient stratification with
distinct immune evasion mechanisms. Nat Cancer. 5:1409–1426. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Wang Y, Wu N, Li J, Liang J, Zhou D, Cao
Q, Li X and Jiang N: The interplay between autophagy and
ferroptosis presents a novel conceptual therapeutic framework for
neuroendocrine prostate cancer. Pharmacol Res. 203:1071622024.
View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Hu Q, Wei W, Wu D, Huang F, Li M, Li W,
Yin J, Peng Y, Lu Y, Zhao Q and Liu L: Blockade of GCH1/BH4 axis
activates ferritinophagy to mitigate the resistance of colorectal
cancer to erastin-induced ferroptosis. Front Cell Dev Biol.
10:8103272022. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Ubellacker JM and Dixon SJ: Prospects for
ferroptosis therapies in cancer. Nat Cancer. 6:1326–1336. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Yan YY, Zhou HM, Shang XG, Xu P, Tang XQ,
Zheng XY, Yu LH, Li CT, Xie T and Lou JS: Dynamic regulators of
ferroptosis: Post-translational modifications in ferroptotic cell
death. Pharmacol Res. 217:1078152025. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Feng S, Rao Z, Zhang J, She X, Chen Y, Wan
K, Li H, Zhao C, Feng Y, Wang G, et al: Inhibition of
CARM1-Mediated methylation of ACSL4 promotes ferroptosis in
colorectal cancer. Adv Sci (Weinh). 10:e23034842023. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Zhang S, Xiao Y, Xie Z, Liu S, Li J, He P,
Lai Y, Yin Y, Lin H, Pan H and Fang C: Jianpi Huayu Jiedu Decoction
prevents the progression of H. pylori and MNU-induced gastric
precancerous lesions by inhibiting ferroptosis and remodeling
linoleic acid metabolism. J Ethnopharmacol. 352:1202172025.
View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Luo G, Jiang X, Hu C, Li L, Yan L, Xiao G,
Duo Y and Zhang X: Artificial intelligence-powered nanomedicine.
Chem Soc Rev. 55:2070–2119. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Hou J, Karin M and Sun B: Targeting
cancer-promoting inflammation - have anti-inflammatory therapies
come of age? Nat Rev Clin Oncol. 18:261–279. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Escuder-Rodríguez JJ, Liang D, Jiang X and
Sinicrope FA: Ferroptosis: Biology and role in gastrointestinal
disease. Gastroenterology. 167:231–249. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Zhu J, Zhang J, Lou Y, Zheng Y, Zheng X,
Cen W, Ye L and Zhang Q: Developing a machine learning-based
prognosis and immunotherapeutic response signature in colorectal
cancer: insights from ferroptosis, fatty acid dynamics, and the
tumor microenvironment. Front Immunol. 15:14164432024. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Wei X, Jiang Y, Chenwu F, Li Z, Wan J, Li
Z, Zhang L, Wang J and Song M: Synergistic
ferroptosis-immunotherapy nanoplatforms: Multidimensional
engineering for tumor microenvironment remodeling and therapeutic
optimization. Nanomicro Lett. 18:562025.PubMed/NCBI
|














