Introduction
Pancreatic cancer ranks as the 12th most common
malignancy and the seventh leading cause of cancer-related deaths
worldwide (1). Its high mortality
has earned it the designation ‘king of cancers’. Due to the absence
of early symptoms and therapy resistance, the 5-year survival rate
for pancreatic cancer remains only ~10%, with a median overall
survival of 9.3 months (2).
Notably, the incidence of pancreatic cancer has increased annually.
It is estimated that pancreatic cancer will become the second
leading cause of cancer death by 2030 in the USA (3). In China, the incidence has risen
sharply from 26.77 thousand cases in 1990 to 114.96 thousand in
2019, and is projected to reach 218.79 thousand by 2030,
representing an approximate two-fold increase (4). Currently, chemotherapy is the most
frequently used approach for most patients with pancreatic cancer,
owing to the frequent presentation of metastatic or unresectable
disease at diagnosis (5). Other
therapeutic approaches including surgery, radiotherapy,
immunotherapy, and targeted therapy, are employed depending on the
cancer stage (6). However, the
effectiveness of all available treatments remains unsatisfactory
(7). Hence, there is an urgent need
to discover novel targets or strategies for pancreatic cancer
treatment.
Acylglycerol kinase (AGK) is a lipid kinase that
catalyzes the phosphorylation of acylglycerol to produce
lysophosphatidic acid (8). Growing
evidence indicates that AGK acts as an oncogene, being upregulated
in various cancers and promoting tumor progression (9). Zhu et al (10) found that AGK upregulation in
nasopharyngeal carcinoma patients is associated with lymph node
metastasis and poor prognosis. In cervical squamous cell carcinoma
cells, high AGK expression enhances epithelial-mesenchymal
transition by inducing hypoxia-inducible factor 1 α expression
(11). AGK also activates the
PI3K/AKT/glycogen synthase kinase-3 beta (GSK3β) signaling pathway,
thereby promoting tumor growth and metastasis in renal cell
carcinoma (12). Overexpression of
AGK leads to constitutive activation of JAK2/STAT3 signaling and
promotes the proliferation of esophageal squamous cell carcinoma
(13). In addition, plasma
membrane-localized AGK has been reported to suppress phosphatase
and tensin homolog (PTEN) phosphorylation, enhancing antitumor
immunity by promoting CD8+ T cell proliferation (14). However, the expression and
biological functions of AGK in pancreatic cancer remain
unclear.
The present study demonstrated that AGK is markedly
upregulated in pancreatic cancer and correlates with shorter
overall survival. Furthermore, it showed that AGK upregulates the
expression of proliferation-related genes, such as MKI67 and
CCNB1, resulting in accelerated cell proliferation.
Importantly, the results indicated that AGK activates the NF-κB
signaling pathway by promoting the phosphorylation of NF-κB p65.
Moreover, AGK upregulation confers resistance to both
chemotherapeutic agents and radiation in human pancreatic cancer
cells. These findings provide new insights into the functions of
AGK in pancreatic cancer and suggest that targeting AGK may
represent a novel therapeutic or diagnostic strategy.
Materials and methods
Cells, tissues and chemicals
The normal human pancreatic ductal epithelial cell
line HPDE6-C7 and four pancreatic cancer cell lines (AsPC-1,
BxPC-3, Capan-2 and PANC-1) were purchased from the American Type
Culture Collection. All cells were cultured in RPMI-1640 medium
(HyClone™; Cytiva) supplemented with 10% fetal bovine serum (FBS;
BioChannel Biological Technology Co., Ltd.), 100 U/ml penicillin,
and 100 µg/ml streptomycin, and maintained at 37°C in a humidified
incubator with 5% CO2. A total of 16 paired pancreatic cancer and
matched adjacent non-cancerous tissue samples (collected ≤2 cm away
from the tumor margin) were collected from patients diagnosed with
pancreatic cancer between January 2025 and June 2025. Of the 16
enrolled patients, nine were male and seven were female, with a
median age of 62 years (age range: 45–78 years). This study was
approved by the Institutional Ethics Committee of Wannan Medical
College (approval no. 245), and written informed consent was
obtained from all participants. Nab-paclitaxel, gemcitabine, and
NF-κB inhibitor
(N4-[2-(4-phenoxyphenyl)ethyl]-1,2-dihydroquinazoline-4,6-diamine,
EVP4593 (QNZ) were purchased from Selleck Chemicals.
Bioinformatics analyses
AGK expression in pancreatic cancer was analyzed
using Gene Expression Profiling Interactive Analysis 2 (GEPIA2), an
online tumor database (version 2.0; http://gepia2.cancer-pku.cn/) that integrates The
Cancer Genome Atlas (TCGA) tumor, TCGA normal and Genotype-Tissue
Expression (GTEx) normal tissue data (http://gepia2.cancer-pku.cn/#analysis) and all
analyses were performed with default parameters. Overall survival
analysis of AGK in pancreatic cancer was also conducted via GEPIA2
(http://gepia2.cancer-pku.cn/#survival) using default
settings. To further assess the prognostic significance of AGK
across tumor stages, a stratified survival analysis was conducted
using the Kaplan-Meier Plotter online tool (https://kmplot.com/analysis/), utilizing integrated
TCGA and Gene Expression Omnibus (GEO) data. Pancreatic cancer
cases were categorized into early-stage (I+II) and advanced-stage
(III+IV) groups based on AJCC criteria. Within each stage subgroup,
patients were dichotomized into high- and low-AGK expression groups
using the median expression value. Kaplan-Meier curves were
plotted, and overall survival differences were evaluated with the
log-rank test.
Correlation analysis between AGK expression and
proliferation-related gene expression in pancreatic cancer was
performed using the GEPIA2 database. For this analysis, the
analysis was restricted to all pancreatic cancer tumor samples
included in the GEPIA2 database, and the expression level of AGK
matched with that of four target proliferation-associated genes
(MKI67, CCNB1, CCND1 and MYC). Pearson correlation coefficient was
calculated to evaluate the association between gene expression
levels, and the visualization results generated by the database
were exported for the present study.
Reverse transcription-quantitative
(RT-q) PCR
Total RNA was extracted from approximately 1×106
cells per sample using RNAiso Plus (Takara Biotechnology Co., Ltd.)
according to the manufacturer's protocol. First-strand cDNA was
synthesized from 1 µg of total RNA using the PrimeScript RT reagent
Kit (Takara Biotechnology Co., Ltd.) strictly following the
manufacturer's instructions. qPCR was performed with the TB Green
Premix Ex Taq II kit (Takara Biotechnology Co., Ltd.) on a
LightCycler 480 real-time PCR system (Roche Diagnostics) in
accordance with the manufacturer's protocol. The PCR cycling
conditions were as follows: initial denaturation at 95°C for 30
sec, followed by 40 cycles of denaturation at 95°C for 5 sec and
annealing/extension at 60°C for 30 sec. The sequences of primers
used for qPCR are listed in Table
I. Relative mRNA levels were calculated using the
2−ΔΔCq method (15), and
data were normalized to the internal reference gene GAPDH
(16). All qPCR experiments were
performed in at least three independent biological replicates.
 | Table I.Primers for reverse
transcription-quantitative PCR |
Table I.
Primers for reverse
transcription-quantitative PCR
| Target gene | Primer | Sequence
(5′-3′) |
|---|
| AGK | Forward |
CTTGACAGGCTGCTCTCCTT |
|
| Reverse |
GGAAGAAAACTACAGCTGGGC |
| MYC | Forward |
GGCTCCTGGCAAAAGGTCA |
|
| Reverse |
CTGCGTAGTTGTGCTGATGT |
| MKI67 | Forward |
CTTTGGGTGCGACTTGACG |
|
| Reverse |
GTCGACCCCGCTCCTTTT |
| CCNB1 | Forward |
AATAAGGCGAAGATCAACATGGC |
|
| Reverse |
TTTGTTACCAATGTCCCCAAGAG |
| GAPDH | Forward |
GCACCGTCAAGGCTGAGAAC |
|
| Reverse |
TGGTGAAGACGCCAGTGGA |
Plasmids, siRNAs and transfection
The full-length coding sequence of human AGK was
amplified by PCR, and cloned into the pcDNA3.1 vector to generate
an N-terminal Flag-tagged AGK overexpression plasmid. For
transfection performed in standard 6-well culture plates, 2 µg of
constructed plasmid (per well) was used for overexpression
experiments, and the final working concentration of siRNA was 50 nM
(per well) for knockdown experiments. Negative control siRNA
(si-NC) and AGK-targeting siRNA (si-AGK) were synthesized by Sangon
Biotech Co., Ltd.. Plasmids were transfected using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) and siRNAs were transfected with
Lipofectamine® RNAiMAX (Invitrogen; Thermo Fisher
Scientific, Inc.), following the manufacturers' protocols. All
transfection reactions were incubated at 37°C in a humidified 5%
CO2 incubator for 4–6 h, before transfection medium was replaced
with fresh complete culture medium. Subsequent downstream
experiments were carried out 48 h after siRNA transfection, and
24–48 h after plasmid transfection. The siRNA sequences are as
follows: si-AGK: 5′-AACAGATGAGGCTACCTTCAG-3′; si-NC:
5′-TTCTCCGAACGTGTCACGT-3′.
Nuclear and cytoplasmic
fractionation
Subcellular fractionation was performed using the
Beyotime Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime
Biotechnology) according to the manufacturer's instructions.
Briefly, harvested cells were washed with ice-cold
phosphate-buffered saline (PBS) and lysed in hypotonic Reagent A
supplemented with protease inhibitors. Following the addition of
Reagent B, cytoplasmic proteins were isolated in the supernatant
after centrifugation (15,000 × g, 5 min, 4°C). The nuclear pellet
was subsequently lysed in high-salt Reagent C (30 min on ice with
intermittent vortexing) and clarified by centrifugation (15,000 ×
g, 10 min, 4°C) to obtain the nuclear fraction. Fraction purity was
verified by western blotting with antibodies against GAPDH (a
cytoplasmic marker) and histone H3 (a nuclear marker).
Western blotting
Western blotting was performed as previously
described (16,17). Total protein was extracted using
ice-cold RIPA lysis buffer (Beyotime Biotechnology) supplemented
with 1% (v/v) 100 mM phenylmethylsulfonyl fluoride and 1% (v/v)
phosphatase inhibitor cocktail (MedChemExpress). Protein
concentration was determined with the bicinchoninic acid protein
assay kit (Thermo Fisher Scientific, Inc.) following the
manufacturer's instructions. Equal amounts of protein (30 µg per
lane) were separated by 10% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis, then transferred onto 0.4 µm polyvinylidene
difluoride membranes (MilliporeSigma). Membranes were blocked with
5% (w/v) non-fat dry milk (Bio-Rad Laboratories, Inc.) in
Tris-buffered saline with 0.1% Tween 20 (TBST) for 1 h at room
temperature. All primary antibodies were diluted and incubated
overnight at 4°C: Anti-Flag (1:1,000; Medical & Biological
Laboratories Co., Ltd.), anti-GAPDH (1:5,000; Proteintech Group,
Inc.), anti-IκBα (1:1,000; Proteintech Group, Inc.), anti-IKKα/β
(1:1,000; MedChemExpress), anti-histone H3 (1:1,000;
MedChemExpress), anti-AGK (1:1,000; Santa Cruz Biotechnology,
Inc.), anti-phospho-p65 (1:1,000; Cell Signaling Technology, Inc.),
anti-phospho-IKKα/β (1:1,000; Cell Signaling Technology, Inc.),
anti-phospho-IκBα (1:1,000; Cell Signaling Technology, Inc.),
anti-p65 (1:1,000; Cell Signaling Technology, Inc.). After washing,
membranes were incubated with HRP-conjugated secondary antibodies
(goat anti-rabbit IgG, 1:5,000; goat anti-mouse IgG, 1:5,000;
Beyotime Biotechnology) for 1 h at room temperature. Protein bands
were visualized using enhanced chemiluminescence visualization
reagent (MilliporeSigma).
Cell growth and viability assay
Cell viability was assessed using the Cell Counting
Kit-8 (CCK-8; Selleck Chemicals), as described previously (18). In brief, cells were seeded in
96-well plates at a density of 2,000 cells per well and cultured at
37°C in a 5% CO2 humidified incubator for 1, 2, and 4 days. Then,
10 µl of CCK-8 reagent was added to each well and incubated for 2 h
at 37°C. Absorbance was measured at 450 nm using a SpectraMax i3
microplate reader (Molecular Devices, LLC.).
Wound healing assay
Wound healing assays were performed as previously
described (19). Briefly, cells in
the logarithmic growth phase were enzymatically detached and
resuspended at 2×106 cells/ml. A total of 2×106 cells (in 1 ml of
suspension) were seeded into each well of a 6-well plate. Then, 1
ml of complete medium (supplemented with 10% FBS) was added to each
well, and the cells were cultured until they reached ~90 confluence
(~18 h). Uniform wounds were created using 10 µl sterile pipette
tips, followed by three washes with PBS. After replacing the medium
with serum-free medium, wound images were captured immediately (T0)
and at 48 h (T48). The percentage of wound closure was calculated
using the formula: [(A0-A48)/A0] × 100%, where A represents the
wound area.
Transwell migration assay
Cell migration was assessed using Transwell chambers
as previously described (20).
Briefly, 2.5×104 cells in serum-free medium were seeded into the
upper chamber of a Transwell insert (8.0 µm pore size; Corning,
Inc.). The lower chamber was filled with medium containing 10% FBS
as a chemoattractant. After incubation for 24 h at 37°C,
non-migrated cells on the upper surface of the membrane were gently
wiped off with cotton swabs. Cells that had migrated to the lower
surface were fixed with 100% methanol for 15 min, stained with 0.1%
crystal violet for 20 min at room temperature (22–24°C), and rinsed
gently with PBS. The membranes were then imaged under
phase-contrast microscopy (Nikon Corporation). Migrated cells were
quantified by counting the number of cells in five randomly
selected fields per membrane at 200× magnification.
Colony formation assay
For assessment of clonogenic ability, transfected
cells (300 cells per well) were seeded in 6-well plates and
cultured in DMEM supplemented with 10% FBS at 37°C in a humidified
atmosphere containing 5% CO2. After 10 days, cells were
washed with ice-cold PBS, fixed in 4% paraformaldehyde for 30 min
at room temperature and stained with 0.1% crystal violet solution
for 30 min at room temperature (22–24°C). After gentle rinsing with
distilled water and air-drying, the number of colonies containing
≥50 cells was counted and representative images were captured.
For evaluation of radiosensitivity, PANC-1 cells
transfected with empty vector (EV) or Flag-AGK overexpression
plasmids were irradiated with X-rays at doses of 0, 2, 4 and 6 Gy.
Cells were then cultured at 37°C in a 5% CO2 humidified incubator
for 14 days to allow colony formation. Colonies were fixed,
stained, and counted as aforementioned. The surviving fraction at
each dose was calculated, and the survival fraction at 2 Gy was
recorded as SF2, a standard indicator of cellular radiosensitivity.
Cell survival curves were fitted using a single-hit multi-target
model in GraphPad Prism 8 (Dotmatics). The sensitization
enhancement ratio (SER) was calculated at the 37% survival fraction
(SER37).
Xenograft models
PANC-1 cells (3×106 cells per injection site) were
inoculated into the right flank of each female BALB/c nude mouse
aged 6–8 weeks. The total number of experimental animals was 10 (5
individuals per group; 2 groups in total), and the average body
weight of mice at inoculation was 18–22 g. All animals were
purchased from GemPharmatech Co., Ltd. Mice were housed in specific
pathogen-free facilities with the following standardized rearing
conditions: ambient temperature 20–24°C, relative humidity 40–60%,
12-h light/dark cycle, with free access to sterile food and water.
When tumors became palpable and reached a volume of ~50
mm3, mice were randomly divided into two groups (n=5 per
group) using a random number table, and intratumorally injected
with indicated siRNAs complexed with in vivo-jetPEI Delivery
Reagent (Polyplus-transfection Inc.) every 7 days. Tumor volume was
calculated using the standard formula: Volume=(length ×
width2)/2, and was measured every 3 days for 3
consecutive weeks. To minimize observation bias, tumor measurements
and assessments were performed by personnel blinded to group
assignments. On Day 21, mice were sacrificed and tumors were
excised for further analysis.
All experimental animals were sacrificed via carbon
dioxide (CO2) inhalation in accordance with the AVMA Guidelines for
the Euthanasia of Animals (2020 Edition; available at: http://www.avma.org/sites/default/files/2020-01/2020-Euthanasia-Final-1-17-20.pdf).
No additional chemical agents were administered to animals prior to
sacrifice. For CO2-induced euthanasia, the volume displacement rate
was set at 40% of the euthanasia chamber volume per minute.
Mortality was confirmed by the absence of spontaneous breathing,
loss of corneal reflex, and no response to toe pinch
stimulation.
Dual-luciferase reporter assay
An NF-κB-driven luciferase reporter plasmid was
purchased from Beyotime Biotechnology. PANC-1 and AsPC-1 cells were
co-transfected with the NF-κB luciferase reporter plasmid, the
internal control Renilla luciferase plasmid and the
indicated experimental plasmids or siRNAs using
Lipofectamine® 2000 Transfection Reagent (Thermo Fisher
Scientific, Inc.). After 48 h of incubation post-transfection,
luciferase activity was measured using the
Dual-Luciferase® Reporter Assay System (Promega
Corporation) according to the manufacturer's instructions. Firefly
luciferase activity was normalized to the Renilla luciferase
activity to calculate the relative NF-κB activity.
Statistical analysis
All experiments were performed in at least three
independent biological replicates. Data are presented as the mean ±
standard deviation (SD). Differences between two groups were
compared using Student's t-test and comparisons among three or more
groups were performed with one-way or two-way ANOVA followed by
Tukey's post hoc test for multiple comparisons. All plots and
graphs were generated using GraphPad Prism version 8.0.2
(Dotmatics).
Results
AGK is upregulated and correlates with
poor prognosis in pancreatic cancer
To investigate the role of AGK in pancreatic cancer,
the present study analyzed its expression levels in pancreatic
cancer tissues and normal tissues based on TCGA and GTEx data using
GEPIA2. AGK expression was markedly higher in tumor tissues
compared to normal tissues (Fig.
1A). Validation in clinical samples showed that AGK expression
was elevated in tumor tissues relative to matched adjacent
non-tumor tissues in 12 of 16 patients (Fig. 1B). Consistently, AGK expression was
also upregulated in various pancreatic cancer cell lines,
particularly in AsPC-1 and PANC-1 cells, relative to the normal
cell line HPDE6-C7 (Fig. 1C).
Survival analysis indicated that patients with high AGK expression
had markedly shorter overall survival (Fig. 1D). These findings demonstrated AGK
overexpression in pancreatic cancer and its potential as a
prognostic marker. To further investigate the clinical relevance of
AGK, the present study performed a stratified survival analysis
based on tumor stage (Fig. 1E). In
early-stage (stage I+II) disease, the prognostic correlation
between high AGK expression and overall survival did not reach
statistical significance. By contrast, in advanced-stage (stage
III+IV) patients, high AGK expression was markedly associated with
shorter overall survival. These results indicate that AGK plays a
more prominent role in driving the progression and aggressiveness
of advanced pancreatic cancer.
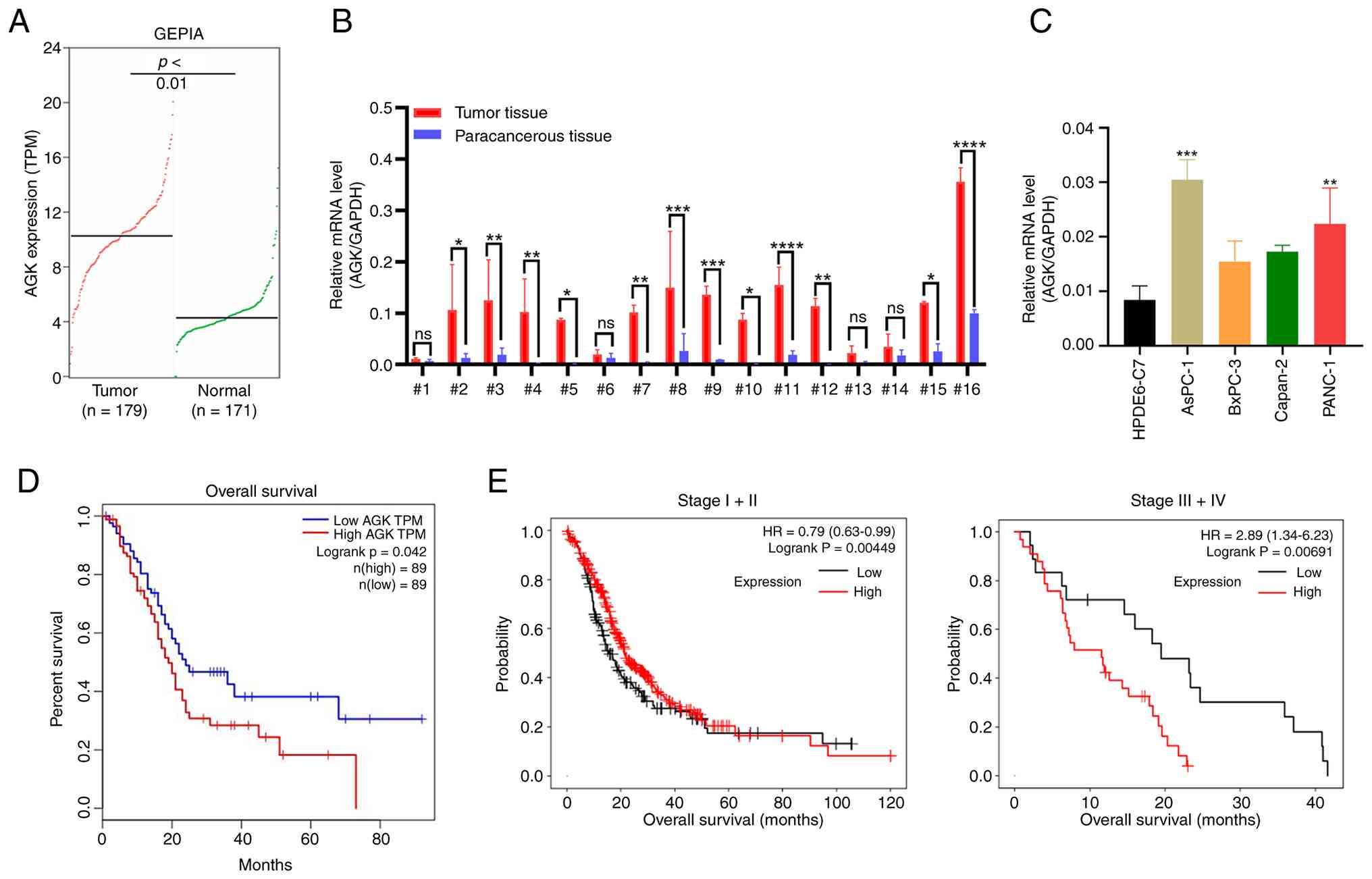 | Figure 1.AGK is upregulated in pancreatic
cancer and correlates with poor prognosis. (A) AGK expression in
PAAD and normal tissues was analyzed using the GEPIA database. (B)
AGK expression were detected in sixteen pairs of tumorous and
adjacent non-tumor tissues by qPCR, with GAPDH as an internal
control. (C) AGK expression was measured in a normal human
pancreatic ductal epithelial cell line (HPDE6-C7) and four
pancreatic cancer cell lines using qPCR. (D) Overall survival
analysis was performed using the GEPIA online database. (E)
Kaplan-Meier overall survival curves of pancreatic cancer patients
stratified by AGK expression level (low vs. high) and tumor stage.
Left panel: Early-stage (Stage I+II) disease; right panel:
Advanced-stage (Stage III+IV) disease. Data are mean ± SD,
*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. AGK,
acylglycerol kinase; PAAD, pancreatic adenocarcinoma; GEPIA, Gene
Expression Profiling Interactive Analysis; qPCR, quantitative PCR;
HR, hazard ratio. |
AGK promotes pancreatic cancer cell
proliferation
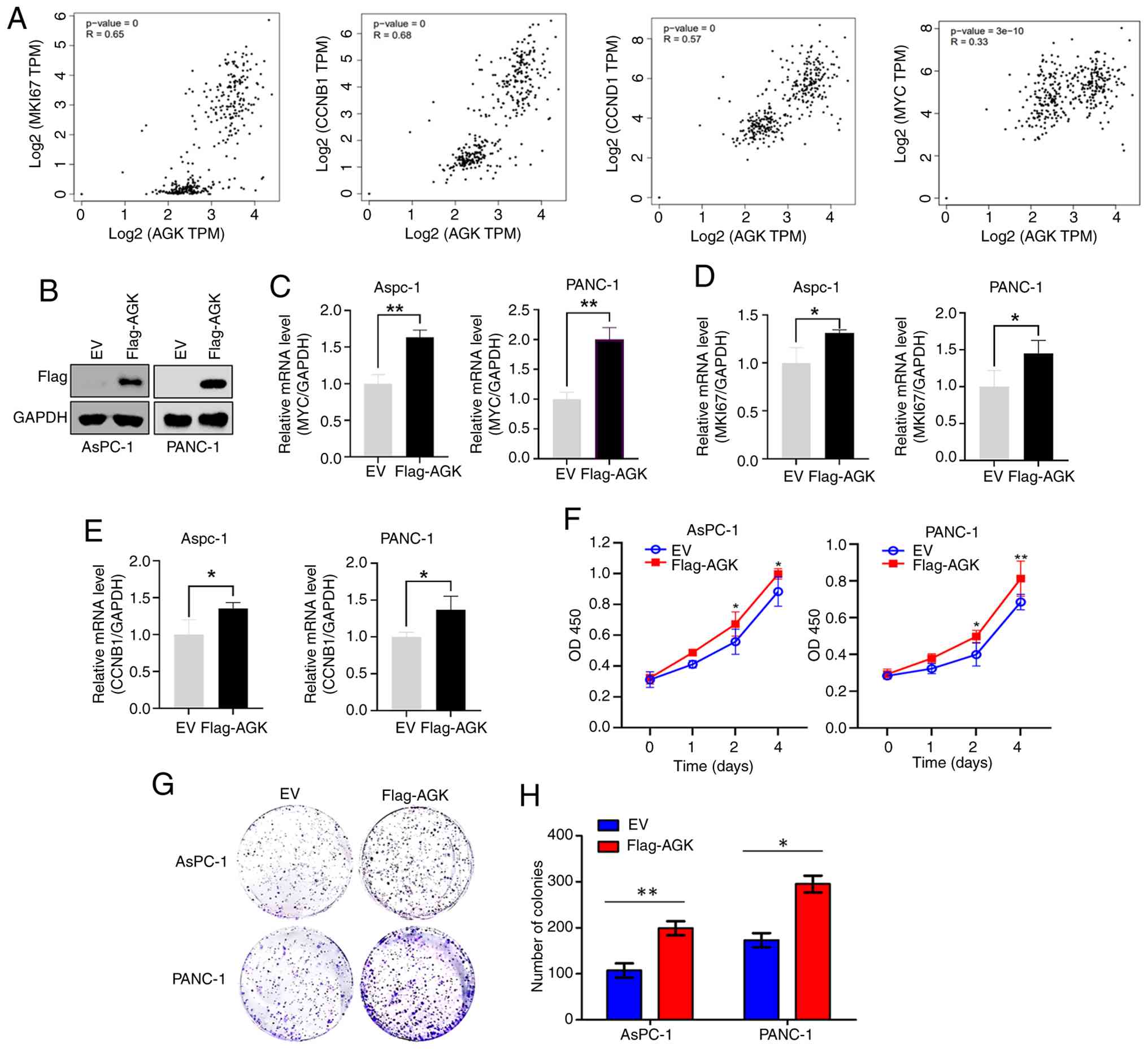
Using GEPIA database, the present study found a
strong positive correlation between AGK expression and that of
proliferation-associated genes, including MKI67, CCNB1,
CCND1 and MYC (Fig. 2A).
To further validate these findings, AsPC-1 and PANC-1 cells were
transfected with either an EV or Flag-tagged AGK-overexpressing
plasmids (Flag-AGK) (Fig. 2B). AGK
overexpression markedly increased the mRNA levels of MYC,
MKI67, and CCNB1 (Fig.
2C-E). Accordingly, CCK-8 assays demonstrated accelerated
proliferation in AGK-overexpressing cells (Fig. 2F). Furthermore, colony formation
assays confirmed an enhanced proliferative capacity following AGK
overexpression (Fig. 2G and H).
Moreover, AGK was knocked down in AsPC-1 and PANC-1 cells. The high
efficiency of knockdown was confirmed by qPCR (Fig. 3A) and western blotting (Fig. 3B). AGK knockdown markedly suppressed
cell proliferation, as evidenced by reduced cell viability
(Fig. 3C) and decreased colony
formation (Fig. 3D). Additionally,
AGK knockdown impaired cell migration in both wound healing and
Transwell assays (Fig. 3E and F).
These in vitro results demonstrate a key role for AGK in
pancreatic cancer cell proliferation and migration.
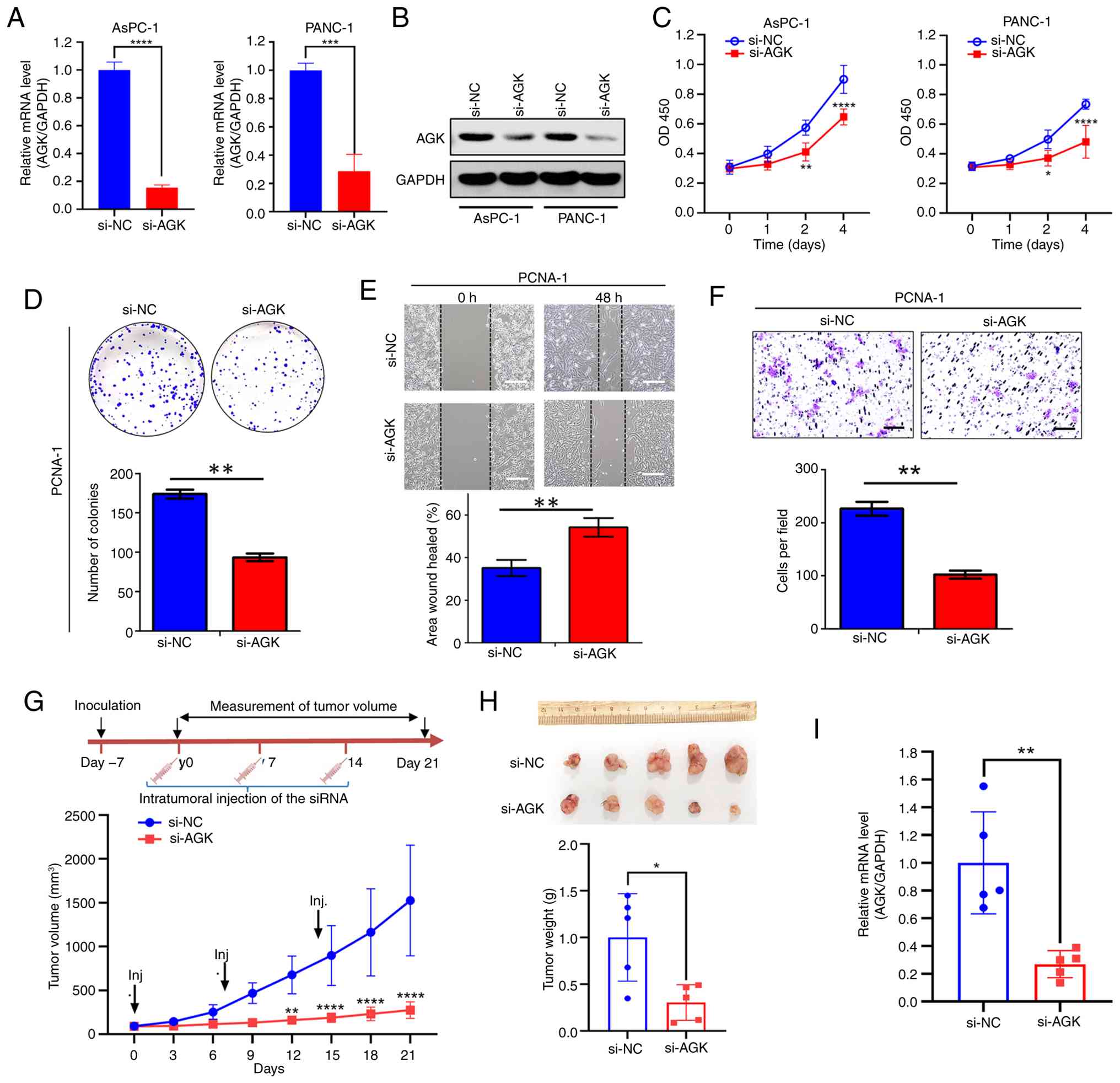 | Figure 3.Knockdown of AGK inhibits cell growth
of pancreatic cancer. AGK knockdown efficiency was validated in
AsPC-1 and PANC-1 cells transfected with control (si-NC) or
AGK-targeting (si-AGK) siRNAs by (A) qPCR and (B) western blot
analyses. (C) Cell viability was measured by CCK-8 assay in AsPC-1
and PANC-1 cells after AGK knockdown. (D) Colony formation assay of
PANC-1 cells transfected with si-NC or si-AGK. Upper:
representative crystal violet staining images; lower:
quantification of colony numbers. (E) Wound healing assay for
migration of PANC-1 cells with or without AGK knockdown. Upper:
Representative wound images at 0 h and 48 h; lower: Quantification
of wound healing percentage. Scale bars, 200 µm. (F) Transwell
invasion assay of PANC-1 cells transfected with si-NC or si-AGK.
Upper: Representative staining images of invaded cells; lower:
Quantification of invaded cells per field. Scale bars, 200 µm. (G)
In vivo tumor growth assay. PANC-1 ×enograft-bearing mice
were intratumorally injected with si-NC or si-AGK on days 0, 7, and
14 (arrows). Tumor volumes were measured every three days. (H)
Tumors were excised and weighed at the endpoint (day 21). (I) AGK
mRNA levels in harvested tumors were analyzed by qPCR. Data are
mean ± SD, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
AGK, acylglycerol kinase; si, short interfering; NC, negative
control; qPCR, quantitative PCR. |
The pro-proliferative role of AGK was further
validated in vivo using a xenograft mouse model. Tumor
growth was markedly suppressed from day 12 onward in mice treated
with AGK siRNA, compared to the control group (Fig. 3G). At the endpoint (Day 21), a
significant decrease in tumor volumes and weight was observed in
the si-AGK group (Fig. 3G and H).
AGK expression in excised tumors was confirmed to be lower in the
si-AGK group (Fig. 3I), supporting
the conclusion that AGK promotes tumor proliferation in
vivo.
AGK exerts its function by regulating
NF-κB signaling
To elucidate the mechanism underlying AGK-mediated
tumor promotion, the present study investigated its effect on the
NF-κB pathway, a key driver of pancreatic cancer proliferation
(21). NF-κB reporter assays in
AsPC-1 and PANC-1 cells showed that AGK overexpression markedly
increased luciferase activity (Fig.
4A), while AGK knockdown markedly decreased it (Fig. 4B), suggesting that AGK modulates-
the activation of the NF-κB pathway. Western blot analysis revealed
that AGK overexpression enhanced the phosphorylation of IKKα/β
(p-IKKα/β) and IκBα (p-IκBα), accompanied by reduced total IκBα
levels (Fig. 4C and D), consistent
with activation of the canonical NF-κB pathway. Subcellular
fractionation assays further revealed that AGK overexpression
enhanced p65 nuclear translocation, evidenced by markedly increased
nuclear p65 and decreased cytoplasmic p65 (Fig. 4E). Moreover, AGK overexpression
increased the phosphorylation of p65 without affecting total p65
levels (Fig. 4F and G), whereas AGK
knockdown blocked the phosphorylation of p65 (Fig. 4H and I). It is well known that
phosphorylation of p65 is a critical regulatory layer for
maximizing NF-κB-driven gene expression (22). Collectively, these data demonstrated
that AGK promotes p65 nuclear translocation and phosphorylation,
resulting in the activation of NF-κB pathway.
![AGK exerts its function by regulating
NF-κB signaling. AsPC-1 and PANC-1 cells harboring NF-κB-driven
luciferase reporter were transfected with (A) AGK-overexpressing
plasmids or (B) AGK-targeting siRNAs for 48 h, followed by
dual-luciferase assay. (C) Representative western blots showing
Flag-AGK, p-IKKα/β, total IKKα/β, p-IκBα, total IκBα, and GAPDH in
AsPC-1 (left) and PANC-1 (right) cells transfected with EV or
Flag-AGK. (D) Quantitative analysis of the normalized ratio of
phosphorylated IKKα/β (p-IKKα/β) to total IKKα/β from panel C. (E)
Representative western blots showing p65 subcellular localization
in cytoplasmic and nuclear fractions of cells transfected with EV
or Flag-AGK. GAPDH (cytosolic) and H3 (nuclear) served as loading
controls. (F) Representative western blotting results of p-p65 and
total p65 in AGK-overexpressing AsPC-1 and PANC-1 cells. (G)
Quantitative analysis of the normalized ratio of phosphorylated p65
(p-p65) to total p65 from panel F. (H) Representative western
blotting results of p-p65 and total p65 in AGK-knockdown AsPC-1 and
PANC-1 cells. (I) Quantitative analysis of the normalized ratio of
phosphorylated p65 (p-p65) to total p65 from panel H. (J) Western
blot showing that p65 knockdown (si-p65) abolishes AGK-induced p65
phosphorylation. (K) Quantitative analysis of the normalized ratio
of phosphorylated p65 (p-p65) to total p65 from panel J. (L) qPCR
analysis showing that AGK-mediated upregulation of MYC mRNA is
dependent on p65. (M) CCK-8 assay demonstrating that p65 knockdown
attenuated AGK-induced cell proliferation. (N) AsPC-1 and (O)
PANC-1 cells overexpressed with AGK were incubated with vehicle or
0.5 µM QNZ for 48 h, followed by CCK-8 assay. Data were presented
as mean ± SD. *P<0.05, **P<0.01, ***P<0.001,
****P<0.0001. AGK, acylglycerol kinase; p-, phosphorylated; EV,
empty vector; si, short interfering; NC, negative control; qPCR,
quantitative PCR; QNZ.,
N4-[2-(4-phenoxyphenyl)ethyl]-1,2-dihydroquinazoline-4,6-diamine.](/article_images/or/56/2/or-56-02-09145-g03.jpg) | Figure 4.AGK exerts its function by regulating
NF-κB signaling. AsPC-1 and PANC-1 cells harboring NF-κB-driven
luciferase reporter were transfected with (A) AGK-overexpressing
plasmids or (B) AGK-targeting siRNAs for 48 h, followed by
dual-luciferase assay. (C) Representative western blots showing
Flag-AGK, p-IKKα/β, total IKKα/β, p-IκBα, total IκBα, and GAPDH in
AsPC-1 (left) and PANC-1 (right) cells transfected with EV or
Flag-AGK. (D) Quantitative analysis of the normalized ratio of
phosphorylated IKKα/β (p-IKKα/β) to total IKKα/β from panel C. (E)
Representative western blots showing p65 subcellular localization
in cytoplasmic and nuclear fractions of cells transfected with EV
or Flag-AGK. GAPDH (cytosolic) and H3 (nuclear) served as loading
controls. (F) Representative western blotting results of p-p65 and
total p65 in AGK-overexpressing AsPC-1 and PANC-1 cells. (G)
Quantitative analysis of the normalized ratio of phosphorylated p65
(p-p65) to total p65 from panel F. (H) Representative western
blotting results of p-p65 and total p65 in AGK-knockdown AsPC-1 and
PANC-1 cells. (I) Quantitative analysis of the normalized ratio of
phosphorylated p65 (p-p65) to total p65 from panel H. (J) Western
blot showing that p65 knockdown (si-p65) abolishes AGK-induced p65
phosphorylation. (K) Quantitative analysis of the normalized ratio
of phosphorylated p65 (p-p65) to total p65 from panel J. (L) qPCR
analysis showing that AGK-mediated upregulation of MYC mRNA is
dependent on p65. (M) CCK-8 assay demonstrating that p65 knockdown
attenuated AGK-induced cell proliferation. (N) AsPC-1 and (O)
PANC-1 cells overexpressed with AGK were incubated with vehicle or
0.5 µM QNZ for 48 h, followed by CCK-8 assay. Data were presented
as mean ± SD. *P<0.05, **P<0.01, ***P<0.001,
****P<0.0001. AGK, acylglycerol kinase; p-, phosphorylated; EV,
empty vector; si, short interfering; NC, negative control; qPCR,
quantitative PCR; QNZ.,
N4-[2-(4-phenoxyphenyl)ethyl]-1,2-dihydroquinazoline-4,6-diamine. |
To determine if AGK-mediated effects depend on
NF-κB/p65, the present study performed a rescue experiment in
PANC-1 cells. Cells were first transfected with control (si-NC) or
p65-specific (si-p65) siRNA, followed by transfection with EV or
Flag-AGK plasmid. Western blot analysis confirmed that AGK
overexpression increased phosphorylated p65 (p-P65) levels in
control cells, whereas si-p65 effectively depleted both total p65
and p-P65 regardless of AGK status (Fig. 4J and K). Accordingly, AGK-induced
upregulation of MYC mRNA (Fig.
4L) and enhancement of cell viability (Fig. 4M) were markedly attenuated upon p65
knockdown. However, AGK overexpression still partially increased
viability in p65-deficient cells, suggesting the involvement of
additional, p65-independent mechanisms. This notion was further
supported by the finding that AGK overexpression partially rescued
the proliferation suppression induced by the NF-κB inhibitor QNZ
(Fig. 4N and O).
AGK confers resistance to chemotherapy
and radiotherapy
The present study next investigated the role of AGK
in mediating chemotherapy or radiation resistance in pancreatic
cancer. Given that nab-paclitaxel and gemcitabine are first-line
chemotherapeutic agents for pancreatic cancer (23), the present study first assessed
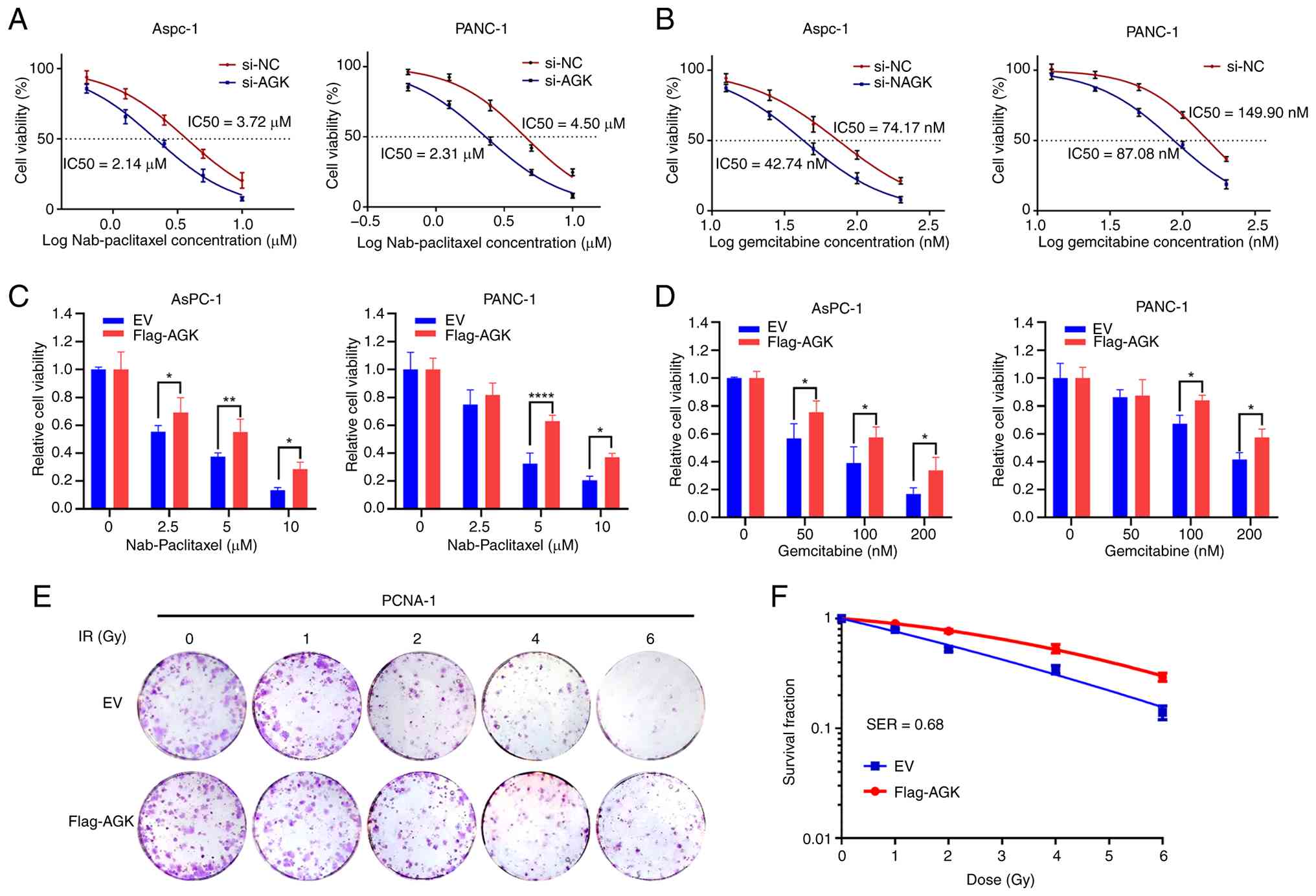
their sensitivity upon modulation of AGK expression in AsPC-1 and
PANC-1 cells. AGK knockdown sensitized pancreatic cancer cells to
both drugs. Specifically, the IC50 of nab-paclitaxel decreased from
~3.72–2.14 µM in AsPC-1 cells and from ~4.50–2.31 µM in PANC-1
cells (Fig. 5A). Similarly, IC50 of
gemcitabine decreased from ~74.17–42.74 nM in AsPC-1 cells and from
~149.90–87.08 nM in PANC-1 cells (Fig.
5B). Conversely, AGK overexpression markedly weakened the
growth-inhibitory effects of both nab-paclitaxel and gemcitabine on
AsPC-1 and PANC-1 cells (Fig. 5C and
D). The present study then evaluated the role of AGK in
radiosensitivity using a colony formation assay. AGK overexpression
markedly reduced radiosensitivity, yielding an SF2 of 0.77 and an
SER37 of 0.68, indicating that AGK overexpression drives
radioresistance in pancreatic cancer cells. (Fig. 5E and F). Collectively, these results
demonstrate that AGK promotes resistance to both chemotherapy and
radiotherapy in pancreatic cancer cells.
Discussion
Accumulating evidence indicates that AGK is
upregulated and promotes tumor progression in various cancers.
However, the signaling pathways induced by AGK vary among different
cancers. AGK triggers the activation of PI3K-AKT signaling pathway
in renal cell carcinoma (12) and
mediates Hippo-YAP1 signaling transduction in gastric cancer
(24). An earlier study also
demonstrated that AGK activates NF-κB signaling pathway by
increasing the phosphorylation of IKK and IκB in hepatocellular
carcinoma (25). The present study
confirmed that AGK promotes NF-κB activation via the canonical
IKK-IκBα-p65 axis in pancreatic cancer. Moreover, the present study
identified AGK as a novel regulator of p65 phosphorylation, a
well-established enhancer of NF-κB activation. These findings
revealed an additional layer of AGK-mediated NF-κB activation
beyond canonical pathway signaling. Although NF-κB/p65 signaling is
critical for AGK-driven proliferation, the partial rescue of cell
viability upon p65 knockdown or treatment with an NF-κB inhibitor
suggests the involvement of complementary, p65-independent
mechanisms. A limitation of the present study is the lack of
systematic assessment of other potential pathways, such as PI3K/AKT
or MAPK. Future work aimed at delineating the full spectrum of
AGK's downstream effectors will be essential to establish a
comprehensive mechanistic model and inform therapeutic
strategies.
Chemoresistance remains a major cause of treatment
failure in pancreatic cancer, for which chemotherapy is the
mainstream treatment for the majority of unresectable cases
(5,26–28).
Oncogenes often contribute to such resistance and AGK has been
reported to drive paclitaxel resistance in nasopharyngeal carcinoma
cells (29) and sunitinib
resistance in renal cell carcinoma (30). Consistent with these findings, the
present data showed that AGK overexpression enhances resistance to
gemcitabine and nab-paclitaxel in pancreatic cancer cells, while
its knockdown markedly sensitizes cells to these agents. Together,
these results suggested that targeting AGK could represent a
promising therapeutic strategy and the development of specific AGK
inhibitors may hold significant potential for novel drug discovery
in pancreatic cancer.
The present study established AGK as an important
oncogenic driver, yet several questions remain. Although NF-κB and
MYC signaling are known to regulate CCND1 and CCNB
expression (31,32), their functional interplay within
AGK-driven oncogenesis requires further investigation; particularly
whether MYC acts as the primary downstream effector. While
AGK expression is known to be transcriptionally regulated by TEAD
and post-transcriptionally modulated by miRNAs (33,34),
its full regulatory network remains incompletely elucidated.
Moreover, the upstream mechanisms driving AGK overexpression in
pancreatic cancer remain to be elucidated. Finally, the clinical
relevance of AGK as a prognostic marker and therapeutic target
warrants validation through large-scale, multicenter studies.
Addressing these questions will be crucial for comprehensively
characterizing AGK's molecular mechanisms and evaluating its
translational potential in pancreatic cancer oncology.
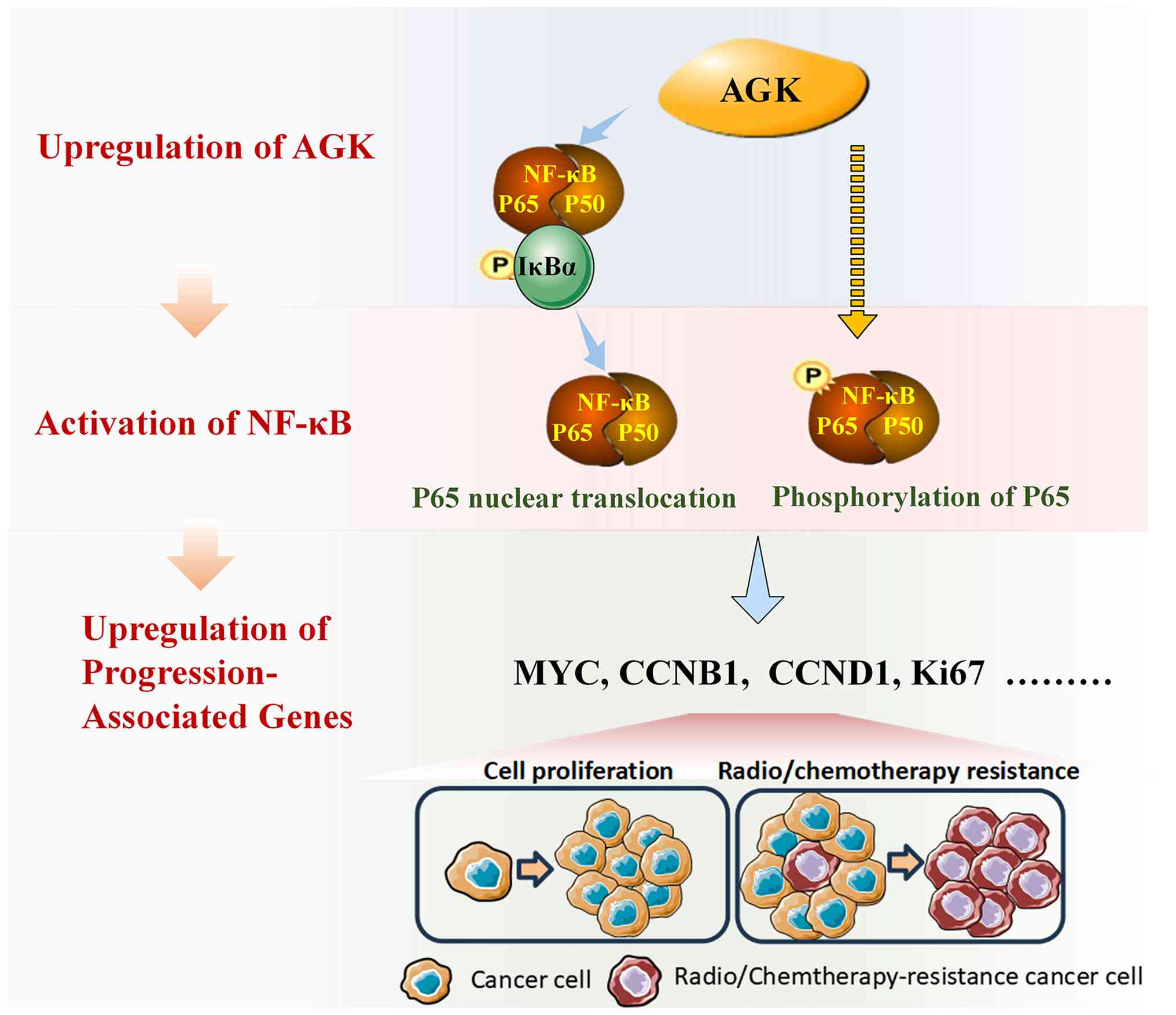
In summary, the present study demonstrated that AGK
is upregulated in pancreatic cancer and correlates with poor
prognosis. Functionally, AGK promotes tumor cell proliferation
in vitro and in vivo. Mechanistically, AGK activates
the NF-κB pathway through two distinct mechanisms. First, it
induces IκBα phosphorylation and subsequent ubiquitin-dependent
degradation, leading to p65 nuclear translocation. Second, it
enhances p65 phosphorylation at Ser536, thereby augmenting its
transcriptional activity. Consequently, the expression of key
pro-proliferative genes (e.g., CCNB1, CCND1, and
MYC), which are regulated by p65, is increased. This
AGK-driven signaling axis promotes tumor growth and confers
resistance to chemotherapy and radiotherapy, likely by reinforcing
cell cycle progression and survival pathways (Fig. 6). Collectively, the findings
established AGK as a key driver of NF-κB-mediated tumor progression
and therapy resistance, highlighting its potential as a novel
therapeutic target in aggressive pancreatic cancer.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural Science Research
Project of Anhui Educational Committee (grant no. 2024AH040240),
Anhui Provincial Natural Science Foundation (grant no.
2308085MH280), the High-Quality Innovation Platform of Science and
Education Innovation Zone in Suzhou Industrial Park-Key Platform
Project (grant no. YZCXPT2023104) and the National College Student
Innovation and Entrepreneurship Training Program (grant no.
202410368075).
Availability of data and materials
The data generated in the present study are included
in the figures and/or tables of this article.
Authors' contributions
Conceptualization was by KH, SL, XX and GC. Data
curation was by QZ, SG, JZ and XM. Formal analysis was by KH, XX
and QZ; Funding acquisition was by GC. Investigation was by KH, QZ
and SG. Methodology was by XX and QZ. Project administration was by
GC and SL. Resources were from KH, XX, XM and JZ. Software was by
SG and FL. Supervision was by GC, XX and SL. Validation was by GC
and SL. Visualization was by KH. Writing the original draft was by
KH and GC. Writing, review and editing was by GC and SL. KH and GC
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board of Wannan Medical College, with approvals granted
under approval no. 245 (medical ethics) and approval no.
WNMC-AWE-2024440 (animal ethics). All patients signed written
informed consent. Animal research was approved by the Ethics
Committee of Wannan Medical College.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Park W, Chawla A and O'Reilly EM:
Pancreatic cancer: A review. JAMA. 326:851–862. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li Q, Feng Z, Miao R, Liu X, Liu C and Liu
Z: Prognosis and survival analysis of patients with pancreatic
cancer: Retrospective experience of a single institution. World J
Surg Oncol. 20:112022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Saad AM, Turk T, Al-Husseini MJ and
Abdel-Rahman O: Trends in pancreatic adenocarcinoma incidence and
mortality in the United States in the last four decades; a
SEER-based study. BMC Cancer. 18:6882018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen J, Chen H, Zhang T, Yin X, Man J,
Yang X and Lu M: Burden of pancreatic cancer along with
attributable risk factors in China from 1990 to 2019, and
projections until 2030. Pancreatology. 22:608–618. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee HS and Park SW: Systemic chemotherapy
in advanced pancreatic cancer. Gut Liver. 10:340–347. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lin QJ, Yang F, Jin C and Fu DL: Current
status and progress of pancreatic cancer in China. World J
Gastroenterol. 21:7988–8003. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu B, Chen Z, Li Z, Zhao X, Zhang W,
Zhang A, Wen L, Wang X, Zhou S and Qian D: Hsp90α promotes
chemoresistance in pancreatic cancer by regulating Keap1-Nrf2 axis
and inhibiting ferroptosis. Acta Biochim Biophys Sin (Shanghai).
57:295–309. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bektas M, Payne SG, Liu H, Goparaju S,
Milstien S and Spiegel S: A novel acylglycerol kinase that produces
lysophosphatidic acid modulates cross talk with EGFR in prostate
cancer cells. J Cell Biol. 169:801–811. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chu B, Hong Z and Zheng X: Acylglycerol
Kinase-targeted therapies in oncology. Front Cell Dev Biol.
9:6591582021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhu Q, Cao SM, Lin HX, Yang Q, Liu SL and
Guo L: Overexpression of acylglycerol kinase is associated with
poorer prognosis and lymph node metastasis in nasopharyngeal
carcinoma. Tumour Biol. 37:3349–3357. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ray U, Roy SS and Chowdhury SR:
Lysophosphatidic acid promotes epithelial to mesenchymal transition
in ovarian cancer cells by repressing SIRT1. Cell Physiol Biochem.
41:795–805. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhu Q, Zhong AL, Hu H, Zhao JJ, Weng DS,
Tang Y, Pan QZ, Zhou ZQ, Song MJ, Yang JY, et al: Acylglycerol
kinase promotes tumour growth and metastasis via activating the
PI3K/AKT/GSK3beta signalling pathway in renal cell carcinoma. J
Hematol Oncol. 13:22020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen X, Ying Z, Lin X, Lin H, Wu J, Li M
and Song L: Acylglycerol kinase augments JAK2/STAT3 signaling in
esophageal squamous cells. J Clin Invest. 123:2576–2589. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hu Z, Qu G, Yu X, Jiang H, Teng XL, Ding
L, Hu Q, Guo X, Zhou Y, Wang F, et al: Acylglycerol kinase
maintains metabolic state and immune responses of CD8+ T cells.
Cell Metab. 30:290–302.e5. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak K and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu X, Wu G, Han K, Cui X, Feng Y, Mei X,
Yang P, You W and Yang Y: Inhibition of OTUB2 suppresses colorectal
cancer cell growth by regulating β-Catenin signaling. Am J Cancer
Res. 13:5382–5393. 2023.PubMed/NCBI
|
|
17
|
Li Q, Zhang L, Sun Y, Du Z, Xu S, Wang X,
Wei S, Tao Y, Li B, Jiang J, et al: p53 Modulates the Gut-Liver
Axis via PI3K/AKT/Wnt Signaling Pathways in Type 2 Diabetes. FASEB
J. 39:e708982025. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang X, Sun T, Fan J, Zuo X and Mao J:
Gastrin-related circRNA_0017065 promotes the proliferation and
metastasis of colorectal cancer through the miR-3174/RBFOX2 axis.
Biol Direct. 19:752024. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang X, Liu J, Wang C, Cheng KK, Xu H, Li
Q, Hua T, Jiang X, Sheng L, Mao J and Liu Z: miR-18a promotes
glioblastoma development by down-regulating ALOXE3-mediated
ferroptotic and anti-migration activities. Oncogenesis. 10:152021.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu X, Wu X, Yang H, Xu Q, Zhang M, Liu X
and Lv K: m6A-mediated upregulation of LINC01003 regulates cell
migration by targeting the CAV1/FAK signaling pathway in glioma.
Biol Direct. 18:272023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Carbone C and Melisi D: NF-κB as a target
for pancreatic cancer therapy. Expert Opin Ther Targets. 16 (Suppl
2):S1–S10. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Viatour P, Merville MP, Bours V and
Chariot A: Phosphorylation of NF-kappaB and IkappaB proteins:
Implications in cancer and inflammation. Trends Biochem Sci.
30:43–52. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Corrie PG, Qian W, Basu B, Valle JW, Falk
S, Lwuji C, Wasan H, Palmer D, Scott-Brown M, Wadsley J, et al:
Scheduling nab-paclitaxel combined with gemcitabine as first-line
treatment for metastatic pancreatic adenocarcinoma. Br J Cancer.
122:1760–1768. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang S, Cao Y, Guo H, Yao Y, Li L, Chen
J, Li J, Xiang X, Deng J and Xiong J: Up-regulated acylglycerol
kinase (AGK) expression associates with gastric cancer progression
through the formation of a novel YAP1-AGK-positive loop. J Cell Mol
Med. 24:11133–11145. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cui Y, Lin C, Wu Z, Liu A, Zhang X, Zhu J,
Wu G, Wu J, Li M, Li J and Song L: AGK enhances angiogenesis and
inhibits apoptosis via activation of the NF-κB signaling pathway in
hepatocellular carcinoma. Oncotarget. 5:12057–12069. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shimoda M, Kubota K, Shimizu T and Katoh
M: Randomized clinical trial of adjuvant chemotherapy with S-1
versus gemcitabine after pancreatic cancer resection. Br J Surg.
102:746–754. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Long J, Zhang Y, Yu X, Yang J, LeBrun DG,
Chen C, Yao Q and Li M: Overcoming drug resistance in pancreatic
cancer. Expert Opin Ther Targets. 15:817–828. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jin L, Qian D, Tang X, Huang Y, Zou J and
Wu Z: SMYD2 imparts gemcitabine resistance to pancreatic
adenocarcinoma cells by upregulating EVI2A. Mol Biotechnol.
66:2920–2933. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhao C, Chen HY, Zhao F, Feng HJ and Su
JP: Acylglycerol kinase promotes paclitaxel resistance in
nasopharyngeal carcinoma cells by regulating FOXM1 via the
JAK2/STAT3 pathway. Cytokine. 148:1555952021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sun Y, Zhu L, Liu P, Zhang H, Guo F and
Jin X: ZDHHC2-Mediated AGK palmitoylation activates AKT-mTOR
signaling to reduce sunitinib sensitivity in renal cell carcinoma.
Cancer Res. 83:2034–2051. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dang CV, O'Donnell KA, Zeller KI, Nguyen
T, Osthus RC and Li F: The c-Myc target gene network. Semin Cancer
Biol. 16:253–264. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dolcet X, Llobet D, Pallares J and
Matias-Guiu X: NF-kB in development and progression of human
cancer. Virchows Arch. 446:475–482. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chi H: miR-194 regulated AGK and inhibited
cell proliferation of oral squamous cell carcinoma by reducing
PI3K-Akt-FoxO3a signaling. Biomed Pharmacother. 71:53–57. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Long Y, Li C and Zhu B: Circ_0008068
facilitates the oral squamous cell carcinoma development by
microRNA-153-3p/acylgycerol kinase (AGK) axis. Bioengineered.
13:13055–13069. 2022. View Article : Google Scholar : PubMed/NCBI
|