Introduction
The deacetylase sirtuin 1 (SIRT1), a class III
histone deacetylase is overexpressed in several types of cancer,
including in tumors with p53 mutation, such as acute lymphoblastic
leukemia (ALL) (1,2) and triple-negative breast cancer
(TNBC) (3,4). The tumor suppressor gene p53 is
mutated in the acute lymphoblastic leukemia cell line, Jurkat
(5,6), as well as in the TNBC cell line,
MDA-MB-468 cells) (7,8), rendering these cancer cell lines as
useful model systems which may be used to study the anticancer
effects of novel drugs on tumors with p53 mutation. A high
expression level of SIRT1 has been found in primary ALL cells from
patients compared to peripheral blood mononuclear cells from
healthy subjects (1). Of note,
Tenovin-6, a selective inhibitor of SIRT1, has been shown to reduce
the growth of primary ALL cells, sensitize ALL cells to etoposide
and cytarabine and to activate the tumor suppressor p53(1). In the same context, it has been shown
that patients with TNBC express high levels of SIRT1 and that this
overexpression is significantly associated with lymph node
metastasis (3). The depletion of
SIRT1 in TNBC cell lines using siRNA has also been shown to
markedly suppress invasiveness, indicating that SIRT1 plays an
oncogenic role in the invasiveness of TNBC (3).
SIRT1 can deacetylate histone proteins (9,10),
as well as a number of non-histone proteins, including p53(11) and p73(12), which has a high degree of
similarity with p53 (13,14). A number of human cancers express
low levels of p73. p73 reactivation and stability in response to
pharmacological tools enables cancer cells to undergo apoptosis via
p53-independent pathways, which renders p73 a potent target for the
anticancer therapy of tumors with p53 mutation, including ALL
(15-17)
and TNBC (e.g., MDA-MB-468 cells) (18,19).
SIRT1 has been shown to interact with p73 and to suppress
p73-dependent transcriptional activity, enabling cancer cells to
escape apoptosis in response to chemotherapy (12). It has been shown that SIRT1
physically interacts with and suppresses the transactivation of the
acetyltransferase p300(20), known
to acetylate p73 in cancer cells (21,22).
Of note, p300 has been shown to acetylate and activate p73 in
response to several anticancer drugs, such as doxorubicin and
cisplatin (23). Thus, targeting
SIRT1 in tumors with p53 mutation, including ALL and TNBC (e.g.,
MDA-MB-468 cells) may be a promising tool for inducing apoptosis
and decreasing tumor resistance to chemotherapy via the
reactivation of p73 through an acetylation process involving
p300.
Thymoquinone (TQ), the bioactive compound of the
volatile oil derived from the seeds of the Nigella sativa
plant, has in vitro and in vivo potent pro-apoptotic
activities against various cancer cells (24-29).
Compared to cancer cells, TQ exerts mild cytotoxic effects on
matched normal cells and tissues, such as normal keratinocytes and
mouse fibroblasts (25,30,31).
TQ has been shown to inhibit the proliferation and induce the
apoptosis of the p53-mutant cell line, Jurkat, through p73
upregulation; however, the TQ-induced signaling pathways leading to
p73 overexpression in ALL remain largely unknown (24,26).
Consequently, the aim of the present study was to evaluate whether
TQ can inhibit SIRT1 expression in cancer cells with p53 mutation,
such as the human ALL cell line, Jurkat, and the human TNBC cell
line, MDA-MB-468 cells, leading to the reactivation and stability
of p73 with subsequent apoptosis.
Materials and methods
Cell culture and treatment
Human T lymphocyte cell line (Jurkat cells) and the
human TNBC cell line (MDA-MB-468 cells) were obtained from the
America Type Culture Collection (ATCC). The Jurkat cells were
maintained in RPMI-1640 (Sigma-Aldrich; Merck KGaA) medium and
MDA-MB-468 cells in Dulbecco's modified Eagle medium (DMEM;
UFC-Biotech) supplemented with 15% (v/v) fetal calf serum (FCS,
Lonza BioWhittaker), 2 mM glutamine, penicillin (100 IU/ml) and
streptomycin (100 µg/ml) (Sigma-Aldrich; Merck KGaA). Both cell
lines were maintained in a humidified incubator containing 5%
CO2 at 37˚C. For all treatments, a 10 mM solution of TQ
(Sigma-Aldrich; Merck KGaA) was prepared in 10% dimethyl sulfoxide
(DMSO; Merck Millipore) and appropriate working concentrations were
prepared with cell culture medium. The final concentration of DMSO
was always <0.1% in both the control and treatment
conditions.
Cell proliferation assay
A colorimetric cell proliferation assay using the
WST-1 Cell Proliferation Reagent kit (Sigma-Aldrich; Merck KGaA)
was used to examine the effects of TQ on the proliferation of
Jurkat cells and MDA-MB-468 cells. Briefly, the cells were seeded
in 96-multi-well plates at a density of 4x104 cells/well
for the Jurkat cells and 104 cells/well for the
MDA-MB-468 cells. Following 24 h of incubation at 37˚C, the cells
were exposed to various concentrations of TQ for 24 h. The cell
proliferation rate then was evaluated through a rapid WST-1
reagent. Following 24 h of incubation, 10 µl of the WST-1 solution
were added followed by incubation for an additional 3 h at 37˚C.
Finally, the absorbance was read at 450 nm using a microplate ELISA
reader (ELx800™; BioTek Instruments, Inc.) and Gen5 software
(BioTek Instruments, Inc.) was used to analyze the results. The
reaction based on the cleavage of the tetrazolium salt WST-1 to
formazan by cellular mitochondrial dehydrogenases. The quantity of
formazan dye in the medium is directly proportional to the number
of viable metabolically active cells. The percentage of cell
viability was calculated by assuming control (untreated) samples as
100% viable.
Apoptosis assay
To examine apoptosis, the Jurkat cells were seeded
in 96-well plates at a density of 4x104 cells/well,
grown for 24 h and exposed to various concentrations of TQ for 24 h
or to 50 µM for 15 min, 30 min, 1 and 3 h. The cell apoptosis rate
was evaluated using the Annexin V Binding Guava Nexin®
assay by capillary cytometry (Guava Easycyte Plus HP system, with
absolute cell count and 6 parameters) according to the
manufacturer's recommendations (Guava Technologies Inc.). Guava
Nexin® Assay utilizes Annexin V-PE.
Western blot analysis
The Jurkat cells were treated with various
concentrations of TQ for 24 h or to 50 µM for 15 min, 30 min, 1 and
3 h. The cells were then harvested, centrifuged at 201 x g for 10
min at room temperature to discard the RPMI medium, washed with
cold phosphate-buffered saline (PBS) and resuspended in RIPA buffer
(25 mM Tris, pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate
and 0.1% SDS; Sigma-Aldrich; Merck KGaA) containing protease
inhibitors. Equal amounts (20 µg) of total protein were separated
on 10-15% polyacrylamide gels and electrophoretically transferred
to a nitrocellulose membrane. After blocking with 5% non-fat dry
milk and Tween-20 in PBS, the nitrocellulose membranes were
incubated, at 4˚C overnight, with either mouse monoclonal
anti-SIRT1 antibody (B-10: cat. no. sc-74504; Santa Cruz
Biotechnology, Inc.; diluted at 1:200), mouse monoclonal anti-p73
antibody (cat. no. 558785; BD Biosciences; diluted at 1 µg/ml),
rabbit polyclonal anti-cleaved caspase-3 (Asp175) antibody (cat.
no. 9661; Cell Signaling Technology, Inc.; diluted at 1:1,000) or
mouse monoclonal anti-β-actin antibody (cat. no. ab8227; Abcam;
diluted at 1:25,000), according to the manufacturer's instructions.
The membranes were then washed 3 times with PBS for 10 min. The
membranes were, thereafter, incubated with the appropriate
horseradish peroxidase-conjugated secondary antibody [Cell
Signaling Technology, Inc.; diluted to 1:10,000 for anti-mouse
antibody cat. no. 7076 and 2:10,000 for anti-rabbit antibody cat.
no. 7074 at room temperature for 1 h and 30 min. The membranes were
then washed with PBS 5 times. Signals were detected by
chemiluminescence using the ECL Plus detection system (Amersham; GE
Healthcare Life Sciences). For the quantification of SIRT1, p73 and
cleaved caspase-3 proteins, images of the western blots were
processed using NIH ImageJ software (Java 8).
Reverse transcription-quantitative PCR
(RT-qPCR)
Cells were treated with various concentrations of TQ
for 24 h. Total RNA was purified and subjected to reverse
transcription using Oligo(dt) (Sigma-Aldrich; Merck KGaA) and
Superscript II reverse transcriptase (Invitrogen; Thermo Fisher
Scientific, Inc.). Quantitative PCR was performed using the
LightCycler 480 SYBR-Green I Master kit (Roche Diagnostics) and the
Mastercycler Realplex apparatus (Eppendorf). The results were
normalized to ribosomal protein L11 (RPL11) mRNA. PCR was performed
with 30 cycles of denaturation for 30 sec at 95˚C; annealing for 45
sec at 60˚C; and extension for 60 sec at 72˚C. The sequences of the
primers for PCR amplification are listed in Table I. Amplicons were size-controlled on
an agarose gel and the purity was assessed by analysis of the
melting curves at the end of the RT-PCR reaction. The expression
level of the target gene in the treated cells was measured relative
to the level observed in the untreated cells and was quantified
using the formula:
2-ΔΔCq (32).
 | Table ISequences of the primers used for PCR
amplification. |
Table I
Sequences of the primers used for PCR
amplification.
| Target gene | Sense sequence
(5'-3') | Antisense sequence
(3'-5') |
|---|
| SIRT1 |
CCGCTTGCTATCATGAAACCA |
TCACAGTCTCCAAGAAGCTCT |
| p300 |
TCCTGGACAGCAGATTGGAG |
CTTGCGCTTCTCTGGATCAG |
| RPL11 |
ATCCTTTGGCATCCGGAGAA |
GTCCAGGCCGTAGATACCAA |
Statistical analysis
All data are presented as the means ± SEM of
triplicates performed for the same experiment or an average of at
least 3 separate experiments. Statistical analysis was performed
using one-way ANOVA followed by Tukey's post hoc test using
GraphPad Prism 6 software (GraphPad Software) and significant
differences were indicated as with values of P<0.05.
Results
TQ induces a concentration-dependent
degradation of SIRT1 associated with p73 upregulation in Jurkat
cells
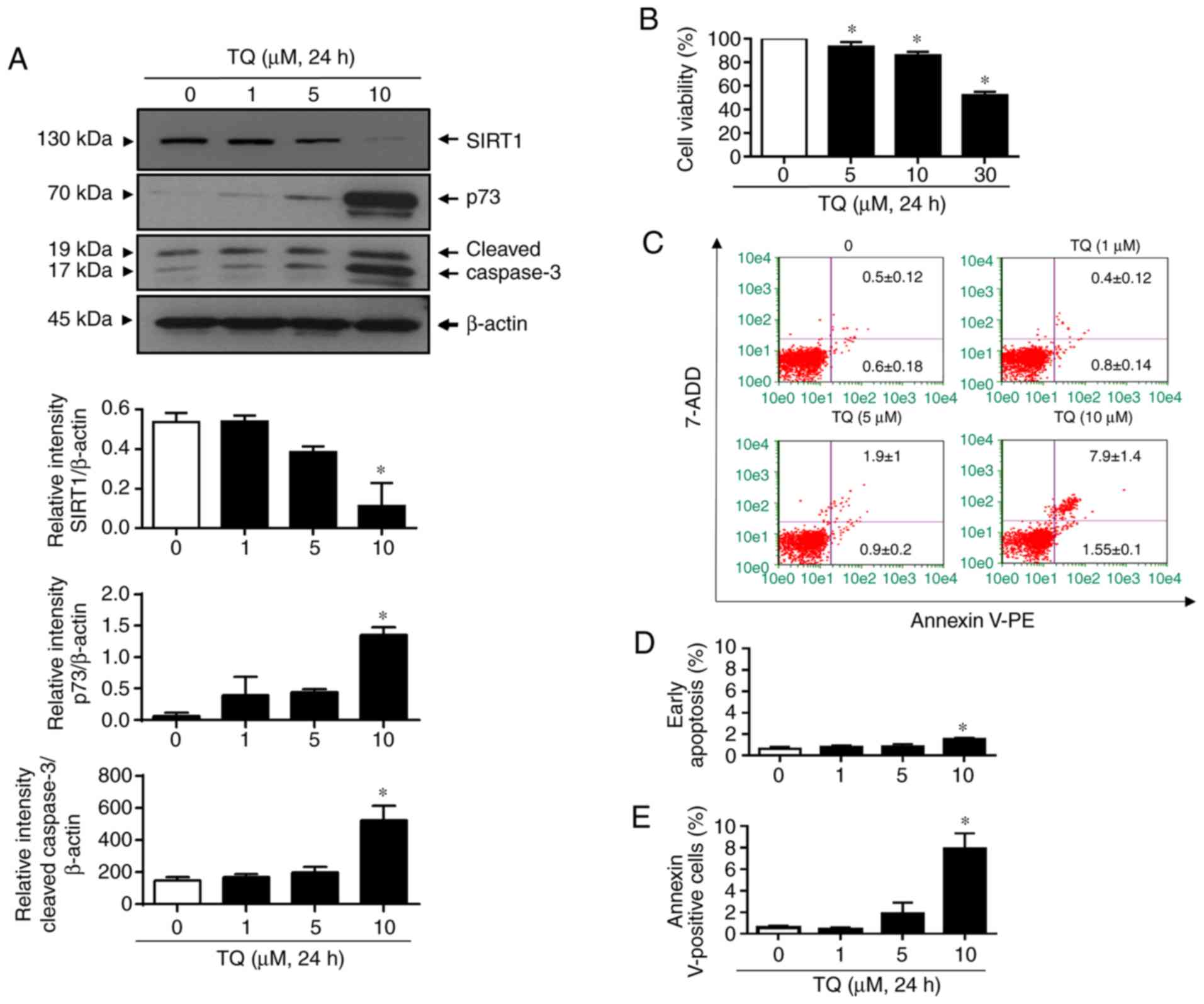
Firstly, the effects of TQ on SIRT1 and p73 protein
expression levels in Jurkat cells were evaluated (Fig. 1A). For this purpose, the cells were
incubated with increasing concentrations of TQ for 24 h. The
results revealed that TQ induced the downregulation of SIRT1 in a
concentration-dependent manner in the Jurkat cells (Fig. 1A). Indeed, treatment with TQ at 5
µM induced a slight decrease in the SIRT1 level and a significant
decrease was detected at 10 µM. The TQ-induced SIRT1 degradation
was associated with the upregulation of p73 and cleaved caspase-3.
Indeed, TQ induced a significant increase in the expression levels
of p73 and cleaved caspase 3 at the concentration of 10 µM
(Fig. 1A). The anti-proliferative
and pro-apoptotic effects of TQ on the Jurkat cells were then
analyzed under the same experimental conditions. Cell proliferation
in response to TQ was decreased in a concentration-dependent manner
(Fig. 1B). TQ significantly
inhibited cell proliferation from the concentration of 5 µM and the
inhibition rate reached approximately 10 and 50% in the Jurkat
cells treated with 10 and 30 µM TQ, respectively (Fig. 1B). Under the same conditions, TQ
was found to induce apoptosis in a dose-dependent manner, which
became significant at the concentration of 10 µM (Fig. 1C-E). Indeed, at the concentration
of 10 µM, approximately 2% of Jurkat cells were in the early
apoptotic stage (Fig. 1C and
D) and 8% were in the late
apoptotic stage (Fig. 1C and
E). Taken together, these results
indicated that TQ induced the downregulation of SIRT1 in Jurkat
cells, which could lead to an upregulation of p73 and cleaved
caspase-3, with subsequent cell proliferation inhibition and
apoptosis induction.
TQ-induced p73 upregulation and
apoptosis are associated with the rapid downregulation of
SIRT1
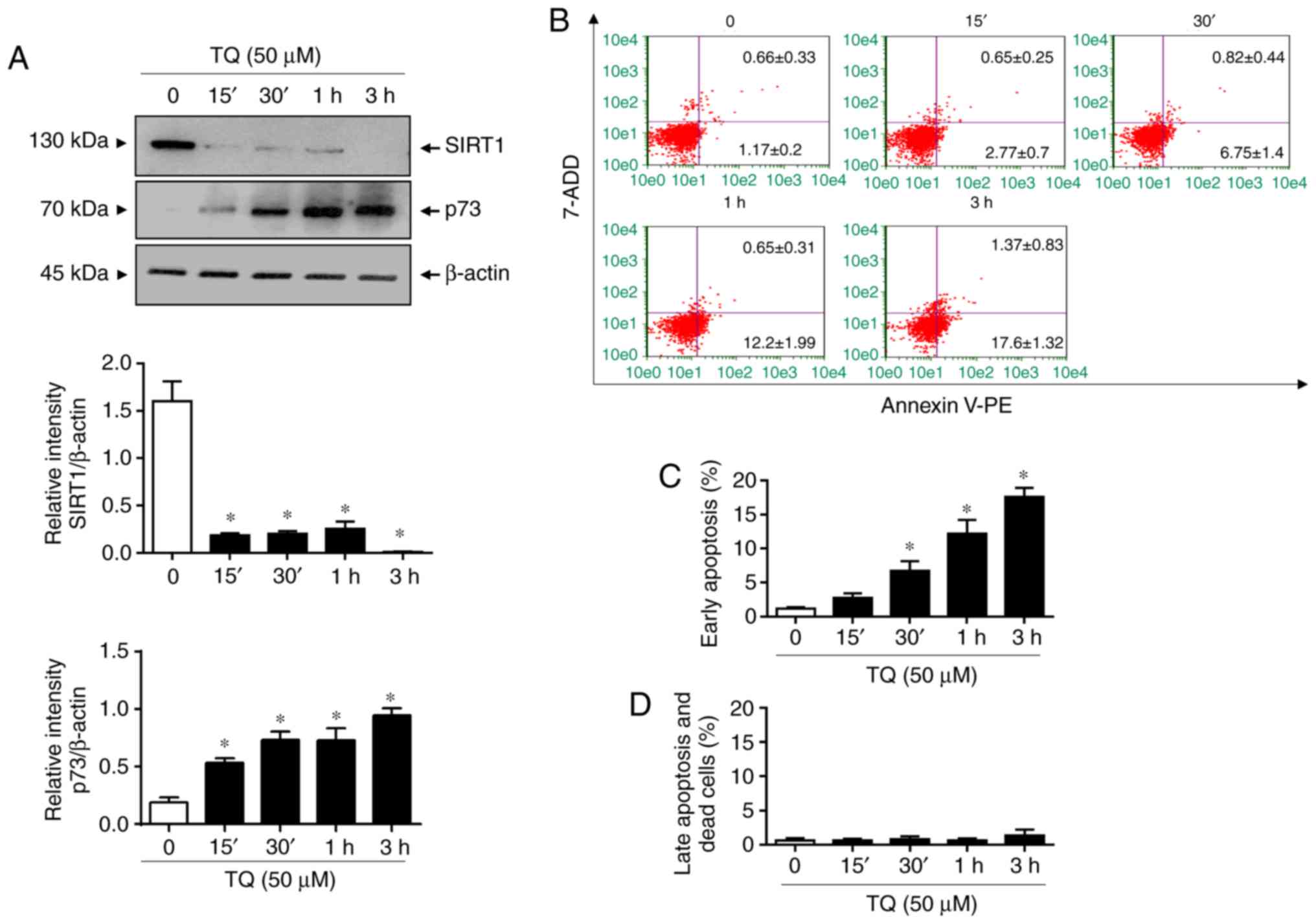
In the next step, a kinetic analysis of TQ on SIRT1
expression in Jurkat cells was performed in order to determine the
chronology of the molecular events induced by TQ leading to the
upregulation of p73 and apoptosis. For this objective, cells were
exposed to 50 µM of TQ, the concentration at which SIRT1 protein
expression was undetectable after 24 h (data not shown). The
time-course effects of TQ on SIRT1 expression in Jurkat cells at 50
µM revealed that SIRT1 expression began to significantly decrease
after 15 min and the loss was almost complete after 3 h of
treatment (Fig. 2A). A significant
decrease in SIRT1 expression observed after 15 min of TQ treatment
and was associated with a significant increase in the expression of
p73 (Fig. 2A). Notably, TQ induced
the apoptosis of Jurkat cells in a time-dependent manner (Fig. 2B-D) following the TQ-induced
deregulation of the SIRT1 and p73 expression levels (Fig. 2A). Indeed, TQ caused a significant
increase in the number of apoptotic cells in the early stage after
30 min (Fig. 2B and C). The percentage of apoptotic cells in
the early stage reached approximately 18% after 3 h (Fig. 2B and C). These findings suggest that the
TQ-induced downregulation of SIRT1 expression is a main event in
the reactivation and stability of the tumor suppressor, p73, with
the subsequent induction of apoptosis.
TQ induces the transcription-dependent
downregulation of SIRT1 in Jurkat cells with p53 mutation
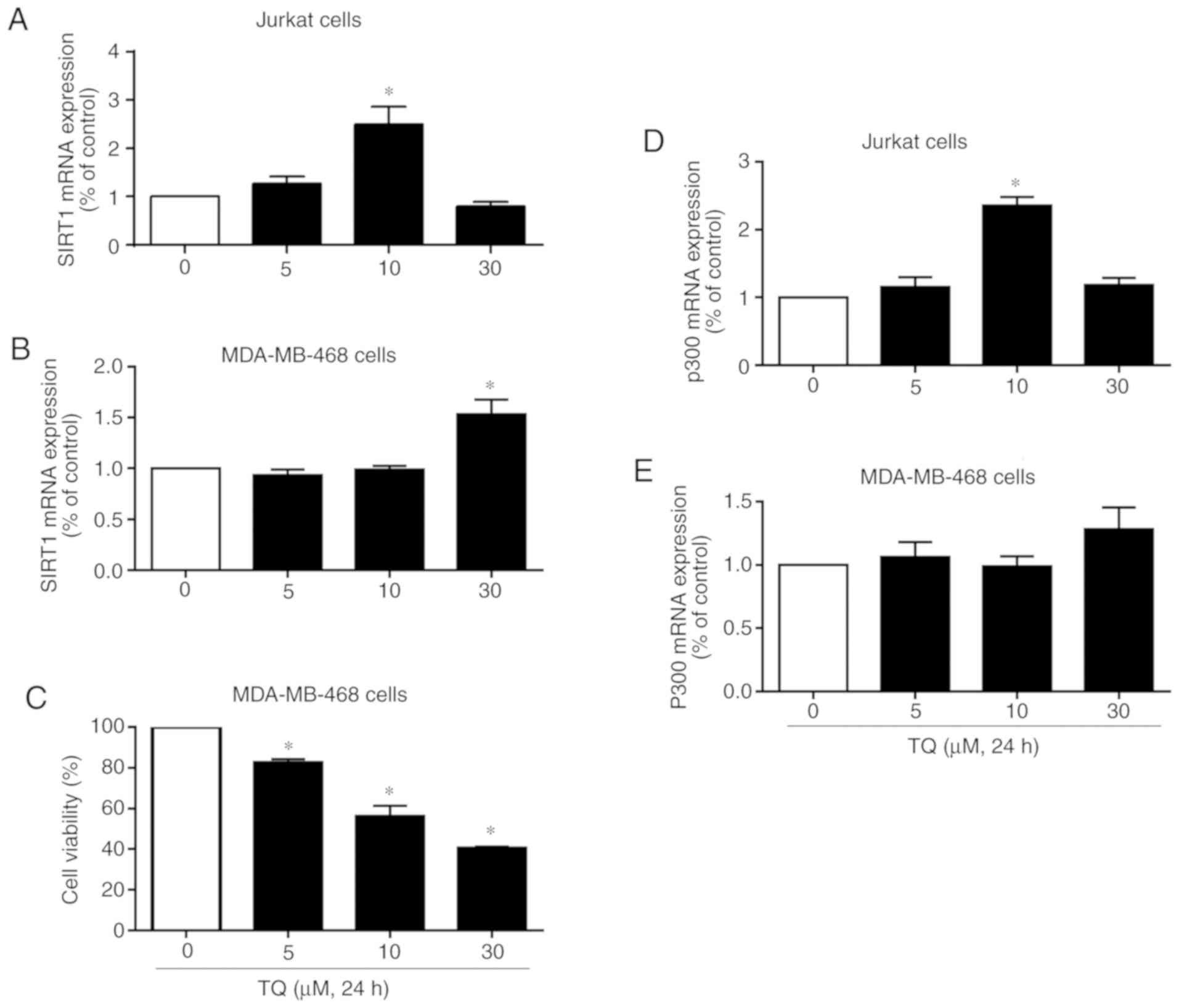
To investigate whether TQ affects SIRT1 expression
also at the transcriptional level, Jurkat cells were exposed to TQ
under the same conditions. As shown in Fig. 3A, the exposure of Jurkat cells to
10 µM of TQ significantly increased SIRT1 mRNA expression. At the
concentration of 30 µM, TQ induced a decrease in SIRT1 mRNA
expression (Fig. 3A), indicating
that the TQ-induced downregulation of SIRT1 expression in Jurkat
cells results from its effects at both the transcriptional and
protein level. The mRNA expression levels of SIRT1 were also
investigated in the human breast cancer cell line, MDA-MB-468
(Fig. 3B). TQ had no effect on
SIRT1 mRNA expression at the concentrations of 5 and 10 µM, whereas
its expression significantly increased following treatment of the
cells with 30 µM TQ (Fig. 3B). The
effects of TQ on the proliferation of MDA-MB-468 cells were also
investigated. TQ significantly inhibited cell proliferation from
the concentration of 5 µM and the inhibition rate reached
approximately 45 and 60% in the MDA-MB-468 cells treated with 10
and 30 µM TQ, respectively (Fig.
3C). Taken together, these findings indicate that the
TQ-induced downregulation of SIRT1 expression in Jurkat cells
results from its effects on both the transcriptional and protein
levels, and that TQ inhibits the proliferation of MDA-MB-468 cells
via a mechanism independent of SIRT1 mRNA expression.
TQ induces the transcriptional
upregulation of the histone acetyltransferase p300 in cancer cells
with p53 mutation
SIRT1 has been shown to physically interact with and
suppress the transactivation of the acetyltransferase,
p300(20). Of note, p300 has been
shown to acetylate and activate p73 in response to treatment with
several anticancer drugs, such as doxorubicin and cisplatin
(23). In the present study, in
order to investigate the mechanisms underlying p73 upregulation
following the decrease in the expression of the deacetylase SIRT1
in response to TQ treatment, the effects of TQ on the expression of
p300 were evaluated in Jurkat and MDA-MB-468 cells. As shown in
Fig. 3D, treatment of the Jurkat
cells with 10 µM TQ significantly increased the transcriptional
levels of p300, as detected by RT-qPCR (Fig. 3D). Indeed, at the concentration of
10 µM, an approximately 2-fold increase in p300 expression was
observed (Fig. 3D), in parallel
with a significant decrease in SIRT1 protein expression and a
significant increase in p73 protein expression, as shown in
Fig. 1A. At 30 µM, only a slight
increase in the level of p300 was found compared to the control
(Fig. 3D). In the MDA-MB-468
cells, TQ had no significant effect on p300 mRNA expression at the
concentrations of 5 and 10 µM, while p300 expression levels began
to increase at the concentration of 30 µM TQ (Fig. 3E). These results indicate that p73
is activated and stabilized in response to TQ in cancer cells with
p53 mutation through the deacetylation/acetylation-dependent
pathway involving the downregulation of SIRT1 protein and the
upregulation of p300, respectively.
Discussion
The deacetylase SIRT1 has been shown to act as a
negative regulator of the function of the tumor suppressors,
p53(33) and p73(12), leading to the inhibition of
apoptosis. SIRT1 has been found to directly bind to p73, reducing
its transcriptional activity via a deacetylation process with the
subsequent inhibition of apoptosis (12). p53 and p73 proteins have a high
degree of similarity in both structure and function (13,14).
Of note, in tumors with p53 mutation, including ALL (1,2,24)
and TNBC (3,4), the upregulation of p73 in response to
anticancer agents leads to the activation of several pro-apoptotic
genes with the subsequent induction of apoptosis. Thus, it is of
interest to identify novel natural compounds that can target
SIRT1/p73 interaction, highlighting new strategies for cancer
therapy. The present study demonstrated that treatment of Jurkat
cells with TQ induced a decrease in SIRT1 protein expression in a
concentration- and time-dependent manner, and that this effect was
associated with an increase in p73 protein expression. The
TQ-induced downregulation of SIRT1 expression was associated with
an increase in cleaved caspase-3 expression and apoptosis. TQ also
induced an increase in the expression of the acetyltransferase,
p300, in Jurkat cells and the human breast cancer cell line,
MDA-MB-468.
SIRT1 can act as either an oncogene or a tumor
suppressor, depending on its cellular targets or specific cancers
(34-36).
Considering the fact that SIRT1 is overexpressed in several tumors
with p53 mutation, including ALL (1) and TNBC (3,4,37),
SIRT1 inhibition holds promise as a novel approach for cancer
therapy in these tumors. The present study demonstrated that SIRT1
protein expression was downregulated in Jurkat cells treated with
TQ in parallel with an increase in p73 protein expression,
indicating that the low expression levels of p73 found in Jurkat
cells may be a result of its degradation through the SIRT1-mediated
deacetylation process; this suggests that TQ may be a novel
potential inhibitor of SIRT1 in cancers with p53 mutation. This
conclusion is supported by ample evidence. A recent study indicated
that the pre-treatment of cancer cells with nicotinamide, an
inhibitor of SIRT1, was able to increase the expression of p73 and
that of pro-apoptotic, Bax (38).
In the same context, it has been shown that the anti-leukemic drug,
arsenic trioxide, induced an upregulation of p73 expression,
leading to the apoptosis of acute promyelocytic leukemia cells via
the inhibition of several oncogenes, including SIRT1(39). The results of the present study are
also in line with previous results, highlighting SIRT1 as an
inhibitor of p73 activity through the deacetylation-mediated
process (12). Indeed, the
knockdown of SIRT1 in HeLa cells using SIRT1 antisense was
previously shown to result in the upregulation of p73 protein and
the induction of apoptosis, while the overexpression of SIRT1
counteracted p73-induced apoptosis, indicating that SIRT1
negatively regulated the expression of p73 and apoptosis (12). This indicates that the knockdown of
SIRT1 mimics the effects of TQ on the expression of p73 and
apoptosis observed in the present study. The present study also
demonstrated that TQ increased the expression of the
acetyltransferase, p300, in Jurkat cells and MDA-MB-468 cells,
suggesting that the upregulation of p73 in response to TQ involves
its acetylation by p300. Of note, the significant increase in the
levels of p300 mRNA in Jurkat cells detected following treatment
with 10 µM TQ was inversely associated with SIRT1 protein
expression and positively with p73 protein expression under the
same conditions, indicating that p73 is activated and stabilized in
response to TQ in Jurkat cells through the
deacetylation/acetylation-dependent pathway involving the
downregulation of SIRT1 and the upregulation of p300, respectively.
This hypothesis is supported by the findings of several previous
studies. Indeed, SIRT1 has been shown to physically interact with
and suppress p300 transactivation (20). Moreover, it has been shown that the
anticancer drug, doxorubicin, increases the expression of p300,
leading to the acetylation of p73 in the human colon cancer cell
line, HCT116(23). This indicates
that the activation and stability of the tumor suppressor, p73,
through the deacetylation/acetylation-dependent pathway, is the
main target in tumors with p53 mutation for natural compounds
exhibiting anticancer activities, including TQ.
In conclusion, the present study demonstrates that
TQ induces the downregulation of the deacetylase SIRT1, with a
coordinated upregulation of the tumor suppressor, p73, most likely
through the acetylation-mediated process. p300 may be the most
likely acetyltransferase associated with the TQ-induced p73
upregulation with the subsequent induction of apoptosis. However,
TQ-induced SIRT1/p300/p73 deregulation warrants further
investigation in order to decipher the chronology of the molecular
events involved, namely SIRT1 downregulation, which triggers the
upregulation of p300, leading to the stability and the reactivation
of p73 followed by apoptosis. The findings of the present study
provide new insight into the regulation of SIRT1/P73 expression
upon treatment with natural anticancer drugs, as it suggests that
the inhibition of SIRT1 by TQ may be a promising tool for cancer
therapy in cancers with p53 mutation.
Acknowledgements
The author would like to acknowledge Mr. Mohammed A.
Hassan for providing technical assistance.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article or are available from the
corresponding author on reasonable request.
Author contributions
MA designed the study, performed the research and
analyzed the data, and wrote the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The author declares that there are no competing
interests.
References
|
1
|
Jin Y, Cao Q, Chen C, Du X, Jin B and Pan
J: Tenovin-6-mediated inhibition of SIRT1/2 induces apoptosis in
acute lymphoblastic leukemia (ALL) cells and eliminates ALL
stem/progenitor cells. BMC Cancer. 15(226)2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Yu W, Li L, Wang G, Zhang W, Xu J and
Liang A: KU70 inhibition impairs both non-homologous end joining
and homologous recombination DNA damage repair through SHP-1
induced dephosphorylation of SIRT1 in T-cell acute lymphoblastic
leukemia (T-ALL) [corrected]. Cell Physiol Biochem. 49:2111–2123.
2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Chung SY, Jung YY, Park IA, Kim H, Chung
YR, Kim JY, Park SY, Im SA, Lee KH, Moon HG, et al: Oncogenic role
of SIRT1 associated with tumor invasion, lymph node metastasis, and
poor disease-free survival in triple negative breast cancer. Clin
Exp Metastasis. 33:179–185. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Sinha S, Patel S, Athar M, Vora J,
Chhabria MT, Jha PC and Shrivastava N: Structure-based
identification of novel sirtuin inhibitors against triple negative
breast cancer: An in silico and in vitro study. Int J Biol
Macromol. 140:454–468. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Cheng J and Haas M: Frequent mutations in
the p53 tumor suppressor gene in human leukemia T-cell lines. Mol
Cell Biol. 10:5502–5509. 1990.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Shan X, Czar MJ, Bunnell SC, Liu P, Liu Y,
Schwartzberg PL and Wange RL: Deficiency of PTEN in Jurkat T cells
causes constitutive localization of Itk to the plasma membrane and
hyperresponsiveness to CD3 stimulation. Mol Cell Biol.
20:6945–6957. 2000.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Shtraizent N, Matsui H, Polotskaia A and
Bargonetti J: Hot spot mutation in TP53 (R248Q) causes oncogenic
gain-of-function phenotypes in a breast cancer cell line derived
from an african american patient. Int J Environ Res Public Health.
13(ijerph13010022)2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Hollestelle A, Nagel JH, Smid M, Lam S,
Elstrodt F, Wasielewski M, Ng SS, French PJ, Peeters JK, Rozendaal
MJ, et al: Distinct gene mutation profiles among luminal-type and
basal-type breast cancer cell lines. Breast Cancer Res Treat.
121:53–64. 2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Imai S, Armstrong CM, Kaeberlein M and
Guarente L: Transcriptional silencing and longevity protein Sir2 is
an NAD-dependent histone deacetylase. Nature. 403:795–800.
2000.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Vaquero A, Scher M, Lee D,
Erdjument-Bromage H, Tempst P and Reinberg D: Human SirT1 interacts
with histone H1 and promotes formation of facultative
heterochromatin. Mol Cell. 16:93–105. 2004.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Vaziri H, Dessain SK, Ng Eaton E, Imai SI,
Frye RA, Pandita TK, Guarente L and Weinberg RA: hSIR2(SIRT1)
functions as an NAD-dependent p53 deacetylase. Cell. 107:149–159.
2001.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Dai JM, Wang ZY, Sun DC, Lin RX and Wang
SQ: SIRT1 interacts with p73 and suppresses p73-dependent
transcriptional activity. J Cell Physiol. 210:161–166.
2007.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Dotsch V, Bernassola F, Coutandin D, Candi
E and Melino G: p63 and p73, the ancestors of p53. Cold Spring Harb
Perspect Biol. 2(a004887)2010.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Marin MC, Jost CA, Irwin MS, DeCaprio JA,
Caput D and Kaelin WG Jr: Viral oncoproteins discriminate between
p53 and the p53 homolog p73. Mol Cell Biol. 18:6316–6324.
1998.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Ahmadianpour MR, Abdolmaleki P, Mowla SJ
and Hosseinkhani S: Gamma radiation alters cell cycle and induces
apoptosis in p53 mutant E6.1 Jurkat cells. Appl Radiat Isot.
71:29–33. 2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ahmadianpour MR, Abdolmaleki P, Mowla SJ
and Hosseinkhani S: Static magnetic field of 6 mT induces apoptosis
and alters cell cycle in p53 mutant Jurkat cells. Electromagn Biol
Med. 32:9–19. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Laumann R, Jucker M and Tesch H: Point
mutations in the conserved regions of the p53 tumour suppressor
gene do not account for the transforming process in the Jurkat
acute lymphoblastic leukemia T-cells. Leukemia. 6:227–228.
1992.PubMed/NCBI
|
|
18
|
Lim LY, Vidnovic N, Ellisen LW and Leong
CO: Mutant p53 mediates survival of breast cancer cells. Br J
Cancer. 101:1606–1612. 2009.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Qiu WG, Polotskaia A, Xiao G, Di L, Zhao
Y, Hu W, Philip J, Hendrickson RC and Bargonetti J: Identification,
validation, and targeting of the mutant p53-PARP-MCM chromatin axis
in triple negative breast cancer. NPJ Breast Cancer.
3:2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Bouras T, Fu M, Sauve AA, Wang F, Quong
AA, Perkins ND, Hay RT, Gu W and Pestell RG: SIRT1 deacetylation
and repression of p300 involves lysine residues 1020/1024 within
the cell cycle regulatory domain 1. J Biol Chem. 280:10264–10276.
2005.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zeng X, Chen L, Jost CA, Maya R, Keller D,
Wang X, Kaelin WG Jr, Oren M, Chen J and Lu H: MDM2 suppresses p73
function without promoting p73 degradation. Mol Cell Biol.
19:3257–3266. 1999.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zeng X, Li X, Miller A, Yuan Z, Yuan W,
Kwok RP, Goodman R and Lu H: The N-terminal domain of p73 interacts
with the CH1 domain of p300/CREB binding protein and mediates
transcriptional activation and apoptosis. Mol Cell Biol.
20:1299–1310. 2000.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Costanzo A, Merlo P, Pediconi N, Fulco M,
Sartorelli V, Cole PA, Fontemaggi G, Fanciulli M, Schiltz L,
Blandino G, et al: DNA damage-dependent acetylation of p73 dictates
the selective activation of apoptotic target genes. Mol Cell.
9:175–186. 2002.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Alhosin M, Abusnina A, Achour M, Sharif T,
Muller C, Peluso J, Chataigneau T, Lugnier C, Schini-Kerth VB,
Bronner C and Fuhrmann G: Induction of apoptosis by thymoquinone in
lymphoblastic leukemia Jurkat cells is mediated by a p73-dependent
pathway which targets the epigenetic integrator UHRF1. Biochem
Pharmacol. 79:1251–1260. 2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Alhosin M, Ibrahim A, Boukhari A, Sharif
T, Gies JP, Auger C and Schini-Kerth VB: Anti-neoplastic agent
thymoquinone induces degradation of α and β tubulin proteins in
human cancer cells without affecting their level in normal human
fibroblasts. Invest New Drugs. 30:1813–1819. 2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ibrahim A, Alhosin M, Papin C, Ouararhni
K, Omran Z, Zamzami MA, Al-Malki AL, Choudhry H, Mély Y, Hamiche A,
et al: Thymoquinone challenges UHRF1 to commit auto-ubiquitination:
A key event for apoptosis induction in cancer cells. Oncotarget.
9:28599–28611. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Qadi SA, Hassan MA, Sheikh RA, Baothman
OA, Zamzami MA, Choudhry H, Al-Malki AL, Albukhari A and Alhosin M:
Thymoquinone-induced reactivation of tumor suppressor genes in
cancer cells involves epigenetic mechanisms. Epigenet Insights.
12(2516865719839011)2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Helmy SA, El-Mesery M, El-Karef A, Eissa
LA and El Gayar AM: Thymoquinone upregulates TRAIL/TRAILR2
expression and attenuates hepatocellular carcinoma in vivo model.
Life Sci. 233(116673)2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zhu WQ, Wang J, Guo XF, Liu Z and Dong WG:
Thymoquinone inhibits proliferation in gastric cancer via the STAT3
pathway in vivo and in vitro. World J Gastroenterol. 22:4149–4159.
2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Gali-Muhtasib HU, Abou Kheir WG, Kheir LA,
Darwiche N and Crooks PA: Molecular pathway for
thymoquinone-induced cell-cycle arrest and apoptosis in neoplastic
keratinocytes. Anticancer Drugs. 15:389–399. 2004.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ivankovic S, Stojkovic R, Jukic M, Milos
M, Milos M and Jurin M: The antitumor activity of thymoquinone and
thymohydroquinone in vitro and in vivo. Exp Oncol. 28:220–224.
2006.PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Langley E, Pearson M, Faretta M, Bauer UM,
Frye RA, Minucci S, Pelicci PG and Kouzarides T: Human SIR2
deacetylates p53 and antagonizes PML/p53-induced cellular
senescence. EMBO J. 21:2383–2396. 2002.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Lin Z and Fang D: The roles of SIRT1 in
cancer. Genes Cancer. 4:97–104. 2013.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Ban J, Aryee DN, Fourtouna A, van der Ent
W, Kauer M, Niedan S, Machado I, Rodriguez-Galindo C, Tirado OM,
Schwentner R, et al: Suppression of deacetylase SIRT1 mediates
tumor-suppressive NOTCH response and offers a novel treatment
option in metastatic Ewing sarcoma. Cancer Res. 74:6578–6588.
2014.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Ming M, Soltani K, Shea CR, Li X and He
YY: Dual role of SIRT1 in UVB-induced skin tumorigenesis. Oncogene.
34:357–363. 2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Liarte S, Alonso-Romero JL and Nicolas FJ:
SIRT1 and estrogen signaling cooperation for breast cancer onset
and progression. Front Endocrinol (Lausanne). 9(552)2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Sharif T, Ahn DG, Liu RZ, Pringle E,
Martell E, Dai C, Nunokawa A, Kwak M, Clements D, Murphy JP, et al:
The NAD(+) salvage pathway modulates cancer cell viability via p73.
Cell Death Differ. 23:669–680. 2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Momeny M, Zakidizaji M, Ghasemi R, Dehpour
AR, Rahimi-Balaei M, Abdolazimi Y, Ghavamzadeh A, Alimoghaddam K
and Ghaffari SH: Arsenic trioxide induces apoptosis in NB-4, an
acute promyelocytic leukemia cell line, through up-regulation of
p73 via suppression of nuclear factor kappa B-mediated inhibition
of p73 transcription and prevention of NF-kappaB-mediated induction
of XIAP, cIAP2, BCL-XL and survivin. Med Oncol. 27:833–842.
2010.PubMed/NCBI View Article : Google Scholar
|