1. Introduction
The investigation of the genetic factors influencing
cancer susceptibility has been increasing in recent years. The
International Human Genome Sequencing Project and the International
HapMap Project have both conducted substantial research into the
location, amount, type and frequency of genetic variants in the
human genome (1-4).
Numerous observational studies examining the association among
polymorphisms in genetic variations and the risk of cancer
development have led to ongoing technology advancements that enable
more rapid and more economical genotyping results (5). A recent genome-wide association study
identified several novel susceptibility loci for prostate cancer
risk, providing new insight into the genetic architecture of this
disease across diverse populations (6). A large cohort study involving
>113,000 women emphasized the contribution of both common and
rare genetic variants to breast cancer susceptibility,
demonstrating the complex genetic architecture underlying breast
cancer risk (7).
Knowledge of the genetic propensity to cancer has
generally improved with the increasing amount of research that has
been performed. The lack of replication has been a major critique
of genetic epidemiology. There have been many ‘false positive’
claims, as evidenced by the failure of numerous research that
attempted to reproduce a statistically significant outcome for a
genetic condition that had already been published (8,9).
Meta- and pooled analyses have been employed to incorporate both
statistically meaningful and non-significant data from several
studies and grade these results according to their precision.
Another significant methodological issue is the scale of these
studies of genetic associations (a proportion of sample size)
(10-12).
The programmed death-ligand 1 (PD-L1), encoded by
the CD274 gene located on chromosome 9p24.1, plays a
critical role in immune regulation by mediating immune checkpoint
pathways that allow tumors to evade immune surveillance. PD-L1
expression is detected in various cells, including
antigen-presenting cells, lymphocytes and epithelial cells, where
its regulated activity helps maintain immune homeostasis during
inflammation (13,14). Toll-like receptors (TLRs),
expressed in immune cells, have been shown to modulate PD-L1
expression by responding to pathogen-associated molecular patterns,
thus influencing the immune microenvironment.
Notably, the aberrant expression of PD-L1 has been
implicated in the pathogenesis of several types of cancer. The
overexpression of PD-L1 in tumors, such as non-small cell lung
cancer is associated with oncogenic mutations, such as those in the
epidermal growth factor receptor (EGFR), highlighting the interplay
between oncogenic signaling pathways and immune evasion mechanisms.
Genetic and epigenetic alterations, including polymorphisms and
mutations in PD-L1 regulatory regions or associated signaling
genes, such as phosphatase and tensin homolog (PTEN) and
anaplastic lymphoma kinase (ALK), may contribute to a
variable PD-L1 expression and consequent differences in tumor
immune escape and patient prognosis (15).
Despite the recognized importance of PD-L1,
comprehensive studies focusing on genetic polymorphisms within the
CD274 gene and their associations with cancer susceptibility
remain limited. Integrating the mechanistic understanding of PD-L1
regulation with genetic variation analyses could provide valuable
insight into individual susceptibility and response to immune
checkpoint therapies.
TLRs, a subtype of non-catalytic receptor that is
widely expressed in APCs and activated by epitope molecular
patterns, have a notable impact on PD-L1 expression (16). The driving oncogenic events in
carcinogenesis may be the cause of PD-L1 overexpression. For
instance, in lung cancer, PD-L1 expression is positively associated
with EGFR mutations in the EGFR (17). Through the unregulated stimulation
of the protein kinase pathway, PD-L1 overexpression is maintained
in enzymatic activity and in PTEN-mutant tumors (18,19).
Through constitutive STAT3 activation, the recombinant genes for
the protein nucleophosmin (NPM) and ALK enhance the expression of
PD-L1 in T-cell lymphoma (20).
Lin et al (21) explored
the genomic and transcriptomic characteristics regulating PD-L1
expression, highlighting the impact of genetic polymorphisms on
PD-L1 and its role across various types of cancer.
Cytochrome P450 (CYP450) enzymes, which include
those from the CYP1, CYP2 and CYP3 families, as well as additional
exogenous and endogenous compounds, metabolize drugs. The key CYP
enzymes involved in the metabolic activation of procarcinogens are
CYP1A1, CYP1A2, CYP1B1, CYP2E1, CYP3A4 and CYP3A5(22).
The majority of CYPP450 enzymes that belong to the
CYP 1, 2, or 3 groups are polymorphic due to gene deletion,
single-nucleotide polymorphisms that occur all alone in
combination, or gene duplications, where mutant alleles result in
discontinued, reduced, modified, or enhanced enzyme activity.
Genetic polymorphisms in CYP enzymes, such as CYP2D6 and CYP2E1
significantly affect enzyme activity and have been linked to
altered cancer susceptibility across diverse populations (23,24).
Phenotyping experiments were utilized to investigate the potential
link between CYP polymorphisms and the risk of developing cancer
before genotyping methods were developed. The delivery of CYP
enzyme substrate and the analysis of the products in plasma and
urine were common procedures used in research. Following the
analysis of the associations among CYP polymorphisms and the risk
of cancer development over the period of a decade in numerous
studies encompassing thousands of patients, the association between
CYP polymorphisms and cancer susceptibility is now understood; the
mechanisms involved include simultaneous genes that code for phase
1 and phase 2 enzymes (25).
Recent studies have shed light on CYP polymorphisms influencing the
risk of cancer development and treatment outcomes. Tan et al
(26) reported on the impact of
CYP2D6 genotypes on breast cancer outcomes and pharmacogenomics,
emphasizing the clinical relevance of CYP2D6 polymorphisms.
Additionally, Jiang et al (27) conducted an updated meta-analysis
linking CYP2E1 polymorphisms with an increased risk of developing
various types of cancer, supporting the role of CYP2E1 genetic
variations in cancer susceptibility.
Numerous biological mechanisms, including bone
metabolism, inborn resistance, proliferation and differentiation
are regulated by the vitamin-D endocrine system (28). Numerous common diseases, such as
cancers, diabetes, cardiac disease, autoimmune disorders, rickets,
as well as other bone diseases, have been firmly related to vitamin
D deficiency by epidemiological and laboratory research (29-32).
The biologically highly active naturally occurring metabolite of
vitamin D, known as 1,25-dihydroxyvitamin D3 (1,25(OH)2D3,
calcitriol), has been found to control the proliferation and
differentiation of a range of cell types, including cancer cells
(33-35).
Research has also demonstrated that angiogenesis, cancer growth and
cell death are all regulated in cancer (36-38).
Previous research has revealed the existence of intracellular
enzyme 25(OH) D3-1-hydroxylase (CYP27B1) activity in a wide range
of cell types, including macrophages, keratinocytes, prostate and
colon cancer cells (39,40). It has been shown that a number of
tissues locally synthesize 1, 25(OH) 2D3. Recent research has
uncovered that vitamin D can be activated via an alternative
pathway by the steroidogenic enzyme CYP11A1. This pathway
complements the classical activation involving CYP27B1 and expands
the understanding of vitamin D metabolism beyond traditional
mechanisms. The CYP11A1-mediated conversion produces novel
secosteroids with potential biological activities relevant to
cellular proliferation, differentiation, and immune modulation in
cancer and other diseases. This alternative pathway adds complexity
to vitamin D regulation and may have implications for cancer
susceptibility and therapy (41,42).
The nucleotide excision repair (NER) system is one
of the main mechanisms by which cells defend themselves from
genotoxic damage, such as that caused by UV radiation and exposure
to chemical carcinogens. Human syndromes, such as trichotillomania,
Cockayne syndrome and xeroderma pigmentosum (XP) are caused by NER
system anomalies (43,44). An exceptionally high sensitivity to
UV light and a comparatively high chance of developing skin cancer
are two features of the heritable human condition known as XP. The
UV sensitivity of patients with XP led to the initial connection
between the illness and DNA repair (45). Based on the ability of distinct
cell types to complement UV sensitivity, eight XP-complementation
groups were identified (46,47),
and the genes encoding the various complementation groups have also
been identified (48-50).
UV radiation plays a dual role in human homeostasis, with both
harmful and beneficial effects that extend beyond simple DNA
damage. Beyond its capacity to induce genotoxic lesions repaired by
the nucleotide excision repair system, UV radiation also plays a
central homeostatic role in neuro-immuno-endocrine regulation.
Recent insights highlight how UV exposure modulates systemic
physiology by affecting neural, immune and endocrine pathways, thus
contributing positively to body regulation and immune surveillance.
This dual nature of UV radiation underscores the complexity of its
impact on health and carcinogenesis, balancing its carcinogenic
potential against vital homeostatic functions (51).
In addition to genetic polymorphisms, epigenetic
modifications, such as DNA methylation play a crucial role in
regulating gene expression and influencing cancer susceptibility.
DNA methylation involves the addition of a methyl group to cytosine
residues, typically at CpG dinucleotides, leading to changes in
chromatin structure and gene activity without altering the DNA
sequence itself. Aberrant DNA methylation patterns can result in
gene silencing or activation that contributes to carcinogenesis,
affecting tumor suppressor genes, oncogenes and DNA repair genes.
The interplay between genetic polymorphisms and epigenetic
alterations is increasingly recognized as fundamental to
understanding the risk of cancer development, progression and the
therapeutic response. Therefore, integrating both genetic and
epigenetic perspectives provides more comprehensive insight into
cancer susceptibility and personalized medicine approaches.
Taken together, the diversity of biological systems,
such as immune surveillance (antigen-presenting cells and
checkpoint proteins), metabolic detoxification (cytochrome P450
enzymes), cellular signaling (vitamin D pathways), DNA repair
(nucleotide excision repair system), and epigenetic regulation (DNA
methylation) forms an intricate network maintaining cell
homeostasis and genomic integrity. Genetic polymorphisms within the
genes governing these systems can significantly alter their
functions, leading to variability in how individuals respond to
environmental exposures, carcinogens and intrinsic cellular
stresses. These variations may influence cancer susceptibility by
affecting immune evasion, the activation or detoxification of
carcinogens, DNA damage repair efficiency, and the epigenetic
regulation of oncogenes and tumor suppressors. Thus, understanding
the complex interplay among these elements is essential for
elucidating the multifactorial nature of cancer risk and
identifying genetic and epigenetic markers for cancer prevention,
diagnosis and personalized therapy.
2. Various genes and genetic
polymorphisms
The following chapter provides an overview of genes
involved in key cellular pathways; it is important to emphasize
that the relevance of these genes to the risk of cancer development
is principally determined by the specific genetic polymorphisms
they harbor. A detailed understanding of the normal function of
each gene provides the necessary foundation to appreciate how
alterations in the genetic sequence, such as single nucleotide
polymorphisms (SNPs), insertions, deletions and copy number
variations, can influence an individual's susceptibility to cancer.
Throughout this section, gene variants with established or
potential links to cancer risk will be highlighted. It should be
noted, however, that some gene polymorphisms discussed in the
subsequent sections affect human health through mechanisms that may
extend beyond cancer alone; these examples are included to
underscore both the complexity and the far-reaching consequences of
genetic variation in human populations. This integrative approach
aims to provide a nuanced perspective, facilitating a comprehensive
understanding of the multifactorial risk factors contributing to
cancer and related diseases.
Within the broad family of CYP enzymes, several
members such as CYP1A1, CYP1A2, CYP1B1, CYP2E1, CYP2D6, CYP3A4 and
CYP3A5 participate in the metabolism of carcinogens and drugs.
Among these, CYP1A1 and CYP2E1 have emerged as particularly
critical, due to their significant roles in activating
procarcinogens commonly found in tobacco smoke and other
environmental toxins. Genetic polymorphisms in these enzymes
influence enzymatic activity and expression levels, thereby
modulating individual susceptibility to various cancers. While all
CYP enzymes contribute to xenobiotic metabolism, the extent of
evidence linking CYP1A1 and CYP2E1 variations to the risk of cancer
development is more substantial, warranting focused research
attention.
CYP1A1
On human chromosome no. 15, the CYP1A1 gene
is only activated in organs other than the liver, such as the
lungs. The enzyme CYP1A1 converts polycyclic aromatic
compounds, which are present in cigarette smoke, into hazardous
arene oxide that can result in DNA mutation and cancer (47,48).
Since CYP1A1 is not expressed in the human liver, it is
dubious whether human studies on animals have any application
(49). There are several
polymorphic CYP1A1 alleles known. A point mutation causes an
MspI restriction fragment-length polymorphism in the 3'
non-coding region (RFLP). The heme-binding domain of CYP1A1
contains the exon 7 polymorphism, which increases the inducibility
of the enzyme. Recent research suggests that the MspI, as
well as exon 7 mutation may increase the risk of developing lung
cancer, although Asians are more likely to be affected. In the 3'
non-coding region of CYP1A1, a second MspI RFLP is
only present in individuals of African origin. Its connection to
the risk of developing lung cancer remains unclear. According to
recent studies, nicotine can boost pulmonary CYP1A1 activity
(50). This research has important
implications for how sensitive humans are to acquiring cancer, even
if it is currently only applicable to rats. First, nicotine most
definitely contributes to the induction of CYP1A1 by
cigarette smoke. Second, the emergence of smoking cessation aids
raises serious concerns about the possibility for nicotine
replacement medications to increase CYP1A1 activity and
hence trigger the bio-activation of carcinogens (51). Last but not least, it remains
unknown how CYP1A1 functions in the presence of nicotine and
an allele variation.
CYP2D6
CYP2D6 (debrisoquine hydroxylase) is
considered to be involved in the metabolism of 25% of all
prescribed medications (52). At
least 29 allelic variations of CYP2D6 have been identified,
and this has been linked to significant inter-individual
variability in drug metabolism (53).
In ~6% of Caucasians, deletions, abnormal splicing
and gene duplication result in the absence of functional
CYP2D6. It is interesting that CYP2D6 cannot be
induced. It is understood that CYP2D6 activates metabolism
(54).
Smoke from cigarettes contains
4-(methylnitrosamine)-1- (3-pyridyl)-1-butanone, which is
considered to cause cancer and is a factor in the development of
human lung adenomas. Extensive metabolizers may be more susceptible
to lung cancer (54).
E-Cadherin
The CDH1 gene, that is found on chromosome
16q22.1, is responsible for producing the 120 kD single
transmembrane glycoprotein known as E-cadherin. One intracellular
and five external domains are present. It converses with catenins.
The development and maintenance of tissue architecture, cell
polarity, intracellular signaling and intercellular adhesion depend
on this protein. It plays a critical role in the formation of
sticky junctions in epithelial cells. The lack of E-cadherin
directly affects essential cellular processes including motility.
Additionally, it has been demonstrated that its expression
decreases typically happen during tissue metastasis. An association
has been found between an increased aggressive behavior and the
decreased expression of E-cadherin. The frequency of OSCC
E-cadherin gene hypermethylation varies between 7 and 46% (55).
PTEN
The tumor-suppressor gene, PTEN, is located
on chromosome 10q23.3. It is anticipated that essential cellular
functions such as survivability, differentiating, proliferating,
apoptosis and invasion are affected by the lack of expression
Ras/phosphoinositide 3-kinase (PI3K)/Akt, which lack control over
the signal transduction that control apoptosis and migration, and
also play a crucial role in the survival, proliferation and
metastasis of tumor cells. Due to mutations or epigenetic
modifications, PTEN is frequently lacking in a variety of
cancer types. Furthermore, it has been demonstrated that
endometrial cancer, gastric cancer, non-small cell lung carcinoma
and cervical cancer all exhibit the methylation of the PTEN
promoter CpG islands (56). The
functions, regulation and implications of PTEN polymorphisms in
cancer were comprehensively reviewed by Song et al (57), detailing the role of PTEN as a key
tumor suppressor. Han et al (58) further performed a meta-analysis of
PTEN mutations, associating genetic alterations with prognosis
across multiple cancer types.
p53
Cell cycle progression, cellular differentiation,
DNA repair and apoptosis are some of the key cell activities that
the TP53 gene, also known as p53, is involved in. It
is located on chromosome 17p13.1. p53 levels increase in response
to endogenous or exogenous stress, which stops the cell cycle and
enables DNA repair. Genomic instability results from p53 loss of
function, which affects how cells react to stress or damage. With a
frequency ranging from 25 to 69%, p53 is mutated in the
majority of human malignancies, including oral tumors (59,60).
In addition to this, p53 frequently exhibits a decrease of function
brought on by epigenetic rather than genetic processes.
Comprehensive analyses of TP53 variations across multiple human
cancers were provided by Bouaoun et al (61), utilizing database and genomic data
to reveal new insight into TP53 mutation patterns. A further
mechanistic understanding of mutant p53 roles in cancer
pathogenesis was provided by Mantovani et al (62), elucidating its function as a cancer
cell guardian.
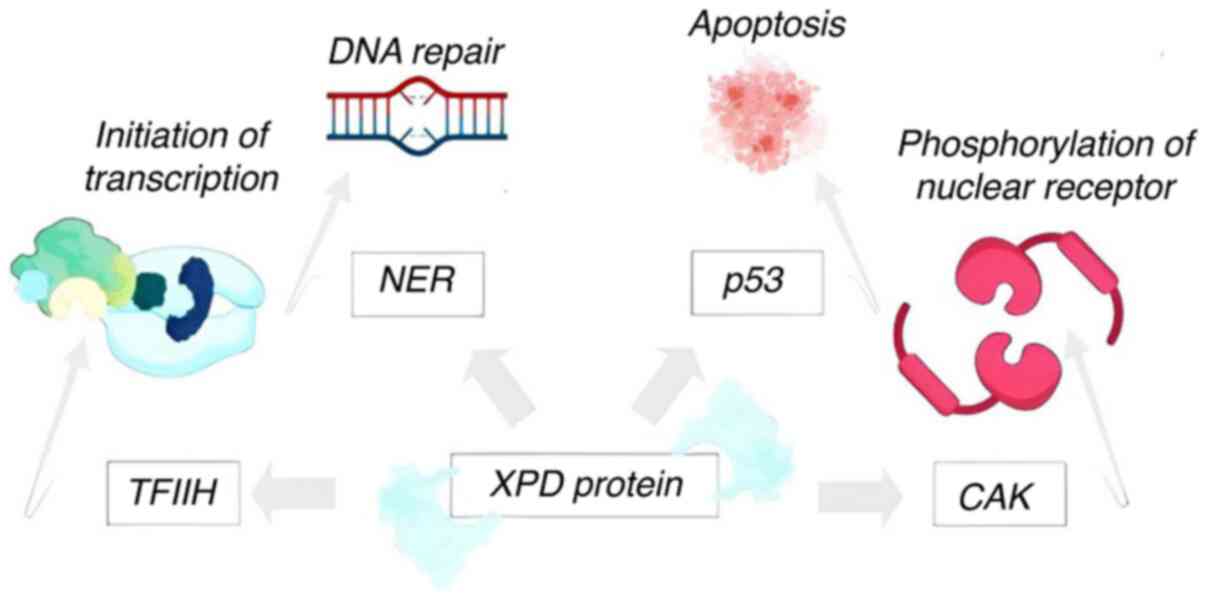
XPD
TFIIH phosphorylates a wide range of substrates,
including nuclear hormone receptors such as RAR or ER,
transcription activators and RNA polymerase II (63). Additionally, TFIIH is
present as a nine-subunit complex, a transcriptionally active core
TFIIH, and a CDK-activating kinase (CAK) complex (Fig. 1). The XPD protein is a component of
all three of these complexes (64). The activity of these complexes can
be decreased by mutations in the XPD gene, which can cause issues
with transcription, the apoptotic response, repair, or, most
likely, hormonal function. All of these deficiencies result in
syndromes that are associated with immature sexual development,
skeletal abnormalities, mental impairment, and, in the case of the
majority of patients with XPD mutations, a high propensity for
cancer. Only a limited amount of research has been performed into
the connection between XPD mutations and DNA repair capacity, as
determined by the biological tests mentioned in the literature. The
Lys/Lys codon 751 XPD genotype was previously linked to a decreased
repair of X-ray-induced DNA damage in a brief study on 31 women at
risk of developing breast cancer (65). More chromatid aberrations are
present in individuals with the wildtype Lys allele than in those
carrying one or more Gln alleles (65).
3. Methods for analyzing genetic
polymorphisms
Association studies are most frequently used to
determine whether polymorphisms contribute to genetic
susceptibility or progression. As a result, the focus is on
variables that determine the effectiveness of associations. If a
specific allele exhibits a greater frequency in cases compared to
controls, that polymorphism is then considered to be linked to the
disease (66). Researchers compare
individuals with extreme phenotypes when analyzing polymorphisms as
contributing factors to the course of disease, rather than diseased
individuals with unaffected controls. There are three possibilities
if a substantial association is found: Either the polymorphism is
at the locus of interest, it is in genetic linkage [namely linkage
disequilibrium (LD)] with the locus, or confounding factors are
involved.
When alleles from two different genetic loci
co-occur more frequently than would be predicted according to their
respective allelic frequencies, a population is considered to
exhibit LD. Possible sources of LD include early mutation, founder
effects and selection. Population admixture, which occurs when
groups that have been separated for a long time unite to form a
hybrid population, is another possible source of LD (67). The resulting LD can be prolonged
beyond distances typically observed in populations with greater
stability, depending on the sort of mixing. The most precise
estimates of LD in an outbred population indicate that LD is
unlikely to span distances >1-2 centimorgans (cM), or ~1-2
million base pairs, whereas LD may occur across 10-fold that
distance in an inbred group, such as the Hutterites (68).
Confounding variables need to be taken into account,
particularly when polymorphisms reported in one study are absent in
another ethnic group. One perplexing aspect is population
stratification. This could be brought on by unequal ethnic
admixture, such as the presence of Caucasians in the gene pool of
an African-American community. Fortunately, this issue can be
resolved by taking great precautions during the analysis or
research plan phases. Association studies based on families are
specifically created to take into account the genetic background
that may add confounding variables, and they include genotyping of
affected people, their parents, and/or their unaffected siblings
(69). These types of
investigations frequently employ the transmissions disparity test
(TDT), as well as the haplo-type relative risk test statistics
(HRR). The TDT compares how frequently each allele is transferred
from a heterozygous parent to a child who has the disease. Similar
to the TDT, the HRR analyses genetic transmission (haplotype) as
compared to allele transmission (70).
The decision of which phenotype to explore is
crucial in genetic polymorphism investigations. In fact, the
presence of numerous phenotypes in the case sample has hampered
much research that analyzing genetic polymorphisms. For instance,
research on asthma has used patient samples from patients with
mild, severe, adult-onset, intrinsic and extrinsic asthma (71). Studying a more precisely defined
intermediate phenotype can significantly enhance studies that aim
to ascertain whether there is a link between a polymorphism and
disease. Total IgE and bronchial hyper responsiveness are examples
of intermediate phenotypes in the case of asthma. Another factor to
take into account is the possibility that the genes involved in
illness progression may not be the same genes responsible for
disease susceptibility. It may be helpful to restrict the sample to
those with a particular stage or severity of disease. In a previous
study, to evaluate potential genes for the disease, the researchers
focused on patients with severe early-onset chronic obstructive
pulmonary disease (71).
The evaluation of DNA polymorphisms close to or
within putative genes is the only application of association
studies. Linkage analysis employing families or affected siblings
is necessary to carry out a genome screen to look for candidate
genes. Although linkage analysis is rigorous and uncovers genes
that significantly affect disease susceptibility, it has very
limited power and will miss genes that merely increase the risk of
mild to moderate disease. For instance, hundreds to thousands of
families would need to be typed if a disease susceptibility allele
increased disease risk by 2-fold relative to the wild-type allele,
which may not be a realistic sample size (72). Although linkage analysis detects
connections over considerably larger genomic regions (thousands of
base pairs), association studies have a stronger power (millions of
base pairs). Including current technology, a genome scan with
association studies would need tens of thousands of markers. The
majority of lung diseases need some type of environmental trigger
before they may become visible. Given that genetic susceptibility
only accounts for a small part of illness variation, failure to
consider environmental factors can drastically degrade gene-finding
research for the majority of complex disorders. Furthermore, genome
screening or association studies carried out on populations that
were neither chosen nor stratified according to their environmental
exposure could only be able to find genes whose environmental
exposure is common in that group. For instance, investigating a
randomly chosen sample of asthmatics from the Midwest region of the
USA would be able to discover genes crucial for regulating the
response to house dust mites, although possibly not with absolute
certainty (73).
Gene-environment interactions can take on many
different shapes, such as distinct exposure risk implications based
on the genotype of an individual or diverse gene risk implications
based on the exposure of an individual. Biological and statistical
interactions are the two main interactions. The coefficient of the
product term of the genetic and environmental risk variables
represents a statistical risk factor interaction, and the
interaction is quantified as a divergence from a multiplicative
model (gene and environment). This approach is arbitrary, relies on
models, and may overlook biological synergy or interaction. The
biologic interaction paradigm states that when two factors cause a
disease to start, they interact. Occasionally, this
co-participation could stand out as a divergence from an additive
model (74).
4. SNPs in the human population
SNPs are single nucleotide variations in the genomic
DNA that occur at different positions in different individuals in a
community (75). Genome-wide
datasets are more frequently used in the drug development process,
as well as to identify molecular pathways and networks underlying
complex disorders (Table I)
(76-79).
In particular, functional pathway analysis of genomic data provides
the possibility of greater capability for discovery and organic
links to biological phenomena. SNPs are the consequence of single
base-pair variations (substitutions or deletions) caused by point
mutations in chromosome sequences, and account for a large portion
of the genetic variation found in the human genome. Finding SNPs in
a genome can be achieved in a variety of laboratories and via
computational mechanisms; however, they all involve comparing the
same DNA segment from various individuals or haplotypes.
 | Table IMethods of selecting SNPs. |
Table I
Methods of selecting SNPs.
| Methods | Advantages | Limitations | (Refs.) |
|---|
| Pathway gene
method | It is simple to
study a subset of SNP based on a description of the pathways
relating to the drug's pharmacokinetics and mechanism of action.
Clinically, general vulnerability is observed in complex genetic
disorders. | The pathway gene
approach will result in fewer false-positive findings than the
genome wide approach because of the disadvantage of multiple
testing. | (76,77) |
| Candidate gene
method | Functional SNPs are
those whose genetic variant affects how a protein functions. This
technique has resulted in the identification of a sizable number of
pertinent SNPs in pharmacogenetics. | As the majority of
complex traits are not considered to be monogenetic, selecting SNPs
using this strategy will frequently result in a limited explanation
of variation in medication response. | (78,79) |
| Genome-wide
method | This strategy may
identify unexpected SNPs linked to drug response. New associations
between SNPs and medication response have been found in genome-wide
association investigations, and complicated features can be
studied, while taking into account polygenetic variation. | In identifying a
related SNP, the discrepancy between type I errors (false positive
findings) and eventually type II faults (false negative results)
may create some issues. | (78,79) |
SNPs can be located using expressive sequence tags,
which are created by single-run sequencing of cDNAs obtained from
various individuals and the assembly of overlapping sequences for
the same region. This allows for the discovery of novel SNPs.
Depending on whether they are located in regions of the genome that
regulate genes, non-coding SNPs can be categorized. A number of
complex disorders may be caused by quantitative discrepancies in
gene products rather than qualitative differences. Based on whether
they alter the amino acid sequences of the protein that the altered
gene encodes, coding SNPs can be categorized. By their impact on
protein structure, modifications that change protein sequences can
be categorized (80).
Genome-wide association studies have emerged as a
crucial method for identifying genes that predispose to complicated
disorders. With the aid of population-based information like as
allele frequency, LD and recombination rates, researchers may
perform genome-wide association analysis on millions of SNP
markers. Some of the discrepancies in association results between
populations for particular traits of interest can be explained by
HapMap data, such as population-specific common variants and LD
blocks (81).
5. VNTR polymorphisms in the human
population
CYP2E1 is a key enzyme found in the microsomal
ethanol oxidation system. It belongs to the CYP superfamily and is
primarily located in the membranes of the endoplasmic reticulum.
CYP2E1 plays a crucial role in the metabolism of various
hydrophobic toxic compounds (82-84).
Additionally, it contributes to the conversion of certain
pro-carcinogens and drugs into highly reactive metabolites.
The activation of N-nitrosamines, which are present
in tobacco smoke, foodstuffs, and certain industrial and endogenous
carcinogens, is facilitated by CYP2E1. Furthermore, this enzyme can
generate highly reactive compounds, such as superoxide anion
radical (O2-), singlet oxygen (O2), hydrogen
peroxide (H2O2) and hydroxyl radical (OH·) by
reducing molecular oxygen. These reactive species are capable of
causing DNA damage and promoting carcinogenesis (85-87).
The human CYP2E1 gene is situated on chromosome 10q26.3 and
consists of 9 exons and 8 introns. The expression of the
CYP2E1 gene can be regulated at multiple levels, including
transcription, translation, mRNA stability and protein degradation.
As with other CYP genes, CYP2E1 exhibits various polymorphic sites
in its 5'-flanking region, introns, and transcribed gene regions.
One notable polymorphism is a variable number tandem repeat (VNTR)
sequence located ~2.0 kilo base pairs upstream of the transcription
start site (88-90).
The activity of the CYP2E1 enzyme exhibits
significant variability among different ethnic groups, influenced
by both environmental and genetic factors, such as polymorphisms.
Polymorphic CYP genes can lead to differences in the ability to
metabolize, detoxify, or activate various substances. Several
studies have indicated that certain polymorphic genes, in
conjunction with alcohol consumption, play a role in the
development of specific types of cancer. Although alcohol itself is
not a carcinogen, it can function as a co-carcinogen by amplifying
the effects of other chemicals that are activated by enzymes, such
as CYP2E1. Research has demonstrated that CYP2E1 is highly
expressed in the liver and pancreas following ethanol ingestion.
The production of acetaldehyde during ethanol oxidation may
directly cause cell damage through the generation of reactive
oxygen species (91-95).
Furthermore, it is widely known that certain tumors
have a hereditary component, and environmental factors associated
with specific habits can increase the risk of tumor occurrence.
6. Using a different approach to identify
human gene polymorphisms
The function of biotransformation enzymes with
reference to occupational health exposure is to provide effective
purification of endogenous or exogenous substances by particular
biochemical pathways. These turn harmful molecules into inactive
compounds, which are then eliminated in urine, preventing the
buildup of metabolites and injury to the human body (96). Although the scanning of specific
gene polymorphisms by molecular biology laboratories is the optimal
approach to determine each the susceptibility of each study
participant, the willingness of study participants to provide the
biosample is essential to move forward with the genetic analysis.
There are some issues, such as the unwillingness of study
participants to consent to venipuncture or, more generally, to the
collection of biopsies, either as they are simply unaccustomed to
the procedure as they consider it to be an invasive and painful
technique, or because they are afraid of the possible outcomes of
the analysis (97). However, the
evaluation of gene polymorphisms does not provide any diagnostic
information regarding the propensity of an individual to develop a
specific disease. In comparison to collecting urine samples,
collecting blood samples may be more difficult. The lack of
language proficiency, the difficulties in communicating, and the
study participants varied cultures, customs, dietary preferences,
and religious beliefs could all play a role in this. To combat this
critical issue and obtain ethnic-specific genotype information
without using laboratory analysis, a publicly available online
library (http://grch37.ensembl.org/Homosapiens/Variation)
appears to contain a tentative catalogue of the majority of
genotype and allele frequencies of several ethnic groups. It is
possible to collect the genetic profiles of many ethnic groups
using this resource, which aids in the prediction and
identification of population-specific susceptibilities in
silico study. This model was created to assess the risk level
of the homozygous variation and heterozygous genotype in relation
to the world population in four macro-groups that includes
Africans, Eastern Asians, South Asians and Europeans. The principal
component analysis statistical technique serves as the foundation
of the model (98). It is intended
to identify the crucial vulnerabilities in the polymorphisms of
genes linked to three main functional biochemical processes,
including detoxification, oxidative stress and DNA repair,
following exposure to the harmful compounds. The SNPs were selected
based on their exposure to harmful and cancer-causing chemicals
that are frequently present in manufacturing plants and shipyards
(99).
7. Benefits of including polymorphisms in
studies on health-related effects
Incorporating polymorphisms opens up a wide range of
intriguing options for investigating the health effects of exposure
to toxins and toxicants in the environment. It is possible to
identify various risk ranges in subgroups of individuals who were
exposed by stratifying a health result or biomarker in accordance
with the relevant genotype (or phenotype) (100), as illustrated in Fig. 2. An implication that corresponds to
the average risk for both fast and slow acetylators is shown by
research evaluating the probability of bladder cancer linked to
exposure to aromatic amines (100). This estimate does not imply that
aromatic amines are as significant etiological factors for
sub-populations or as powerful carcinogens as a stratified study
might. For routine exposures, to food ingredients or carbon
emission, for example, whose relation to a disease consequence is
normally small, effect dilution may be especially crucial. Second,
proof of impact change by genotype provides insight into the
fundamental biologic mechanism of cytotoxicity or carcinogenicity
when substrates or targets of possible genetic variants are
identified as likely causal agents (101). Lipopolysaccharide (LPS), a
constituent of particulates in rural regions and sometimes known as
an endotoxin, may have a negative impact on lung function metrics.
Kelada et al (101)
demonstrated that the response to LPS varied by TLR4 genotype. The
TLR4, which is encoded by TLR4, binds LPS and begins a signaling
pathway that causes lung inflammation. According to their findings,
although individuals with the mutated TLR4 genotype may be more
susceptible to an inflammatory process, they may not be as
susceptible to an inflammation of the lungs brought on by LPS.
These results may contribute to answering the difficult question of
whether particulates matter component(s) is/are accountable for the
multitude of documented health consequences, particularly in rural
areas where LPS levels are substantial. The development of drugs or
dietary interventions that postpone the start or progression of
disease may be made possible by the increased understanding of
pathologic pathways gained via combined epidemiological and
toxicological investigations. Oltipraz [OPZ;
5-(2-pyrazinyl)-4-methyl-1, 2-dithiole-3-thione] is an example of a
medication that activates phase II XMEs, specifically the GSTs
(102). Aflatoxin B1 can cause
liver cancer in rats, according to early research. Research has
also revealed that administering OPZ to patients significantly
improved the clearance of a phase II substance known as
aflatoxin-mercapturic acids (103). Research suggests that OPZ may
function by competitively inhibiting CYP1A2, preventing the
activation of aflatoxin. Ultimately, hypothesis-based
epidemiological research and the knowledge of aflatoxin
biotransformation routes from investigations on human tissue grown
in vitro and animal models has helped to establish a
chemoprevention method for aflatoxin-induced hepatocellular
carcinoma. Studies on the consequences of exposure to controlled
environmental pollutants that take genetic sensitivities into
consideration will increase our understanding of the range of human
genetic variation in response to these pollutants. Genes that may
be associated with susceptibility should be included in studies
designed to examine the effects of these substances. By replacing
the standard default assumptions (i.e., uncertainty factor of 10)
with more accurate estimates of human variability, the risk
assessment may be improved. As a result, acceptable exposure levels
may be redefined, improving overall public health protection and
industry regulation. Although this benefit has been promoted for
some time, there is still no concrete illustration of how it may be
achieved, particularly in light of the myriad social, legal, or
ethical issues that surround use of genetic data (104). Preventive efforts on those who
are genetically sensitive to disease has started in the
environmental health field, with a focus on the intrinsically
complicated ethical, legal and social issues (105).
8. Synopsis
Polymorphisms and DNA methylation both play critical
roles in the development of cancer. Polymorphisms are DNA sequence
variations that occur when a single nucleotide in the DNA sequence
is altered. These variations can lead to changes in the function of
the gene or the expression of the gene product, which can result in
cancerous changes to cells. Epigenetic changes, such as DNA
methylation are biochemical processes wherein enzymes add a methyl
group to the DNA strand at 5th position of the cytosine to be
precise. These methylation sites can control gene expression by
either silencing a gene or increasing its activity depending upon
the number of methylation sites present. DNA methylation can also
alter splicing patterns, downregulate or ‘turn off’ gene
expression, which can lead to cancerous changes in cells. Overall,
both polymorphisms and DNA methylation function as key regulators
of gene expression and can therefore exert significant effects on
the development of cancer (106).
However, they do differ in their mechanisms of action.
The role of genetic polymorphisms within different
genes in the development and risk of cancer development is a
complex and multifactorial process. While genetic polymorphisms can
contribute to the susceptibility of an individual to cancer, they
are not the sole determinant. Environmental factors, lifestyle
choices and other non-genetic factors also play a crucial role.
Numerous studies have identified specific genetic polymorphisms
that are associated with an increased risk of developing cancer.
For example, variations in the BRCA1 and BRCA2 genes
have been linked to an increased risk of developing breast and
ovarian cancer, while variations in the TP53 gene have been
linked to an increased risk of developing several types of cancer,
including breast, ovarian and colorectal cancer (107,108). However, it is important to note
that the presence of a genetic polymorphism does not necessarily
mean that an individual will develop cancer. A number of
individuals with these genetic variations never develop cancer, and
numerous individuals without these genetic variations do develop
cancer.
Emerging evidence demonstrates that tumors possess
the capacity not only to modulate their local microenvironment, but
also to dysregulate systemic body homeostasis through
neuroendocrine pathways. This tumor-driven autoregulation
interferes with normal physiological processes, such as metabolism,
immune function and stress response, effectively hijacking the
neuroendocrine system of the body to support cancer progression and
evade host defenses. Understanding this intricate interplay reveals
cancer as a systemic disease with extensive body-wide impacts,
highlighting potential therapeutic targets to restore homeostatic
balance and counter tumor-driven systemic disruption (109).
The study of genetic polymorphisms and cancer risk
is an active area of research, and new discoveries are continuously
being made. As the understanding of the genetic basis of cancer
increases, it is likely that the identification of novel genetic
variations that are associated with an increased risk of cancer
development will be achieved, and the development of novel
strategies for the prevention and treatment of cancer may be
possible.
9. Limitations and future challenges
While the present review comprehensively covers the
role of genetic polymorphisms across various genes associated with
susceptibility to cancer, several limitations should be
acknowledged. First, the heterogeneity of study designs, sample
sizes and populations in the original research contributes to
variability and potential inconsistency in reported associations. A
number of genetic association studies face challenges, such as
population stratification, limited replication and publication
bias, which affect the strength and generalizability of
conclusions. Second, the complex interplay between genetic
polymorphisms, epigenetic modifications and environmental factors
remains incompletely understood, limiting the ability to fully
elucidate causal mechanisms. Third, the functional characterization
of numerous polymorphisms is still lacking, impeding translation to
clinical applications. Finally, rapidly evolving genomic
technologies and the emergence of multi-omics approaches
necessitate continuous updates to maintain a current and holistic
perspective on genetic risks in cancer. Future studies
incorporating large, well-characterized cohorts with integrated
genomic, epigenetic, and environmental data are essential to
overcome these limitations and advance personalized cancer
prevention and therapy.
10 Conclusion
In conclusion, DNA methylation and genetic
polymorphisms both have a major impact on the occurrence and risk
of developing cancer. Genetic polymorphisms are differences in DNA
sequence that may have an impact on gene expression and function,
possibly causing malignant cell alterations. Contrarily, DNA
methylation is an epigenetic change that can influence gene
expression by activating or silencing genes, having an effect on
the onset of cancer. New findings are continuously being made as a
result of ongoing research into genetic variants and the risk of
developing cancer. Additional genetic variants linked to the risk
of developing cancer will probably be discovered as the knowledge
of the genetic basis of cancer increases. This knowledge could
potentially lead to the development of personalized strategies for
cancer prevention and treatment based on the unique genetic makeup
of an individual.
Acknowledgements
The authors gratefully acknowledge the Galgotias
University (Greater Noida, India) for providing the academic
resources and valuable technical assistance during the preparation
of the present review.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Author's contributions
RM and NK conducted a significant portion of the
literature search and drafted the manuscript. GS contributed to
editing specific sections of the manuscript. SPS edited and
proofread the manuscript. AKJ contributed to the initial conception
and scope of the review, provided critical review and feedback on
the manuscript. All authors have read and approved the final
version of the manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lander ES, Linton LM, Birren B, Nusbaum C,
Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al:
Initial sequencing and analysis of the human genome. Nature.
409:860–921. 2001.PubMed/NCBI View
Article : Google Scholar
|
|
2
|
Venter JC, Adams MD, Myers EW, Li PW,
Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al:
The sequence of the human genome. Science. 291:1304–1351.
2001.PubMed/NCBI View Article : Google Scholar
|
|
3
|
International Human Genome Sequencing
Consortium: Finishing the euchromatic sequence of the human genome.
Nature. 431:931–945. 2004.PubMed/NCBI View Article : Google Scholar
|
|
4
|
International HapMap Consortium: The
international HapMap project. Nature. 426:789–796. 2003.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Lin BK, Clyne M, Walsh M, Gomez O, Yu W,
Gwinn M and Khoury MJ: Tracking the epidemiology of human genes in
the literature: The HuGE published literature database. Am J
Epidemiol. 164:1–4. 2006.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Schumacher FR, Al Olama AA, Berndt SI,
Benlloch S, Ahmed M, Saunders EJ, Dadaev T, Leongamornlert D,
Anokian E, Cieza-Borrella C, et al: Association analyses of more
than 140,000 men identify 63 new prostate cancer susceptibility
loci. Nat Genet. 50:928–936. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Breast Cancer Association Consortium.
Dorling L, Carvalho S, Allen J, González-Neira A, Luccarini C,
Wahlström C, Pooley KA, Parsons MT, Fortuno C, et al: Breast cancer
risk genes-association analysis in more than 113,000 women. N Engl
J Med. 384:428–439. 2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ioannidis JP, Ntzani EE, Trikalinos TA and
Contopoulos-Ioannidis DG: Replication validity of genetic
association studies. Nat Genet. 29:306–309. 2001.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Morgan TM, Krumholz HM, Lifton RP and
Spertus JA: Nonvalidation of reported genetic risk factors for
acute coronary syndrome in a large-scale replication study. JAMA.
297:1551–1561. 2007.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Dickersin K and Berlin JA: Meta-analysis:
State-of-the-science. Epidemiol Rev. 14:154–176. 1992.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Mosteller F and Colditz GA: Understanding
research synthesis (meta-analysis). Annu Rev Public Health.
17:1–23. 1996.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Morris RD: Meta-analysis in cancer
epidemiology. Environ Health Perspect. 102 (Suppl 8):S61–S66.
1994.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Chen J, Jiang CC, Jin L and Zhang XD:
Regulation of PD-L1: A novel role of pro-survival signalling in
cancer Ann. Oncol. 27:409–416. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Bardhan K, Anagnostou T and Boussiotis VA:
The PD1:PD-L1/2 pathway from discovery to clinical implementation.
Front Immunol. 7(550)2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wang Q, Lin W, Tang X, Li S, Guo L, Lin Y
and Kwok HF: The roles of microRNAs in regulating the expression of
PD-1/PD-L1 immune checkpoint. Int J Mol Sci.
18(2540)2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Keir ME, Butte MJ, Freeman GJ and Sharpe
AH: PD-1 and its ligands in tolerance and immunity. Annu Rev
Immunol. 26:677–704. 2008.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Tang Y, Fang W, Zhang Y, Hong S, Kang S,
Yan Y, Chen N, Zhan J, He X, Qin T, et al: The association between
PD-L1 and EGFR status and the prognostic value of PD-L1 in advanced
non-small cell lung cancer patients treated with EGFR-TKIs.
Oncotarget. 6:14209–14219. 2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Cretella D, Digiacomo G, Giovannetti E and
Cavazzoni A: PTEN Alterations as a Potential Mechanism for Tumor
Cell Escape from PD-1/PD-L1 Inhibition. Cancers (Basel).
11(1318)2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Suzuki A, Nakano T, Mak TW and Sasaki T:
Portrait of PTEN: Messages from mutant mice. Cancer Sci.
99:209–213. 2008.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Marzec M, Zhang Q, Goradia A, Raghunath
PN, Liu X, Paessler M, Wang HY, Wysocka M, Cheng M, Ruggeri BA and
Wasik MA: Oncogenic kinase NPM/ALK induces through STAT3 expression
of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad
Sci USA. 105:20852–20857. 2008.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Lin H, Wei S, Hurt EM, Green MD, Zhao L,
Vatan L, Szeliga W, Herbst R, Harms PW, Fecher LA, et al: Host
expression of PD-L1 determines efficacy of PD-L1 pathway
blockade-mediated tumor regression. J Clin Invest. 128:805–815.
2018.PubMed/NCBI View Article : Google Scholar : Erratum in: J Clin
Invest 128: 1708, 2018.
|
|
22
|
McDonnell AM and Dang CH: . Basic review
of the cytochrome p450 system. J Adv Pract Oncol. 4:263–268.
2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chan CWH, Li C, Xiao EJ, Li M, Phiri PGM,
Yan T and Chan JYW: Association between genetic polymorphisms in
cytochrome P450 enzymes and survivals in women with breast cancer
receiving adjuvant endocrine therapy: a systematic review and
meta-analysis. Expert Rev Mol Med. 24(e1)2022.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Datkhile KD, Durgawale PP, Gudur RA, Gudur
AK and Patil SR: CYP2D6 and CYP2E1 gene polymorphisms and their
association with cervical cancer susceptibility: A hospital based
case-control study from South-Western Maharashtra. Asian Pac J
Cancer Prev. 23:2591–2597. 2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Agundez JAG: Cytochrome P450 gene
polymorphism and cancer. Curr Drug Metab. 5:211–224.
2004.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Tan EY, Bharwani L, Chia YH, Soong RCT,
Lee SSY, Chen JJC and Chan PMY: . Impact of cytochrome P450 2D6
polymorphisms on decision-making and clinical outcomes in adjuvant
hormonal therapy for breast cancer. World J Clin Oncol. 13:712–724.
2022.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Jiang O, Zhou R, Wu D, Liu Y, Wu W and
Cheng N: CYP2E1 polymorphisms and colorectal cancer risk: a HuGE
systematic review and meta-analysis. Tumor Biol. 34:1215–1224.
2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Holick MF: Sunlight and vitamin D for bone
health and prevention of autoimmune diseases, cancers, and
cardiovascular disease. Am J Clin Nutr. 80 (6 Suppl):1678S–1688S.
2004.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Mathieu C and Badenhoop K: Vitamin D and
type 1 diabetes mellitus: State of the art. Trends Endocrinol
Metab. 16:261–266. 2005.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Misra M, Pacaud D, Petryk A,
Collett-Solberg PF and Kappy M: Drug and Therapeutics Committee of
the Lawson Wilkins Pediatric Endocrine Society. Vitamin D
deficiency in children and its management: Review of current
knowledge and recommendations. Pediatrics. 122:398–417.
2008.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Handono K, Sidarta YO, Pradana BA, Nugroho
RA, Hartono IA, Kalim H and Endharti AT: Vitamin D prevents
endothelial damage induced by increased neutrophil extracellular
traps formation in patients with systemic lupus erythematosus. Acta
Med Indones. 46:189–198. 2014.PubMed/NCBI
|
|
32
|
Van Belle TL, Vanherwegen AS, Feyaerts D,
De Clercq P, Verstuyf A, Korf H, Gysemans C and Mathieu C:
1,25-Dihydroxyvitamin D3 and its analog TX527 promote a stable
regulatory T cell phenotype in T cells from type 1 diabetes
patients. PLoS One. 9(e109194)2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Reddy KK: Reply to Glossmann: Vitamin D
compounds and oral supplementation methods. J Invest Dermatol.
133(2649)2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Takahashi H, Hatta Y, Iriyama N, Hasegawa
Y, Uchida H, Nakagawa M, Makishima M, Takeuchi J and Takei M:
Induced differentiation of human myeloid leukemia cells into M2
macrophages by combined treatment with retinoic acid and
1α,25-dihydroxyvitamin D3. PLoS One. 9(e113722)2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhang Z, Zhang H, Hu Z, Wang P, Wan J and
Li B: Synergy of 1,25-dihydroxyvitamin D3 and carboplatin in growth
suppression of SKOV-3 cells. Oncol Lett. 8:1348–1354.
2014.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Henry HL: Regulation of vitamin D
metabolism. Best Pract Res Clin Endocrinol Metab. 25:531–541.
2011.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Hossein-nezhad A and Holick MF: Vitamin D
for health: A global perspective. Mayo Clin Proc. 88:720–755.
2013.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Cantorna MT, Zhu Y, Froicu M and Wittke A:
Vitamin D status, 1,25-dihydroxyvitamin D3, and the immune system.
Am J Clin Nutr. 80 (6 Suppl):1717S–1720S. 2004.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Weinstein SJ, Purdue MP, Smith-Warner SA,
Mondul AM, Black A, Ahn J, Huang WY, Horst RL, Kopp W, Rager H, et
al: Serum 25-hydroxyvitamin D, vitamin D binding protein and risk
of colorectal cancer in the prostate, lung, colorectal and ovarian
cancer screening trial. Int J Cancer. 136:E654–E664.
2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Slominski AT, Kim TK, Janjetovic Z,
Slominski RM, Li W, Jetten AM, Indra AK, Mason RS and Tuckey RC:
Biological effects of CYP11A1-derived vitamin D and lumisterol
metabolites in the skin. J Invest Dermatol. 144:2145–2161.
2024.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Slominski AT, Tuckey RC, Jenkinson C, Li W
and Jetten AM: Alternative pathways for vitamin D metabolism. In:
Hewison M, Bouillon R, Giovanucci E, Goltzman D, Meyer M and Welsh
J (eds.), Feldman and Pike's Vitamin D: Volume One: Biochemistry,
Physiology and Diagnostics. 5th edition. Academic Press, pp85-109,
2024.
|
|
42
|
Hoeijmakers JH: Genome maintenance
mechanisms for preventing cancer. Nature. 411:366–374.
2001.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Stary A and Sarasin A: The genetics of the
hereditary xeroderma pigmentosum syndrome. Biochimie. 84:49–60.
2002.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Cleaver JE: Defective repair replication
of DNA in xeroderma pigmentosum. Nature. 218:652–656.
1968.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Kraemer KH, Levy DD, Parris CN, Gozukara
EM, Moriwaki S, Adelberg S and Seidman MM: Xeroderma pigmentosum
and related disorders: Examining the linkage between defective DNA
repair and cancer. J Invest Dermatol. 103 (5 Suppl):96S–101S.
1994.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Wood RD: DNA damage recognition during
nucleotide excision repair in mammalian cells. Biochimie. 81:39–44.
1999.PubMed/NCBI View Article : Google Scholar
|
|
47
|
de Boer J and Hoeijmakers JH: Nucleotide
excision repair and human syndromes. Carcinogenesis. 21:453–460.
2000.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Friedberg EC, Feaver WJ and Gerlach VL:
The many faces of DNA polymerases: Strategies for mutagenesis and
for mutational avoidance. Proc Natl Acad Sci USA. 97:5681–5683.
2000.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Whitlock JP Jr: Induction of cytochrome
P4501A1. Annu Rev Pharmacol Toxicol. 39:103–125. 1999.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Iba MM, Scholl H, Fung J, Thomas PE and
Alam J: Induction of pulmonary CYP1A1 by nicotine. Xenobiotica.
28:827–843. 1998.PubMed/NCBI View Article : Google Scholar
|
|
51
|
San Jose C, Cabanillas A, Benitez J,
Carrillo JA, Jimenez M and Gervasini G: CYP1A1 gene polymorphisms
increase lung cancer risk in a high-incidence region of Spain: A
case control study. BMC Cancer. 10(463)2010.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Johansson I, Xanthopoulou EM, Zhou Y,
Sanchez-Spitman A, van der Lee M, Wollmann BM, Størset E, Swen JJ,
Guchelaar HJ, Molden E, et al: Improved prediction of CYP2D6
catalyzed drug metabolism by taking variant substrate specificities
and novel polymorphic haplotypes into account. Clin Pharmacol Ther.
118:218–231. 2025.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Wolf CR and Smith G: Cytochrome P450
CYP2D6. In: Metabolic Polymorphisms and Susceptibility to Cancer.
Vol 148. International Agency for Research on Cancer, Lyon,
pp209-229, 1999.
|
|
54
|
Crespi CL, Penman BW, Gelboin HV and
Gonzalez FJ: A tobacco smoke-derived nitrosamine,
4-(methylnitrosamino)-1-(3- pyridyl)-1-butanone, is activated by
multiple human cytochrome P450s including the polymorphic human
cytochrome P4502D6,. Carcinogenesis. 12:1197–1201. 1991.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Lorenzo-Pouso AI, Silva FFE, Pérez-Jardón
A, Chamorro-Petronacci CM, Oliveira-Alves MG,
Álvarez-Calderón-Iglesias Ó, Caponio VCA, Pinti M, Perrotti V and
Pérez-Sayáns M: Overexpression of E-cadherin is a favorable
prognostic biomarker in oral squamous cell carcinoma: A systematic
review and meta-analysis. Biology (Basel). 12(239)2023.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Chen CY, Chen J, He L and Stiles BL: PTEN:
Tumor Suppressor and Metabolic Regulator. Front Endocrinol
(Lausanne). 9(338)2018.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Song MS, Salmena L and Pandolfi PP: The
functions and regulation of the PTEN tumour suppressor. Nat Rev Mol
Cell Biol. 13:283–296. 2012.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Han F, Hu R, Yang H, Liu J, Sui J, Xiang
X, Wang F, Chu L and Song S: PTEN gene mutations correlate
to poor prognosis in glioma patients: a meta-analysis. Onco Targets
Ther. 9:3485–3492. 2016.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Aubrey BJ, Kelly GL, Janic A, Herold MJ
and Strasser A: How does p53 induce apoptosis and how does this
relate to p53-mediated tumour suppression? Cell Death Differ.
25:104–113. 2018.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Vaddavalli P and Schumacher B: The p53
network: Cellular and systemic DNA damage responses in cancer and
aging. Trends Genet. 38:598–612. 2022.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Bouaoun L, Sonkin D, Ardin M, Hollstein M,
Byrnes G, Zavadil J and Olivier M: TP53 variations in human
cancers: New lessons from the IARC TP53 database and genomics data.
Hum Mutat. 37:865–876. 2016.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Mantovani F, Collavin L and Del Sal G:
Mutant p53 as a guardian of the cancer cell. Cell Death Differ.
26:199–212. 2019.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Keriel A, Stary A, Sarasin A,
Rochette-Egly C and Egly JM: XPD mutations prevent TFIIH-dependent
transactivation by nuclear receptors and phosphorylation of
RARalpha. Cell. 109:125–135. 2002.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Coin F, Marinoni JC, Rodolfo C, Fribourg
S, Pedrini AM and Egly JM: Mutations in the XPD helicase gene
result in XP and TTD phenotypes, preventing interaction between XPD
and the p44 subunit of TFIIH. Nat Genet. 20:184–188.
1998.PubMed/NCBI View
Article : Google Scholar
|
|
65
|
Lunn RM, Helzlsouer KJ, Parshad R, Umbach
DM, Harris EL, Sanford KK and Bell DA: XPD polymorphisms: Effects
on DNA repair proficiency. Carcinogenesis. 21:551–555.
2000.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Mohamed NS, Ali Albsheer MM, Abdelbagi H,
Siddig EE, Mohamed MA, Ahmed AE, Omer RA, Muneer MS, Ahmed A, Osman
HA, et al: Genetic polymorphism of the N-terminal region in
circumsporozoite surface protein of Plasmodium falciparum field
isolates from Sudan. Malar J. 18(333)2019.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Ju HL, Kang JM, Moon SU, Kim JY, Lee HW,
Lin K, Sohn WM, Lee JS, Kim TS and Na BK: Genetic polymorphism and
natural selection of Duffy binding protein of Plasmodium vivax
Myanmar isolates. Malar J. 11:1–110. 2012.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Thompson EE, Sun Y, Nicolae D and Ober C:
Shades of gray: A comparison of linkage disequilibrium between
Hutterites and Europeans. Genet Epidemiol. 34:133–139.
2010.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Tibayrenc M: Human genetic diversity and
the epidemiology of parasitic and other transmissible diseases. Adv
Parasitol. 64:377–422. 2007.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Guo CY, DeStefano AL, Lunetta KL, Dupuis J
and Cupples LA: Expectation maximization algorithm based haplotype
relative risk (EM-HRR): Test of linkage disequilibrium using
incomplete case-parents trios. Hum Hered. 59:125–135.
2005.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Silverman EK, Chapman HA, Drazen JM, Weiss
ST, Rosner B, Campbell EJ, O'Donnell WJ, Reilly JJ, Ginns L,
Mentzer S, et al: Genetic epidemiology of severe, early-onset
chronic obstructive pulmonary disease. Risk to relatives for
airflow obstruction and chronic bronchitis. Am J Respir Crit Care
Med. 157:1770–1778. 1998.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Karahalil B, Bohr VA and Wilson DM III:
Impact of DNA polymorphisms in key DNA base excision repair
proteins on cancer risk. Hum Exp Toxicol. 31:981–1005.
2012.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Bookman EB, McAllister K, Gillanders E,
Wanke K, Balshaw D, Rutter J, Reedy J, Shaughnessy D, Agurs-Collins
T, Paltoo D, et al: Gene-environment interplay in common complex
diseases: Forging an integrative model-recommendations from an NIH
workshop. Genet Epidemiol. 35:217–225. 2011.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Daly AK: Pharmacogenetics and human
genetic polymorphisms. Biochem J. 429:435–449. 2010.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Mathur R, Rana BS and Jha AK: Single
nucleotide polymorphism (SNP). In: Vonk J, Shackelford T (eds).
Encyclopedia of Animal Cognition and Behavior. Springer, Cham,
pp1-4, 2018.
|
|
76
|
Ando Y, Saka H, Ando M, Sawa T, Muro K,
Ueoka H, Yokoyama A, Saitoh S, Shimokata K and Hasegawa Y:
Polymorphisms of UDP-glucuronosyltransferase gene and irinotecan
toxicity: A pharmacogenetic analysis. Cancer Res. 60:6921–6926.
2000.PubMed/NCBI
|
|
77
|
Mallal S, Phillips E, Carosi G, Molina JM,
Workman C, Tomazic J, Jägel-Guedes E, Rugina S, Kozyrev O, Cid JF,
et al: HLA-B*5701 screening for hypersensitivity to abacavir. N
Engl J Med. 358:568–579. 2008.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Halder I and Shriver MD: Measuring and
using admixture to study the genetics of complex diseases. Hum
Genomics. 1:52–62. 2003.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Hu D and Ziv E: Confounding in genetic
association studies and its solutions. Methods Mol Biol. 448:31–39.
2008.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Hettiarachchi G and Komar AA: GWAS to
identify SNPs associated with common diseases and individual risk:
Genome wide association studies (GWAS) to identify SNPs associated
with common diseases and individual risk. In: Sauna ZE,
Kimchi-Sarfaty C (eds). Single Nucleotide Polymorphisms. Springer,
Cham, pp51-76, 2022.
|
|
81
|
Uffelmann E, Huang QQ, Munung NS, De Vries
J, Okada Y, Martin AR, Martin HC, Lappalainen T and Posthuma D:
Genome-wide association studies. Nat Rev Methods Primers.
1(59)2021.
|
|
82
|
Umeno M, McBride OW, Yang CS, Gelboin HV
and Gonzalez FJ: Human ethanol-inducible P450IIE1: Complete gene
sequence, promoter characterization, chromosome mapping, and
cDNA-directed expression. Biochemistry. 27:9006–9013.
1988.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Guengerich FP, Kim DH and Iwasaki M: Role
of human cytochrome P-450 IIE1 in the oxidation of many low
molecular weight cancer suspects. Chem Res Toxicol. 4:168–179.
1991.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Nakajima T and Aoyama T: Polymorphism of
drug-metabolizing enzymes in relation to individual susceptibility
to industrial chemicals. Ind Health. 38:143–152. 2000.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Wang AH, Sun CS, Li LS, Huang JY and Chen
QS: Relationship of tobacco smoking CYP1A1 GSTM1 gene polymorphism
and esophageal cancer in Xi'an. World J Gastroenterol. 8:49–53.
2002.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Bartsch H, Nair U, Risch A, Rojas M,
Wikman H and Alexandrov K: Genetic polymorphism of CYP genes, alone
or in combination, as a risk modifier of tobacco-related cancers.
Cancer Epidemiol Biomarkers Prev. 9:3–28. 2000.PubMed/NCBI
|
|
87
|
Itoga S, Nomura F, Makino Y, Tomonaga T,
Shimada H, Ochiai T, Iizasa T, Baba M, Fujisawa T and Harada S:
Tandem repeat polymorphism of the CYP2E1 gene: An association study
with esophageal cancer and lung cancer. Alcohol Clin Exp Res. 26 (8
Suppl):15S–19S. 2002.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Danko IM and Chaschin NA: Association of
CYP2E1 gene polymorphism with predisposition to cancer development.
Exp Oncol. 27:248–256. 2005.PubMed/NCBI
|
|
89
|
Hayashi S, Watanabe J and Kawajiri K:
Genetic polymorphisms in the 5'-flanking region change
transcriptional regulation of the human cytochrome P450IIE1 gene. J
Biochem. 110:559–565. 1991.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Watanabe M: Polymorphic CYP genes and
disease predisposition-what have the studies shown so far? Toxicol
Lett. 102-103:167–171. 1998.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Yang B, O'Reilly DA, Demaine AG and
Kingsnorth AN: Study of polymorphisms in the CYP2E1 gene in
patients with alcoholic pancreatitis. Alcohol. 23:91–97.
2001.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Yu SZ, Huang XE, Koide T, Cheng G, Chen
GC, Harada K, Ueno Y, Sueoka E, Oda H, Tashiro F, et al: Hepatitis
B and C viruses infection, lifestyle and genetic polymorphisms as
risk factors for hepatocellular carcinoma in Haimen, China. Jpn J
Cancer Res. 93:1287–1292. 2002.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Munaka M, Kohshi K, Kawamoto T, Takasawa
S, Nagata N, Itoh H, Oda S and Katoh T: Genetic polymorphisms of
tobacco- and alcohol-related metabolizing enzymes and the risk of
hepatocellular carcinoma. J Cancer Res Clin Oncol. 129:355–360.
2003.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Norton ID, Apte MV, Haber PS, McCaughan
GW, Pirola RC and Wilson JS: Cytochrome P4502E1 is present in rat
pancreas and is induced by chronic ethanol administration. Gut.
42:426–430. 1998.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Catanzaro I, Naselli F, Saverini M,
Giacalone A, Montalto G and Caradonna F: Cytochrome P450 2E1
variable number tandem repeat polymorphisms and health risks: A
genotype-phenotype study in cancers associated with drinking and/or
smoking. Mol Med Rep. 6:416–420. 2012.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Christiani DC, Mehta AJ and Yu CL: Genetic
susceptibility to occupational exposures. Occup Environ Med.
65:430–436, 436, 397. 2008.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Pizzino G, Bitto A, Interdonato M, Galfo
F, Irrera N, Mecchio A, Pallio G, Ramistella V, De Luca F, Minutoli
L, et al: Oxidative stress and DNA repair and detoxification gene
expression in adolescents exposed to heavy metals living in the
Milazzo-Valle del Mela area (Sicily, Italy). Redox Biol. 2:686–693.
2014.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Kringel D, Lippmann C, Parnham MJ, Kalso
E, Ultsch A and Lötsch J: A machine-learned analysis of human gene
polymorphisms modulating persisting pain points to major roles of
neuroimmune processes. Eur J Pain. 22:1735–1756. 2018.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Pandiyan A, Lari S, Vanka J, Kumar BS,
Ghosh S, Jee B and Jonnalagadda PR: Genetic polymorphism in
xenobiotic metabolising genes and increased oxidative stress among
pesticides exposed agricultural workers diagnosed with cancers.
Asian Pac J Cancer Prev. 24:3795–3804. 2023.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Rothman N, Wacholder S, Caporaso NE,
Garcia-Closas M, Buetow K and Fraumeni JF Jr: The use of common
genetic polymorphisms to enhance the epidemiologic study of
environmental carcinogens. Biochim Biophys Acta. 1471:C1–C10.
2001.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Kelada SN, Eaton DL, Wang SS, Rothman NR
and Khoury MJ: The role of genetic polymorphisms in environmental
health. Environ Health Perspect. 111:1055–1064. 2003.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Carr BA and Franklin MR: Drug-metabolizing
enzyme induction by 2,2'-dipyridyl, 1,7-phenanthroline,
7,8-benzoquinoline and oltipraz in mouse. Xenobiotica. 28:949–956.
1998.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Langouët S, Coles B, Morel F, Becquemont
L, Beaune P, Guengerich FP, Ketterer B and Guillouzo A: Inhibition
of CYP1A2 and CYP3A4 by oltipraz results in reduction of aflatoxin
B1 metabolism in human hepatocytes in primary culture. Cancer Res.
55:5574–5579. 1995.PubMed/NCBI
|
|
104
|
Kensler TW, Egner PA, Wang JB, Zhu YR,
Zhang BC, Lu PX, Chen JG, Qian GS, Kuang SY, Jackson PE, et al:
Chemoprevention of hepatocellular carcinoma in aflatoxin endemic
areas. Gastroenterology. 127 (5 Suppl 1):S310–S318. 2004.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Schulte P and Howard J: Genetic
susceptibility and the setting of occupational health standards.
Annu Rev Public Health. 32:149–159. 2011.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Naselli F, Catanzaro I, Bellavia D, Perez
A, Sposito L and Caradonna F: Role and importance of polymorphisms
with respect to DNA methylation for the expression of CYP2E1
enzyme. Gene. 536:29–39. 2014.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Sharma S, Sambyal V, Guleria K, Manjari M,
Sudan M, Uppal MS, Singh NR, Bansal D and Gupta A: TP53
polymorphisms in sporadic North Indian breast cancer patients.
Asian Pac J Cancer Prev. 15:6871–6879. 2014.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Petrucelli N, Daly MB and Pal T: BRCA1-
and BRCA2-associated hereditary breast and ovarian cancer. 1998 Sep
4 [Updated 2025 Mar 20]. In: Adam MP, Feldman J, Mirzaa GM, Pagon
RA, Wallace SE and Amemiya A (eds). GeneReviews®
[Internet]. Seattle (WA): University of Washington, Seattle,
1993.
|
|
109
|
Slominski RM, Raman C, Chen JY and
Slominski AT: How cancer hijacks the body's homeostasis through the
neuroendocrine system. Trends Neurosci. 46:263–275. 2023.PubMed/NCBI View Article : Google Scholar
|