Mitochondria are the powerhouses of mammalian cells,
providing the energy and materials required for cellular function.
Mitochondria utilize pyruvate, products from glycolysis, amino acid
metabolism and fatty acid oxidation to transfer electrons through
the electron transport chain, synthesizing energy in the form of
adenosine triphosphate (ATP). Mitochondrial dysfunction causes
bioenergetic imbalance and increases oxidative stress, which paves
the way for pathogenesis to occur. Multiple diseases arise from
mitochondrial damage or are a consequence of impaired mitochondria
due to tissue injury. Aging and degenerative diseases are common
examples of conditions involving mitochondrial dysfunction. While
genetic modifications and the reduction of telomere length are
definite hallmarks of aging, impaired mitochondrial function and
the accumulation of mitochondrial DNA (mtDNA) mutations critically
determine healthy aging outcomes and contribute to longevity
(1). Degenerative diseases, such
as brain, muscle and macular degeneration are consequences of
mitochondrial dysfunction. Modifications in mitochondrial membrane
potential, reduced calcium homeostasis and decreased ATP production
are associated with mitochondrial dynamic dysregulation and
increased levels of reactive oxygen species (ROS). This leads to
protein degradation, the loss of intercellular interactions and
eventually, apoptosis (2-5).
For instance, in the immune system, reduced mitochondrial
respiration and metabolism are associated with immune paralysis
following septic shock (6,7). In metabolic syndromes, such as type 2
diabetes, mitochondrial oxidative stress promotes glucose
intolerance and excessive lipid accumulation in adipocytes, and
drives systemic inflammation, which further accelerates insulin
resistance (8-10).
Mitochondria have also been an unrecognized factor in the
progression of skin diseases (11). In pulmonary diseases derived from
smoking, cigarette smoke directly blocks complexes I and II, causes
mitochondrial depolarisation through altered mitophagy, and induces
mitochondrial ROS generation. This consequently results in the
apoptosis and necrosis of pulmonary endothelial cells, contributing
to the development of chronic and fibrotic conditions (12-14).
In addition, mitochondria exhibiting a low oxygen consumption rate,
high levels of mtDNA damage and ROS production are observed in the
vascular endothelial cells of preterm infants with bronchopulmonary
dysplasia and adverse outcomes (15). Mitochondria are also a key site for
calcium homeostasis, as they buffer cytoplasmic calcium ions
through interactions with the endoplasmic reticulum (16). The disruption of calcium balance
may interfere with mitochondrial respiration and ATP production,
leading to the development of pathological conditions and diseases,
such as neuron degeneration, cancers, diabetes, and muscular and
cardiac dystrophy (17-19).
Thus, the ability of cells to maintain competent mitochondrial
function and capacity is key to preventing disease progression and
negative outcomes.
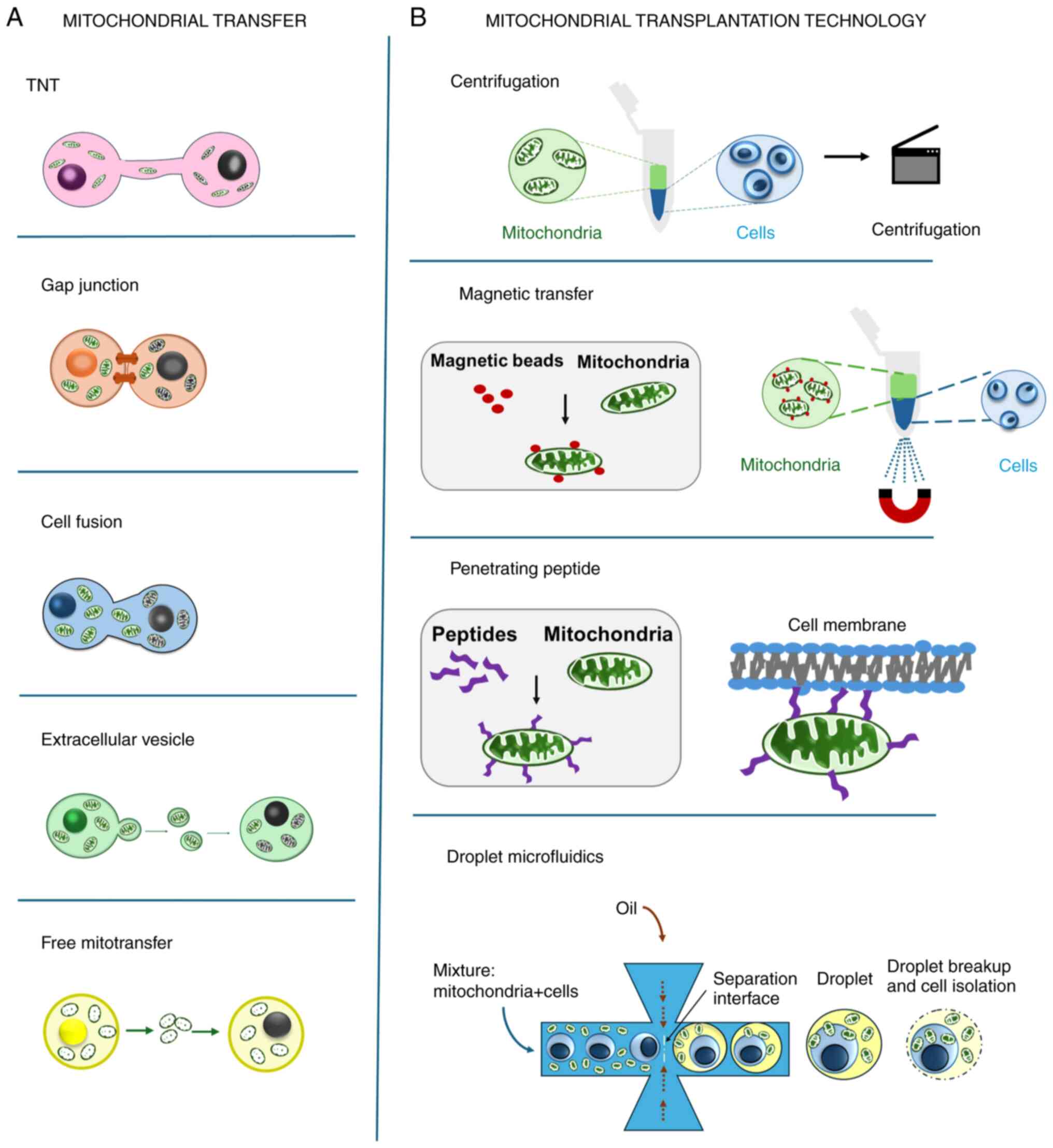
Mitochondrial transfer refers to the process through
which cells exchange mitochondria, serving as a form of cell-cell
communication. This transfer may involve the sharing of damaged
mitochondria to signal neighboring cells about oxidative damage,
while surrounding cells offer functional mitochondria to stressed
cells. Receiving mitochondria supports cells in recovering from
oxidative stress, improving cellular respiration and ATP
production, and consequently, reversing the effects of cellular
dysfunction and tissue damage (20-22).
The mechanisms of intercellular mitochondrial transfer have been
well established and are illustrated in Fig. 1A. The application of mitochondrial
transfer has been transformed into mitochondrial transplantation
therapy (Fig. 1B), in which
exogenous mitochondria are isolated and artificially transferred
into cells or tissues. Mitochondrial therapy, which encompasses
both mitochondrial transfer and transplantation, is effective in
improving cell metabolism, restoring cells from stress and
preventing tissue degeneration (23-25).
Early clinical trials of mitochondrial therapy have been conducted
in ischemic diseases, demonstrating that the injection of exogenous
mitochondria effectively improves the conditions of patients and
reduces the recovery time (26,27).
These data indicate that mitochondrial therapy holds potential for
future applications. However, the autologous transfer of
mitochondria may be invasive and non-accessible in a number of
cases; thus, a source of qualified mitochondrial donors is
critical.
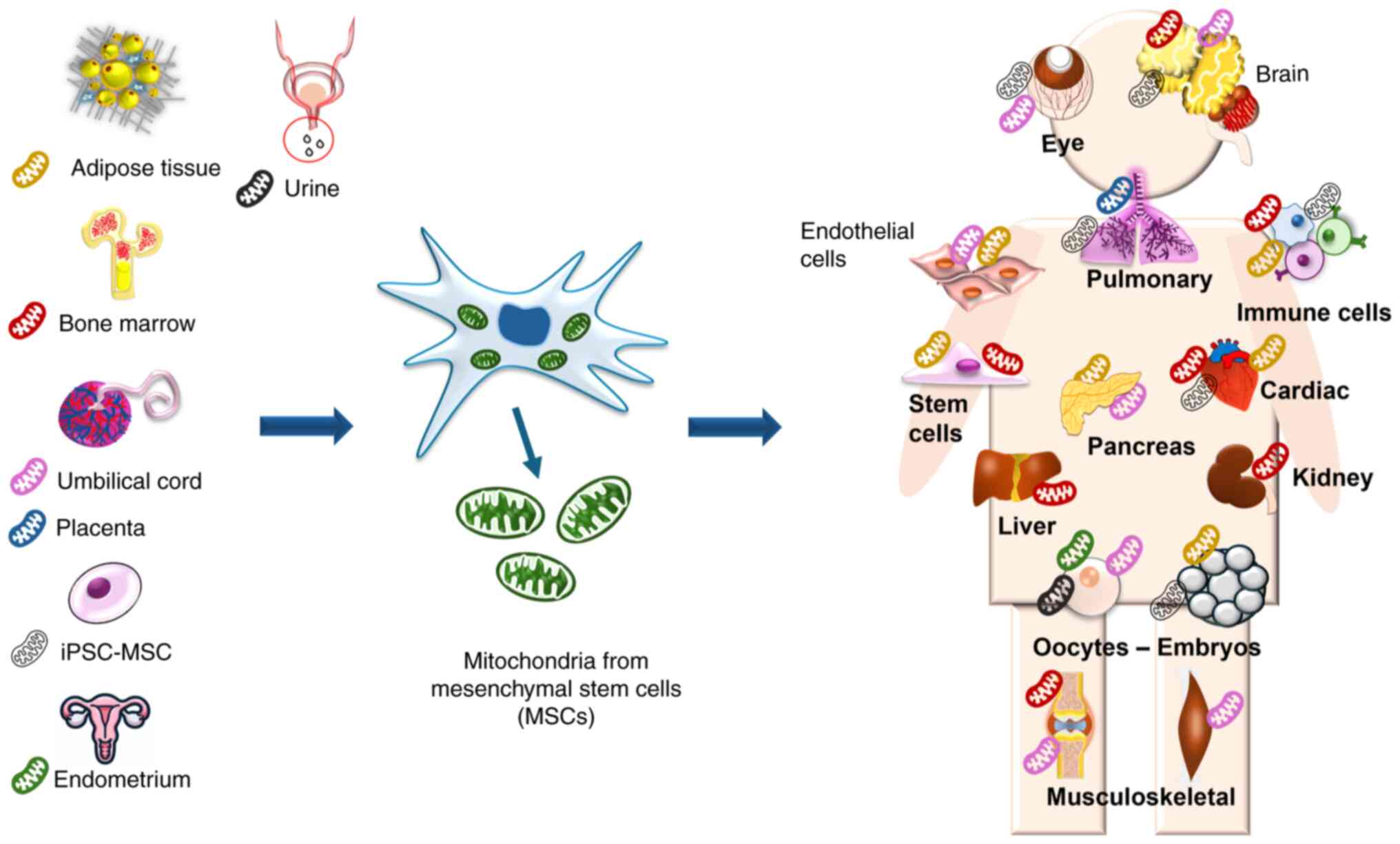
Mesenchymal stem cells (MSCs) feature high
plasticity and proliferation, which demonstrates their inherent
mitochondrial capacity. The ability to acquire, handle and expand
MSCs in large quantities in culture, along with a stable and
reliable assessment of mitochondrial quality, may further
facilitate access to mitochondrial transplantation in various
clinical settings, rendering MSCs promising for such therapeutic
purposes. The use of MSCs as a mitochondrial source has been shown
in in vitro and in vivo models of different diseases,
indicating that mitochondria isolated from MSCs are efficient and
safe for translational studies (Fig.
2) (28-30).
The present review summarizes data from numerous studies in the
literature on mitochondrial transfer using MSCs as mitochondrial
donors. Detailed descriptions of the mechanisms underlying
mitochondrial therapy are also included. The present review also
provides an update on the broad application of mitochondrial
donation in various pathological conditions and diseases,
indicating that MSCs from different origins are valuable sources
for mitochondrial collection and transplantation technology.
Asthma is a chronic condition involving a persistent
inflammatory disorder in the airways (31). Mitochondrial dysfunction causes
oxidative stress and reduced ATP synthase activity, leading to
apoptosis and damage to airway epithelial cells, which results in
allergic responses and inflammation (32,33).
Through tunneling nanotubes (TNTs), mitochondria transferred from
MSCs have been shown to protect the human bronchial epithelium cell
line (BEAS-2B) against mitochondrial dysfunction and apoptosis
induced by cobalt(II) chloride (CoCl2), which has been
used as a hypoxia-mimicking agent (34). Connexin43 (Cx43), a transmembrane
protein encoded by the GJA1 gene, is essential for TNT formation
and induced pluripotent stem cell (iPSC)-MSC-mediated mitochondrial
transfer. This transfer alleviates the symptoms of allergic airway
inflammation by recovering mitochondrial function in the bronchi
and inhibiting programmed cell death in the lungs (34). The positive effects of
mitochondrial transfer have also been reported when inducing airway
inflammation by ovalbumin, specifically via the restoration of
mitochondrial membrane potential (MMP) and the inhibition of
caspase-3 and -9(34).
The dysfunction of mitochondria, which is associated
with changes in mitochondrial morphology and homeostasis and the
disruption of MMP in lung cells, is one of the main causes of
pulmonary fibrosis (PF) (35). ROS
overproduction in the mitochondria is associated with mitochondrial
dysfunction, leading to lung damage and inflammatory responses,
which contributes to the pathogenesis of PF (36). Increasing mitochondrial biogenesis
within human MSCs (hMSCs) using pioglitazone and iron oxide
nanoparticles has been shown to enhance mitochondrial transfer to
mouse alveolar epithelial cells (TC-1) damaged by bleomycin (BLM),
a treatment that induces mtDNA strand breaks and mitochondrial
respiratory chain dysfunction (37). TC-1 cells damaged by BLM exhibit
restored ATP levels, reduced intracellular ROS levels and recover
MMP when they receive functional mitochondria from hMSCs (37).
In the model of chronic obstructive pulmonary
disease (COPD), mitochondrial dysfunction due to oxidative stress
contributes to tissue inflammation and remodeling (38). Oxidative stress accelerates the
inflammatory process by stimulating neutrophils and macrophages
(39), leading to apoptosis,
autophagy and cellular senescence in the airways (38). Smoking is a well-known contributing
factor to the development of COPD and airway structure remodeling.
Cigarette smoke medium (CSM) has been shown to alter mitochondrial
metabolic regulation, leading to glycolysis and ROS production
(40,41). It was previously demonstrated that
the transfer of mitochondria from iPSC-MSCs reduced intracellular
ROS levels and prevented MMP impairment in airway smooth muscle
cells induced by CSM, significantly decreasing apoptosis by 50%
(42). Antoehr study demonstrated
that BEAS-2B cells treated with 2% CSM for 24 h induced significant
inflammatory responses and oxidative stress without leading to cell
death, and also received mitochondria from bone marrow-derived MSCs
(BM-MSCs) (43). Mitochondrial
transfer from BM-MSCs to BEAS-2B cells treated with CSM showed
recovery in intracellular ATP levels (43). This evidence indicated that
transferring mitochondria from MSCs can restore mitochondrial
dysfunction in the respiratory system by preventing oxidative
stress, reducing epithelial cell death, and airway inflammation,
and is a potential supportive method in pulmonary diseases.
In cardiac tissue, damage to cardiac muscles can
initiate numerous cardiac issues. For example, progressive
myocardial damage can be caused by the use of anthracyclines,
including doxorubicin, which are commonly used as drugs for the
treatment of cancer. Mitochondrial biogenesis inhibition has been
shown to contribute to this damage (44). The overproduction of ROS caused by
doxorubicin induces mitochondrial dysfunction through the
impairment of MMP, the opening of mitochondrial permeability
transition pore (mPTP), and the alteration of mitochondrial
morphology in human vascular endothelial cells (45). Mitochondrial dysfunction caused by
doxorubicin has a significant effect on cardiomyocytes as
mitochondria comprise 40% of the volume of each cardiomyocyte, and
the majority of energy in these cells is generated through
mitochondrial respiration (46).
In this context, the supplementation of fresh mitochondria could be
beneficial. Evidence has indicated that the injection of autologous
mitochondria isolated from skeletal muscles into the ischemic
hearts of pediatric patients effectively helped them separate from
extracorporeal membrane oxygenation (27). However, isolating mitochondria from
skeletal muscles may be invasive and impossible in some cases.
Thus, a readily available source of healthy mitochondria for
transplantation is expected to advance current treatment. As such,
MSCs appear as a potential source for mitochondria isolation due to
their rapid proliferation. Several studies have demonstrated that
MSCs may be a safe source for mitochondrial transfer in
cardiovascular conditions (Table
I). A previous study demonstrated that functional mitochondrial
transfer by iPSC-MSCs protected against doxorubicin-induced damage
in cardiomyocytes from neonatal mice, resulting in a marked
recovery of the mitochondrial oxygen consumption rate (OCR), ATP
production, and optimal mitochondrial respiration in cardiomyocytes
(47). It was noted that this
effect was distinct from the paracrine activity of MSCs and
involved mitochondrial Rho GTPase 1 (Miro1), an outer mitochondrial
membrane protein, in the formation of TNTs (47). In another study, the
transplantation of mitochondria derived from MSCs reduced apoptosis
and ROS levels, while increasing ATP production in cardiomyocytes
treated with doxorubicin (48).
Mitochondrial dysfunction is also a key event in
simulated ischemia/reperfusion injury (IRI) (49). Abnormal mitochondrial morphology
and irregularities in mitochondrial quality control have been
identified as cellular mechanisms leading to IRI (50). Mitochondrial ROS production is a
primary target of damage caused by hypoxia (51). In an ischemic model, MSCs were
shown to reduce the death of H9c2 cardiomyocytes when co-cultured
under hypoxic conditions for 150 min via the transfer of healthy
mitochondria (52). In another
study, BM-MSCs delivered functional mitochondria to injured H9c2
rat ventricular cells, which were cultured in a medium deprived of
serum and glucose and incubated under hypoxic conditions for 12 h
(53). Co-culturing injured H9c2
cells with BM-MSCs showed significant downregulation of Bax
expression and an upregulation of B-cell lymphoma (Bcl-2)
expression (53). Furthermore,
BM-MSCs also decreased the caspase-3 levels induced by SI/R in H9c2
cells, as this enzyme is an executor of the apoptosis process
(53). However, when BM-MSCs were
treated with latrunculin A (LatA), which is known to inhibit TNT
formation, apoptosis increased (53), suggesting that TNT formation is
critical. In another study, when co-culturing human adipose-derived
stem cells (hADSCs) with rat cardiomyocytes for 24 h under hypoxic
conditions, functional ADSC-derived mitochondria enhanced the OCR
of rat cardiomyocytes. This effect was shown to be independent of
the paracrine effects from ADSCs (54). On the other hand, mitochondria
released by damaged somatic cells, such as hydrogen peroxide
(H2O2)-treated RL14 cardiomyocytes or human
umbilical vein endothelial cells (HUVECs), activate MSCs to
transfer healthy mitochondria toward dying cells (55). Heme oxygenase-1 (HO-1) is a
rate-limiting enzyme in heme metabolism and exerts a wide range of
anti-inflammatory, anti-apoptotic, and immunoregulatory effects in
various diseases (56).
Co-culturing MSCs with RL4 cells damaged by
H2O2 and HUVECs leads to the increased
expression of HO-1 and enhances HO-1 enzyme activity in MSCs
(55). These data indicate that
MSCs are promising sources of mitochondria that can be used for
mitochondrial transplantation technologies.
Osteoarthritis (OA) is a common musculoskeletal
disorder related to the aging process, which is characterized by
cartilage degradation, joint pain, and impaired function (57). Chondrocytes play a functional role
in the synthesis, deposition and formation of the extracellular
matrix of cartilage tissue (57).
The mitochondria of chondrocytes in the OA model exhibit a reduced
activity of mitochondrial respiratory chain (MRC) enzymes (complex
I, II, III, IV and ATP synthase), along with decreased MMP
(58). Furthermore, mitochondria
exhibiting a swollen morphology and loss of cristae regularity have
also been observed in chondrocytes in models of OA (58). Mitochondrial dysfunction and energy
metabolism disruption markedly contribute to the pathogenesis of
OA, including cartilage degeneration, increased synovial
inflammation and the calcification of the cartilage matrix
(59). The abnormal alteration in
the metabolic process of cartilage cells is a response to
inflammation and is correlated with the process of cartilage
degeneration (60). In a previous
study, the bilateral anterior cruciate ligament and medial meniscus
were resected in 8-week-old rats to induce OA pathogenesis
(61). Functional mitochondria
from BM-MSCs rescued OA chondrocytes from mitochondrial dysfunction
by restoring the normal morphology of mitochondria, MMP, MRCI, II,
and III activity, citrate synthase, and ATP content (61). Another study indicated that
mitochondrial dysfunction in chondrocytes in OA is characterized by
a reduced type II collagen secretion (58). It should be noted that type II
collagen and proteoglycans are key components of cartilage tissue
(62). OA chondrocytes co-cultured
with MSCs showed a significant increase in the concentration of
type II collagen and proteoglycan compared to the OA chondrocytes
group (61). Another study
indicated that inhibiting mitochondria in chondrocytes with agents
such as rotenone or oligomycin promoted MSCs to transfer functional
mitochondria to chondrocytes with mitochondrial dysfunction
(63). Although chondrocytes adapt
to low oxygen conditions, a number of their functions are still
oxygen-dependent (64). Thus,
culture conditions, such as hyperoxia and hyperglycemia, affected
the mitochondrial transfer from MSCs to chondrocytes because
chondrocyte homeostasis responds to the low oxygen levels and
nutrient-deficient conditions in vivo (63).
Tendon injuries often occur in the severely hypoxic
zone, characterized by mitochondrial dysfunction and increased
oxidative stress (65).
Mitochondrial dysfunction is characterized by an increase in ROS,
decreased superoxide dismutase activity, and a reduced number of
mitochondria, all of which contribute to tendon pathology (66). In a previous study on a murine
model of supraspinatus tendinopathy, mitochondrial dysfunction led
to the altered expression of genes related to morphology and
abnormal mitochondrial quantity associated with the development of
tendinitis (67). Another study
indicated that H2O2 activated the apoptotic
pathway by inducing oxidative stress and the depolarization of MMP
in mouse models of Achilles tendinopathy (68). Tenomodulin (TNMD) is a marker
specific to tendons and contributes to the durability and aging of
tendon tissue at the tissue level (69). TNMD is associated with the
structural and functional properties of collagen 1 (COL1) (69). A reduction in the levels of
tenomodulin is a sign of pathogenesis. Mitochondrial transfer from
BM-MSCs to H2O2-damaged tenocytes triggered
anti-apoptotic mechanisms and restored mitochondrial function by
recovering MMP and ATP levels (70). Functional mitochondria derived from
MSCs enhanced the expression of TNMD and collagen 1 and reduced the
expression of matrix metalloproteinase 1 in tenocytes treated with
tumor necrosis factor-α (TNF-α) (71). Moreover, functional mitochondria
from MSCs also reduced levels of pro-inflammatory cytokines, such
as interleukin (IL)-1β and IL-6(70), demonstrating an anti-inflammatory
effect.
Mitochondria contribute to several key functions for
the survival of muscle cells, such as ATP production, substrate
biosynthesis, metabolism and participation in the regulation of
apoptosis (72). Skeletal muscle
atrophy is characterized by a reduction in muscle mass and strength
and is associated with mitochondrial dysfunction as mitochondria
occupy a significant portion of muscle cell volume (72,73).
The reduction in mitochondrial mass, MMP, ATP production and
mitochondrial glucose metabolism activity are characteristic
features of muscle atrophy (74).
ROS overproduction related to mitochondrial dysfunction is the main
mechanism associated with muscle atrophy (75-77).
Moreover, mitochondrial dysfunction has also been implicated in the
pathology of muscular dystrophy (78). The restoration of mitochondrial
bioenergetics has been shown to ameliorate the progression of
muscular dystrophy in mice (79).
Dexamethasone is an agent that induces muscular atrophy through a
process of pyroptosis, a form of programmed cell death involving
inflammation as the initial process (80). A previous study demonstrated that
mitochondrial transplantation derived from umbilical cord-derived
MSCs (UC-MSCs) promoted myofiber hypertrophy and reduced
intracellular lactate concentration in dexamethasone-induced
atrophied muscles (81). This also
contributed to enhancing the expression of muscle-specific markers,
such as desmin and activated adenosine monophosphate-activated
protein kinase, which aided the functional recovery of atrophied
muscles (81).
These data indicate that the use of mitochondria
isolated from MSCs may be beneficial in different types of
pathology in the musculoskeletal system. The addition of MSC
mitochondria aids in the restoration of various cell types, the
construction of different functional parts, and the reduction of
tissue destruction caused by immune responses to stress.
Ischemic stroke is associated with a reduction in
cerebral blood flow, leading to severe oxygen and glucose
deprivation (OGD) in neurons (82-84).
OGD has been shown to cause neuronal mitochondrial dysfunction,
such as reduced mitochondrial complex I activity, MMP collapse,
decreased ATP synthesis and increased oxidative stress (85). Transient focal cerebral ischemia is
characterized by irreversible neuronal loss, leading to persistent
neurological impairment in individuals (86). The in vitro model of brain
ischemia is a state of OGD closely related to oxidative stress due
to the overproduction of ROS (87). In a previous study, mitochondrial
damage in astrocytes caused by OGD, which resulted in fragmented
mitochondria, stimulated the transfer of functional mitochondria
from multipotent MSCs (MMSCs) to astrocytes (88). The functional mitochondria from
MMSCs not only restored bioenergetics, but also normalized the
proliferation of neuron-like ρ0 PC12 pheochromocytoma cells,
which carried damaged mitochondrial DNA and generated most of their
energy through anaerobic glycolysis (88). Injured astrocytes received more
functional mitochondria from MMSCs when MMSCs overexpressed
Miro1(88). Furthermore, BM-MSCs
also transferred functional mitochondria to VSC4.1 motor neurons
injured by OGD (89). BM-MSCs
exhibited an increased CD157 expression when co-cultured with
VSC4.1 motor neurons injured by OGD, an effect that enhanced the
mitochondrial transfer capacity of BM-MSCs (89). Mitochondria derived from BM-MSCs
could alleviate apoptosis and inflammation in damaged VSC4.1 motor
neurons and promoted the outgrowth of VSC4.1 motor neurons, but
only affected their length without changing their number (89). The mitochondrial transfer from
iPSC-MSCs increased intracellular ATP levels in PC12 cells
following CoCl2-induced hypoxic damage (90). Furthermore, the mitochondrial
transfer from MSCs improved mitochondrial respiration, ATP
production, and the basal metabolic rate in neurons after
H2O2-induced injury (91).
Spinal cord injury (SCI) is a severe type of central
nervous system injury and is associated with oxidative stress
caused by the excessive production of ROS (92). Mitochondrial dysfunction related to
mitochondrial homeostasis leads to the inactivation of
ATP-dependent ion pumps and is closely associated with the
activation of cell death (93).
Mitochondrial quality control and the mitochondria-related process
of ferroptosis play a crucial in SCI (94). Ferroptosis is an iron-dependent
cell death process that occurs due to the accumulation of damaged
phospholipids on the cell membrane and is associated with SCI
(95-97).
In a previous study, UC-MSCs were shown to transfer mitochondria to
HT22 neuronal cells under the stimulation of the ferroptosis
inducer Rsl3 when co-cultured (98). UC-MSCs helped to restore
mitochondrial mass, promoted mitochondrial fusion, restored MMP,
and balanced intracellular ROS and ATP levels in ferroptosis
neurons (98). Furthermore,
mitochondrial transfer from UC-MSCs reduced ferroptosis by
decreasing the total intracellular iron content and free
Fe2+ levels in neurons undergoing ferroptosis (98). Inhibiting ferroptosis using
dihydroorotate dehydrogenase in injured neurons has been shown to
normalize mitochondrial morphology and MMP (96).
Parkinson's disease (PD) is a condition related to
the degeneration of the nervous system of the brain, and damaged
mitochondria are one of the causes of PD (99,100). Mitochondrial dysfunction has been
identified as a major factor contributing to the death of
dopaminergic (DA) neurons (101).
The dysfunction of mitochondrial respiratory chain complex I is a
major cause of PD pathogenesis (102). The impairment of mitophagy and
oxidative stress are hallmarks of PD (103). Mitochondria derived from UC-MSCs
reduce DA cell death caused by 1-methyl-4-phenylpyridinium,
6-hydroxydopamine and rotenone (104). Furthermore, mitochondria derived
from UC-MSCs also reduce the mRNA expression of TNF-α and nitric
oxide synthase, and decrease the expression of IL-6 and IL-8
induced by lipopolysaccharide (LPS) in murine microglia BV2 cells
(104). The reduced expression of
pro-inflammatory cytokines has been shown to alleviate oxidative
stress, inflammation and normalize mitochondrial dysfunction in a
mouse model of PD (105). In a
previous study, astrocytes derived from iPSC-MSCs provided
functional mitochondria to DA neurons damaged by rotenone, a
compound that inhibits complex I (106). Notably, the neuroprotective
effect of astrocytes was not due to paracrine activity, but
primarily to the transfer of mitochondria to DA neurons (106). The transfer of mitochondria from
iPSC-MSCs-derived astrocytes restored ATP levels impaired by
rotenone in DA neurons (106).
In addition, cisplatin is a chemotherapeutic agent
used to treat cancer; however, it is known to cause morphological
changes and mitochondrial dysfunction in mouse neurons (107). Cisplatin has been shown to cause
excessive ROS production and alter the mitochondrial content of
cells (108). The donation of
MSCs was shown to normalize the membrane potential of neural stem
cells (NSCs) treated with cisplatin, which had shown a marked
decrease in basal respiratory capacity, ATP levels, and MMP
(109). Furthermore, excessive
expression of Miro1 in MSCs increased mitochondrial transfer and
promoted the survival of NSCs (109). In summary, these findings
indicate that the transfer of functional mitochondria from MSCs can
improve mitochondrial dysfunction and neuronal survival under
various pathological conditions in brain tissue (106).
Mitochondrial damage and abnormalities in
mitochondrial homeostasis are among the causes of ocular diseases
(110,111). The risk of mitochondrial damage
increases when the cornea is exposed to ultraviolet rays (112). Corneal endothelial cells (CECs)
play a crucial role in maintaining the transparency of the cornea;
therefore, a decline in their function will negatively impact
vision (110). Metabolic activity
and mitochondrial respiration, such as the tricarboxylic acid cycle
and acetyl coenzyme A-related enzymes, are closely related to the
survival and function of CECs (113). Therefore, the addition of
functional mitochondria is expected to support the recovery of eye
disorders where impaired mitochondria have been identified to play
a role. In a model of human corneal endothelial cells (hCECs)
damaged by rotenone, the inhibition of mitochondrial complex I and
increased ROS production were shown to promote the uptake of MSC
mitochondria through TNT, whereas healthy hCECs could not
accelerate similar effects (111,114). Rotenone-induced ROS promote
NF-κB, which enhances TNT formation via the upregulation of
TNF-α-induced protein 2 (TNFαip2) (114). The underlying mechanism has been
previously reported (115). The
contribution of functional mitochondria from MSCs improves basal
respiration, ATP production, and the expression of mitochondrial
component proteins, such as complex I, mitochondrial matrix
components and the mitochondrial membrane (111). The transfer of functional
mitochondria from MSCs is the primary mechanism for protecting CECs
from the effects of rotenone, although the paracrine effects of
MSCs are also a contributing factor (114). However, the transferred
mitochondria are either digested by lysosomes in the host cell or
excreted after 8 days (111),
suggesting that the therapeutic effect of exogenous mitochondria
may be limited to the short term in CECs.
Mitochondria play a crucial role in the functional
activity of renal tubular epithelial cells (TECs) (125,126), and the disruption in
mitochondrial activities has been shown to cause the aging of renal
TECs, leading to renal complications and pathogenesis (127-129).
Hyperglycemia causes mitochondrial dysfunction, characterized by a
reduction in intracellular ATP and MMP, changes in mitochondrial
morphology, and the excessive production of ROS in renal TECs,
which contribute to diabetic nephropathy (130). A recent study revealed that high
glucose levels increased oxidatively modified proteins in
mitochondria and intracellular ROS in TECs (131). Streptozotocin (STZ) is an agent
that causes alterations in MMP and mitochondrial NAD and NADH
enzyme activity, leading to the inhibition of ATP synthesis, and
thereby inducing oxidative stress and apoptosis in pancreatic
β-cells (132). The use of STZ to
model diabetes has been shown to lead to cellular metabolic changes
and mitochondrial dysfunction in renal cells (133). In that context, mitochondrial
transfer from BM-MSCs was previously been shown to inhibit
apoptosis and ROS production in damaged proximal TECs induced by
STZ (29). This was achieved by
promoting the expression of mitochondrial superoxide dismutase 2
and Bcl-2, which are involved in regulating the process of
apoptosis of proximal TECs (29).
Furthermore, BM-MSCs-derived functional mitochondria improved the
expression of megalin, a surface receptor protein in TECs that
participates in the reabsorption of proteins from urine (134), as well as sodium-glucose
cotransporter-2 (SGLT2), a member of the glucose transporter family
in proximal TECs (29). Since high
glucose levels have been shown to reduce the expression of megalin
(which is also associated with glucose transport via SGLT)
(135), the recovery of megalin
and SGLT2 is a sign of improved TEC function.
Non-alcoholic steatohepatitis (NASH) has been shown
to be a result of dysfunctional mitochondria, which includes
alterations of enzymatic activity, changes in mitochondrial shape
and quantity, and disrupted calcium regulation (136,137). In the NASH disease model,
impaired mitochondria facilitate hepatic lipid accumulation by
promoting ROS production and lipid peroxidation processes (138). High-fat diets over a long period
of time have been shown to cause mitochondrial deformation and
alter the levels of proteins involved in mitochondrial dynamics
(139). It was previously shown
that when fatty hepatocytes were co-cultured with BM-MSCs,
mitochondrial transfer was observed through cell-to-cell contact,
which promoted lipolytic activities via enhancing oxidative
capacity and mitochondrial biogenesis (140,141). Mitochondria from BM-MSCs restored
several mitochondrial abnormalities in the hepatocytes of mice fed
a high-fat diet, including MMP, mtDNA copy number, OCR and ATP
levels (140). Functional
mitochondrial-derived BM-MSCs also restored impaired calcium
activity in steatotic cells (140), an effect which is important for
glucose production and lipogenesis (142).
Severe acute pancreatitis (SAP) is an inflammation
of the pancreas that can lead to mortality. SAP is often linked to
smoking, gallstone disease, alcohol consumption, obesity, diabetes,
and hyperlipidemia (143). The
excessive accumulation of intracellular ROS is associated with
pyroptosis in pancreatic cells during acute inflammation (144). The dysregulation of the mPTP
caused by excessive intracellular calcium levels results in changes
in MMP, which is associated with the progression of SAP (145). The transfer of mitochondria via
extracellular vesicles (EVs) from hypoxia (5%
O2)-treated hUC-MSCs (hypo-EVs) was previously shown to
restore ATP levels and OCR, and to maintain relatively stable MMP
in sodium taurocholate-induced damaged rat pancreatic acinar cells
(PACs) (146). EVs carrying
mitochondria also promoted the normalization of glycolytic activity
in PACs (146).
Allogeneic islet transplantation provides a
promising opportunity for a limited group of patients with type I
diabetes (147). Allogeneic
transplantation of human islets requires short-term cultivation to
ensure safety assessments (148).
However, islet functions are influenced by various environmental
conditions during this period, which may induce oxidative stress
and inflammation (148). The
insulin-secreting ability of pancreatic islet β-cells is associated
with mitochondrial mass and respiratory capacity (149,150). It is also primarily regulated by
mitochondrial ATP production in response to extracellular glucose
levels (150). ADSCs have been
shown to transfer mitochondria through extracellular vesicles to
mouse islet cells exposed to hypoxia (1% oxygen), resulting in
enhanced OCR and insulin secretion of islet cells when stimulated
with 20 mM glucose (151). These
data suggest that mitochondrial transfer can also be used as a
supportive technique in optimizing islet pre-transplantation
conditions.
Mitochondrial myopathy, encephalomyopathy with
lactic acidosis, and stroke-like episodes (MELAS) is the acronym
for a clinical subgroup of mtDNA diseases affecting several body
systems (152). MELAS is often a
result of point mutations in the mtDNA and is thus inheritable from
the affected mother (152). The
transition from adenine to guanine at nucleotide 3243 in the mtDNA
of the MT-LT1 gene encoding tRNAleu (UUR), is among
the most frequently observed pathogenic mutations in individuals
with MELAS (153). This
alteration leads to the impaired translation and synthesis of ETC
protein subunits, resulting in reduced mitochondrial energy
production (154). Patients with
MELAS have fibroblasts (MELAS fibroblasts) with a high mutation
burden, featuring mitochondrial dysfunction and increased ROS
production (155). It was
previously shown that Wharton's jelly-derived MSCs (WJMSCs)
transferred mitochondria via TNTs into MELAS fibroblasts
pre-treated with rotenone, resulting in an increased protein
expression of the respiratory complexes, which suppressed ROS
production and enhanced MMP (155). Furthermore, this also enhanced
the proliferation of fibroblasts and reduced the expression of
cleaved caspase-3 and TUNEL, thereby suppressing apoptosis
(155). The results of another
study suggested that highly purified MSCs have a greater ability to
transfer mitochondria into MELAS neurons compared to traditional
MSCs (156). Furthermore,
co-culturing with RECs or MSCs significantly enhanced the viability
of MELAS neurons (156).
Myoclonus epilepsy with ragged red fiber syndrome
(MERRF) is a result of a transition from adenine to guanine at
nucleotide 8344 (mt8344A>G) within the mitochondrial
tRNALys coding gene (157). This mutation has been shown to
increase ROS production and oxidative stress, along with
compromised mitochondrial bioenergetics (157). MERRF cybrids, which were created
by fusing ρ0 cells lacking mtDNA with human
platelets in a medium devoid of pyruvate and uridine, were unable
to survive in a galactose-added and glucose-free medium. This
suggests that the mt8344A>G mutation leads to mitochondrial
dysfunction (158). The MERRF
mutation impairs the translation of 13 proteins encoded by mtDNA
and produces abnormal translation products (159). The transfer of mitochondria from
WJMSCs into MERRF cybrids decreases intracellular ROS,
re-establishes MMP, and enables the growth of MERRF cells in
glucose-free environments (158).
In clinical applications, hematopoietic stem cells
(HSCs) were utilized for mitochondrial transplantation into the
HSCs of patients with large-scale mtDNA deletion, such as Pearson
or Kearns-Sayre syndromes (160,161). Recipient HSCs were then infused
back into the patients, improving their quality of life (160,161). This evidence suggests a novel
therapeutic application of mitochondrial transplantation for
currently incurable diseases. While MSCs have not yet been used in
such clinical trials, they can be collected from multiple sources
and easily expanded. Their mitochondria vary in both morphology and
functional capacity, and can be selected to suit HSC biology,
making them a potential source for mitochondrial transplantation
into HSCs for similar cases. Although replacing or supplementing
defective mitochondria in all body systems appears challenging and
warrants further research, mitochondrial transplantation holds
therapeutic potential in mitochondrial pathogenesis.
Stem cell therapies have often been ineffective due
to low cell survival and engraftment (162,163). The excessive production of ROS in
damaged tissues usually decreases the viability of transplanted
MSCs (164). Additionally, MSC
functional characteristics may be compromised with prolonged
isolation and in vitro cultivation, or due to the age of the
donor or disease conditions (165,166). In this context, it has been shown
that healthy hMSCs could donate mitochondria to those pre-treated
with H2O2 through TNT formation (167). The treatment of hMSCs with
mesenchymal stem cell adjuvant has been shown to enhance their
mitochondrial transfer into other
H2O2-induced damaged hMSCs, resulting in a
significant reduction in intracellular ROS levels, maintenance of
MMP, and an increase in TNT length and formation (167). Mitochondrial Drp 1
phosphorylation (Drp1 S616) is involved in mitochondrial fission
and fragmentation (168). It was
previously demonstrated that functional mitochondria derived from
healthy hMSCs reduced mitochondrial fragmentation and Drp1 S616
phosphorylation in damaged hMSCs (167). The mitochondria derived from
ADSCs supplied ATP to fulfill metabolic needs, thereby enhancing
the overall metabolic activity of the recipient ADSCs through
glycolysis (169). Moreover,
ADSCs that received donated mitochondria showed enhanced
proliferation and mobility in serum starvation conditions,
demonstrating enhanced stress tolerance, anti-aging capabilities
and multidirectional differentiation potential in ADSCs treated
with doxorubicin (169). It
should be noted that mitochondria can be artificially introduced
into BM-MSCs in vitro, and within certain limits, the
addition of extracellular mitochondria could enhance the capacity
of the recipient cells to take up more mitochondria (170). Receiving mitochondria also
increases BM-MSC proliferation and migration, promoting cells to
enter the S and G2/M phases, and resisting the replicative
senescence of BM-MSCs (170). In
addition, mitochondrial transfer also enhances the osteogenic
differentiation potential and mitochondrial functions, including
OCR and ATP production in BM-MSCs (170).
A recent study demonstrated that the transfer of
mitochondria from MSCs to endothelial cells, either by direct
cell-to-cell contact or artificial transplantation, significantly
enhanced the engraftment ability of the cells in the in vivo
environment (171). The
stimulation of these effects was shown to be related to mitophagy;
however, the precise mechanism remained unknown. These data
suggested that boosting mitochondrial activity by mitochondrial
transfer and donation can also support cell survival in an in
vivo environment, proposing a potential application for
mitochondrial therapy in cell transplantation technology.
MSCs possess the capacity to modulate immune
responses, particularly those of T-cells, indicating their
therapeutic potential in treating autoimmune disorders, preventing
graft rejection and managing graft vs. host disease (GVHD)
(172). One mechanism of
immunoregulation by MSCs may be attributed to their ability to
transfer mitochondria to immune cells, regulating cell function and
consequent immune outcomes. It was previously demonstrated that
CD3+ T-cells receiving mitochondria from UC-MSCs
increased the expression of CD25 (a factor for T-cell activation)
and FoxP3 [a marker of T-regulatory cell (Treg) differentiation],
compared to CD3+ T-cells not receiving mitochondria from
UC-MSCs (173). Since Tregs
participate in maintaining tolerance and preventing autoimmunity,
they are involved in various pathological conditions, including
GVHD (174). Treg cell
differentiation induced by mitochondria transferred from UC-MSCs
enhanced the ability to inhibit the proliferation of peripheral
blood mononuclear cells (173).
As previously demonstrated, mitochondrial transfer also markedly
improved the survival rate of mice with GVHD, while decreasing
tissue inflammation (173). The
key strategy employed by Tregs to preserve immune homeostasis is
the inhibition of conventional T-cell (Tconv) proliferation
(175). A previous study
demonstrated that ADSCs transferred mitochondria to Tregs during
co-culture, 85% of which remained functional, allowing Tregs to
efficiently inhibit the proliferation of Tconvs (176). The insufficient presence of HLA
class II antigens on ASCs restricted the uptake of mitochondria by
>80% (176). Thus,
mitochondrial transfer from MSCs supports T-cells in regulating
uncontrolled immunity.
Diabetes mellitus is chiefly characterized by
increased macrophage infiltration in the kidneys. Implementing
strategies to prevent the pro-inflammatory (M1) phenotype or to
shift them to an anti-inflammatory (M2) state has been proposed to
help reduce kidney injury in diabetic mice (180). Metabolic and physiological
changes in mitochondria are essential for regulating macrophage
polarization, proliferation and survival (181). While M1 macrophages utilize
glycolysis for energy, M2 macrophages depend on mitochondrial
oxidative phosphorylation (182,183). Mitochondrial dysfunction in
macrophages derived from diabetic mice has been shown to lead to M1
polarization and resulted in subsequent inflammation (184). In that context, in another study,
mitochondria transferred from MSCs to RAW264.7 macrophages
pre-treated with high glucose helped the cells to reduce ROS
production and restore MMP and ATP levels by regulating gene
expression of the glycolytic and citric acid cycles (185). RAW264.7 macrophages induced by
high glucose that took up mitochondria exhibited lower levels of
cytokine secretion, such as IL-1β and TNF-α, two cytokines which
are critical for mitigating inflammation (185).
Alveolar macrophages (AMs), found in the lung lumen
adjacent to the epithelium, function to phagocytose debris and help
resolve inflammation (186).
Dysfunctional AMs with an irregular mitochondrial function
frequently contribute to severe inflammation in the lungs (186). It has been shown that MSCs
transferring mitochondria to macrophages can enhance their
phagocytosis, which may uncover a mechanism behind the
antimicrobial effects of BM-MSCs (187). Furthermore, mitochondrial
transfer from MSCs can boost the anti-inflammatory and
antibacterial functions of macrophages by promoting their
differentiation into the M2 phenotype (188). As previously demonstrated, when
MSCs and primary human monocyte-derived macrophages (MDMs) were
co-cultured for 4 h, the mitochondrial transfer from BM-MSCs
through TNTs enhanced the ability of macrophages to kill up to 80%
of extracellular Escherichia coli bacteria (187). Notably, macrophages co-cultured
with MSCs exhibited a substantial and consistent enhancement of
both OCR and ATP production (187). This was consistent with a report
that macrophages receiving mitochondria from exosomes secreted by
MSCs in the co-culture settings significantly increased OCR and
reduced proton leak (189). This
functioned as a survival strategy in response to oxidative stress,
where donated mitochondria were re-utilized through a fusion
process to enhance the bioenergetics of macrophages (189). Exosomes derived from ADSCs could
transfer mitochondrial components to murine alveolar macrophages,
which then fused with the macrophage mitochondria (190). Murine alveolar macrophages in
acute lung injury exhibited an increased number of mitochondria,
improved mitochondrial morphology, and increased membrane potential
after receiving mitochondria from ADSCs (190). Moreover, mitochondria played a
critical role in the metabolic programming of immune cells and in
reshaping cellular phenotypes and function (191). The transfer of mitochondria via
ADSC-derived exosomes caused LPS-stimulated macrophages to
transition from a pro-inflammatory M1 phenotype to an
anti-inflammatory M2 phenotype (190). Furthermore, mitochondria have
been shown to be transferred from MSCs to macrophages via EVs,
which boosted oxidative phosphorylation (OXPHOS), leading to
enhanced phagocytosis of macrophages (192). These data demonstrate that MSCs
employ multiple mechanisms to transfer mitochondria to immune
cells, which often have high mobility in circulation. Thus,
questions regarding the driving factors for mitochondrial transfer
from MSCs to immune cells warrant future research, potentially
opening a potential opportunity for therapeutic application.
The reproductive system consists of various organs
and structures that are distinct in males and females. Different
cell compartments in the reproductive system are responsible for
specific functions that support the formation and production of
offspring. The two key cell types directly responsible for
fertilization are sperm and eggs. While mitochondria are crucial
for sperm maturation, survival, and mobility, oocyte mitochondria
are critical for fertilization and embryonic development, which are
also majorly inherited by the fetus (193). Thus, both mtDNA content and
mitochondrial function are essential for successful fertilization,
pregnancy and live births. Mitochondria in oocytes produce energy
mainly via OXPHOS, as glycolysis is only unblocked at the early
stage of blastocyst formation. ATP production is associated with
egg quality, as aged and poor-quality eggs have significantly lower
mtDNA content and produce much less ATP compared to normal and
healthy oocytes (194). This
could potentially result in low or no fertilization (195,196). Aging is one of the major
contributing factors to reduced female fertility, as it directly
affects mtDNA replication and mutation, and changes mitochondrial
metabolism (197).
Mitochondria can be isolated from healthy donors
and directly injected into the eggs or embryos of unhealthy
individuals who may carry mutations interfering with mitochondrial
function. The technology is effective in supplementing oocytes with
healthy mitochondria to improve oocyte quality and fertilization
rate, and to increase chances of live births and offspring with
normal phenotypes, providing hope to infertile women and those with
inherited diseases (198).
Multiple studies have been conducted on both animal and human
models, aiming to examine the applicability of mitochondria
isolated from various cell types to identify the most suitable
source of mitochondria specifically for oocyte improvement; a
number of these sources were from MSCs of different origins. Even
though the current technology focuses on the autologous grafting of
mitochondria due to concerns about mtDNA and nuclear DNA mismatch
(199), the therapy has yielded
positive results in assisting cases of aged and low-quality
oocytes. For example, in animal models, for instance, the
mitochondria isolated from MSCs derived from UC-MSCs were directly
injected into metaphase I oocytes of aged mice, which consequently
resulted in improved mitochondrial function and development of
early embryos (200). On the
other hand, mitochondria derived from ADSCs were shown to be
effective in improving oocyte competence, fertilization rate, and
blastocyst development in both aged-MII and cryopreserved oocytes
(201,202). MSCs (EnMSCs) were also a source
of cells that have been tested in mice. The increase in oocyte
quality and maturation associated with an enhanced birth rate and
embryonic development suggested that mitochondrial transfer from
EnMSCs was effective in rescuing infertility due to the poor
quality of germinal vesicle oocytes (203). Additionally, mitochondria
isolated from iPSCs were also shown to be a good source for
mitochondrial transfer due to their compatibility with the
mitochondrial morphology in oocytes and embryos (204). Furthermore, advancements have
been made to bring the technology of mitochondrial therapy to
humans. Particularly, mitochondria from urine-derived MSCs and
oogonial stem cells were isolated for use in aged human oocytes.
The results revealed an improvement in pregnancy rates, embryonic
development and live birth rates (205-209).
These data highlight the promising potential of mitochondrial
transplantation technology in supporting the reproductive system,
particularly in addressing female infertility. Future research is
also anticipated to advance mitochondrial therapy in the male
reproductive system.
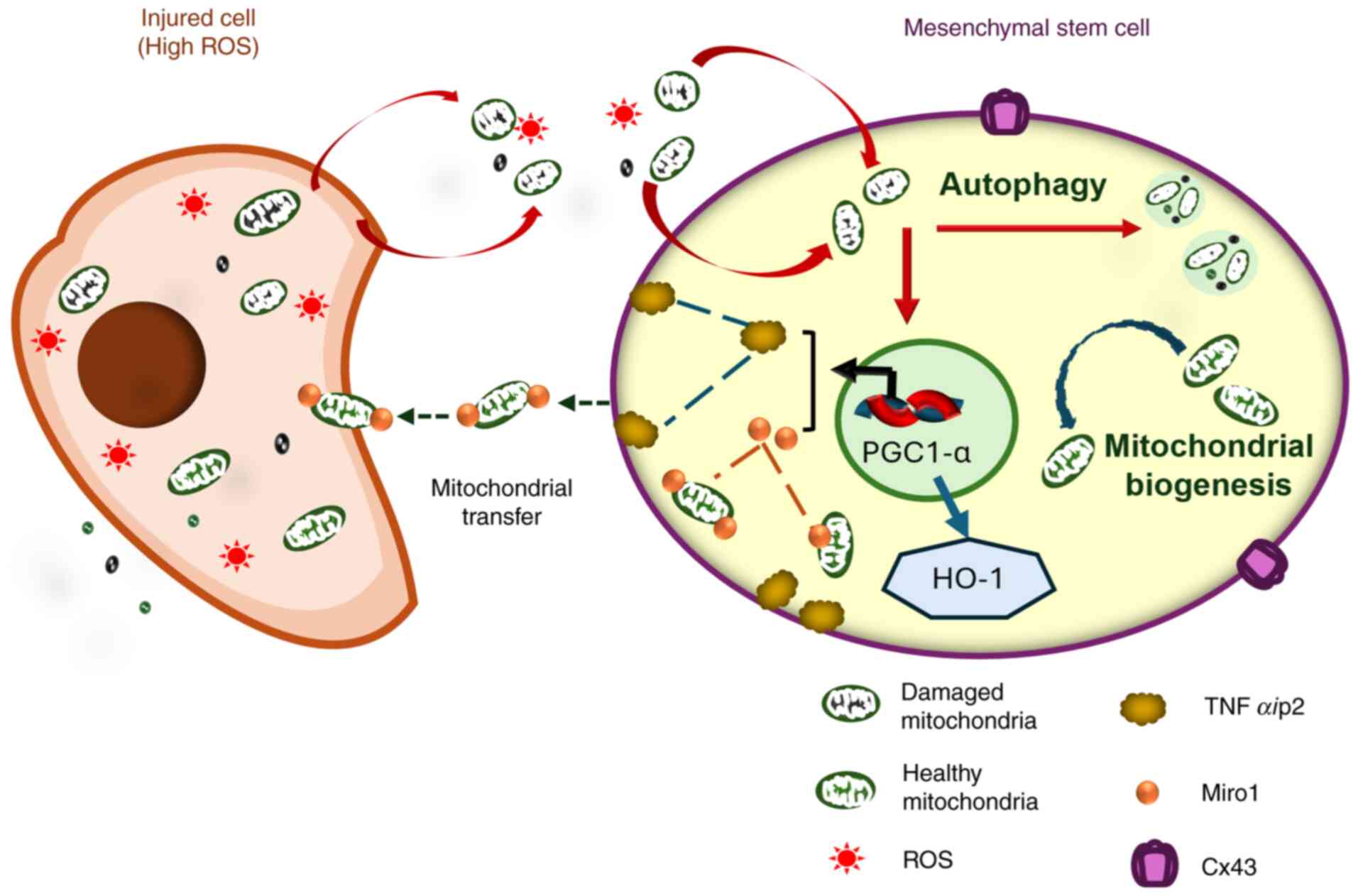
The molecular mechanism of TNT formation involves
Miro1, an outer mitochondrial membrane protein essential for
mitochondrial transport and donation (217,218). Following the increased levels of
Miro1 induced by oxidative stress, MSCs enhance their capacity for
mitochondrial transfer through TNTs (47,55,88,219). Miro1 deletion has been
proven to inhibit MSC mitochondrial transfer and impair the
microtubule movement of donated mitochondria (217,218). It should be noted that the
expression of Miro1 and TNFαip2 are interrelated in
mitochondrial transfer (219).
TNFαip2 is a marker of TNT formation that regulates
intercellular interactions (114). TNF-α-induced oxidative stress
causes significantly high TNFαip2 expression (220). When TNFαip2 is deleted in
MSCs, the overexpression of Miro1 also decreases
mitochondrial transfer, and the control of ROS levels is impaired
both in vitro and in vivo (217). NF-κB and TNFαip2
overexpression increase ROS levels, which subsequently leads to
TNTs formation, and NAC reverses this process by reducing ROS
(47,114,221). Furthermore, there is an
association between the phosphorylation of NF-κB and TNT
formation inhibition (114,221,222). The expression of TNFαip2
is directly related to mitochondrial transfer capacity between
cells through TNTs (219,220). The N- and C-terminal region of
the TNFaip2 protein have different contributions to the remodeling
of the MSC's plasma membrane to form TNTs (223).
Additionally, after receiving stimulation from
injured cell signals, MSCs also promote cell-to-cell interactions
and mitochondrial transfer capacity. Connexin 43 (Cx43) has been
shown to be related to the transfer of mitochondria between cells
(63,224). ROS overproduction caused by LPS
in injured cells promotes the mitochondrial transfer from MSCs
through Cx43 regulation (214).
Cx43 knockdown in MSCs interrupts the mitochondrial transfer from
MSCs to injured chondrocytes (63,225). In the event that MSCs upregulate
Cx43 expression by iron oxide nanoparticles, the mitochondrial
transfer ability of MSCs will be enhanced (226). On the other hand, various
mechanisms can be utilized by other cells, particularly cancer
cells, to steal mitochondria from their neighboring cells in the
microenvironment (227-231);
however, these mechanisms have not been implicated in the
MSC-mediated mitochondrial rescue pathway. Questions remain as to
whether these mechanisms exist between MSCs and other cells.
There are two separate fates for donated exogenous
mitochondria once internalized inside recipient cells: Integration
into the host mitochondrial network or degradation through
mitophagy and autophagy processes. Within the host cells, exogenous
mitochondria can fuse with the host mitochondrial network, and this
fusion can occur via different mechanisms across various types of
cells (232-234).
This process is associated with the expression of mitofusin (MNF)1,
MNF2 and optic atrophy 1 (OPA1) (235). Under oxidative stress conditions,
cells downregulate the expression of these mitochondrial fusion
proteins, particularly MFN1 and OPA1(232). In the majority of cases, this
downregulation is affected by the presence of exogenous
mitochondria. A previous study demonstrated that following
exogenous mitochondrial transplantation, the recipient cells
contained nearly 12% mtDNA from donor cells after 24 h (171). Notably, this exogenous mtDNA
cannot be detected on day 7. Lysosomes of host cells degrade parts
of exogenous mitochondria (232,233,235). On the other hand, it has been
shown that at ~6-12 h following transplantation, exogenous
mitochondria enter the autophagy process in host cells (171). There is evidence to indicate that
donated mitochondria from MSCs inside endothelial cells undergo
mitophagy mediated by the PINK1-Parkin pathway, which facilitates
biogenesis in the recipient cells and enhances their engraftment
in vivo (171). Future
research is warranted to elucidate the precise mechanisms through
which host cells take up and regulate the use of exogenous
mitochondria within their cytoplasm.
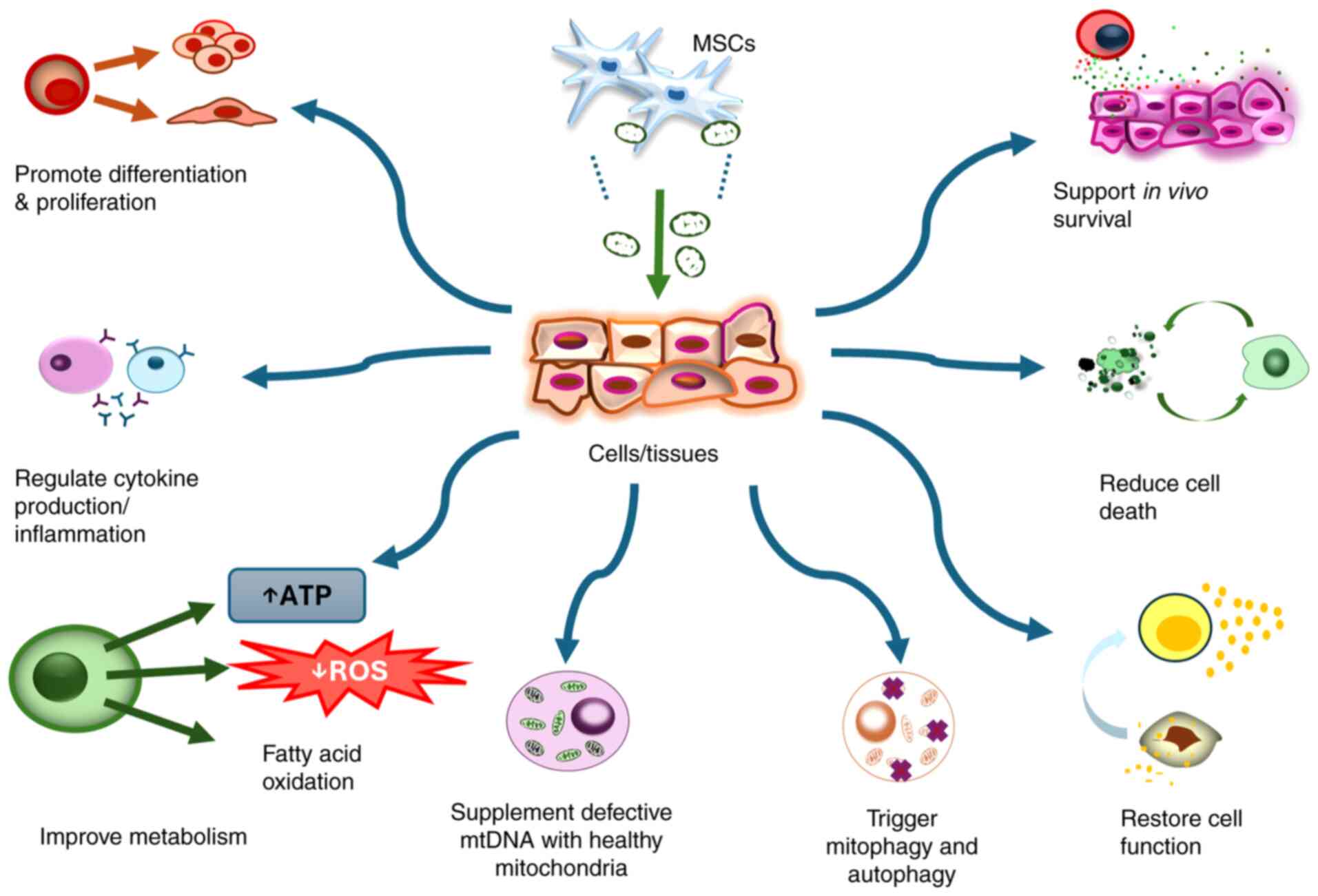
Mitochondrial transfer and transplantation appear
to be an innovative and promising tool for targeting
mitochondria-related pathological conditions (25,236-238).
However, identifying a qualified and ready-to-use source of
mitochondria has been a challenge for clinical translation.
Difficulties in obtaining mitochondrial sources can limit the
reproducibility of the approach, increase the need for invasive
procedures in patients, and hinder the quality control of the
transplanted mitochondria. In clinical trials, ready-to-use
mitochondria may help to improve the feasibility and efficacy of
their application, ensuring timely treatment (239). A recent study demonstrated that
storable human mitochondria, as a ready source of exogenous
mitochondria, are effective in a mouse model of Leigh syndrome
(240). Thus, a stable and
quality source of mitochondria is key to the success of
mitochondrial transplantation. In this context, MSCs have emerged
as potential donors of mitochondria in mitochondrial therapy, and
preclinical data indicate that their use is effective across a
range of cell types, diseases and conditions. A summary of
recipient cell types receiving mitochondrial transfer from MSCs is
presented in Table I. However,
although the exact mechanisms underlying tissue regeneration
through mitochondrial transfer are not yet fully understood, it is
proposed to involve several rescuing effects (Fig. 4).
Despite this promising application, it should be
noted that mitochondrial transfer and transplantation may also pose
significant health risks in the case of misuse. Mitochondrial
therapy, whether utilizing mitochondria from MSCs or other cell
types (both autologous or allogenic), may encounter various
challenges, including the acceleration of the immune response and
the promotion of tumor metastasis (241-243).
For instance, allogenic mtDNA has been shown to accelerate the
innate immune response in mice (244), and even in cases of autologous
transplantation, an mtDNA mutation in the cells of the donor could
elicit an immune response that is dependent on host major
histocompatibility complex (245). Although allogenic mitochondrial
transplantation has been shown in several studies, it is important
to understand the mechanism and effects of the treatment, and
screening for mtDNA is crucial to ensure safety in clinical
applications. In addition, several ethical questions have also been
raised, primarily in the field of assisted reproductive technology,
concerning the retention of exogenous mitochondria and the
potential for mtDNA/nuclear DNA mismatch within host cells
(243,246,247). Given the relevance of these
issues in other clinical applications, further research is required
to clarify the mechanisms of exogenous mitochondrial interaction
with the host cells and determine the effective dosages for
different cases. Furthermore, optimizing the procedures and routes
of application for specific pathological conditions is crucial to
enhance the accessibility of this technique in diverse clinical
settings.
Although challenges persist, progress has been
achieved to improve the safety and broad application of
mitochondrial transfer and transplantation. Firstly, research has
focused on optimizing the cellular uptake of exogenous mitochondria
through various techniques of artificial transplantation (Fig. 1B). These technologies focus on
improving mitochondrial permeabilization across the cell membranes
by utilizing both mechanical forces and penetrating peptides. While
none of these methods are currently suitable for clinical
application, ongoing efforts are focused on refining existing
techniques and developing new materials and technologies. Current
approaches, however, have successfully achieved mitochondrial
transplantation into isolated cells. This success holds promise for
improving cell function in therapeutic areas, such as stem cell and
immune cell therapies. Furthermore, research is being conducted to
understand the mechanism of intercellular mitochondrial transfer,
aiming to enhance the rescue of damaged cells in in vivo
settings (248). For instance,
stimulating mitochondrial transfer from MSCs, such as enhancing the
formation of TNT, increasing the secretion of EVs, and promoting
cell fusion, could be advantageous for multiple applications. Most
importantly, future research is required to verify the
effectiveness of different stem cell types and their culture
conditions, while clarifying and defining appropriate mitochondrial
quality measures. With the current progress, it is expected that
MSCs will provide a cell source for mitochondrial transplantation,
offering a new approach to regenerative medicine and adding an
essential element to stem cell therapy.
Not applicable.
Funding: No funding was received.
Not applicable.
CMT was involved in the conceptualization of the
study, in the preparation of the figures, and in the writing of the
original draft of the manuscript. VTTN was involved in the
conceptualization of the study, in the preparation of the figures
visualization, in the writing of the original draft of the
manuscript, and in the writing, reviewing and editing of the
manuscript. Both authors have read and confirmed the final
manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
During the preparation of this work, AI tools were
used to improve the readability and language of the manuscript or
to generate images, and subsequently, the authors revised and
edited the content produced by the AI tools as necessary, taking
full responsibility for the ultimate content of the present
manuscript.
|
1
|
Amorim JA, Coppotelli G, Rolo AP, Palmeira
CM, Ross JM and Sinclair DA: Mitochondrial and metabolic
dysfunction in ageing and age-related diseases. Nat Rev Endocrinol.
18:243–258. 2022.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Hyatt H, Deminice R, Yoshihara T and
Powers SK: Mitochondrial dysfunction induces muscle atrophy during
prolonged inactivity: A review of the causes and effects. Arch
Biochem Biophys. 662:49–60. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Dagda RK: Role of mitochondrial
dysfunction in degenerative brain diseases, an overview. Brain Sci.
8(178)2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kaarniranta K, Uusitalo H, Blasiak J,
Felszeghy S, Kannan R, Kauppinen A, Salminen A, Sinha D and
Ferrington D: Mechanisms of mitochondrial dysfunction and their
impact on age-related macular degeneration. Prog Retin Eye Res.
79(100858)2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zilio E, Piano V and Wirth B:
Mitochondrial dysfunction in spinal muscular atrophy. Int J Mol
Sci. 23(10878)2022.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Weiss SL, Zhang D, Bush J, Graham K, Starr
J, Murray J, Tuluc F, Henrickson S, Deutschman CS, Becker L, et al:
Mitochondrial dysfunction is associated with an immune paralysis
phenotype in pediatric sepsis. Shock. 54:285–293. 2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
McBride MA, Owen AM, Stothers CL,
Hernandez A, Luan L, Burelbach KR, Patil TK, Bohannon JK, Sherwood
ER and Patil NK: The metabolic basis of immune dysfunction
following sepsis and trauma. Front Immunol. 11(1043)2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Prasun P: Mitochondrial dysfunction in
metabolic syndrome. Biochim Biophys Acta Mol Basis Dis.
1866(165838)2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Bhatti JS, Bhatti GK and Reddy PH:
Mitochondrial dysfunction and oxidative stress in metabolic
disorders-a step towards mitochondria based therapeutic strategies.
Biochim Biophys Acta Mol Basis Dis. 1863:1066–1077. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Montgomery MK: Mitochondrial dysfunction
and diabetes: Is mitochondrial transfer a friend or foe? Biology
(Basel). 8(33)2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Feichtinger RG, Sperl W, Bauer JW and
Kofler B: Mitochondrial dysfunction: A neglected component of skin
diseases. Exp Dermatol. 23:607–614. 2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Mizumura K, Cloonan SM, Nakahira K,
Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko
GR, et al: Mitophagy-dependent necroptosis contributes to the
pathogenesis of COPD. J Clin Invest. 124:3987–4003. 2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Li X, Zhang W, Cao Q, Wang Z, Zhao M, Xu L
and Zhuang Q: Mitochondrial dysfunction in fibrotic diseases. Cell
Death Discov. 6(80)2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ryter SW, Rosas IO, Owen CA, Martinez FJ,
Choi ME, Lee CG, Elias JA and Choi AMK: Mitochondrial dysfunction
as a pathogenic mediator of chronic obstructive pulmonary disease
and idiopathic pulmonary fibrosis. Ann Am Thorac Soc. 15 (Suppl
4):S266–S272. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Kandasamy J, Olave N, Ballinger SW and
Ambalavanan N: Vascular endothelial mitochondrial function predicts
death or pulmonary outcomes in preterm infants. Am J Respir Crit
Care Med. 196:1040–1049. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Rizzuto R, De Stefani D, Raffaello A and
Mammucari C: Mitochondria as sensors and regulators of calcium
signalling. Nat Rev Mol Cell Biol. 13:566–578. 2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Matuz-Mares D, González-Andrade M,
Araiza-Villanueva MG, Vilchis-Landeros MM and Vázquez-Meza H:
Mitochondrial calcium: Effects of its imbalance in disease.
Antioxidants (Basel). 11(801)2022.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Belosludtsev KN, Talanov EY, Starinets VS,
Agafonov AV, Dubinin MV and Belosludtseva NV: Transport of
Ca2+ and Ca2+-dependent permeability
transition in rat liver mitochondria under the
streptozotocin-induced type I diabetes. Cells.
8(1014)2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Sundaramoorthy P, Sim JJ, Jang YS, Mishra
SK, Jeong KY, Mander P, Chul OB, Shim WS, Oh SH, Nam KY and Kim HM:
Modulation of intracellular calcium levels by calcium lactate
affects colon cancer cell motility through calcium-dependent
calpain. PLoS One. 10(e0116984)2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Spees JL, Olson SD, Whitney MJ and Prockop
DJ: Mitochondrial transfer between cells can rescue aerobic
respiration. Proc Natl Acad Sci USA. 103:1283–1288. 2006.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Rodriguez AM, Nakhle J, Griessinger E and
Vignais ML: Intercellular mitochondria trafficking highlighting the
dual role of mesenchymal stem cells as both sensors and rescuers of
tissue injury. Cell Cycle. 17:712–721. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Plotnikov EY, Babenko VA, Silachev DN,
Zorova LD, Khryapenkova TG, Savchenko ES, Pevzner IB and Zorov DB:
Intercellular transfer of mitochondria. Biochemistry (Mosc).
80:542–548. 2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Hayashida K, Takegawa R, Shoaib M, Aoki T,
Choudhary RC, Kuschner CE, Nishikimi M, Miyara SJ, Rolston DM,
Guevara S, et al: Mitochondrial transplantation therapy for
ischemia reperfusion injury: A systematic review of animal and
human studies. J Transl Med. 19(214)2021.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Yang C, Yokomori R, Chua LH, Tan SH, Tan
DQ, Miharada K, Sanda T and Suda T: Mitochondria transfer mediates
stress erythropoiesis by altering the bioenergetic profiles of
early erythroblasts through CD47. J Exp Med.
219(e20220685)2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Liu D, Gao Y, Liu J, Huang Y, Yin J, Feng
Y, Shi L, Meloni BP, Zhang C, Zheng M and Gao J: Intercellular
mitochondrial transfer as a means of tissue revitalization. Signal
Transduct Target Ther. 6(65)2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Emani SM, Piekarski BL, Harrild D, Del
Nido PJ and McCully JD: Autologous mitochondrial transplantation
for dysfunction after ischemia-reperfusion injury. J Thorac
Cardiovasc Surg. 154:286–289. 2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Guariento A, Piekarski BL, Doulamis IP,
Blitzer D, Ferraro AM, Harrild DM, Zurakowski D, Del Nido PJ,
McCully JD and Emani SM: Autologous mitochondrial transplantation
for cardiogenic shock in pediatric patients following
ischemia-reperfusion injury. J Thorac Cardiovasc Surg.
162:992–1001. 2021.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Michaeloudes C, Li X, Mak JCW and Bhavsar
PK: Study of mesenchymal stem cell-mediated mitochondrial transfer
in in vitro models of oxidant-mediated airway epithelial and smooth
muscle cell injury. Methods Mol Biol. 2269:93–105. 2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Konari N, Nagaishi K, Kikuchi S and
Fujimiya M: Mitochondria transfer from mesenchymal stem cells
structurally and functionally repairs renal proximal tubular
epithelial cells in diabetic nephropathy in vivo. Sci Rep.
9(5184)2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Moschoi R, Imbert V, Nebout M, Chiche J,
Mary D, Prebet T, Saland E, Castellano R, Pouyet L, Collette Y, et
al: Protective mitochondrial transfer from bone marrow stromal
cells to acute myeloid leukemic cells during chemotherapy. Blood.
128:253–264. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Lambrecht BN and Hammad H: The immunology
of asthma. Nat Immunol. 16:45–56. 2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Chan TK, Tan WSD, Peh HY and Wong WSF:
Aeroallergens induce reactive oxygen species production and DNA
damage and dampen antioxidant responses in bronchial epithelial
cells. J Immunol. 199:39–47. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhao L, Gao J, Chen G, Huang C, Kong W,
Feng Y and Zhen G: Mitochondria dysfunction in airway epithelial
cells is associated with type 2-low asthma. Front Genet.
14(1186317)2023.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Yao Y, Fan XL, Jiang D, Zhang Y, Li X, Xu
ZB, Fang SB, Chiu S, Tse HF, Lian Q and Fu QL: Connexin 43-mediated
mitochondrial transfer of iPSC-MSCs alleviates asthma inflammation.
Stem Cell Reports. 11:1120–1135. 2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Malsin ES and Kamp DW: The mitochondria in
lung fibrosis: Friend or foe? Transl Res. 202:1–23. 2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Takahashi M, Mizumura K, Gon Y, Shimizu T,
Kozu Y, Shikano S, Iida Y, Hikichi M, Okamoto S, Tsuya K, et al:
Iron-dependent mitochondrial dysfunction contributes to the
pathogenesis of pulmonary fibrosis. Front Pharmacol.
12(643980)2022.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Huang T, Lin R, Su Y, Sun H, Zheng X,
Zhang J, Lu X, Zhao B, Jiang X, Huang L, et al: Efficient
intervention for pulmonary fibrosis via mitochondrial transfer
promoted by mitochondrial biogenesis. Nat Commun.
14(5781)2023.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Li CL, Liu JF and Liu SF: Mitochondrial
dysfunction in chronic obstructive pulmonary disease: Unraveling
the molecular nexus. Biomedicines. 12(814)2024.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Canton M, Sánchez-Rodríguez R, Spera I,
Venegas FC, Favia M, Viola A and Castegna A: Reactive oxygen
species in macrophages: Sources and targets. Front Immunol.
12(734229)2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Aridgides DS, Mellinger DL, Armstrong DA,
Hazlett HF, Dessaint JA, Hampton TH, Atkins GT, Carroll JL and
Ashare A: Functional and metabolic impairment in cigarette
smoke-exposed macrophages is tied to oxidative stress. Sci Rep.
9(9624)2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Tulen CBM, Wang Y, Beentjes D, Jessen PJJ,
Ninaber DK, Reynaert NL, van Schooten FJ, Opperhuizen A, Hiemstra
PS and Remels AHV: Dysregulated mitochondrial metabolism upon
cigarette smoke exposure in various human bronchial epithelial cell
models. Dis Model Mech. 15(dmm049247)2022.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Li X, Michaeloudes C, Zhang Y, Wiegman CH,
Adcock IM, Lian Q, Mak JCW, Bhavsar PK and Chung KF: Mesenchymal
stem cells alleviate oxidative stress-induced mitochondrial
dysfunction in the airways. J Allergy Clin Immunol.
141:1634–1645.e5. 2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Li X, Zhang Y, Yeung SC, Liang Y, Liang X,
Ding Y, Ip MS, Tse HF, Mak JC and Lian Q: Mitochondrial transfer of
induced pluripotent stem cell-derived mesenchymal stem cells to
airway epithelial cells attenuates cigarette smoke-induced damage.
Am J Respir Cell Mol Biol. 51:455–465. 2014.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Wu BB, Leung KT and Poon EN:
Mitochondrial-targeted therapy for doxorubicin-induced
cardiotoxicity. Int J Mol Sci. 23(1912)2022.PubMed/NCBI View Article : Google Scholar
|
|
45
|
He H, Wang L, Qiao Y, Zhou Q, Li H, Chen
S, Yin D, Huang Q and He M: Doxorubicin induces endotheliotoxicity
and mitochondrial dysfunction via ROS/eNOS/NO pathway. Front
Pharmacol. 10(1531)2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Tang H, Tao A, Song J, Liu Q, Wang H and
Rui T: Doxorubicin-induced cardiomyocyte apoptosis: Role of
mitofusin 2. Int J Biochem Cell Biol. 88:55–59. 2017.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Zhang Y, Yu Z, Jiang D, Liang X, Liao S,
Zhang Z, Yue W, Li X, Chiu SM, Chai YH, et al: iPSC-MSCs with high
intrinsic MIRO1 and sensitivity to TNF-α yield efficacious
mitochondrial transfer to rescue anthracycline-induced
cardiomyopathy. Stem Cell Reports. 7:749–763. 2016.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Jin N, Zhang M, Zhou L, Jin S, Cheng H, Li
X, Shi Y, Xiang T, Zhang Z, Liu Z, et al: Mitochondria
transplantation alleviates cardiomyocytes apoptosis through
inhibiting AMPKα-mTOR mediated excessive autophagy. FASEB J.
38(e23655)2024.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Tanaka-Esposito C, Chen Q and Lesnefsky
EJ: Blockade of electron transport before ischemia protects
mitochondria and decreases myocardial injury during reperfusion in
aged rat hearts. Transl Res. 160:207–216. 2012.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Huang J, Li R and Wang C: The role of
mitochondrial quality control in cardiac ischemia/reperfusion
injury. Oxid Med Cell Longev. 2021(5543452)2021.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Bouhamida E, Morciano G, Perrone M, Kahsay
AE, Della Sala M, Wieckowski MR, Fiorica F, Pinton P, Giorgi C and
Patergnani S: The interplay of hypoxia signaling on mitochondrial
dysfunction and inflammation in cardiovascular diseases and cancer:
From molecular mechanisms to therapeutic approaches. Biology
(Basel). 11(300)2022.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Cselenyák A, Pankotai E, Horváth EM, Kiss
L and Lacza Z: Mesenchymal stem cells rescue cardiomyoblasts from
cell death in an in vitro ischemia model via direct cell-to-cell
connections. BMC Cell Biol. 11(29)2010.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Han H, Hu J, Yan Q, Zhu J, Zhu Z, Chen Y,
Sun J and Zhang R: Bone marrow-derived mesenchymal stem cells
rescue injured H9c2 cells via transferring intact mitochondria
through tunneling nanotubes in an in vitro simulated
ischemia/reperfusion model. Mol Med Rep. 13:1517–1524.
2016.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Mori D, Miyagawa S, Kawamura T, Yoshioka
D, Hata H, Ueno T, oda K, Kuratani T, Oota M, Kawai K, et al:
Mitochondrial transfer induced by adipose-derived mesenchymal stem
cell transplantation improves cardiac function in rat models of
ischemic cardiomyopathy. Cell Transplant.
32(9636897221148457)2023.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Mahrouf-Yorgov M, Augeul L, Da Silva CC,
Jourdan M, Rigolet M, Manin S, Ferrera R, Ovize M, Henry A, Guguin
A, et al: Mesenchymal stem cells sense mitochondria released from
damaged cells as danger signals to activate their rescue
properties. Cell Death Differ. 24:1224–1238. 2017.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Zhang ZH, Zhu W, Ren HZ, Zhao X, Wang S,
Ma HC and Shi XL: Mesenchymal stem cells increase expression of
heme oxygenase-1 leading to anti-inflammatory activity in treatment
of acute liver failure. Stem Cell Res Ther. 8(70)2017.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Tang Q, Zheng G, Feng Z, Chen Y, Lou Y,
Wang C, Zhang X, Zhang Y, Xu H, Shang P and Liu H: Trehalose
ameliorates oxidative stress-mediated mitochondrial dysfunction and
ER stress via selective autophagy stimulation and autophagic flux
restoration in osteoarthritis development. Cell Death Dis.
8(e3081)2017.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Liu H, Li Z, Cao Y, Cui Y, Yang X, Meng Z
and Wang R: Effect of chondrocyte mitochondrial dysfunction on
cartilage degeneration: A possible pathway for osteoarthritis
pathology at the subcellular level. Mol Med Rep. 20:3308–3316.
2019.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Sanchez-Lopez E, Coras R, Torres A, Lane
NE and Guma M: Synovial inflammation in osteoarthritis progression.
Nat Rev Rheumatol. 18:258–275. 2022.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Zheng L, Zhang Z, Sheng P and Mobasheri A:
The role of metabolism in chondrocyte dysfunction and the
progression of osteoarthritis. Ageing Res Rev.
66(101249)2021.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Wang R, Maimaitijuma T, Ma YY, Jiao Y and
Cao YP: Mitochondrial transfer from bone-marrow-derived mesenchymal
stromal cells to chondrocytes protects against cartilage
degenerative mitochondrial dysfunction in rats chondrocytes. Chin
Med J (Engl). 134:212–218. 2020.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Wu Z, Korntner SH, Mullen AM and Zeugolis
DI: Collagen type II: From biosynthesis to advanced biomaterials
for cartilage engineering. Biomater Biosyst.
4(100030)2021.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Fahey M, Bennett M, Thomas M, Montney K,
Vivancos-Koopman I, Pugliese B, Browning L, Bonassar LJ and Delco
M: Mesenchymal stromal cells donate mitochondria to articular
chondrocytes exposed to mitochondrial, environmental, and
mechanical stress. Sci Rep. 12(21525)2022.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Michelacci YM, Baccarin RYA and Rodrigues
NNP: Chondrocyte homeostasis and differentiation: Transcriptional
control and signaling in healthy and osteoarthritic conditions.
Life (Basel). 13(1460)2023.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Fearon U, Canavan M, Biniecka M and Veale
DJ: Hypoxia, mitochondrial dysfunction and synovial invasiveness in
rheumatoid arthritis. Nat Rev Rheumatol. 12:385–397.
2016.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Zhang X, Eliasberg CD and Rodeo SA:
Mitochondrial dysfunction and potential mitochondrial protectant
treatments in tendinopathy. Ann N Y Acad Sci. 1490:29–41.
2021.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Zhang X, Wada S, Zhang Y, Chen D, Deng XH
and Rodeo SA: Assessment of mitochondrial dysfunction in a murine
model of supraspinatus tendinopathy. J Bone Joint Surg Am.
103:174–183. 2021.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Liu YC, Wang HL, Huang YZ, Weng YH, Chen
RS, Tsai WC, Yeh TH, Lu CS, Chen YL, Lin YW, et al: Alda-1, an
activator of ALDH2, ameliorates Achilles tendinopathy in cellular
and mouse models. Biochem Pharmacol. 175(113919)2020.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Dex S, Alberton P, Willkomm L, Söllradl T,
Bago S, Milz S, Shakibaei M, Ignatius A, Bloch W, Clausen-Schaumann
H, et al: Tenomodulin is required for tendon endurance running and
collagen I fibril adaptation to mechanical load. EBioMedicine.
20:240–254. 2017.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Wei B, Ji M, Lin Y, Wang S, Liu Y, Geng R,
Hu X, Xu L, Li Z, Zhang W and Lu J: Mitochondrial transfer from
bone mesenchymal stem cells protects against tendinopathy both in
vitro and in vivo. Stem Cell Res Ther. 14(104)2023.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Lee JM, Hwang JW, Kim MJ, Jung SY, Kim KS,
Ahn EH, Min K and Choi YS: Mitochondrial transplantation modulates
inflammation and apoptosis, alleviating tendinopathy both in vivo
and in vitro. Antioxidants (Basel). 10(696)2021.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Kubat GB, Bouhamida E, Ulger O, Turkel I,
Pedriali G, Ramaccini D, Ekinci O, Ozerklig B, Atalay O, Patergnani
S, et al: Mitochondrial dysfunction and skeletal muscle atrophy:
Causes, mechanisms, and treatment strategies. Mitochondrion.
72:33–58. 2023.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Hyatt HW and Powers SK: Mitochondrial
dysfunction is a common denominator linking skeletal muscle wasting
due to disease, aging, and prolonged inactivity. Antioxidants
(Basel). 10(588)2021.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Matsumoto C, Sekine H, Nahata M, Mogami S,
Ohbuchi K, Fujitsuka N and Takeda H: Role of mitochondrial
dysfunction in the pathogenesis of cisplatin-induced myotube
atrophy. Biol Pharm Bull. 45:780–792. 2022.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Shen S, Liao Q, Liu J, Pan R, Lee SMY and
Lin L: Myricanol rescues dexamethasone-induced muscle dysfunction
via a sirtuin 1-dependent mechanism. J Cachexia Sarcopenia Muscle.
10:429–444. 2019.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Yang B, Yang X, Sun X, Shi J, Shen Y and
Chen R: IL-6 deficiency attenuates skeletal muscle atrophy by
inhibiting mitochondrial ROS production through the upregulation of
PGC-1 α in septic mice. Oxid Med Cell Longev.
2022(9148246)2022.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Xiao Y, Karam C, Yi J, Zhang L, Li X, Yoon
D, Wang H, Dhakal K, Ramlow P, Yu T, et al: ROS-related
mitochondrial dysfunction in skeletal muscle of an ALS mouse model
during the disease progression. Pharmacol Res. 138:25–36.
2018.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Moore TM, Lin AJ, Strumwasser AR, Cory K,
Whitney K, Ho T, Ho T, Lee JL, Rucker DH, Nguyen CQ, et al:
Mitochondrial dysfunction is an early consequence of partial or
complete dystrophin loss in mdx mice. Front Physiol.
11(690)2020.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Mohiuddin M, Choi JJ, Lee NH, Jeong H,
Anderson SE, Han WM, Aliya B, Peykova TZ, Verma S, García AJ, et
al: Transplantation of muscle stem cell mitochondria rejuvenates
the bioenergetic function of dystrophic muscle. bioRxiv:
2020.04.17.017822, 2020.
|
|
80
|
Wang L, Jiao XF, Wu C, Li XQ, Sun HX, Shen
XY, Zhang KZ, Zhao C, Liu L, Wang M, et al: Trimetazidine
attenuates dexamethasone-induced muscle atrophy via inhibiting
NLRP3/GSDMD pathway-mediated pyroptosis. Cell Death Discov.
7(251)2021.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Kim MJ, Lee JM, Min K and Choi YS:
Xenogeneic transplantation of mitochondria induces muscle
regeneration in an in vivo rat model of dexamethasone-induced
atrophy. J Muscle Res Cell Motil. 45:53–68. 2024.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Xu S, Li Y, Chen JP, Li DZ, Jiang Q, Wu T
and Zhou XZ: Oxygen glucose deprivation/re-oxygenation-induced
neuronal cell death is associated with Lnc-D63785 m6A methylation
and miR-422a accumulation. Cell Death Dis. 11(816)2020.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Juntunen M, Hagman S, Moisan A, Narkilahti
S and Miettinen S: In vitro oxygen-glucose deprivation-induced
stroke models with human neuroblastoma cell- and induced
pluripotent stem cell-derived neurons. Stem Cells Int.
2020(8841026)2020.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Liu F, Lu J, Manaenko A, Tang J and Hu Q:
Mitochondria in ischemic stroke: New insight and implications.
Aging Dis. 9:924–937. 2018.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Norat P, Soldozy S, Sokolowski JD, Gorick
CM, Kumar JS, Chae Y, Yağmurlu K, Prada F, Walker M, Levitt MR, et
al: Mitochondrial dysfunction in neurological disorders: Exploring
mitochondrial transplantation. NPJ Regen Med. 5(22)2020.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Babenko VA, Silachev DN, Zorova LD,
Pevzner IB, Khutornenko AA, Plotnikov EY, Sukhikh GT and Zorov DB:
Improving the post-stroke therapeutic potency of mesenchymal
multipotent stromal cells by cocultivation with cortical neurons:
The role of crosstalk between cells. Stem Cells Transl Med.
4:1011–1020. 2015.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Jian Z, Ding S, Deng H, Wang J, Yi W, Wang
L, Zhu S, Gu L and Xiong X: Probenecid protects against
oxygen-glucose deprivation injury in primary astrocytes by
regulating inflammasome activity. Brain Res. 1643:123–129.
2016.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Babenko VA, Silachev DN, Popkov VA, Zorova
LD, Pevzner IB, Plotnikov EY, Sukhikh GT and Zorov DB: Miro1
enhances mitochondria transfer from multipotent mesenchymal stem
cells (MMSC) to neural cells and improves the efficacy of cell
recovery. Molecules. 23(687)2018.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Li J, Li H, Cai S, Bai S, Cai H and Zhang
X: CD157 in bone marrow mesenchymal stem cells mediates
mitochondrial production and transfer to improve neuronal apoptosis
and functional recovery after spinal cord injury. Stem Cell Res
Ther. 12(289)2021.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Yang Y, Ye G, Zhang YL, He HW, Yu BQ, Hong
YM, You W and Li X: Transfer of mitochondria from mesenchymal stem
cells derived from induced pluripotent stem cells attenuates
hypoxia-ischemia-induced mitochondrial dysfunction in PC12 cells.
Neural Regen Res. 15:464–472. 2020.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Tseng N, Lambie SC, Huynh CQ, Sanford B,
Patel M, Herson PS and Ormond DR: Mitochondrial transfer from
mesenchymal stem cells improves neuronal metabolism after oxidant
injury in vitro: The role of Miro1. J Cereb Blood Flow Metab.
41:761–770. 2021.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Alizadeh A, Dyck SM and Karimi-Abdolrezaee
S: Traumatic spinal cord injury: An overview of pathophysiology,
models and acute injury mechanisms. Front Neurol.
10(282)2019.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Schmidt J and Quintá HR: Mitochondrial
dysfunction as a target in spinal cord injury: Intimate correlation
between pathological processes and therapeutic approaches. Neural
Regen Res. 18:2161–2166. 2023.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Rong Y, Fan J, Ji C, Wang Z, Ge X, Wang J,
Ye W, Yin G, Cai W and Liu W: USP11 regulates autophagy-dependent
ferroptosis after spinal cord ischemia-reperfusion injury by
deubiquitinating beclin 1. Cell Death Differ. 29:1164–1175.
2022.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Li D, Lu X, Xu G, Liu S, Gong Z, Lu F, Xia
X, Jiang J, Wang H, Zou F and Ma X: Dihydroorotate dehydrogenase
regulates ferroptosis in neurons after spinal cord injury via the
P53-ALOX15 signaling pathway. CNS Neurosci Ther. 29:1923–1939.
2023.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Stockwell BR: Ferroptosis turns 10:
Emerging mechanisms, physiological functions, and therapeutic
applications. Cell. 185:2401–2421. 2022.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Yao S, Pang M, Wang Y, Wang X, Lin Y, Lv
Y, Xie Z, Hou J, Du C, Qiu Y, et al: Mesenchymal stem cell
attenuates spinal cord injury by inhibiting mitochondrial quality
control-associated neuronal ferroptosis. Redox Biol.
67(102871)2023.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Jain R, Begum N, Tryphena KP, Singh SB,
Srivastava S, Rai SN, Vamanu E and Khatri DK: Inter and
intracellular mitochondrial transfer: Future of mitochondrial
transplant therapy in Parkinson's disease. Biomed Pharmacother.
159(114268)2023.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Poewe W, Seppi K, Tanner CM, Halliday GM,
Brundin P, Volkmann J, Schrag AE and Lang AE: Parkinson disease.
Nat Rev Dis Primers. 3(17013)2017.PubMed/NCBI View Article : Google Scholar
|
|
101
|
van der Bliek AM, Sedensky MM and Morgan
PG: Cell biology of the mitochondrion. Genetics. 207:843–871.
2017.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Ahn EH, Lei K, Kang SS, Wang ZH, Liu X,
Hong W, Wang YT, Edgington-Mitchell LE, Jin L and Ye K:
Mitochondrial dysfunction triggers the pathogenesis of Parkinson's
disease in neuronal C/EBPβ transgenic mice. Mol Psychiatry.
26:7838–7850. 2021.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Xiao B, Kuruvilla J and Tan EK: Mitophagy
and reactive oxygen species interplay in Parkinson's disease. NPJ
Parkinsons Dis. 8(135)2022.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Eo H, Yu SH, Choi Y, Kim Y, Kang YC, Lee
H, Kim JH, Han K, Lee HK, Chang MY, et al: Mitochondrial
transplantation exhibits neuroprotective effects and improves
behavioral deficits in an animal model of Parkinson's disease.
Neurotherapeutics. 21(e00355)2024.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Thakur P and Nehru B: Inhibition of
neuroinflammation and mitochondrial dysfunctions by carbenoxolone
in the rotenone model of Parkinson's disease. Mol Neurobiol.
51:209–219. 2015.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Cheng XY, Biswas S, Li J, Mao CJ,
Chechneva O, Chen J, Li K, Li J, Zhang JR, Liu CF and Deng WB:
Human iPSCs derived astrocytes rescue rotenone-induced
mitochondrial dysfunction and dopaminergic neurodegeneration in
vitro by donating functional mitochondria. Transl Neurodegener.
9(13)2020.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Chiu GS, Maj MA, Rizvi S, Dantzer R,
Vichaya EG, Laumet G, Kavelaars A and Heijnen CJ: Pifithrin-μ
prevents cisplatin-induced chemobrain by preserving neuronal
mitochondrial function. Cancer Res. 77:742–752. 2017.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Kleih M, Böpple K, Dong M, Gaißler A,
Heine S, Olayioye MA, Aulitzky WE and Essmann F: Direct impact of
cisplatin on mitochondria induces ROS production that dictates cell
fate of ovarian cancer cells. Cell Death Dis.
10(851)2019.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Boukelmoune N, Chiu GS, Kavelaars A and
Heijnen CJ: Mitochondrial transfer from mesenchymal stem cells to
neural stem cells protects against the neurotoxic effects of
cisplatin. Acta Neuropathol Commun. 6(139)2018.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Méthot S, Proulx S, Brunette I and
Rochette PJ: Rescuing cellular function in Fuchs endothelial
corneal dystrophy by healthy exogenous mitochondrial
internalization. Sci Rep. 13(3380)2023.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Jiang D, Chen FX, Zhou H, Lu YY, Tan H, Yu
SJ, Yuan J, Liu H, Meng W and Jin ZB: Bioenergetic crosstalk
between mesenchymal stem cells and various ocular cells through the
intercellular trafficking of mitochondria. Theranostics.
10:7260–7272. 2020.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Vallabh NA, Romano V and Willoughby CE:
Mitochondrial dysfunction and oxidative stress in corneal disease.
Mitochondrion. 36:103–113. 2017.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Numa K, Ueno M, Fujita T, Ueda K, Hiramoto
N, Mukai A, Tokuda Y, Nakano M, Sotozono C, Kinoshita S and Hamuro
J: Mitochondria as a Platform for dictating the cell fate of
cultured human corneal endothelial cells. Invest Ophthalmol Vis
Sci. 61(10)2020.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Jiang D, Gao F, Zhang Y, Wong DS, Li Q,
Tse HF, Xu G, Yu Z and Lian Q: Mitochondrial transfer of
mesenchymal stem cells effectively protects corneal epithelial
cells from mitochondrial damage. Cell Death Dis.
7(e2467)2016.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Hanna SJ, McCoy-Simandle K, Leung E, Genna
A, Condeelis J and Cox D: Tunneling nanotubes, a novel mode of
tumor cell-macrophage communication in tumor cell invasion. J Cell
Sci. 132(jcs223321)2019.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Wang L, Klingeborn M, Travis AM, Hao Y,
Arshavsky VY and Gospe SM III: Progressive optic atrophy in a
retinal ganglion cell-specific mouse model of complex I deficiency.
Sci Rep. 10(16326)2020.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Yu AK, Song L, Murray KD, van der List D,
Sun C, Shen Y, Xia Z and Cortopassi GA: Mitochondrial complex I
deficiency leads to inflammation and retinal ganglion cell death in
the Ndufs4 mouse. Hum Mol Genet. 24:2848–2860. 2015.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Warwick AM, Bomze HM, Wang L, Hao Y,
Stinnett SS and Gospe SM III: Hypoxia-mediated rescue of retinal
ganglion cells deficient in mitochondrial complex I is independent
of the hypoxia-inducible factor pathway. Sci Rep.
14(24114)2024.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Jiang D, Xiong G, Feng H, Zhang Z, Chen P,
Yan B, Chen L, Gandhervin K, Ma C, Li C, et al: Donation of
mitochondria by iPSC-derived mesenchymal stem cells protects
retinal ganglion cells against mitochondrial complex I
defect-induced degeneration. Theranostics. 9:2395–2410.
2019.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Navneet S, Wilson K and Rohrer B: Muller
glial cells in the macula: Their activation and cell-cell
interactions in age-related macular degeneration. Invest Ophthalmol
Vis Sci. 65(42)2024.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Xue B, Xie Y, Xue Y, Hu N, Zhang G, Guan H
and Ji M: Involvement of P2X7 receptors in retinal
ganglion cell apoptosis induced by activated Müller cells. Exp Eye
Res. 153:42–50. 2016.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Hu X, Zhao GL, Xu MX, Zhou H, Li F, Miao
Y, Lei B, Yang XL and Wang Z: Interplay between Muller cells and
microglia aggravates retinal inflammatory response in experimental
glaucoma. J Neuroinflammation. 18(303)2021.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Qiu AW, Bian Z, Mao PA and Liu QH: IL-17A
exacerbates diabetic retinopathy by impairing Müller cell function
via Act1 signaling. Exp Mol Med. 48(e280)2016.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Natoli R, Fernando N, Madigan M, Chu-Tan
JA, Valter K, Provis J and Rutar M: Microglia-derived IL-1β
promotes chemokine expression by Müller cells and RPE in focal
retinal degeneration. Mol Neurodegener. 12(31)2017.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Marques E, Alves Teixeira M, Nguyen C,
Terzi F and Gallazzini M: Lipocalin-2 induces mitochondrial
dysfunction in renal tubular cells via mTOR pathway activation.
Cell Rep. 42(113032)2023.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Chen Y, Yang Y, Liu Z and He L:
Adiponectin promotes repair of renal tubular epithelial cells by
regulating mitochondrial biogenesis and function. Metabolism.
128(154959)2022.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Ji X, Yang X, Gu X, Chu L, Sun S, Sun J,
Song P, Mu Q, Wang Y, Sun X, et al: CUL3 induces mitochondrial
dysfunction via MRPL12 ubiquitination in renal tubular epithelial
cells. FEBS J. 290:5340–5352. 2023.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Cai F, Li D, Xie Y, Wang X, Ma H, Xu H,
Cheng J, Zhuang H and Hua ZC: Sulfide:quinone oxidoreductase
alleviates ferroptosis in acute kidney injury via ameliorating
mitochondrial dysfunction of renal tubular epithelial cells. Redox
Biol. 69(102973)2024.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Xiong YB, Huang WY, Ling X, Zhou S, Wang
XX, Li XL and Zhou LL: Mitochondrial calcium uniporter promotes
kidney aging in mice through inducing mitochondrial
calcium-mediated renal tubular cell senescence. Acta Pharmacol Sin.
45:2149–2162. 2024.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Wang J, Yue X, Meng C, Wang Z, Jin X, Cui
X, Yang J, Shan C, Gao Z, Yang Y, et al: Acute hyperglycemia may
induce renal tubular injury through mitophagy inhibition. Front
Endocrinol (Lausanne). 11(536213)2020.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Aluksanasuwan S, Plumworasawat S, Malaitad
T, Chaiyarit S and Thongboonkerd V: High glucose induces
phosphorylation and oxidation of mitochondrial proteins in renal
tubular cells: A proteomics approach. Sci Rep.
10(5843)2020.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Nahdi AMTA, John A and Raza H: Elucidation
of molecular mechanisms of streptozotocin-induced oxidative stress,
apoptosis, and mitochondrial dysfunction in Rin-5F pancreatic
β-cells. Oxid Med Cell Longev. 2017(7054272)2017.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Bhargava P and Schnellmann RG:
Mitochondrial energetics in the kidney. Nat Rev Nephrol.
13:629–646. 2017.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Kapetanaki S, Kumawat AK, Persson K and
Demirel I: TMAO suppresses megalin expression and albumin uptake in
human proximal tubular cells via PI3K and ERK signaling. Int J Mol
Sci. 23(8856)2022.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Peruchetti DB, Silva-Aguiar RP, Siqueira
GM, Dias WB and Caruso-Neves C: High glucose reduces
megalin-mediated albumin endocytosis in renal proximal tubule cells
through protein kinase B O-GlcNAcylation. J Biol Chem.
293:11388–11400. 2018.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Kupriyanova Y, Zaharia OP, Bobrov P,
Karusheva Y, Burkart V, Szendroedi J, Hwang JH and Roden M: GDS
group. Early changes in hepatic energy metabolism and lipid content
in recent-onset type 1 and 2 diabetes mellitus. J Hepatol.
74:1028–1037. 2021.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Tilg H, Moschen AR and Roden M: NAFLD and
diabetes mellitus. Nat Rev Gastroenterol Hepatol. 14:32–42.
2017.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Nassir F and Ibdah JA: Role of
mitochondria in nonalcoholic fatty liver disease. Int J Mol Sci.
15:8713–8742. 2014.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Zheng P, Ma W, Gu Y, Wu H, Bian Z, Liu N,
Yang D and Chen X: High-fat diet causes mitochondrial damage and
downregulation of mitofusin-2 and optic atrophy-1 in multiple
organs. J Clin Biochem Nutr. 73:61–76. 2023.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Bi Y, Guo X, Zhang M, Zhu K, Shi C, Fan B,
Wu Y, Yang Z and Ji G: Bone marrow derived-mesenchymal stem cell
improves diabetes-associated fatty liver via mitochondria
transformation in mice. Stem Cell Res Ther. 12(602)2021.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Hsu MJ, Karkossa I, Schäfer I, Christ M,
Kühne H, Schubert K, Rolle-Kampczyk UE, Kalkhof S, Nickel S, Seibel
P, et al: Mitochondrial transfer by human mesenchymal stromal cells
ameliorates hepatocyte lipid load in a mouse model of NASH.
Biomedicines. 8(350)2020.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Gaspers LD, Pierobon N and Thomas AP:
Intercellular calcium waves integrate hormonal control of glucose
output in the intact liver. J Physiol. 597:2867–2885.
2019.PubMed/NCBI View Article : Google Scholar
|
|
143
|
Barreto SG, Habtezion A, Gukovskaya A,
Lugea A, Jeon C, Yadav D, Hegyi P, Venglovecz V, Sutton R and
Pandol SJ: Critical thresholds: key to unlocking the door to the
prevention and specific treatments for acute pancreatitis. Gut.
70:194–203. 2021.PubMed/NCBI View Article : Google Scholar
|
|
144
|
Liu W, Ren Y, Wang T, Wang M, Xu Y, Zhang
J, Bi J, Wu Z, Zhang Y and Wu R: Blocking CIRP protects against
acute pancreatitis by improving mitochondrial function and
suppressing pyroptosis in acinar cells. Cell Death Discov.
10(156)2024.PubMed/NCBI View Article : Google Scholar
|
|
145
|
Mukherjee R, Mareninova OA, Odinokova IV,
Huang W, Murphy J, Chvanov M, Javed MA, Wen L, Booth DM, Cane MC,
et al: Mechanism of mitochondrial permeability transition pore
induction and damage in the pancreas: Inhibition prevents acute
pancreatitis by protecting production of ATP. Gut. 65:1333–1346.
2016.PubMed/NCBI View Article : Google Scholar
|
|
146
|
Hu Z, Wang D, Gong J, Li Y, Ma Z, Luo T,
Jia X, Shi Y and Song Z: MSCs deliver hypoxia-treated mitochondria
reprogramming acinar metabolism to alleviate severe acute
pancreatitis injury. Adv Sci (Weinh). 10(e2207691)2023.PubMed/NCBI View Article : Google Scholar
|
|
147
|
Shapiro AM, Pokrywczynska M and Ricordi C:
Clinical pancreatic islet transplantation. Nat Rev Endocrinol.
13:268–277. 2017.PubMed/NCBI View Article : Google Scholar
|
|
148
|
Kin T, Senior P, O'Gorman D, Richer B,
Salam A and Shapiro AMJ: Risk factors for islet loss during culture
prior to transplantation. Transpl Int. 21:1029–1035.
2008.PubMed/NCBI View Article : Google Scholar
|
|
149
|
Jimenez-Sánchez C, Brun T and Maechler P:
Mitochondrial carriers regulating insulin secretion profiled in
human islets upon metabolic stress. Biomolecules.
10(1543)2020.PubMed/NCBI View Article : Google Scholar
|
|
150
|
Zhang K, Bao R, Huang F, Yang K, Ding Y,
Lauterboeck L, Yoshida M, Long Q and Yang Q: ATP synthase
inhibitory factor subunit 1 regulates islet β-cell function via
repression of mitochondrial homeostasis. Lab Invest. 102:69–79.
2022.PubMed/NCBI View Article : Google Scholar
|
|
151
|
Rackham CL, Hubber EL, Czajka A, Malik AN,
King AJF and Jones PM: Optimizing beta cell function through
mesenchymal stromal cell-mediated mitochondria transfer. Stem
Cells. 38:574–584. 2020.PubMed/NCBI View Article : Google Scholar
|
|
152
|
Shoop WK, Lape J, Trum M, Powell A,
Sevigny E, Mischler A, Bacman SR, Fontanesi F, Smith J, Jantz D, et
al: Efficient elimination of MELAS-associated m.3243G mutant
mitochondrial DNA by an engineered mitoARCUS nuclease. Nat Metab.
5:2169–2183. 2023.PubMed/NCBI View Article : Google Scholar
|
|
153
|
Ikeda T, Osaka H, Shimbo H, Tajika M,
Yamazaki M, Ueda A, Murayama K and Yamagata T: Mitochondrial DNA
3243A>T mutation in a patient with MELAS syndrome. Hum Genome
Var. 5(25)2018.PubMed/NCBI View Article : Google Scholar
|
|
154
|
El-Hattab AW, Adesina AM, Jones J and
Scaglia F: MELAS syndrome: Clinical manifestations, pathogenesis,
and treatment options. Mol Genet Metab. 116:4–12. 2015.PubMed/NCBI View Article : Google Scholar
|
|
155
|
Lin TK, Chen SD, Chuang YC, Lan MY, Chuang
JH, Wang PW, Hsu TY, Wang FS, Tsai MH, Huang ST, et al:
Mitochondrial transfer of wharton's jelly mesenchymal stem cells
eliminates mutation burden and rescues mitochondrial bioenergetics
in rotenone-stressed MELAS fibroblasts. Oxid Med Cell Longev.
2019(9537504)2019.PubMed/NCBI View Article : Google Scholar
|
|
156
|
Liu L, Yang J, Otani Y, Shiga T, Yamaguchi
A, Oda Y, Hattori M, Goto T, Ishibashi S, Kawashima-Sonoyama Y, et
al: MELAS-derived neurons functionally improve by mitochondrial
transfer from highly purified mesenchymal stem cells (REC). Int J
Mol Sci. 24(17186)2023.PubMed/NCBI View Article : Google Scholar
|
|
157
|
Tuppen HA, Blakely EL, Turnbull DM and
Taylor RW: Mitochondrial DNA mutations and human disease. Biochim
Biophys Acta. 1797:113–128. 2010.PubMed/NCBI View Article : Google Scholar
|
|
158
|
Chuang YC, Liou CW, Chen SD, Wang PW,
Chuang JH, Tiao MM, Hsu TY, Lin HY and Lin TK: Mitochondrial
transfer from wharton's jelly mesenchymal stem cell to MERRF cybrid
reduces oxidative stress and improves mitochondrial bioenergetics.
Oxid Med Cell Longev. 2017(5691215)2017.PubMed/NCBI View Article : Google Scholar
|
|
159
|
Capristo M, Del Dotto V, Tropeano CV,
Fiorini C, Caporali L, La Morgia C, Valentino ML, Montopoli M,
Carelli V and Maresca A: Rapamycin rescues mitochondrial
dysfunction in cells carrying the m.8344A > G mutation in the
mitochondrial tRNALys. Mol Med. 28(90)2022.PubMed/NCBI View Article : Google Scholar
|
|
160
|
Jacoby E, Blumkin M, Anikster Y,
Varda-Bloom N, Pansheen J, Bar Yoseph O, Gruber N, Lahav E, Besser
MJ, Schachter J, et al: First-in-human mitochondrial augmentation
of hematopoietic stem cells in pearson syndrome. Blood.
132(1024)2018.
|
|
161
|
Jacoby E, Bar-Yosef O, Gruber N, Lahav E,
Varda-Bloom N, Bolkier Y, Bar D, Blumkin MB, Barak S, Eisenstein E,
et al: Mitochondrial augmentation of hematopoietic stem cells in
children with single large-scale mitochondrial DNA deletion
syndromes. Sci Transl Med. 14(eabo3724)2022.PubMed/NCBI View Article : Google Scholar
|
|
162
|
Fu Y, Ni J and Chen J, Ma G, Zhao M, Zhu
S, Shi T, Zhu J, Huang Z, Zhang J and Chen J: Dual-functionalized
MSCs that express CX3CR1 and IL-25 exhibit enhanced therapeutic
effects on inflammatory bowel disease. Mol Ther. 28:1214–1228.
2020.PubMed/NCBI View Article : Google Scholar
|
|
163
|
Van Nguyen TT, Vu NB and Van Pham P:
Mesenchymal stem cell transplantation for ischemic diseases:
Mechanisms and challenges. Tissue Eng Regen Med. 18:587–611.
2021.PubMed/NCBI View Article : Google Scholar
|
|
164
|
Zhu XY, Lerman A and Lerman LO: Concise
review: Mesenchymal stem cell treatment for ischemic kidney
disease. Stem Cells. 31:1731–1736. 2013.PubMed/NCBI View Article : Google Scholar
|
|
165
|
Baldari S, Di Rocco G, Piccoli M, Pozzobon
M, Muraca M and Toietta G: Challenges and strategies for improving
the regenerative effects of mesenchymal stromal cell-based
therapies. Int J Mol Sci. 18(2087)2017.PubMed/NCBI View Article : Google Scholar
|
|
166
|
Hu C and Li L: Preconditioning influences
mesenchymal stem cell properties in vitro and in vivo. J Cell Mol
Med. 22:1428–1442. 2018.PubMed/NCBI View Article : Google Scholar
|
|
167
|
Li CJ, Chen PK, Sun LY and Pang CY:
Enhancement of mitochondrial transfer by antioxidants in human
mesenchymal stem cells. Oxid Med Cell Longev.
2017(8510805)2017.PubMed/NCBI View Article : Google Scholar
|
|
168
|
Lee DS and Kim JE: PDI-mediated
S-nitrosylation of DRP1 facilitates DRP1-S616 phosphorylation and
mitochondrial fission in CA1 neurons. Cell Death Dis.
9(869)2018.PubMed/NCBI View Article : Google Scholar
|
|
169
|
Yao X, Ma Y, Zhou W, Liao Y, Jiang Z, Lin
J, He Q, Wu H, Wei W, Wang X, et al: In-cytoplasm mitochondrial
transplantation for mesenchymal stem cells engineering and tissue
regeneration. Bioeng Transl Med. 7(e10250)2021.PubMed/NCBI View Article : Google Scholar
|
|
170
|
Guo Y, Chi X, Wang Y, Heng BC, Wei Y,
Zhang X, Zhao H, Yin Y and Deng X: Mitochondria transfer enhances
proliferation, migration, and osteogenic differentiation of bone
marrow mesenchymal stem cell and promotes bone defect healing. Stem
Cell Res Ther. 11(245)2020.PubMed/NCBI View Article : Google Scholar
|
|
171
|
Lin RZ, Im GB, Luo AC, Zhu Y, Hong X,
Neumeyer J, Tang HW, Perrimon N and Melero-Martin JM: Mitochondrial
transfer mediates endothelial cell engraftment through mitophagy.
Nature. 629:660–668. 2024.PubMed/NCBI View Article : Google Scholar
|
|
172
|
Akhter W, Nakhle J, Vaillant L, Garcin G,
Le Saout C, Simon M, Crozet C, Djouad F, Jorgensen C, Vignais ML
and Hernandez J: Transfer of mesenchymal stem cell mitochondria to
CD4+ T cells contributes to repress Th1 differentiation
by downregulating T-bet expression. Stem Cell Res Ther.
14(12)2023.PubMed/NCBI View Article : Google Scholar
|
|
173
|
Court AC, Le-Gatt A, Luz-Crawford P, Parra
E, Aliaga-Tobar V, Bátiz LF, Contreras RA, Ortúzar MI, Kurte M,
Elizondo-Vega R, et al: Mitochondrial transfer from MSCs to T cells
induces Treg differentiation and restricts inflammatory response.
EMBO Rep. 21(e48052)2020.PubMed/NCBI View Article : Google Scholar
|
|
174
|
Romano M, Tung SL, Smyth LA and Lombardi
G: Treg therapy in transplantation: A general overview. Transpl
Int. 30:745–753. 2017.PubMed/NCBI View Article : Google Scholar
|
|
175
|
Li LZ, Zhang Z and Bhoj VG: Conventional T
cell therapies pave the way for novel Treg therapeutics. Cell
Immunol. 359(104234)2021.PubMed/NCBI View Article : Google Scholar
|
|
176
|
Piekarska K, Urban-Wójciuk Z, Kurkowiak M,
Pelikant-Małecka I, Schumacher A, Sakowska J, Spodnik JH,
Arcimowicz Ł, Zielińska H, Tymoniuk B, et al: Mesenchymal stem
cells transfer mitochondria to allogeneic Tregs in an HLA-dependent
manner improving their immunosuppressive activity. Nat Commun.
13(856)2022.PubMed/NCBI View Article : Google Scholar
|
|
177
|
Espinosa-Carrasco G, Le Saout C, Fontanaud
P, Stratmann T, Mollard P, Schaeffer M and Hernandez J:
CD4+ T helper cells play a key role in maintaining
diabetogenic CD8+ T cell function in the pancreas. Front
Immunol. 8(2001)2018.PubMed/NCBI View Article : Google Scholar
|
|
178
|
Luz-Crawford P, Hernandez J, Djouad F,
Luque-Campos N, Caicedo A, Carrère-Kremer S, Brondello JM, Vignais
ML, Pène J and Jorgensen C: Mesenchymal stem cell repression of
Th17 cells is triggered by mitochondrial transfer. Stem Cell Res
Ther. 10(232)2019.PubMed/NCBI View Article : Google Scholar
|
|
179
|
Lazarevic V, Glimcher LH and Lord GM:
T-bet: A bridge between innate and adaptive immunity. Nat Rev
Immunol. 13:777–789. 2013.PubMed/NCBI View Article : Google Scholar : Ji L, Chen Y, Wang
H, Zhang W, He L, Wu J and Liu Y: Overexpression of Sirt6 promotes
M2 macrophage transformation, alleviating renal injury in diabetic
nephropathy. Int J Oncol 55: 103-115, 2019.
|
|
180
|
Xu L, Yan X, Zhao Y, Wang J, Liu B, Yu S,
Fu J, Liu Y and Su J: Macrophage polarization mediated by
mitochondrial dysfunction induces adipose tissue inflammation in
obesity. Int J Mol Sci. 23(9252)2022.PubMed/NCBI View Article : Google Scholar
|
|
181
|
Ran L, Zhang S, Wang G, Zhao P, Sun J,
Zhou J, Gan H, Jeon R, Li Q, Herrmann J and Wang F: Mitochondrial
pyruvate carrier-mediated metabolism is dispensable for the
classical activation of macrophages. Nat Metab. 5:804–820.
2023.PubMed/NCBI View Article : Google Scholar
|
|
182
|
De Santa F, Vitiello L, Torcinaro A and
Ferraro E: The role of metabolic remodeling in macrophage
polarization and its effect on skeletal muscle regeneration.
Antioxid Redox Signal. 30:1553–1598. 2019.PubMed/NCBI View Article : Google Scholar
|
|
183
|
Yuan Y, Chen Y, Peng T, Li L, Zhu W, Liu
F, Luo R, Cheng J, Liu J and Lu Y: Mitochondrial ROS-induced
lysosomal dysfunction impairs autophagic flux and contributes to M1
macrophage polarization in a diabetic condition. Clin Sci (Lond).
133:1759–1777. 2019.PubMed/NCBI View Article : Google Scholar
|
|
184
|
Yuan Y, Yuan L, Li L, Liu F, Liu J, Chen
Y, Cheng J and Lu Y: Mitochondrial transfer from mesenchymal stem
cells to macrophages restricts inflammation and alleviates kidney
injury in diabetic nephropathy mice via PGC-1α activation. Stem
Cells. 39:913–928. 2021.PubMed/NCBI View Article : Google Scholar
|
|
185
|
Hussell T and Bell TJ: Alveolar
macrophages: Plasticity in a tissue-specific context. Nat Rev
Immunol. 14:81–93. 2014.PubMed/NCBI View Article : Google Scholar
|
|
186
|
Jackson MV, Morrison TJ, Doherty DF,
McAuley DF, Matthay MA, Kissenpfennig A, O'Kane CM and
Krasnodembskaya AD: Mitochondrial transfer via tunneling nanotubes
is an important mechanism by which mesenchymal stem cells enhance
macrophage phagocytosis in the in vitro and in vivo models of ARDS.
Stem Cells. 34:2210–2223. 2016.PubMed/NCBI View Article : Google Scholar
|
|
187
|
Jackson MV and Krasnodembskaya AD:
Analysis of mitochondrial transfer in direct Co-cultures of human
monocyte-derived macrophages (MDM) and mesenchymal stem cells
(MSC). Bio Protoc. 7(e2255)2017.PubMed/NCBI View Article : Google Scholar
|
|
188
|
Phinney DG, Di Giuseppe M, Njah J, Sala E,
Shiva S, St Croix CM, Stolz DB, Watkins SC, Di YP, Leikauf GD, et
al: Mesenchymal stem cells use extracellular vesicles to outsource
mitophagy and shuttle microRNAs. Nat Commun. 6(8472)2015.PubMed/NCBI View Article : Google Scholar
|
|
189
|
Xia L, Zhang C, Lv N, Liang Z, Ma T, Cheng
H, Xia Y and Shi L: AdMSC-derived exosomes alleviate acute lung
injury via transferring mitochondrial component to improve
homeostasis of alveolar macrophages. Theranostics. 12:2928–2947.
2022.PubMed/NCBI View Article : Google Scholar
|
|
190
|
West AP, Khoury-Hanold W, Staron M, Tal
MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff
DA, et al: Mitochondrial DNA stress primes the antiviral innate
immune response. Nature. 520:553–557. 2015.PubMed/NCBI View Article : Google Scholar
|
|
191
|
Morrison TJ, Jackson MV, Cunningham EK,
Kissenpfennig A, McAuley DF, O'Kane CM and Krasnodembskaya AD:
Mesenchymal stromal cells modulate macrophages in clinically
relevant lung injury models by extracellular vesicle mitochondrial
transfer. Am J Respir Crit Care Med. 196:1275–1286. 2017.PubMed/NCBI View Article : Google Scholar
|
|
192
|
Kang MH, Kim YJ and Lee JH: Mitochondria
in reproduction. Clin Exp Reprod Med. 50:1–11. 2023.PubMed/NCBI View Article : Google Scholar
|
|
193
|
Dadarwal D, Pfeifer L, Cervantes M, Adams
GP and Singh J: Effect of maternal age on ATP content and
distribution of mitochondria in bovine oocytes. PLoS One.
19(e0302444)2024.PubMed/NCBI View Article : Google Scholar
|
|
194
|
Zhao J and Li Y: Adenosine triphosphate
content in human unfertilized oocytes, undivided zygotes and
embryos unsuitable for transfer or cryopreservation. J Int Med Res.
40:734–739. 2012.PubMed/NCBI View Article : Google Scholar
|
|
195
|
May-Panloup P, Chrétien MF, Jacques C,
Vasseur C, Malthièry Y and Reynier P: Low oocyte mitochondrial DNA
content in ovarian insufficiency. Hum Reprod. 20:593–597.
2005.PubMed/NCBI View Article : Google Scholar
|
|
196
|
Zhang D, Keilty D, Zhang ZF and Chian RC:
Mitochondria in oocyte aging: Current understanding. Facts Views
Vis Obgyn. 9:29–38. 2017.PubMed/NCBI
|
|
197
|
Cozzolino M, Marin D and Sisti G: New
frontiers in IVF: mtDNA and autologous germline mitochondrial
energy transfer. Reprod Biol Endocrinol. 17(55)2019.PubMed/NCBI View Article : Google Scholar
|
|
198
|
Takeda K: Functional consequences of
mitochondrial mismatch in reconstituted embryos and offspring. J
Reprod Dev. 65:485–489. 2019.PubMed/NCBI View Article : Google Scholar
|
|
199
|
Tang S, Yang N, Yu M, Wang S, Hu X, Ni H
and Cai W: Noninvasive autologous mitochondria transport improves
the quality and developmental potential of oocytes from aged mice.
F S Sci. 3:310–321. 2022.PubMed/NCBI View Article : Google Scholar
|
|
200
|
Wang ZB, Hao JX, Meng TG, Guo L, Dong MZ,
Fan LH, Ouyang YC, Wang G, Sun QY, Ou XH and Yao YQ: Transfer of
autologous mitochondria from adipose tissue-derived stem cells
rescues oocyte quality and infertility in aged mice. Aging (Albany
NY). 9:2480–2488. 2017.PubMed/NCBI View Article : Google Scholar
|
|
201
|
Kankanam Gamage US, Hashimoto S, Miyamoto
Y, Nakano T, Yamanaka M, Koike A, Satoh M and Morimoto Y:
Mitochondria transfer from adipose stem cells improves the
developmental potential of cryopreserved oocytes. Biomolecules.
12(1008)2022.PubMed/NCBI View Article : Google Scholar
|
|
202
|
Zhang Q, Hao JX, Liu BW, Ouyang YC, Guo
JN, Dong MZ, Wang ZB, Gao F and Yao YQ: Supplementation of
mitochondria from endometrial mesenchymal stem cells improves
oocyte quality in aged mice. Cell Prolif. 56(e13372)2023.PubMed/NCBI View Article : Google Scholar
|
|
203
|
Zhang C, Tao L, Yue Y, Ren L, Zhang Z,
Wang X, Tian J and An L: Mitochondrial transfer from induced
pluripotent stem cells rescues developmental potential of in vitro
fertilized embryos from aging females†. Biol Reprod. 104:1114–1125.
2021.PubMed/NCBI View Article : Google Scholar
|
|
204
|
Tilly J and Woods D: Compositions and
methods for autologous germline mitochondrial energy transfer.
United States Patent Number. 8,642,329, 2014.
|
|
205
|
Fakih MH, Shmoury ME, Szeptycki J, Cruz
DBD, Lux C, Verjee S, Burgess CM, Cohn GM and Casper RF: The
AUGMENTSM treatment: Physician reported outcomes of the
initial global patient experience. JFIV Reprod Med Genet. 3:1–7.
2015.
|
|
206
|
Oktay K, Baltaci V, Sonmezer M, Turan V,
Unsal E, Baltaci A, Aktuna S and Moy F: Oogonial precursor
cell-derived autologous mitochondria injection to improve outcomes
in women with multiple IVF failures due to low oocyte quality: A
clinical translation. Reprod Sci. 22:1612–1617. 2015.PubMed/NCBI View Article : Google Scholar
|
|
207
|
Jiang Z, Shi C, Han H, Fu M, Zhu H, Han T,
Fei J, Huang Y, Jin Z, He J, et al: Autologous non-invasively
derived stem cells mitochondria transfer shows therapeutic
advantages in human embryo quality rescue. Biol Res.
56(60)2023.PubMed/NCBI View Article : Google Scholar
|
|
208
|
Morimoto Y, Gamage USK, Yamochi T, Saeki
N, Morimoto N, Yamanaka M, Koike A, Miyamoto Y, Tanaka K, Fukuda A,
et al: Mitochondrial transfer into human oocytes improved embryo
quality and clinical outcomes in recurrent pregnancy failure cases.
Int J Mol Sci. 24(2738)2023.PubMed/NCBI View Article : Google Scholar
|
|
209
|
Acquistapace A, Bru T, Lesault PF, Figeac
F, Coudert AE, le Coz O, Christov C, Baudin X, Auber F, Yiou R, et
al: Human mesenchymal stem cells reprogram adult cardiomyocytes
toward a progenitor-like state through partial cell fusion and
mitochondria transfer. Stem Cells. 29:812–824. 2011.PubMed/NCBI View Article : Google Scholar
|
|
210
|
Jiao H, Jiang D, Hu X, Du W, Ji L, Yang Y,
Li X, Sho T, Wang X, Li Y, et al: Mitocytosis, a migrasome-mediated
mitochondrial quality-control process. Cell. 184:2896–2910.e13.
2021.PubMed/NCBI View Article : Google Scholar
|
|
211
|
Sadeghsoltani F, Avci ÇB, Hassanpour P,
Haiaty S, Rahmati M, Mota A, Rahbarghazi R, Nemati M, Mahdipour M,
Talebi M, et al: Autophagy modulation effect on homotypic transfer
of intracellular components via tunneling nanotubes in mesenchymal
stem cells. Stem Cell Res Ther. 15(189)2024.PubMed/NCBI View Article : Google Scholar
|
|
212
|
Dai L, Wu Z, Yin L, Cheng L, Zhou Q and
Ding F: Exogenous functional mitochondria derived from bone
mesenchymal stem cells that respond to ROS can rescue neural cells
following ischemic stroke. J Inflamm Res. 17:3383–3395.
2024.PubMed/NCBI View Article : Google Scholar
|
|
213
|
Mistry JJ, Marlein CR, Moore JA, Hellmich
C, Wojtowicz EE, Smith JGW, Macaulay I, Sun Y, Morfakis A,
Patterson A, et al: ROS-mediated PI3K activation drives
mitochondrial transfer from stromal cells to hematopoietic stem
cells in response to infection. Proc Natl Acad Sci USA.
116:24610–24619. 2019.PubMed/NCBI View Article : Google Scholar
|
|
214
|
Li Y, Wang Y, Yang W, Wu Z, Ma D, Sun J,
Tao H, Ye Q, Liu J, Ma Z, et al: ROS-responsive exogenous
functional mitochondria can rescue neural cells post-ischemic
stroke. Front Cell Dev Biol. 11(1207748)2023.PubMed/NCBI View Article : Google Scholar
|
|
215
|
Saito K, Zhang Q, Yang H, Yamatani K, Ai
T, Ruvolo V, Baran N, Cai T, Ma H, Jacamo R, et al: Exogenous
mitochondrial transfer and endogenous mitochondrial fission
facilitate AML resistance to OxPhos inhibition. Blood Adv.
5:4233–4255. 2021.PubMed/NCBI View Article : Google Scholar
|
|
216
|
Ahmad T, Mukherjee S, Pattnaik B, Kumar M,
Singh S, Kumar M, Rehman R, Tiwari BK, Jha KA, Barhanpurkar AP, et
al: Miro1 regulates intercellular mitochondrial transport &
enhances mesenchymal stem cell rescue efficacy. EMBO J.
33:994–1010. 2014.PubMed/NCBI View Article : Google Scholar
|
|
217
|
Novak J, Nahacka Z, Oliveira GL, Brisudova
P, Dubisova M, Dvorakova S, Miklovicova S, Dalecka M, Puttrich V,
Grycova L, et al: The adaptor protein Miro1 modulates horizontal
transfer of mitochondria in mouse melanoma models. Cell Rep.
44(115154)2025.PubMed/NCBI View Article : Google Scholar
|
|
218
|
Barutta F, Corbetta B, Bellini S, Gambino
R, Bruno S, Kimura S, Hase K, Ohno H and Gruden G: Protective
effect of mesenchymal stromal cells in diabetic nephropathy: The In
vitro and In vivo role of the M-Sec-tunneling nanotubes. Clin Sci
(Lond). 138:1537–1559. 2024.PubMed/NCBI View Article : Google Scholar
|
|
219
|
Barutta F, Kimura S, Hase K, Bellini S,
Corbetta B, Corbelli A, Fiordaliso F, Barreca A, Papotti MG,
Ghiggeri GM, et al: Protective role of the M-Sec-tunneling nanotube
system in podocytes. J Am Soc Nephrol. 32:1114–1130.
2021.PubMed/NCBI View Article : Google Scholar
|
|
220
|
Kastl L, Sauer SW, Ruppert T, Beissbarth
T, Becker MS, Süss D, Krammer PH and Gülow K: TNF-α mediates
mitochondrial uncoupling and enhances ROS-dependent cell migration
via NF-κB activation in liver cells. FEBS Lett. 588:175–183.
2014.PubMed/NCBI View Article : Google Scholar
|
|
221
|
Wang K, Zhou L, Mao H, Liu J, Chen Z and
Zhang L: Intercellular mitochondrial transfer alleviates pyroptosis
in dental pulp damage. Cell Prolif. 56(e13442)2023.PubMed/NCBI View Article : Google Scholar
|
|
222
|
Kimura S, Yamashita M, Yamakami-Kimura M,
Sato Y, Yamagata A, Kobashigawa Y, Inagaki F, Amada T, Hase K,
Iwanaga T, et al: Distinct roles for the N- and C-terminal regions
of M-Sec in plasma membrane deformation during tunneling nanotube
formation. Sci Rep. 6(33548)2016.PubMed/NCBI View Article : Google Scholar
|
|
223
|
Gao C, Dai Y, Spezza PA, Boasiako P, Tang
A, Rasquinha G, Zhong H, Shao B, Liu Y, Shi PA, et al:
Megakaryocytes transfer mitochondria to bone marrow mesenchymal
stromal cells to lower platelet activation. J Clin Invest.
135(e189801)2025.PubMed/NCBI View Article : Google Scholar
|
|
224
|
Irwin RM, Thomas MA, Fahey MJ, Mayán MD,
Smyth JW and Delco ML: Connexin 43 regulates intercellular
mitochondrial transfer from human mesenchymal stromal cells to
chondrocytes. Stem Cell Res Ther. 15(359)2024.PubMed/NCBI View Article : Google Scholar
|
|
225
|
Huang T, Zhang T, Jiang X, Li A, Su Y,
Bian Q, Wu H, Lin R, Li N, Cao H, et al: Iron oxide nanoparticles
augment the intercellular mitochondrial transfer-mediated therapy.
Sci Adv. 7(eabj0534)2021.PubMed/NCBI View Article : Google Scholar
|
|
226
|
Qiao X, Huang N, Meng W, Liu Y, Li J, Li
C, Wang W, Lai Y, Zhao Y, Ma Z, et al: Beyond mitochondrial
transfer, cell fusion rescues metabolic dysfunction and boosts
malignancy in adenoid cystic carcinoma. Cell Rep.
43(114652)2024.PubMed/NCBI View Article : Google Scholar
|
|
227
|
Powell AE, Anderson EC, Davies PS, Silk
AD, Pelz C, Impey S and Wong MH: Fusion between intestinal
epithelial cells and macrophages in a cancer context results in
nuclear reprogramming. Cancer Res. 71:1497–1505. 2011.PubMed/NCBI View Article : Google Scholar
|
|
228
|
Nahacka Z, Novak J, Zobalova R and Neuzil
J: Miro proteins and their role in mitochondrial transfer in cancer
and beyond. Front Cell Dev Biol. 10(937753)2022.PubMed/NCBI View Article : Google Scholar
|
|
229
|
Zhang H, Yu X, Ye J, Li H, Hu J, Tan Y,
Fang Y, Akbay E, Yu F, Weng C, et al: Systematic investigation of
mitochondrial transfer between cancer cells and T cells at
single-cell resolution. Cancer Cell. 41:1788–1802.e10.
2023.PubMed/NCBI View Article : Google Scholar
|
|
230
|
Goliwas KF, Libring S, Berestesky E,
Gholizadeh S, Schwager SC, Frost AR, Gaborski TR, Zhang J and
Reinhart-King CA: Mitochondrial transfer from cancer-associated
fibroblasts increases migration in aggressive breast cancer. J Cell
Sci. 136(jcs260419)2023.PubMed/NCBI View Article : Google Scholar
|
|
231
|
Xie Q, Zeng J, Zheng Y, Li T, Ren J, Chen
K, Zhang Q, Xie R, Xu F and Zhu J: Mitochondrial transplantation
attenuates cerebral ischemia-reperfusion injury: Possible
involvement of mitochondrial component separation. Oxid Med Cell
Longev. 2021(1006636)2021.PubMed/NCBI View Article : Google Scholar
|
|
232
|
Peruzzotti-Jametti L, Bernstock JD, Willis
CM, Manferrari G, Rogall R, Fernandez-Vizarra E, Williamson JC,
Braga A, van den Bosch A, Leonardi T, et al: Neural stem cells
traffic functional mitochondria via extracellular vesicles. PLoS
Biol. 19(e3001166)2021.PubMed/NCBI View Article : Google Scholar
|
|
233
|
Zheng D, Zhou H, Wang H, Zhu Y, Wu Y, Li
Q, Li T and Liu L: Mesenchymal stem cell-derived microvesicles
improve intestinal barrier function by restoring mitochondrial
dynamic balance in sepsis rats. Stem Cell Res Ther.
12(299)2021.PubMed/NCBI View Article : Google Scholar
|
|
234
|
Cowan DB, Yao R, Thedsanamoorthy JK,
Zurakowski D, del Nido PJ and McCully JD: Transit and integration
of extracellular mitochondria in human heart cells. Sci Rep.
7(17450)2017.PubMed/NCBI View Article : Google Scholar
|
|
235
|
Fu A: Mitotherapy as a novel therapeutic
strategy for mitochondrial diseases. Curr Mol Pharmacol. 13:41–49.
2020.PubMed/NCBI View Article : Google Scholar
|
|
236
|
Chen J, Zhong J, Wang LL and Chen YY:
Mitochondrial transfer in cardiovascular disease: From mechanisms
to therapeutic implications. Front Cardiovasc Med.
8(771298)2021.PubMed/NCBI View Article : Google Scholar
|
|
237
|
Wang ZH, Chen L, Li W, Chen L and Wang YP:
Mitochondria transfer and transplantation in human health and
diseases. Mitochondrion. 65:80–87. 2022.PubMed/NCBI View Article : Google Scholar
|
|
238
|
Walker M, Levitt MR, Federico EM, Miralles
FJ, Levy SH, Lynne Prijoles K, Winter A, Swicord JK and Sancak Y:
Autologous mitochondrial transplant for acute cerebral ischemia:
Phase 1 trial results and review. J Cereb Blood Flow Metab: Dec 4,
2024 (Epub ahead of print).
|
|
239
|
Nakai R, Varnum S, Field RL, Shi H, Giwa
R, Jia W, Krysa SJ, Cohen EF, Borcherding N, Saneto RP, et al:
Mitochondria transfer-based therapies reduce the morbidity and
mortality of Leigh syndrome. Nat Metab. 6:1886–1896.
2024.PubMed/NCBI View Article : Google Scholar
|
|
240
|
Li C, Cheung MKH, Han S, Zhang Z, Chen L,
Chen J, Zeng H and Qiu J: Mesenchymal stem cells and their
mitochondrial transfer: A double-edged sword. Biosci Rep.
39(BSR20182417)2019.PubMed/NCBI View Article : Google Scholar
|
|
241
|
Hosseinian S, Ali Pour P and Kheradvar A:
Prospects of mitochondrial transplantation in clinical medicine:
Aspirations and challenges. Mitochondrion. 65:33–44.
2022.PubMed/NCBI View Article : Google Scholar
|
|
242
|
Caicedo A, Aponte PM, Cabrera F, Hidalgo C
and Khoury M: Artificial mitochondria transfer: Current challenges,
advances, and future applications. Stem Cells Int.
2017(7610414)2017.PubMed/NCBI View Article : Google Scholar
|
|
243
|
Ishikawa K, Toyama-Sorimachi N, Nakada K,
Morimoto M, Imanishi H, Yoshizaki M, Sasawatari S, Niikura M,
Takenaga K, Yonekawa H and Hayashi J: The innate immune system in
host mice targets cells with allogenic mitochondrial DNA. J Exp
Med. 207:2297–2305. 2010.PubMed/NCBI View Article : Google Scholar
|
|
244
|
Deuse T, Hu X, Agbor-Enoh S, Koch M,
Spitzer MH, Gravina A, Alawi M, Marishta A, Peters B,
Kosaloglu-Yalcin Z, et al: De novo mutations in mitochondrial DNA
of iPSCs produce immunogenic neoepitopes in mice and humans. Nat
Biotechnol. 37:1137–1144. 2019.PubMed/NCBI View Article : Google Scholar
|
|
245
|
Klopstock T, Klopstock B and Prokisch H:
Mitochondrial replacement approaches: Challenges for clinical
implementation. Genome Med. 8(126)2016.PubMed/NCBI View Article : Google Scholar
|
|
246
|
Yamada Y, Ito M, Arai M, Hibino M,
Tsujioka T and Harashima H: Challenges in promoting mitochondrial
transplantation therapy. Int J Mol Sci. 21(6365)2020.PubMed/NCBI View Article : Google Scholar
|
|
247
|
Yuan J, Chen F, Jiang D, Xu Z, Zhang H and
Jin ZB: ROCK inhibitor enhances mitochondrial transfer via
tunneling nanotubes in retinal pigment epithelium. Theranostics.
14:5762–5777. 2024.PubMed/NCBI View Article : Google Scholar
|
|
248
|
Chi A, Yang B, Dai H, Li X, Mo J, Gao Y,
Chen Z, Feng X, Ma M, Li Y, et al: Stem Leydig cells support
macrophage immunological homeostasis through mitochondrial transfer
in mice. Nat Commun. 15(2120)2024.PubMed/NCBI View Article : Google Scholar
|
|
249
|
Játiva S, Calle P, Torrico S, Muñoz Á,
García M, Martinez I, Sola A and Hotter G: Mitochondrial
transplantation enhances phagocytic function and decreases lipid
accumulation in foam cell macrophages. Biomedicines.
10(329)2022.PubMed/NCBI View Article : Google Scholar
|