Introduction
Diabetes, a class of metabolic diseases that are
prevalent worldwide affecting 422 million people in 2014 according
to the World Health Organization (1), is characterized by hyperglycemia and
can cause severe damage to different organs, particularly the eyes,
kidneys, nerves, heart and blood vessels. According to the American
Diabetes Association, the disease can be classified into two main
types, including type 1 and type 2 diabetes (2). Type 1 diabetes results from a
cell-mediated autoimmune attack on β cells, whereas type 2 diabetes
is a low-grade, chronic inflammatory disease that shares a common
final pathway with type 1 diabetes, in which activation of the
nuclear factor κB (NF-κB) signaling pathway causes a reduction in
pancreatic β cells (3–10).
Zinc participates in numerous biological processes,
and is essential for the correct processing, storage, secretion,
and function of insulin in pancreatic β cells (11–13).
Zinc flux is controlled by two types of transporters: The zinc
transporter (ZnT; also known as SLC30A) family, and the Zrt- and
Irt-like protein (ZIP) family. Typically, the ZnT family regulates
Zn2+ efflux out of the cytoplasm, while the ZIP family
regulates Zn2+ influx into the cytoplasm. Although 10
ZnTs and 14 ZIPs have been identified in mammals, ZnT8 is
exclusively expressed in the pancreatic islet and serves a major
role in Zn2+ efflux from cytoplasm to insulin secretory
granules, which is indispensable for insulin crystallization and
hexamerization (14–16).
Alterations to ZnT8 expression may affect diabetes
pathogenesis. For instance, ZnT8 deletion leads to impaired insulin
secretion from pancreatic β cells (17,18). In
addition, the expression of ZnT8 has been demonstrated to be
decreased in the β cells of diabetic mice (19). A ZnT8 (or SLC30A8) gene polymorphism
has been identified, and is associated with type 2 diabetes
(20–22). Furthermore, an autoantibody to ZnT8
is widely considered as a biomarker of autoimmune diabetes
(23,24). There is also evidence that zinc has
an anti-inflammatory effect, and that ZnT8 expression can be
downregulated by proinflammatory cytokines, which are expressed at
high levels in type 2 diabetes (25–27).
Tumor necrosis factor α-induced protein-3 (TNFAIP3)
is a zinc finger and ubiquitin-editing protein that is able to
inhibit or promote the NF-κB signaling cascade by attaching to the
K48-linked or K63-linked polyubiquitin chains, respectively
(28,29). Although TNFAIP3 was originally
characterized as an inhibitor of tumor necrosis factor
(TNF)-induced apoptosis, it is best characterized as an inhibitor
of NF-κB activation (30). Notably,
a number of studies have shown the association of TNFAIP3 with
common inflammatory and immune diseases, including type 1 diabetes
(31–36). Thus, it is possible that TNFAIP3
participates in the pathogenesis of type 1 and type 2 diabetes.
Indeed, there is certain evidence indicating this role of TNFAIP3,
since TNFAIP3 expression has been found to inhibit hallmarks of
diabetes in a number of models, including transplanted islets
(37,38), endothelial cells exposed to high
glucose (39) and
streptozotocin-induced diabetes mice (40).
Based on the aforementioned previous findings, we
conclude that cytokines may alter insulin secretion through the
induction of apoptosis in pancreatic β cells and through the
inhibition of insulin secretion via alterations to ZnT8 expression.
Therefore, the aim of the present study was to assess whether the
expression of TNFAIP3 is altered in patients with diabetes.
Furthermore, the study investigated whether proinflammatory
cytokines are able to alter the expression of ZnT8 in islet cells,
and whether the expression of TNFAIP3 can protect against
cytokine-induced ZnT8 alterations. The observations of the present
study may reveal a novel view on how proinflammatory cytokines
inhibit insulin secretion besides apoptosis of β cells, and how
TNFAIP3 exerts a beneficial effect on insulin secretion besides its
anti-apoptosis effect, which will also provide a potential
conception in the prevention and therapy of diabetes.
Materials and methods
NIT-1 cell culture
NIT-1 cells (murine) were purchased from Focusbio
(Guangzhou, China) and were cultured in a 5% CO2
atmosphere at 37°C in low-glucose Dulbeccos modified Eagle's medium
(DMEM) supplemented with 15% heat-inactivated fetal bovine serum
(FBS), 100 U/ml penicillin and 100 g/ml streptomycin. All the
aforementioned reagents mentioned were purchased from GE Healthcare
Life Sciences (Shanghai, China).
Cytokine stimulation
For stimulation assays, cells were plated
(106 cells/well) in 6-well plates (Sigma-Aldrich, St.
Louis, MO, USA). After 48 h of culturing, the culture medium was
changed to serum-reduced culture medium (DMEM and 1% FBS),
containing 1% FBS supplemented with no cytokines (control), with 10
ng/ml recombinant murine TNF-α or with 5 ng/ml recombinant murine
interleukin-1β (IL-1β; ProSpec, Ness Ziona, Israel). The cells were
stimulated with cytokines for 6, 24 or 48 h, and then collected by
centrifugation at 200 × g for 5 min at 4°C after digestion with
pancreatin digestion (Sigma-Aldrich). Each assay was performed in
triplicate.
Adenoviral transfection of NIT-1
cells
The adenovirus expressing human TNFAIP3 and enhanced
green fluorescent protein (Ad-TNFAIP3-EGFP; cat. no. V494-20), and
the control adenovirus expressing EGFP (Ad-EGFP; cat. no. V493-20)
were purchased from Wuhan GenSil Biotechnology (Wuhan, China).
Adenovirus-mediated gene transfection was performed following the
manufacturers protocol. Briefly, NIT-1 cells were plated in 6-well
plates (106 cells/well). At 1 day after cell adhesion, 1
ml DMEM with Ad-EGFP or Ad-TNFAIP3-EGFP virus [multiplicity of
infection (MOI), 200] was added to each well, and the cells were
infected for 24 h. The MOI was selected based on the lowest
toxicity by viral infection. Following infection, the cells were
cultured for an additional 24 h before being used in further
experiments. TNFAIP3 and indicator protein EGFP were co-expressed,
and the latter was verified using fluorescence microcopy, while
transfection was detected by western blot analysis.
Determination of NF-κB activation
NF-κB activity was assessed using an NF-κB
Activation and Nuclear Translocation Assay kit (Beyotime Institute
of Biotechnology, Haimen, China) according to the manufacturers
protocol. Briefly, the NF-κB major subunit, p65, was labeled with
Cy3 and the nucleus was stained with DAPI. After washing and
fixing, cytokine-treated NIT-1 cells were incubated with blocking
buffer to block non-specific epitopes. Subsequently, cells were
incubated with rabbit anti-mouse NF-κB p65 antibody at 4°C
overnight, followed by incubation with a Cy3-conjugated secondary
antibody for 1 h at room temperature and DAPI for 5 min prior to
fluorescence microcopy. All the reagents used in this experiment
were included in the kit.
Western blot analysis
Expression of proteins in cells was determined by
western blot analysis. GAPDH was used as an internal control. Three
rabbit anti-mouse/human polyclonal antibodies for GAPDH (cat. no.
sc-25778; 1:400 dilution), TNFAIP3 (cat. no. 23456-1-AP; 1:1,000
dilution) and ZnT8 (cat. no. R12-3525; 1:1,000 dilution) were
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA),
Proteintech Group Inc. (Wuhan, China) and Assay Biotechnology
Company, Inc. (Sunnyvale, CA, USA), respectively. Polyvinylidene
difluoride (PVDF) membranes were purchased from EMD Millipore
(Billerica, MA, USA). All other reagents were purchased from
Beyotime Institute of Biotechnology. Briefly, harvested NIT-1 cells
were lysed and extracted by a standard protocol with cell lysis
buffer, and the protein levels were quantified using a Enhanced BCA
Protein Assay kit. Proteins were separated by 10% SDS-PAGE and then
transferred to PVDF membranes. Subsequent to blocking with
QuickBlock blocking buffer for 10 min at 25°C followed by three
washes for 10 min with western wash buffer, the membranes were
incubated overnight at 4°C with polyclonal rabbit anti-TNFAIP3,
anti-ZnT8 or anti-GAPDH antibodies. Subsequent to washing three
times for 10 min with western wash buffer, the membranes were
incubated with goat anti-rabbit IgG secondary antibodies conjugated
to horseradish peroxidase (cat. no. A0208; 1:1,000 dilution), and
an enhanced chemiluminescence detection system (BeyoECL Plus; cat.
no. P0018; Beyotime Institute of Biotechnology) were used for
detection.
Statistical analysis
Data are presented as the raw values or as the mean
± standard deviation. IBM SPSS software (version 19.0; IBM Corp.,
Armonk, NY, USA) was used to perform statistical analyses using
one-way analysis of variance, Kruskal-Wallis test or Mann-Whitney U
test in order to assess differences, as appropriate. A P-value
(two-tailed) of ≤0.05 was considered to indicate a statistically
significant difference.
Results
Cytokine-induced alterations in
TNFAIP3 and ZnT8 expression and NF-κB activation
Previous studies have demonstrated the expression of
TNFAIP3 (41,42) and ZnT8 (43,44) in
several pancreatic β cell lines or primary islet cells; however, to
date, no such reports concerning their expression in
insulin-producing NIT-1 cells exist. Therefore, the present study
investigated whether TNFAIP3 and ZnT8 were expressed in NIT-1
cells, and then examined whether their expression was altered by
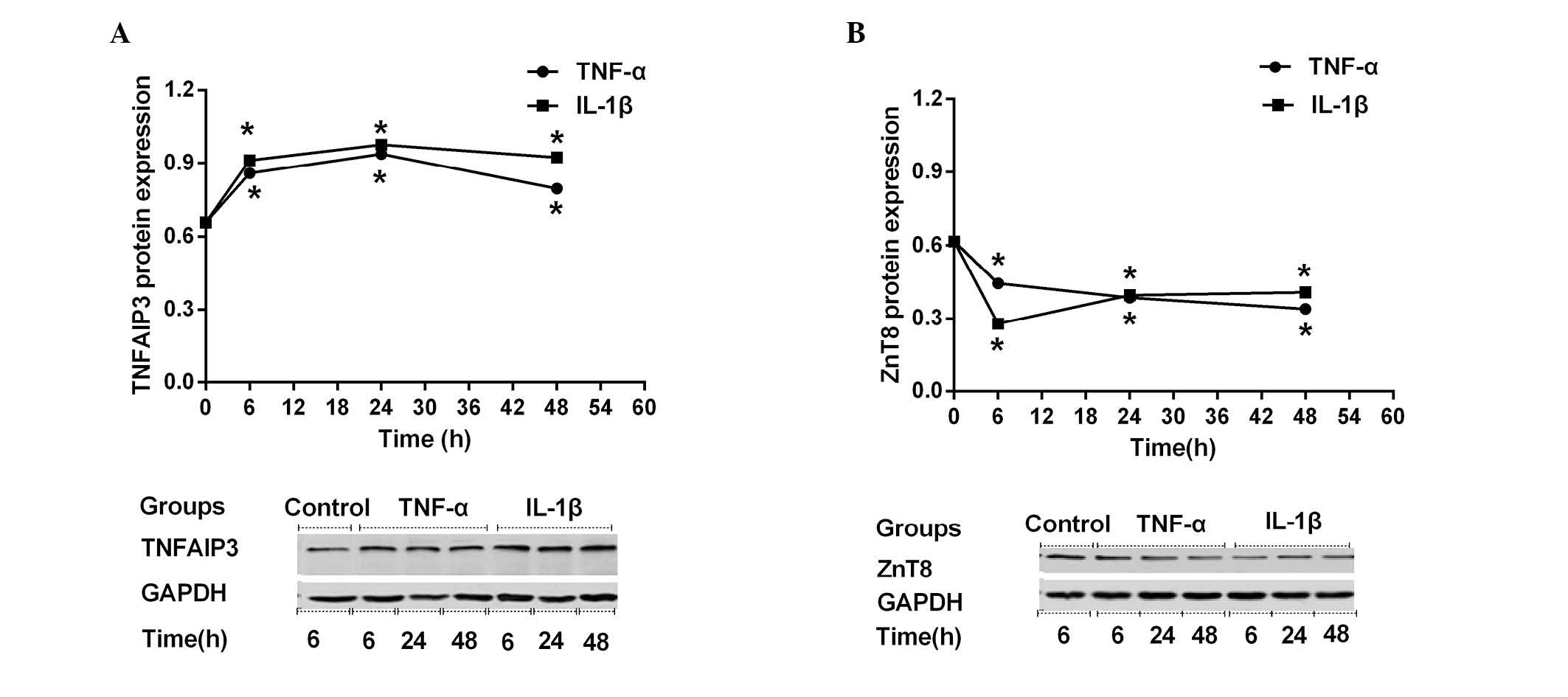
stimulation with TNF-α or IL-1β. As shown in Fig. 1, TNFAIP3 (Fig. 1A) and ZnT8 (Fig. 1B) were both expressed in NIT-1 cells,
as assessed by western blot analysis. NIT-1 cells demonstrated
similar TNFAIP3 expression-time curves (0, 6, 24 or 48 h) when
stimulated by TNF-α and IL-1β. During the first 6 h of cytokine
stimulation (0–6 h), TNFAIP3 expression increased sharply. During
the subsequent 18 h (6–24 h), the upregulation slowed, while
TNFAIP3 expression began to slowly decline during the subsequent 24
h (24–48 h), despite still being higher compared with prior to
stimulation (Fig. 1A). Stimulated
TNFAIP3 expression levels at all time points (6, 24 and 48 h) were
significantly altered compared with the control (all P<0.001).
In contrast to TNFAIP3 expression, ZnT8 expression declined rapidly
during the first 6 h of cytokine stimulation. During the subsequent
42 h, ZnT8 expression decreased or recovered slowly in the TNF-α or
IL-1β groups, respectively (Fig.
1B). In conclusion, stimulated ZnT8 expression levels at all
time points (6, 24 and 48 h) were significantly altered compared
with the control (all P<0.001).
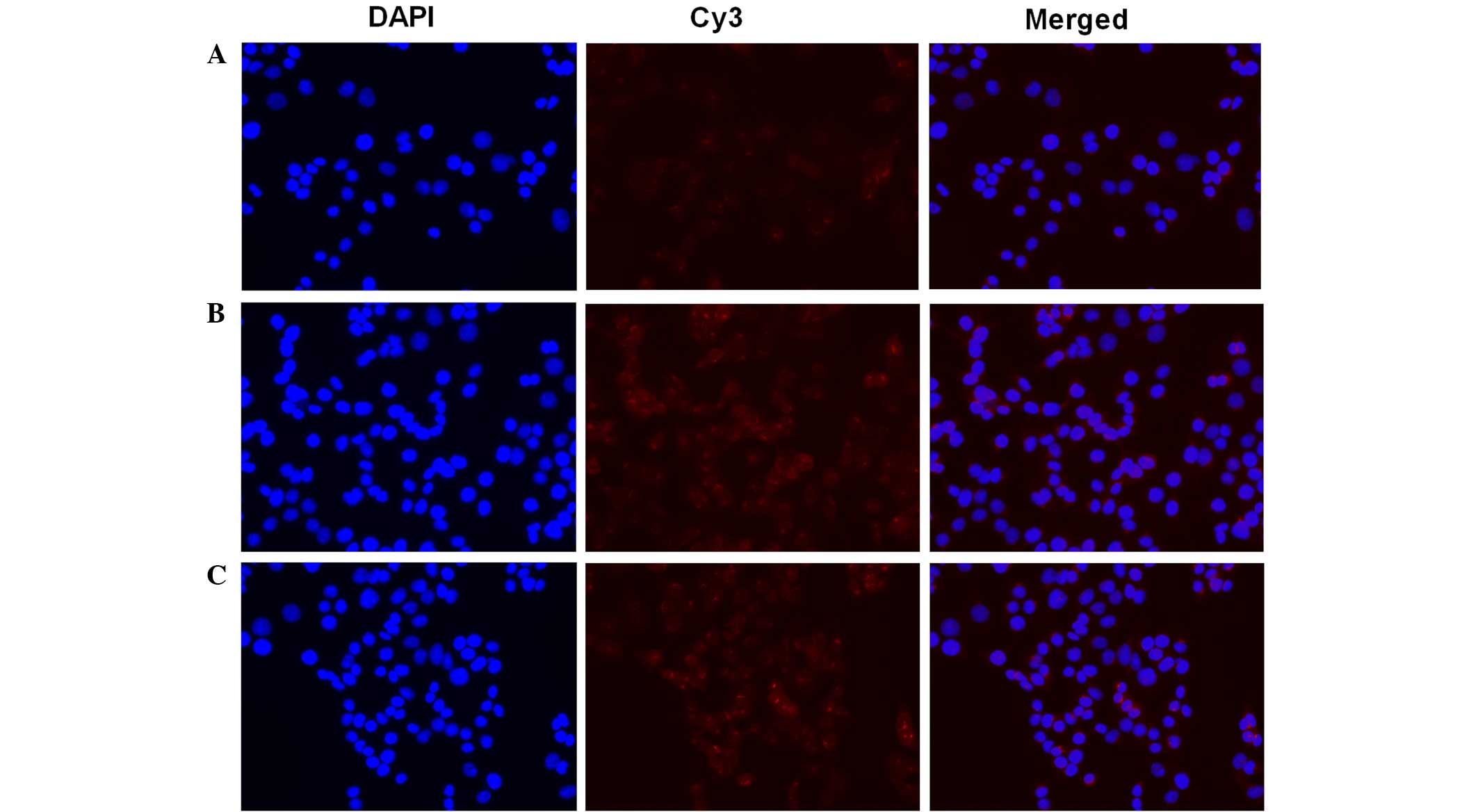
Furthermore, the present study examined the NF-κB
activation in NIT-1 cells subsequent TNF-α and IL-1β stimulation
(Fig. 2). In resting cells, NF-κB is
know to be inactivated through it interaction with IκB. After cells
are stimulated, NF-κB is released and translocates into the nucleus
(45). In order to assess NF-κB
activation, an NF-κB activation and Nuclear Translocation Assay kit
was used in the current study. The cellular p65 subunit of NF-κB
was identified using anti-p65 antibodies, which were then labeled
by Cy3-coupled secondary antibodies (red fluorescence). Nuclear DNA
of the same group was stained with DAPI (blue fluorescence) as an
indicator. Merged images demonstrated the positional association
between p65 and the nucleus. Compared with the control (Fig. 2A), the TNF-α (Fig. 2B) and IL-1β (Fig. 2C) stimulation resulted in assembly of
the p65 subunit in and around the nucleus, which indicated NF-κB
activation.
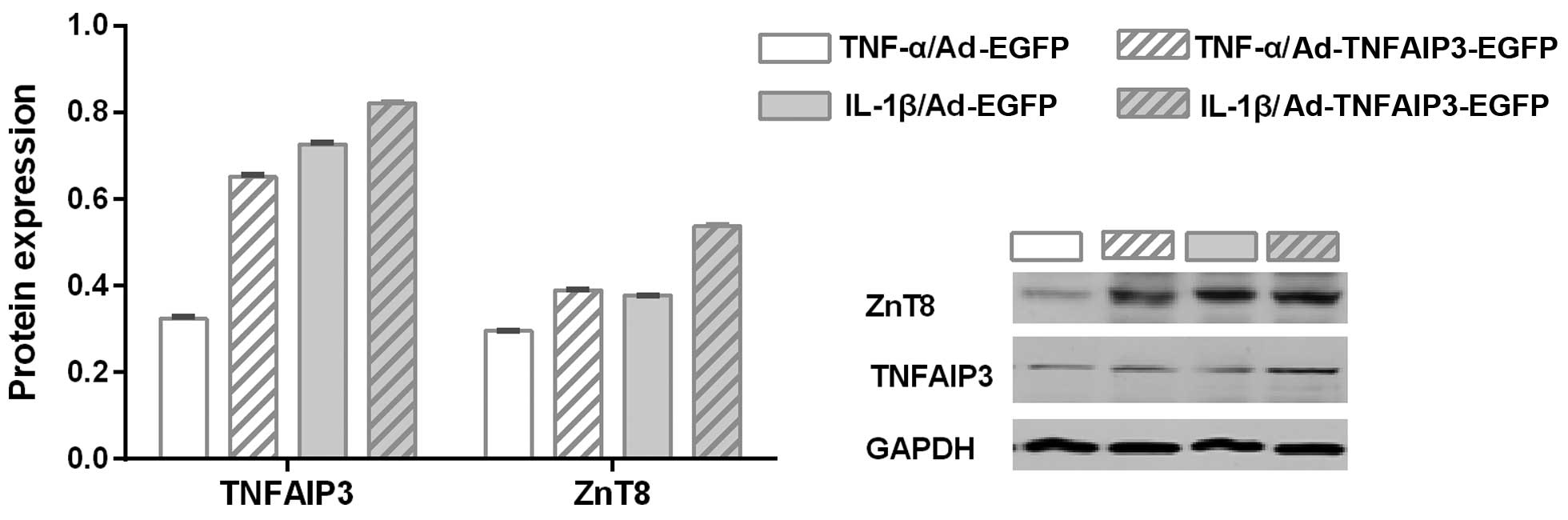
TNFAIP3 overexpression protects ZnT8
from cytokine-induced downregulation
As previously discussed, stimulation with the TNF-α
and IL-1β inflammatory cytokines resulted in NF-κB activation and
ZnT8 downregulation. It was therefore hypothesized that TNFAIP3, an
inhibitor of NF-κB activation (30),
may reverse the low expression of ZnT8 in NIT-1 cells exposed to
TNF-α and IL-1β. Therefore, the influence of TNFAIP3 overexpression
(using a TNFAIP3 overexpressing adenovirus) on the suppression of
ZnT8 expression resulting from cytokine intervention was
investigated. As shown in Fig. 3,
when stimulated by the same cytokine, the experimental group
(transfected with an Ad-TNFAIP3-EGFP adenovirus) presented higher
expression of TNFAIP3 compared with the control group (transfected
with an Ad-EGFP adenovirus), indicating successful transfection
(P<0.001 for TNF-α and IL-1β). Similarly, the experimental group
with high TNFAIP3 expression was shown to express a greater level
of ZnT8 when compared with the control group (P<0.001 for TNF-α
and IL-1β). These findings suggest that TNFAIP3 overexpression
significantly suppresses cytokine-induced ZnT8 downregulation.
Discussion
The results of the present study, in agreement with
the findings of previous studies (27,46),
indicated that insulin secretion can be inhibited by
proinflammatory cytokines causing dysregulation of β cells via ZnT8
downregulation, and a decrease in β cell number by affecting
apoptosis. The current study showed that proinflammatory cytokines,
TNF-α and IL-1β, downregulate ZnT8 protein expression in NIT-1
cells. This effect was previously observed in MIN6 cells (27), indicating that decreased ZnT8
expression may contribute to impaired β cell function and insulin
secretion. ZnT8 is predominantly expressed in pancreatic islets,
and is responsible for transporting zinc from the cytoplasm into
insulin secretory vesicles, where insulin is stored as a solid
hexamer bound with two Zn2+ ions (15). Thus, Zn2+ is essential for
the storage, crystallization and secretion of insulin. In addition,
a previous study observed that TNF-α and IL-1β were higher in the
plasma of diabetes patients (47).
It is well established that numerous cytokines, including TNF-α,
IL-1β and interferon-γ, contribute to β cell dysfunction and death,
via activation of the NF-κB signaling pathway (48–50),
thus promoting the immune system activity and the transcription of
proinflammatory and cell survival genes (51). This mechanism has been previously
verified using therapeutic drugs (52) and using a mouse allogeneic pancreatic
islet graft experiment (53).
Furthermore, studies of the human genome have identified numerous
genes associated with the pathophysiology of autoimmune and
inflammatory diseases, of which numerous are involved in the NF-κB
signaling pathway (33,35). Therefore, NF-κB serves a major role
in cytokine-dependent β cell dysfunction, and may represent a
target for therapeutics to improve islet function during
inflammatory insults.
Inappropriate activation of NF-κB signaling pathway
is a key event in the progressive loss of β cells in autoimmune
diabetes and type 2 diabetes (54–56).
TNFAIP3, an immediate early response gene, serves a critical role
in the negative regulation of the NF-κB signaling pathway, as well
as in β cell protection, through its actions as a dual
ubiquitin-editing protein (49,57). The
mechanism through which TNFAIP3 turns off inflammation signaling
has previously been described as the disruption of ubiquitin enzyme
complexes (58); in cells expressing
deficient or mutant TNFAIP3 protein, there is defective removal of
Lys63-linked ubiquitin from ubiquitin enzyme complexes following
stimulation with TNF (59). Our
previous study found that the plasma levels of TNF-α and IL-1β were
significantly increased, and TNFAIP3 mRNA and protein expression
levels were significant decreased in the peripheral blood
mononuclear cells of diabetes patients (60). The decrease in TNFAIP3 mRNA and
protein expression levels is consistent with other inflammatory
diseases. For instance, TNFAIP3 expression is decreased in systemic
lupus erythematosus patients, and the expression level of TNFAIP3
negatively correlates with the systemic lupus erythematosus disease
activity index (61).
TNFAIP3 is known for its potent anti-apoptotic
activity, particularly in β cells. Apoptosis is the main form of
β-cell death in the two forms of diabetes. Previous research
suggested that the mechanisms leading to nutrient- and
cytokine-induced β cell death in type 2 and type 1 diabetes,
respectively, share the activation of a final common pathway
involving IL-1β, NF-κB and Fas (50). TNFAIP3 is one of the anti-apoptotic
genes induced by cytokines through NF-κB in cultured human islets
(62) and INS-1E cells (42); induction of protective genes, such as
TNFAIP3, leads to islet survival and function (37). The results of the present study
provide a novel role for TNFAIP3 in the protection of normal β cell
functions via ZnT8 downregulation. This was supported by the data
obtained through TNFAIP3 overexpression in vitro in islet
(NIT-1) cells. Furthermore, inflammatory cytokines were found to
downregulate ZnT8 protein expression in NIT-1 cells. Notably,
TNFAIP3 overexpression protected against cytokine-induced
downregulation of ZnT8. This role is in addition to the
anti-apoptotic activity of TNFAIP3.
To the best of our knowledge, this is the first
study to examine the influence of TNFAIP3 overexpression on
cytokine-induced ZnT8 inhibition. One limitation of the current
study is that single nucleotide polymorphism of TNFAIP3 was not
considered, which may influence its affinity with targets. Future
studies will further analyze whether single nucleotide
polymorphisms of TNFAIP3 alter the effect of TNFAIP3 on ZnT8
expression, and a gene knockout model will be investigated.
In conclusion, the results of the present study
revealed that TNFAIP3 overexpression was able to protect ZnT8 from
cytokine-induced downregulation. Thus, it is hypothesized that
cytokines may not only lead to the death of β cells, but also alter
their normal function. Notably, TNFAIP3 serves a positive role
through its anti-apoptotic effects and its ability to maintain
normal β cell function. Therefore, TNFAIP3 may serve a pivotal role
in the pathogenesis of diabetes and may offer a novel therapeutic
target for the prevention and treatment of diabetes.
Acknowledgements
The study was partly supported by a grant from the
Scientific and Technological Plans of Chongqing (no. cstc2014yykfA
110004). The authors would like to thank all their colleagues for
their help in the experiments.
Glossary
Abbreviations
Abbreviations:
|
ZnT8
|
zinc transporter 8
|
|
TNFAIP3
|
tumor necrosis factor α-induced
protein-3
|
|
GAPDH
|
glyceraldehyde phosphate
dehydrogenase
|
|
ZIP
|
Zrt- and Irt-like protein
|
|
TNF
|
tumor necrosis factor
|
|
NF-κB
|
nuclear factor κB
|
|
IL-1β
|
interleukin-1β
|
|
DMEM
|
Dulbeccos modified Eagle's medium
|
|
FBS
|
fetal bovine serum
|
References
|
1
|
Global Report on Diabetes. World Health
Organization. Geneva, Swirzerland: 2016.PubMed/NCBI
|
|
2
|
American Diabetes Association: Diagnosis
and classification of diabetes mellitus. Diabetes Care. 33(Suppl
1): S62–S69. 2010.PubMed/NCBI
|
|
3
|
Velloso LA, Eizirik DL and Cnop M: Type 2
diabetes mellitus-an autoimmune disease? Nat Rev Endocrinol.
9:750–755. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Itariu BK and Stulnig TM: Autoimmune
aspects of type 2 diabetes mellitus - A mini-review. Gerontology.
60:189–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Richardson VR, Smith KA and Carter AM:
Adipose tissue inflammation: Feeding the development of type 2
diabetes mellitus. Immunobiology. 218:1497–1504. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Akash MS, Rehman K and Chen S: Role of
inflammatory mechanisms in pathogenesis of type 2 diabetes
mellitus. J Cell Biochem. 114:525–531. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sell H, Habich C and Eckel J: Adaptive
immunity in obesity and insulin resistance. Nat Rev Endocrinol.
8:709–716. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Odegaard JI and Chawla A: Connecting type
1 and type 2 diabetes through innate immunity. Cold Spring Harb
Perspect Med. 2:a0077242012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Guzmán-Flores JM and López-Briones S:
Cells of innate and adaptive immunity in type 2 diabetes and
obesity. Gac Med Mex. 148:381–389. 2012.(In Spanish). PubMed/NCBI
|
|
10
|
King GL: The role of inflammatory
cytokines in diabetes and its complications. J Periodontol.
79(Suppl 8): 1527–1534. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li YV: Zinc and insulin in pancreatic
beta-cells. Endocrine. 45:175–189. 2014. View Article : Google Scholar
|
|
12
|
Cousins RJ, Liuzzi JP and Lichten LA:
Mammalian zinc transport, trafficking and signals. J Biol Chem.
281:24085–24089. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kambe T, Yamaguchi-Iwai Y, Sasaki R and
Nagao M: Overview of mammalian zinc transporters. Cell Mol Life
Sci. 61:49–68. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chimienti F, Devergnas S, Pattou F, Schuit
F, Garcia-Cuenca R, Vandewalle B, Kerr-Conte J, Van Lommel L,
Grunwald D, Favier A and Seve M: In vivo expression and functional
characterization of the zinc transporter ZnT8 in glucose-induced
insulin secretion. J Cell Sci. 119:4199–4206. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chimienti F, Devergnas S, Favier A and
Seve M: Identification and cloning of a beta-cell-specific zinc
transporter, ZnT-8, localized into insulin secretory granules.
Diabetes. 53:2330–2337. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lemaire K, Ravier MA, Schraenen A,
Creemers JW, Van de Plas R, Granvik M, Van Lommel L, Waelkens E,
Chimienti F, Rutter GA, et al: Insulin crystallization depends on
zinc transporter ZnT8 expression, but is not required for normal
glucose homeostasis in mice. Proc Natl Acad Sci USA.
106:14872–14877. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pound LD, Sarkar SA, Benninger RK, Wang Y,
Suwanichkul A, Shadoan MK, Printz RL, Oeser JK, Lee CE, Piston DW,
et al: Deletion of the mouse Slc30a8 gene encoding zinc
transporter-8 results in impaired insulin secretion. Biochem J.
421:371–376. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wijesekara N, Dai FF, Hardy AB, Giglou PR,
Bhattacharjee A, Koshkin V, Chimienti F, Gaisano HY, Rutter GA and
Wheeler MB: Beta cell-specific Znt8 deletion in mice causes marked
defects in insulin processing, crystallisation and secretion.
Diabetologia. 53:1656–1668. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tamaki M, Fujitani Y, Uchida T, Hirose T,
Kawamori R and Watada H: Downregulation of ZnT8 expression in
pancreatic β-cells of diabetic mice. Islets. 1:124–128. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jing YL, Sun QM, Bi Y, Shen SM and Zhu DL:
SLC30A8 polymorphism and type 2 diabetes risk: Evidence from 27
study groups. Nutr Metab Cardiovasc Dis. 21:398–405. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang Q, Yin JY, Dai XP, Wu J, Chen X,
Deng CS, Yu M, Gong ZC, Zhou HH and Liu ZQ: Association analysis of
SLC30A8 rs13266634 and rs16889462 polymorphisms with type 2
diabetes mellitus and repaglinide response in Chinese patients. Eur
J Clin Pharmacol. 66:1207–1215. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen G, Xu Y, Lin Y, Lai X, Yao J, Huang
B, Chen Z, Huang H, Fu X, Lin L, et al: Association study of
genetic variants of 17 diabetes-related genes/loci and
cardiovascular risk and diabetic nephropathy in the Chinese She
population. J Diabetes. 5:136–145. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sørgjerd EP, Skorpen F, Kvaløy K,
Midthjell K and Grill V: Prevalence of ZnT8 antibody in relation to
phenotype and SLC30A8 polymorphism in adult autoimmune diabetes:
Results from the HUNT study, Norway. Autoimmunity. 46:74–79. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Skärstrand H, Lernmark A and Vaziri-Sani
F: Antigenicity and epitope specificity of ZnT8 autoantibodies in
type 1 diabetes. Scand J Immunol. 77:21–29. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vasto S, Mocchegiani E, Malavolta M,
Cuppari I, Listì F, Nuzzo D, Ditta V, Candore G and Caruso C: Zinc
and inflammatory/immune response in aging. Ann N Y Acad Sci.
1100:111–122. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Egefjord L, Jensen JL, Bang-Berthelsen CH,
Petersen AB, Smidt K, Schmitz O, Karlsen AE, Pociot F, Chimienti F,
Rungby J and Magnusson NE: Zinc transporter gene expression is
regulated by pro-inflammatory cytokines: A potential role for zinc
transporters in beta-cell apoptosis? BMC Endocr Disord. 9:72009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
El Muayed M, Billings LK, Raja MR, Zhang
X, Park PJ, Newman MV, Kaufman DB, OHalloran TV and Lowe WL Jr:
Acute cytokine-mediated downregulation of the zinc transporter ZnT8
alters pancreatic beta-cell function. J Endocrinol. 206:159–169.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen ZJ: Ubiquitination in signaling to
and activation of IKK. Immunol Rev. 246:95–106. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pickart CM and Eddins MJ: Ubiquitin:
Structures, functions, mechanisms. Biochim Biophys Acta.
1695:55–72. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vereecke L, Beyaert R and van Loo G: The
ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of
immunopathology. Trends Immunol. 30:383–391. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hughes LB, Reynolds RJ, Brown EE, Kelley
JM, Thomson B, Conn DL, Jonas BL, Westfall AO, Padilla MA, Callahan
LF, et al: Most common single-nucleotide polymorphisms associated
with rheumatoid arthritis in persons of European ancestry confer
risk of rheumatoid arthritis in African Americans. Arthritis Rheum.
62:3547–3553. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tejasvi T, Stuart PE, Chandran V, Voorhees
JJ, Gladman DD, Rahman P, Elder JT and Nair RP: TNFAIP3 gene
polymorphisms are associated with response to TNF blockade in
psoriasis. J Invest Dermatol. 132:593–600. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fung EY, Smyth DJ, Howson JM, Cooper JD,
Walker NM, Stevens H, Wicker LS and Todd JA: Analysis of 17
autoimmune disease-associated variants in type 1 diabetes
identifies 6q23/TNFAIP3 as a susceptibility locus. Genes Immun.
10:188–191. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Graham RR, Cotsapas C, Davies L, Hackett
R, Lessard CJ, Leon JM, Burtt NP, Guiducci C, Parkin M, Gates C, et
al: Genetic variants near TNFAIP3 on 6q23 are associated with
systemic lupus erythematosus. Nat Genet. 40:1059–1061. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Adrianto I, Wen F, Templeton A, Wiley G,
King JB, Lessard CJ, Bates JS, Hu Y, Kelly JA, Kaufman KM, et al:
Association of a functional variant downstream of TNFAIP3 with
systemic lupus erythematosus. Nat Genet. 43:253–258. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Eyre S, Hinks A, Bowes J, Flynn E, Martin
P, Wilson AG, Morgan AW, Emery P, Steer S, Hocking LJ, et al:
Overlapping genetic susceptibility variants between three
autoimmune disorders: Rheumatoid arthritis, type 1 diabetes and
coeliac disease. Arthritis Res Ther. 12:R1752010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang H, Ferran C, Attanasio C, Calise F
and Otterbein LE: Induction of protective genes leads to islet
survival and function. J Transplant. 2011:1418982011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Grey ST, Longo C, Shukri T, Patel VI,
Csizmadia E, Daniel S, Arvelo MB, Tchipashvili V and Ferran C:
Genetic engineering of a suboptimal islet graft with A20 preserves
beta cell mass and function. J Immunol. 170:6250–6256. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hou CL, Zhang W, Wei Y, Mi JH, Li L, Zhou
ZH, Zeng W and Ying DJ: Zinc finger protein A20 overexpression
inhibits monocyte homing and protects endothelial cells from injury
induced by high glucose. Genet Mol Res. 10:1050–1059. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yu LY, Lin B, Zhang ZL and Guo LH: Direct
transfer of A20 gene into pancreas protected mice from
streptozotocin-induced diabetes. Acta Pharmacol Sin. 25:721–726.
2004.PubMed/NCBI
|
|
41
|
Tan BM, Zammit NW, Yam AO, Slattery R,
Walters SN, Malle E and Grey ST: Baculoviral inhibitors of
apoptosis repeat containing (BIRC) proteins fine-tune TNF-induced
nuclear factor κB and c-Jun N-terminal kinase signalling in mouse
pancreatic beta cells. Diabetologia. 56:520–532. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ortis F, Pirot P, Naamane N, Kreins AY,
Rasschaert J, Moore F, Théâtre E, Verhaeghe C, Magnusson NE,
Chariot A, et al: Induction of nuclear factor-kappaB and its
downstream genes by TNF-alpha and IL-1beta has a pro-apoptotic role
in pancreatic beta cells. Diabetologia. 51:1213–1225. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fu Y, Tian W, Pratt EB, Dirling LB, Shyng
SL, Meshul CK and Cohen DM: Down-regulation of ZnT8 expression in
INS-1 rat pancreatic beta cells reduces insulin content and
glucose-inducible insulin secretion. PLoS One. 4:e56792009.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Huang L: Zinc and its transporters,
pancreatic beta-cells, and insulin metabolism. Vitam Horm.
95:365–390. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shembade N, Ma A and Harhaj EW: Inhibition
of NF-kappaB signaling by A20 through disruption of ubiquitin
enzyme complexes. Science. 327:1135–1139. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Feve B and Bastard JP: The role of
interleukins in insulin resistance and type 2 diabetes mellitus.
Nat Rev Endocrinol. 5:305–311. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Reinehr T, Karges B, Meissner T, Wiegand
S, Stoffel-Wagner B, Holl RW and Woelfle J: Inflammatory markers in
obese adolescents with type 2 diabetes and their relationship to
hepatokines and adipokines. J Pediatr. S0022-3476(16): 00277–8.
2016.
|
|
48
|
Liuwantara D, Elliot M, Smith MW, Yam AO,
Walters SN, Marino E, McShea A and Grey ST: Nuclear factor-kappaB
regulates beta-cell death: A critical role for A20 in beta-cell
protection. Diabetes. 55:2491–2501. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Melloul D: Role of NF-kappaB in beta-cell
death. Biochem Soc Trans. 36:334–339. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen
S and Eizirik DL: Mechanisms of pancreatic beta-cell death in type
1 and type 2 diabetes: Many differences, few similarities.
Diabetes. 54(Suppl 2): S97–S107. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Baltimore D: NF-κB is 25. Nat Immunol.
12:683–685. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liu XH, Wang YP, Wang LX, Chen Z, Liu XY
and Liu LB: Exendin-4 protects murine MIN6 pancreatic β-cells from
interleukin-1β-induced apoptosis via the NF-κB pathway. J
Endocrinol Invest. 36:803–811. 2013.PubMed/NCBI
|
|
53
|
Eldor R, Abel R, Sever D, Sadoun G, Peled
A, Sionov R and Melloul D: Inhibition of nuclear factor-κB
activation in pancreatic β-cells has a protective effect on
allogeneic pancreatic islet graft survival. PLoS One. 8:e569242013.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Donath MY and Shoelson SE: Type 2 diabetes
as an inflammatory disease. Nat Rev Immunol. 11:98–107. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kiechl S, Wittmann J, Giaccari A, Knoflach
M, Willeit P, Bozec A, Moschen AR, Muscogiuri G, Sorice GP, Kireva
T, et al: Blockade of receptor activator of nuclear factor-κB
(RANKL) signaling improves hepatic insulin resistance and prevents
development of diabetes mellitus. Nat Med. 19:358–363. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kim HS, Han MS, Chung KW, Kim S, Kim E,
Kim MJ, Jang E, Lee HA, Youn J, Akira S and Lee MS: Toll-like
receptor 2 senses beta-cell death and contributes to the initiation
of autoimmune diabetes. Immunity. 27:321–333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Coornaert B, Carpentier I and Beyaert R:
A20: Central gatekeeper in inflammation and immunity. J Biol Chem.
284:8217–8221. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wertz IE, Newton K, Seshasayee D, Kusam S,
Lam C, Zhang J, Popovych N, Helgason E, Schoeffler A, Jeet S, et
al: Phosphorylation and linear ubiquitin direct A20 inhibition of
inflammation. Nature. 528:370–375. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhou Q, Wang H, Schwartz DM, Stoffels M,
Park YH, Zhang Y, Yang D, Demirkaya E, Takeuchi M, Tsai WL, et al:
Loss-of-function mutations in TNFAIP3 leading to A20
haploinsufficiency cause an early-onset autoinflammatory disease.
Nat Genet. 48:67–73. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Cheng L, Zhang D, Jiang Y, Deng W, Wu Q,
Jiang X and Chen B: Decreased A20 mRNA and protein expression in
peripheral blood mononuclear cells in patients with type 2 diabetes
and latent autoimmune diabetes in adults. Diabetes Res Clin Pract.
106:611–616. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Li D, Wang L, Fan Y, Song L, Guo C, Zhu F,
Zhang L and Shi Y: Down-regulation of A20 mRNA expression in
peripheral blood mononuclear cells from patients with systemic
lupus erythematosus. J Clin Immunol. 32:1287–1291. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sarkar SA, Kutlu B, Velmurugan K,
Kizaka-Kondoh S, Lee CE, Wong R, Valentine A, Davidson HW, Hutton
JC and Pugazhenthi S: Cytokine-mediated induction of anti-apoptotic
genes that are linked to nuclear factor kappa-B (NF-kappaB)
signalling in human islets and in a mouse beta cell line.
Diabetologia. 52:1092–1101. 2009. View Article : Google Scholar : PubMed/NCBI
|