Introduction
Pulmonary embolism (PE) is a frequent clinical
syndrome with abnormal pulmonary circulation, which is
predominantly caused by the blockage of pulmonary and/or bronchial
arteries by thrombus shedding (1).
Due to the sudden disease onset, PE is difficult to treat and
usually results in the patient succumbing to the disease (2,3).
Homocysteine (Hcy) is an intermediate product of methionine
metabolism, and is an independent risk factor for cardiovascular
diseases (4,5). Previous studies reported that serum Hcy
levels are markedly elevated in patients with PE, which is
implicated in the disease pathogenesis (6–8).
Vessel wall injury is one of the important factors
for PE, as the pathological changes in endothelial cells may result
in the formation of thrombosis (9,10). It
has been demonstrated that high concentrations of Hcy induce
apoptosis in endothelial cells (11,12), in
which mitochondria are involved. Cytochrome c oxidase (COX)
is a key enzyme in mitochondrial function (13–15).
Enhanced COX activity may increase intracellular reactive oxygen
species (ROS) levels, resulting in cellular apoptosis (16–21).
Based on these reports, it would be of interest to define the
association between Hcy, COX, and ROS in apoptosis, specifically in
the context of PE pathogenesis.
In the present study, the effects of Hcy on
endothelial cells and the associated molecular mechanisms were
investigated.
Materials and methods
Patients
A total of 10 patients with PE, 4 males and 6
females, aged 50–67 years (with an average age of 60.4 years), were
included in this study. These PE cases were diagnosed based on the
clinical manifestations and the results from radiological and
laboratory tests, in line with the standard literature (22). Furthermore, a further 30 healthy
subjects, 15 males and 15 females, aged 30–88 years (with an
average age of 56.5 years), were used as the control group. All
subjects were recruited from the Laiwu City People's Hospital
(Laiwu, China). Prior written and informed consent was obtained
from each patient and the study was approved by the ethics review
board of the Laiwu City People's Hospital.
Measurement of Hcy concentration
Blood samples were obtained from the PE patients and
control subjects, and the concentration of Hcy was measured.
Firstly, the column was washed with 95% methanol for 30 min,
followed by 5% methanol for 30 min. The column was then
equilibrated with the mobile phase, 0.05 M
KH2PO4 (pH 2.1):acetonitrile (93:7). For
fluorescence detection, the excitation wavelength was set as 385
nm, and the emission wavelength was set as 515 nm. A 10 µl sample
was injected, with a detection duration of 10 min. The
concentration was calculated using the peak area as the response
value, and the results were normalized with the total protein
concentration in cardiomyocytes.
Cell culture
Human umbilical vein endothelial cells (HUVECs) were
obtained from Beijing Yuhengfeng Biotech (Beijing, China). The
cells were cultured in Endothelial Cell Medium (Beijing Yuhengfeng
Biotech) supplemented with 10% fetal bovine serum (FBS; Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 37°C in an
incubator containing 5% CO2. The control group was free
from intervention; the folic acid group was treated with 100 µmol/l
folic acid; the Hcy group was treated with 1 mM Hcy; and the Hcy +
folic acid group was treated with 1 mM Hcy and 100 µmol/l folic
acid.
High-performance liquid chromatography
(HPLC)
Hcy concentration was determined with HPLC. For
serum sample preparation, anticoagulant blood (10 ml) was
centrifuged at 200 × g at 4°C for 15 min, and the supernatant was
used for measurement. For cell sample preparation, the cells were
collected and washed with phosphate-buffered saline (PBS).
Following the addition of 100 µl 1% sodium dodecyl sulfate (SDS),
the cells were lysed with an ultrasonic cell disruptor, and
centrifuged at 12,000 × g at 4°C for 10 min, and the supernatant
was used for measurement. Prior to the measurements, protein
concentrations were determined using a BCA kit (cat. no. BCA1;
Sigma-Aldrich, St. Louis, MO, USA). Standard substance
concentration series were set as 3.125, 6.25, 12.5, 25, 50, and 100
µM. For HPLC detection, 10 µl tris (2-carboxyethyl) phosphine
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) was added into 90
µl protein sample, followed by vortex for 1 min and incubation at
4°C for 30 min. Subsequently, 100 µl methanol was added, and the
sample was vortexed for 1 min and centrifuged at 20,000 × g for 10
min. A total of 150 µl supernatant was collected into an EP tube,
and 20 µl 1.55 M NaOH, 250 µl borate buffer (containing 0.125 M
H3BO3 and 4 mM ethylenediaminetetraacetic
acid, pH 9.5), and 10 µl SBD-F (10 mg/ml in borate buffer;
Sigma-Aldrich) were added to the sample prior to vortex for 1 min
and incubation at 60°C for 60 min. Following centrifugation at
10,000 rpm for 10 min, 200 µl supernatant was collected and
subjected to HPLC detection.
Flow cytometry
Cellular apoptosis was detected by flow cytometry
and Annexin-V staining. HUVECs were first incubated with Hcy at 0,
0.01, 0.1, and 1 mM, respectively, for 24 h. The cells
(3×106 cells) were then trypsinized, and collected
through centrifugation at 200 × g at 4°C for 5 min. Following
washing with PBS, the cells were incubated with 100 µl
Annexin-V-FLUOS reagent (Roche Molecular Systems, Inc., Branchburg,
NJ, USA) supplemented with 2% propidium iodide (PI) at room
temperature for 10–15 min, and the fluorescence was detected by
flow cytometry (FACScan; BS Biosciences, Franklin Lakes, NJ, USA).
The apoptosis rate was calculated as the percentage of Annexin-V
and PI double positive cells out of the total cells. Experiments
were repeated in triplicate.
Mitochondrial isolation
Cardiomyocytes (2.5×106 cells; purchased
from Dashou Biotechnology Company, Chengdu, China), obtained from
heart tissues in rats within 3 days of birth, were cultured in T-25
flasks at 37°C for 5 days containing Hepes-buffered Dulbecco's
modified Eagle's medium (Gibco; Thermo Fisher Scientific, Inc.)
with 10% FBS. The cells were then digested with trypsin and
collected into EP tubes. A total of 1 ml mitochondrial separation
medium buffer (containing 0.21 M mannitol, 0.07 M sucrose, 10 mM
Tris base and 1 mM EGTA; pH 7.4) was added to re-suspend the cells,
and the cell suspension was homogenized 200 times in a Dounce
homogenizer (Active Motif, San Diego, CA, USA) on ice. Cell
homogenates were transferred into a new 2 ml EP tube, and
centrifuged at 600 × g for 5 min at 4°C. The supernatant was
collected into an 1.5 ml EP tube, and centrifuged at 1,000 × g for
5 min at 4°C. Subsequently, the supernatant was collected into
another 1.5 ml EP tube, and subjected to centrifugation at 7,000 ×
g for 10 min at 4°C. This time, the supernatant was discarded, and
1 ml mitochondrial separation medium buffer was used to re-suspend
the precipitate, followed by centrifugation at 7,000 × g for 10 min
at 4°C. The supernatant was then discarded, and the precipitate was
re-suspended with 1 ml MSTE. Following centrifugation at 10,000 × g
for 10 min at 4°C, the mitochondria were obtained.
COX activity assay
A total of 40 µl 1% SDS was added into the
mitochondria in the EP tube on ice. Following vortexing 6 times
within 30 min, the mitochondria were centrifuged at 12,000 × g for
10 min at 4°C. The supernatant was collected into a new EP tube and
contained the mitochondrial proteins. The protein concentration was
determined using the DAC protein assay reagent (Bio-Rad
Laboratories, Inc.), and the standard curve was obtained with
bovine serum albumin in 1% SDS. COX activity was determined with a
COX activity assay kit (cat. no. GMS100014.3.1; Shanghai Genmed
Gene Pharmaceutical Technology Co., Ltd., Shanghai, China),
according to the manufacturer's protocol.
Intracellular ROS level
measurement
The cells (2×106 cells)were trypsinized
and collected by centrifugation at 300 × g at 4°C for 5 min. An
intracellular ROS red fluorescence determination kit (cat. no.
GMS10111.1; Shanghai Genmed Gene Pharmaceutical Technology Co.,
Ltd.) was used to stain the cells. Fluorescence was detected using
a microplate reader (iMark; Bio-Rad Laboratories, Inc.), with an
excitation wavelength of 540 nm and an emission wavelength of 590
nm. The intracellular ROS levels were expressed as relative
fluorescence unit values.
Western blot analysis
Protein expression levels were assessed by western
blot analysis. Total proteins were extracted with a lysis buffer
(Sigma-Aldrich). The protein concentration was measured with a DAC
protein assay reagent. A total of 30 µg protein sample was
separated by 12% SDS-PAGE, and then transferred onto the
polyvinylidene difluoride (PVDF) membrane. The membrane was blocked
with non-fat milk at room temperature for 1 h. Subsequently, goat
anti-rat COX 17 primary polyclonal antibody (1:1,000 dilution; cat.
no. sc-27533; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) was
added to the membrane at incubated at 4°C overnight. The secondary
horseradish peroxidase-labeled rabbit anti-goat immunoglobulin G
(1:2,000 dilution; cat. no. A0208; Beyotime Institute of
Biotechnology, Haimen, China) antibody was then added for
incubation at room temperature for 1 h. Expression levels were
quantified using Quantity One software (version 4.62; Bio-Rad
Laboratories, Inc.). GAPDH was used as the internal reference.
Statistical analysis
Data were expressed as means ± standard deviation.
SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA) was used for the
statistical analyses. A t-test was performed for the group
comparison. P<0.05 was considered to indicate a statistically
significant result.
Results
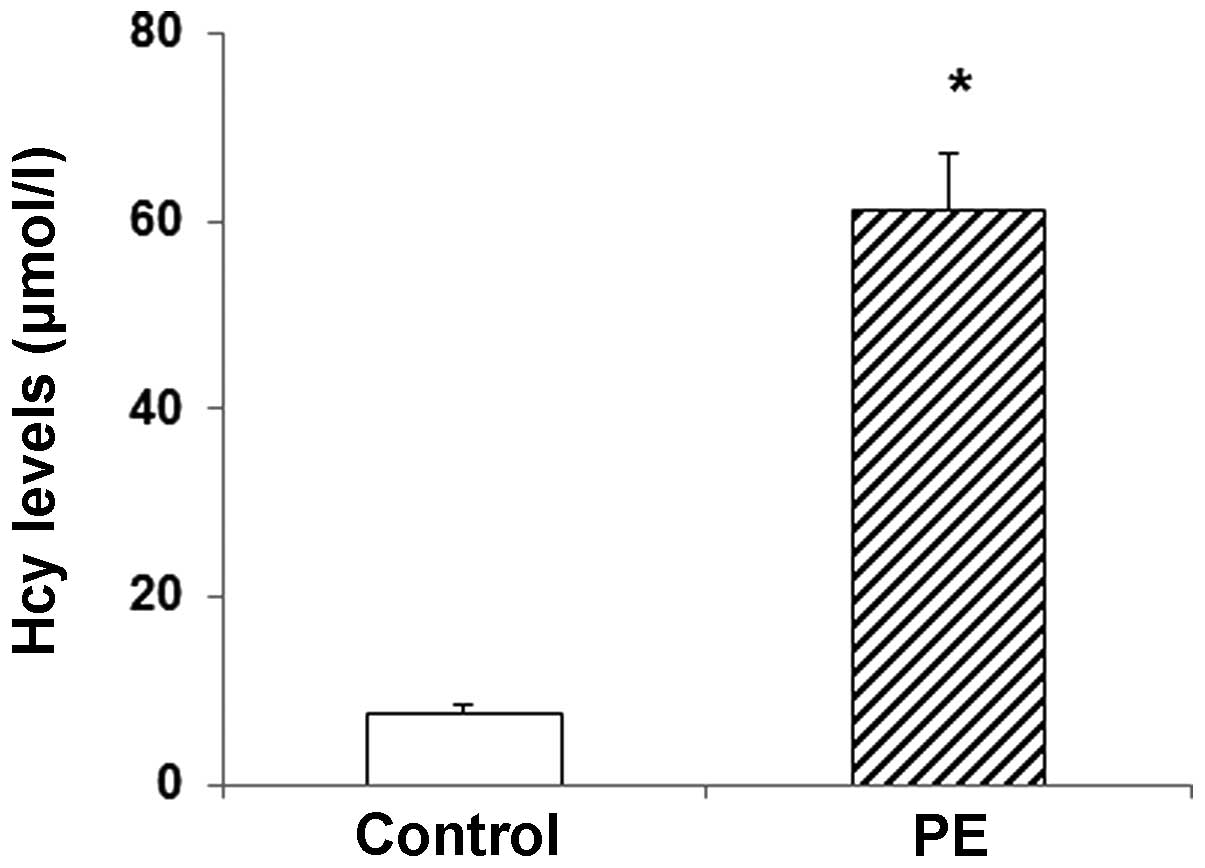
Serum Hcy levels were elevated in
patients with PE
The serum Hcy levels were determined in patients
with PE and healthy controls. The results obtained from the HPLC
demonstrated that, compared with the control group, the serum Hcy
levels were significantly elevated in the PE group (P<0.05;
Fig. 1). These results suggest that
the serum Hcy levels may be an indicator for PE.
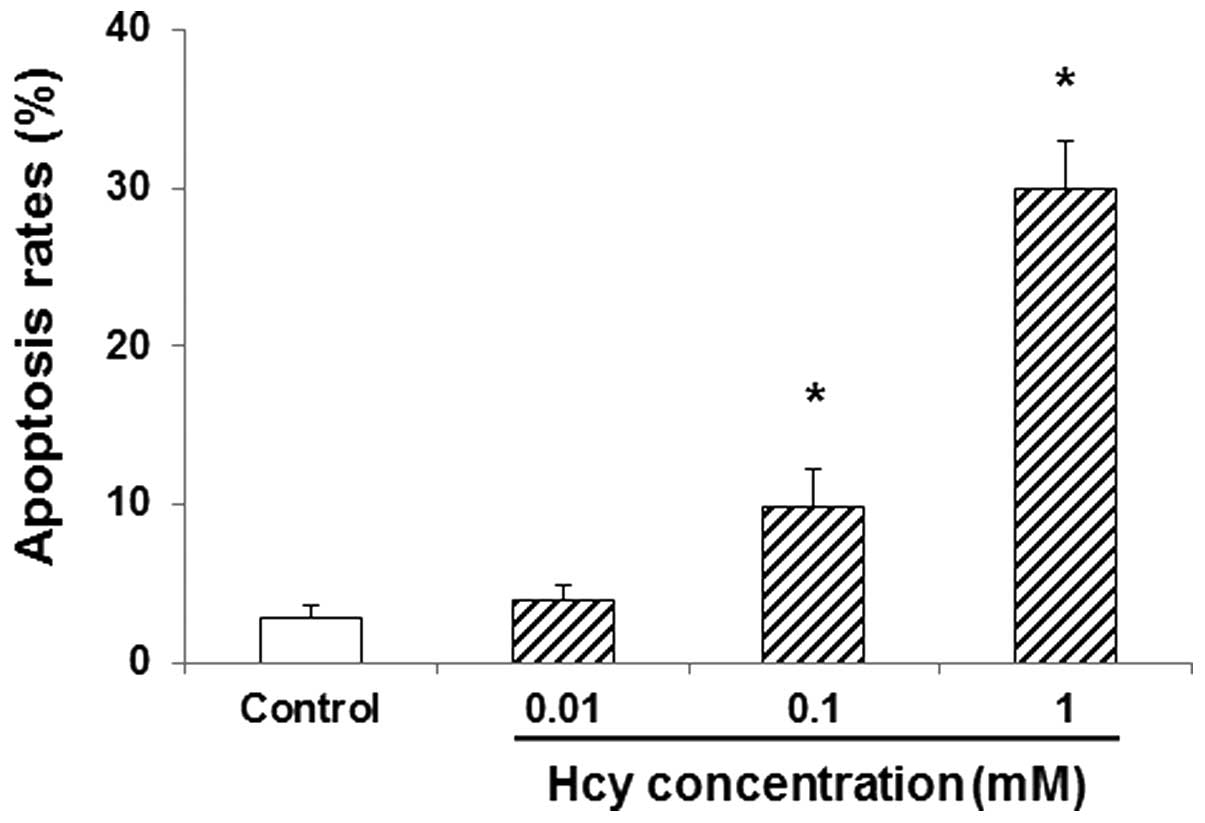
Hcy induces apoptosis in endothelial
cells
The effects of Hcy on apoptosis in endothelial cells
were subsequently investigated. HUVECs were first incubated with
Hcy at indicated concentrations (0, 0.01, 0.1 and 1 mM) for 24 h,
and then the apoptotic rates were detected by flow cytometry.
Compared with the control group, treatment with 0.01 mM Hcy
marginally increased the apoptosis rate in HUVECs (P>0.05;
Fig. 2). Furthermore, at
concentrations of 0.1 and 1 mM, Hcy significantly increased the
apoptosis rate in HUVECs, compared with the control group
(P<0.05; Fig. 2). These results
suggest that Hcy may induce apoptosis in HUVECs, which may
contribute to the pathogenesis of PE.
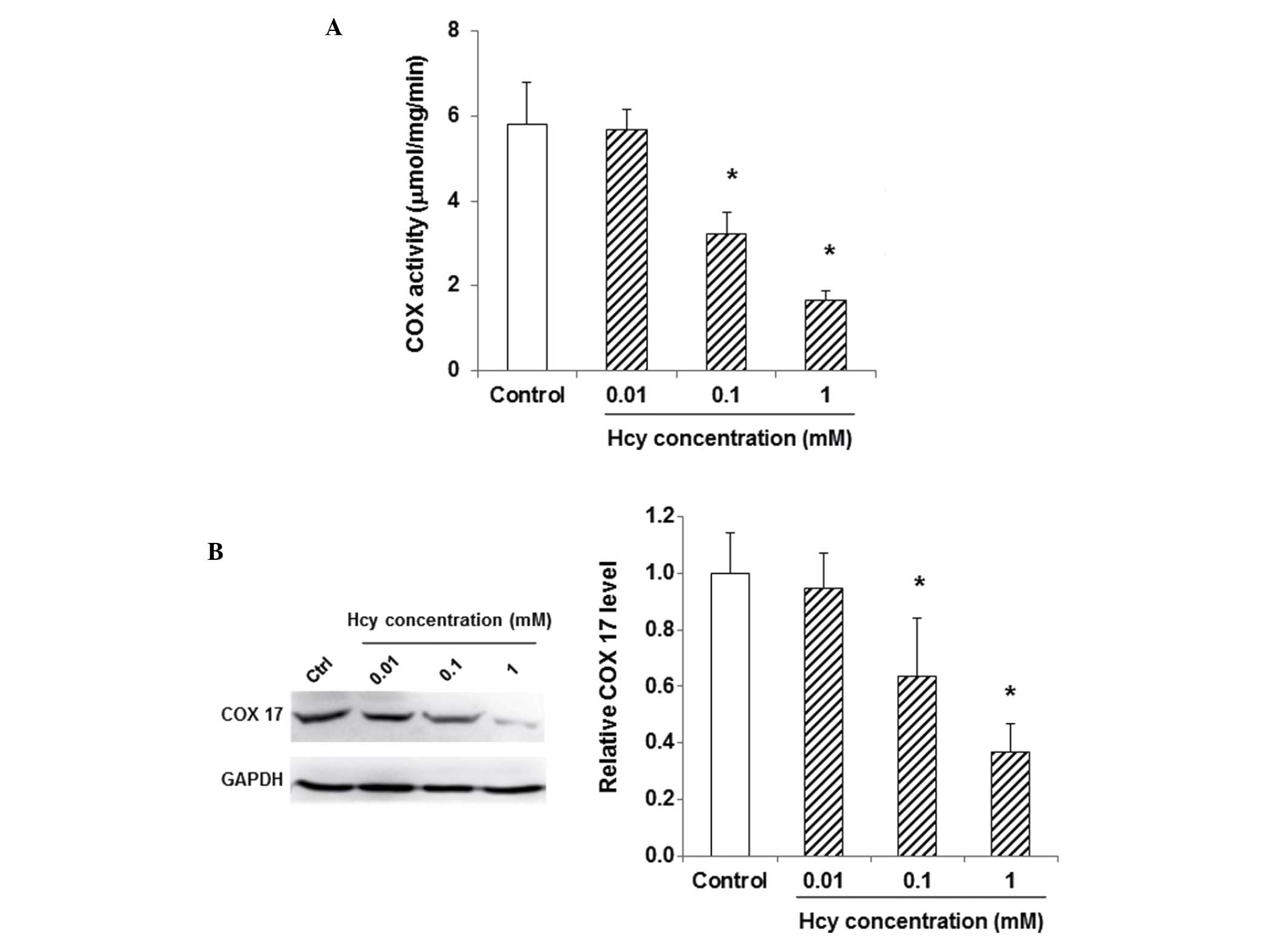
Hcy decreases COX activity and
inhibits COX 17 expression
To investigate the effects of Hcy on COX activity
levels in endothelial cells, HUVECs were first treated with Hcy (0,
0.01, 0.1, and 1 mM) for 24 h, and then COX activity was assessed
using the COX activity assay kit. Compared with the control group,
Hcy decreased COX activity levels in HUVECs (for 0.01 mM,
P>0.05; for 0.1 and 1 mM, P<0.05; Fig. 3A). In addition, the expression levels
of COX 17 in these cells were detected by western blot analysis. As
shown in Fig. 3B, 0.01 mM Hcy
marginally decreased the protein expression levels of COX 17,
whereas 0.1 and 1 mM Hcy significantly decreased the expression
levels of COX 17 (P<0.05). These results suggest that Hcy could
decrease COX activity and inhibit COX 17 expression in endothelial
cells.
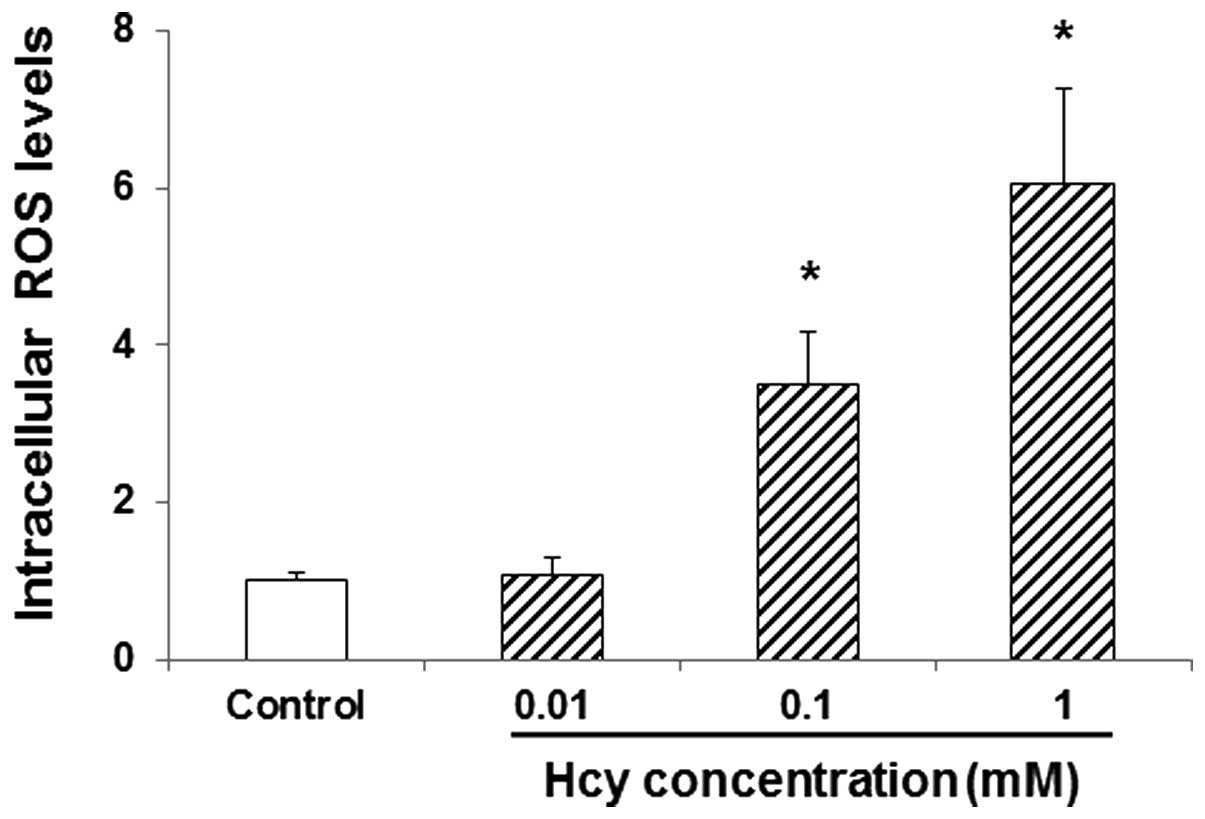
Hcy increases intracellular ROS levels
in endothelial cells
The effects of Hcy on the intracellular ROS levels
in endothelial cells were then investigated. HUVECs were incubated
with 0, 0.01, 0.1, and 1 mM Hcy for 24 h, and the intracellular ROS
levels were determined. Compared with the control group, 0.01 mM
Hcy marginally increased the intracellular ROS levels in HUVECs
(P>0.05; Fig. 4). Furthermore,
0.1 and 1 mM Hcy significantly elevated the intracellular ROS
levels in these cells (P<0.05; Fig.
4). These results suggest that Hcy could increase the
intracellular ROS levels in endothelial cells, which may be
associated with its effects on apoptosis in these cells.
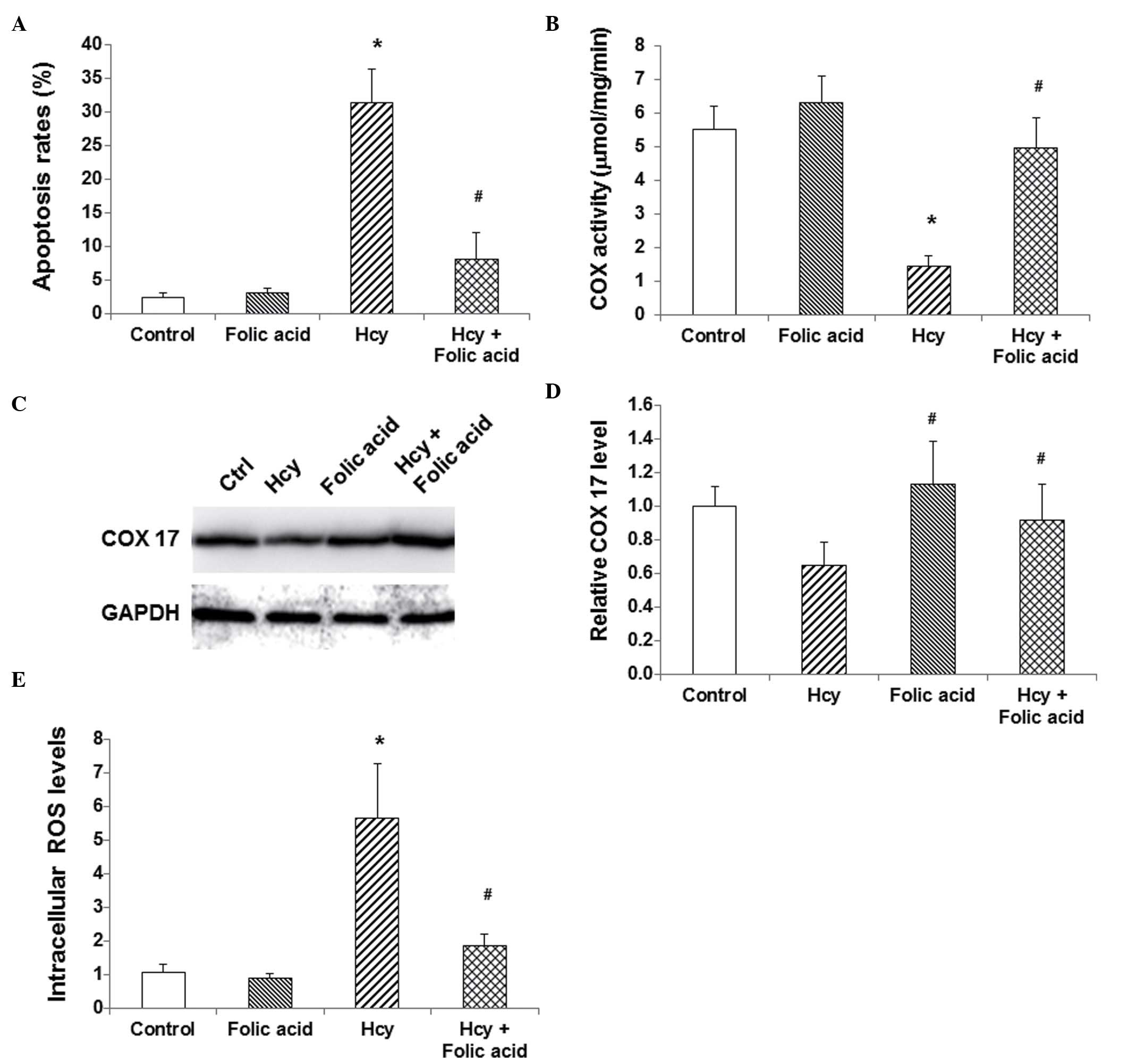
Folic acid alleviates the
physiopathological changes induced by Hcy
The effects of folic acid on Hcy-induced
pathological changes in endothelial cells were then investigated.
HUVECs were treated with Hcy and/or folic acid for 24 h, and then
the apoptosis levels, the COX activity and COX 17 expression
levels, and the intracellular ROS levels were evaluated. Compared
with the control group, folic acid treatment alone did not alter
the apoptosis rate, COX activity and COX 17 expression, or the
intracellular ROC levels in HUVECs (P>0.05; Fig. 5). However, treatment with folic acid
inhibited the effects of Hcy in these cells. Compared with the Hcy
group, folic acid treatment significantly decreased the apoptosis
rate, inhibited the Hcy-induced COX activity decline and COX 17
expression downregulation, and decreased intracellular ROS levels
in HUVECs (P<0.05; Fig. 5). These
results suggest that folic acid could alleviate the
physiopathological changes induced by Hcy in endothelial cells.
Discussion
PE is an acute pathological syndrome with a
relatively high mortality (2,3). It has
been reported that the serum Hcy levels are elevated in patients
with PE, which is an independent risk factor for cardiovascular
diseases, and is an important factor for the occurrence of PE
(4–8). In the present study, the effects of Hcy
on PE pathogenesis and the molecular mechanisms underlying these
effects were investigated.
COX 17 is an important factor for COX activity, and
is responsible for the transition of Cu2+ to the COX
subunit (23,24). The results of the present study
demonstrated that 1 mM Hcy significantly decreased COX activity,
and downregulated the expression of COX 17 in HUVECs. Further
studies are required in order to elucidate the underlying
mechanisms. Decreased COX activity levels may lead to the elevation
of intracellular ROS levels, further inducing apoptosis (16,17). The
results showed that 1 mM Hcy significantly increased the
intracellular ROS levels and the apoptosis rate in endothelial
cells.
Within cells, Hcy may be methylated into methionine
when catalyzed with methionine synthase (MS), which involves folic
acid (3,4). In the present study, folic acid was
demonstrated to inhibit Hcy in endothelial cells. Moreover,
treatment with folic acid treatment alleviated the
physiopathological changes induced by Hcy in these cells.
In conclusion, the results demonstrated that Hcy
inhibited COX activity and decreased the expression levels of COX
17, thereby increasing intracellular ROS levels, which eventually
resulted in increased apoptosis rates in endothelial cells.
However, treatment with folic acid was able to alleviate the
physiopathological changes induced by Hcy, restoring the COX
activity and COX 17 expression levels, inhibiting intracellular
ROS, and reducing the apoptosis rate in the endothelial cells.
These findings suggest that the Hcy concentration in blood is
important for the prevention of pulmonary embolism, which provides
theoretical basis for the disease prevention in clinic. Further
studies are required in order to explore the mechanisms underlying
Hcy-induced COX activity decline.
Acknowledgements
The present study was supported by a grant from the
National Natural Science Foundation (grant no. 81201494).
References
|
1
|
de Miguel-Díez J, Jiménez-García R, de
Andrés A López, Hernández-Barrera V, Carrasco-Garrido P, Monreal M,
Jiménez D, Jara-Palomares L and Álvaro-Meca A: Analysis of
environmental risk factors for pulmonary embolism: A case-crossover
study (2001-2013). Eur J Intern Med. 31:55–61. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Heit JA: The epidemiology of venous
thromboembolism in the community: Implications for prevention and
management. J Thromb Thrombolysis. 21:23–29. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tapson VF: Acute pulmonary embolism. N
Engl J Med. 358:1037–1052. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen P, Poddar R, Tipa EV, Dibello PM,
Moravec CD, Robinson K, Green R, Kruger WD, Garrow TA and Jacobsen
DW: Homocysteine metabolism in cardiovascular cells and tissues:
Implications for hyperhomocysteinemia and cardiovascular disease.
Adv Enzyme Regul. 39:93–109. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jeremy JY, Shukla N, Angelini GD, Day A,
Wan IY, Talpahewa SP and Ascione R: Sustained increases of plasma
homocysteine, copper and serum ceruloplasmin after coronary artery
bypass grafting. Ann Thorac Surg. 74:1553–1557. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Karalezli A, Parlak ES, Kanbay A, Senturk
A and Hasanoglu HC: Homocysteine and serum-lipid levels in
pulmonary embolism. Clin Appl Thromb Hemost. 17:E186–E189. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Köktürk N, Kanbay A, Aydoğdu M, Özyılmaz
E, Bukan N and Ekim N: Hyperhomocysteinemia prevalence among
patients with venous thromboembolism. Clin Appl Thromb Hemost.
17:487–493. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Okumus G, Kiyan E, Arseven O, Tabak L,
DizKucukkaya R, Unlucerci Y, Abaci N, Unaltuna NE and Issever H:
Hereditary thrombophilic risk factors and venous thromboembolism in
Istanbul, Turkey: The role in different clinical manifestations of
venous thromboembolism. Clin Appl Thromb Hemost. 14:168–173. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
SallesCrawley II, Monkman JH, Ahnström J,
Lane DA and Crawley JT: Vessel wall BAMBI contributes to hemostasis
and thrombus stability. Blood. 123:2873–2881. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ren M, Li R, Luo M, Chen N, Deng X, Yan K,
Zeng M and Wu J: Endothelial cells but not platelets are the major
source of Toll-like receptor 4 in the arterial thrombosis and
tissue factor expression in mice. Am J Physiol Regul Integr Comp
Physiol. 307:R901–R907. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mansoor MA, Bergmark C, Haswell SJ, Savage
IF, Evans PH, Berge RK, Svardal AM and Kristensen O: Correlation
between plasma total homocysteine and copper in patients with
peripheral vascular disease. Clin Chem. 46:385–391. 2000.PubMed/NCBI
|
|
12
|
Shukla N, Angelini GD and Jeremy JY:
Interactive effects of homocysteine and copper on angiogenesis in
porcine isolated saphenous vein. Ann Thorac Surg. 84:43–49. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Garnier A, Fortin D, Deloménie C, Momken
I, Veksler V and Ventura-Clapier R: Depressed mitochondrial
transcription factors and oxidative capacity in rat failing cardiac
and skeletal muscles. J Physiol. 551:491–501. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cawthon D, McNew R, Beers KW and Bottje
WG: Evidence of mitochondrial dysfunction in broilers with
pulmonary hypertension syndrome (Ascites): Effect of t-butyl
hydroperoxide on hepatic mitochondrial function, glutathione and
related thiols. Poult Sci. 78:114–124. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cawthon D, Beers K and Bottje WG: Electron
transport chain defect and inefficient respiration may underlie
pulmonary hypertension syndrome (ascites)-associated mitochondrial
dysfunction in broilers. Poult Sci. 80:474–484. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Suter M, Remé C, Grimm C, Wenzel A,
Jäättela M, Esser P, Kociok N, Leist M and Richter C: Age-related
macular degeneration. The lipofusion component
N-retinyl-N-retinylidene ethanolamine detaches proapoptotic
proteins from mitochondria and induces apoptosis in mammalian
retinal pigment epithelial cells. J Biol Chem. 275:39625–39630.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Srinivasan S and Avadhani NG: Cytochrome c
oxidase dysfunction in oxidative stress. Free Radic Biol Med.
53:1252–1263. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Leadsham JE, Sanders G, Giannaki S, Bastow
EL, Hutton R, Naeimi WR, Breitenbach M and Gourlay CW: Loss of
cytochrome c oxidase promotes RAS-dependent ROS production from the
ER resident NADPH oxidase, Yno1p, in yeast. Cell Metab. 18:279–286.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zou H, Li Y, Liu X and Wang X: An APAF-1.
cytochrome c multimeric complex is a functional apoptosome that
activates procaspase-9. J Biol Chem. 274:11549–11556. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu Y, Benedict MA, Ding L and Núñez G:
Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated
caspase-9 activation and apoptosis. EMBO J. 18:3586–3595. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Saleh A, Srinivasula SM, Acharya S, Fishel
R and Alnemri ES: Cytochrome c and dATP-mediated oligomerization of
Apaf-1 is a prerequisite for procaspase-9 activation. J Biol Chem.
274:17941–17945. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hassanin IM, Shahin AY, Badawy MS and
Karam K: D-dimer testing versus multislice computed tomography in
the diagnosis of postpartum pulmonary embolism in symptomatic
high-risk women. Int J Gynaecol Obstet. 115:200–201. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Glerum DM, Shtanko A and Tzagoloff A:
Characterization of COX17, a yeast gene involved in copper
metabolism and assembly of cytochrome oxidase. J Biol Chem.
271:14504–14509. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Takahashi Y, Kako K, Kashiwabara S,
Takehara A, Inada Y, Arai H, Nakada K, Kodama H, Hayashi J, Baba T
and Munekata E: Mammalian copper chaperone Cox17p has an essential
role in activation of cytochrome C oxidase and embryonic
development. Mol Cell Biol. 22:7614–7621. 2002. View Article : Google Scholar : PubMed/NCBI
|