Introduction
Chronic myeloid leukemia (CML) is a type of white
blood cell cancer characterized by increased proliferation of
granulocytes, which accounts for 20% of all leukemias in adults
(1). Patients with CML usually show
symptoms such as enlarged spleen and liver (2). CML is the first cancer that was found
to be associated with a genetic abnormality, the Philadelphia
chromosome, which results from chromosome translocation (3,4). The
causes of CML are difficult to identify, although some
environmental stimuli, for example, exposure to ionizing radiation,
appear to be risk factors (5).
Imatinib and other tyrosine kinase inhibitors have been applied
clinically in CML treatment strategies (6). Second generation inhibitors for
imatinib-resistant patients (7,8) and stem
cell transplantation treatments (9)
are also being investigated.
The two vital factors involved in the Philadelphia
chromosome are the proto-oncogene ABL and the breakpoint
cluster region BCR. The two genes participate in the
activation of several signaling pathways resulting in the altered
adhesion, mitogenic activation and inhibition of apoptosis of CML
cells (10). Signal transducers and
activators of transcription (STAT) are important factors among
these pathways, which are activated in blood cells from acute
myeloid leukemia and CML (11).
Among the STAT family members, STAT3 has been demonstrated to
function in CML via the Janus activated kinase (JAK)/STAT pathway
(12) and helps to mediate the
anticancer activity of certain phytochemicals in the combination
therapy of CML (13). Furthermore,
its contribution to the regulation of the migration and viability
of CML cells is indicated by molecular target studies (14,15).
However, the detailed composition of the STAT3 pathway remains
unclear, particularly for the downstream factors of STAT3.
A previous study demonstrated the interactions
between STAT3 and superior cervical ganglia protein 10-like protein
(SCLIP, or SCG10-like protein) in the breast cancer cell line MCF-7
(16). Therefore, the present study
focused on the roles of SCLIP in the CML cell line K562, with the
aim of identifying the downstream factors of STAT3 to reveal the
roles of this pathway in the regulation of CML. First, the
relationship between STAT3 and SCLIP was analyzed. Then the
knockdown of SCLIP was conducted in the CML cell line K562 using
specific small interfering RNA (siRNA) and cell viability and
apoptosis were compared between transfected and untreated cells.
Also, the expression changes of two possible downstream factors of
SCLIP were examined. The results should provide a more
comprehensive understanding of the CML-related STAT3-SCLIP
pathway.
Materials and methods
Cell culture
The leukemia model cell line K562 (Sigma-Aldrich;
Merck Millipore, Darmstadt, Germany) was cultured in Roswell Park
Memorial Institute-1640 medium (Sigma-Aldrich) supplemented with 2
mM glutamine (Sigma-Aldrich), 10% fetal bovine serum (HyClone FBS;
GE Healthcare Life Sciences, Logan, UT, USA), 100 U/ml penicillin
and 100 mg/ml streptomycin (Beyotime Institute of Biotechnology,
Shanghai, China). The cells were maintained in a humidified
atmosphere with 5% CO2 at 37°C. The medium was changed
weekly and cells were plated at a density of 1×105
cells/ml. To explore the correlation between phosphorylated STAT3
(p-STAT3) and SCLIP, cells (1×106) were seeded onto
6-well plates. With different treatment, cells were divided into
three groups: Cells without treatment, cells treated with JSI-124
(1 µM; La Jolla, CA, USA) for 24 h and cells treated with
recombinant interleukin-21 (10 ng/ml; Sigma-Aldrich) for 24 h.
Cell transfection
Human SCLIP-specific double-strand siRNA
(SCLIP-siRNA) and the negative control siRNA were purchased from
Ambion (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Cell
transfection was performed using Lipofectamine® RNAiMAX
Reagent (Ambion) according to the manufacturer's protocol. Prior to
transfection, the K562 cells were cultured and seeded to become 60%
confluent in 96-well plates. The SCLIP-siRNA (1 pmol for the
SCLIP-siRNA group) or the negative control siRNA (1 pmol for the
siRNA control group) was diluted and added to the corresponding
wells. The cells were then incubated for 1 day at room temperature.
The transfection efficiency was determined using a
fluorescein-labeled siRNA. Untransfected cells served as the
control group.
Cell proliferation and viability
assays
Cell proliferation and viability assays were
conducted with Cell Proliferation kit I (Roche Life Science,
Branford, CT, USA) according to the manufacturer's protocol.
Specifically, cells were added to 96-well plates at a concentration
of 2×104 cells/ml and cultured for 3 days.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT; 5 mg/ml)
diluted in phosphate-buffered saline (PBS) was added to each well
and incubated with the cells for 4 h. The cells were then
centrifuged at 80 × g for 10 min at 4°C, collected and resuspended
in dimethylsulfoxide. The plates were shaken until the crystals
dissolved. The absorbance was determined at 490 nm using a
spectrophotometer at 0 h, 72 h and 7 days after transfection. All
assays were performed in triplicate.
Cell apoptosis assays
Cell apoptosis assays were conducted by flow
cytometry (FCM) using Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI). An Annexin V-FITC/PI Apoptosis
Detection kit (Beijing Solarbio Science & Technology Co., Ltd.,
Beijing, China) was used and the experiment was performed according
to the manufacturer's protocol. Specifically, the cell
concentration was adjusted to 5×105 cells/ml, and the
cells were centrifuged at 200 × g for 5 min at room temperature and
collected. After washing with cold PBS 3 times, the cells were
resuspended in 200 µl 1X Binding Buffer, 10 µl Annexin V-FITC and
10 µl PI and then incubated in the dark for 15 min at room
temperature. Prior to detection by FCM, another 300 µl 1X Binding
Buffer was added. Detection was performed at 0 h, 72 h and 7 days
after transfection. The Annexin V-positive and PI negative cells
located in quadrant 3 (Q3) of the plot were considered to be
apoptotic cells and were used for comparison among groups.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The expression levels of various mRNAs were detected
by RT-qPCR using the following primers: STAT3, forward:
5′-GCCAGAGAGCCAGGAGCA-3′ and reverse:
5′-TGAAGCTGACCCAGGTAGCGCTGC-3′; SCLIP, forward:
5′-AGGAGTTATCTGTGCTGTCGC-3′ and reverse:
5′-TGGTAGATGGTGTTCGGGTG-3′; Bcl-2, forward:
5′-AAGAGCAGACGGATGGAAAAAGG-3′ and reverse:
5′-GGGCAAAGAAATGCAAGTGAATG-3′; and cyclin E1, forward:
5′-GCAAGCCTCGGATTATTGCA-3′ and reverse:
5′-CCTCTCTATTTGCCCAGCTCAGTA-3′. GAPDH was used as the
internal control, with the following primer sequences: Forward:
5′-GAGTCAACGGATTTGGTCGT-3′ and reverse: 5′-GACAAGCTTCCCGTTCTCAG-3′
The total RNA of cells was isolated with TRIzol (Invitrogen; Thermo
Fisher Scientific, Inc.) as well as DNase I (Promega Corporation,
Madison, WI, USA) and complementary DNA synthesis was performed
using a PrimerScript RT Reagent kit (Takara Biotechnology Co.,
Ltd., Dalian, China) according to the manufacturer's protocol. qPCR
was conducted in a 20-µl reaction system using a LightCycler 96
Real-Time PCR System (Roche Life Science, Basel, Switzerland) with
the following cycling procedures: Pre-denaturation at 94°C for 2
min, followed by 40 cycles comprising denaturation at 94°C for 1
min, annealing at 60°C for 1 min and extension at 72°C for 1 min,
and then a final extension step at 72°C for 7 min. All the
experiments were performed in triplicate. The data were calculated
using the 2−ΔΔCq method (17).
Western blot analysis
The cells were harvested and washed twice with cold
PBS. Protein samples were extracted with cell lysis buffer
containing 2% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium
dodecyl sulfate (SDS), 100 µg/ml phenylmethylsulfonyl fluoride and
10 µg/ml leupeptin. Equal amounts of protein (20 µg) from each
group were separated by 12% SDS-polyacrylamide gel electrophoresis
and transferred to a polyvinylidene fluoride membrane. The blot was
blocked with 5% skimmed milk in Tris-buffered saline Tween-20
(TBST) buffer (pH 8.0) overnight at 4°C and then incubated with
specific primary antibodies for phosphorylated (p)-STAT3 (1:500;
cat. no. sc-135649), SCLIP (1:500; cat. no. sc-85907) or β-actin
(1:1,000; cat. no. sc-130656) (Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA) for 2 h at room temperature. The membrane was then
washed with TBST and incubated with horseradish
peroxidase-conjugated goat anti-rabit IgG (1:5,000; cat. no.
sc-2004) and donkey anti-goat IgG (1:5,000; cat. no. sc-2020)
secondary antibodies (Santa Cruz Biotechnology, Inc.) for 2 h at
room temperature. Positive signals were detected using an enhanced
chemiluminescence reagent (Amersham ECL Detection kit; GE
Healthcare Life Sciences) and then analyzed using a Kodak Digital
Imaging System (DC120; Kodak, Rochester, NY, USA). β-actin was used
as the internal reference.
Statistical analysis
Differences between groups were examined using the
unpaired Student's t-test. Data are presented as the mean ±
standard deviation of three independent experiements. P<0.05 was
considered to indicate a statistically significant difference. All
data analyses were performed using Statistical Package for Social
Sciences (SPSS) version 19 software (IBM SPSS, Armonk, NY,
USA).
Results
SCLIP is a downstream molecule of
STAT3
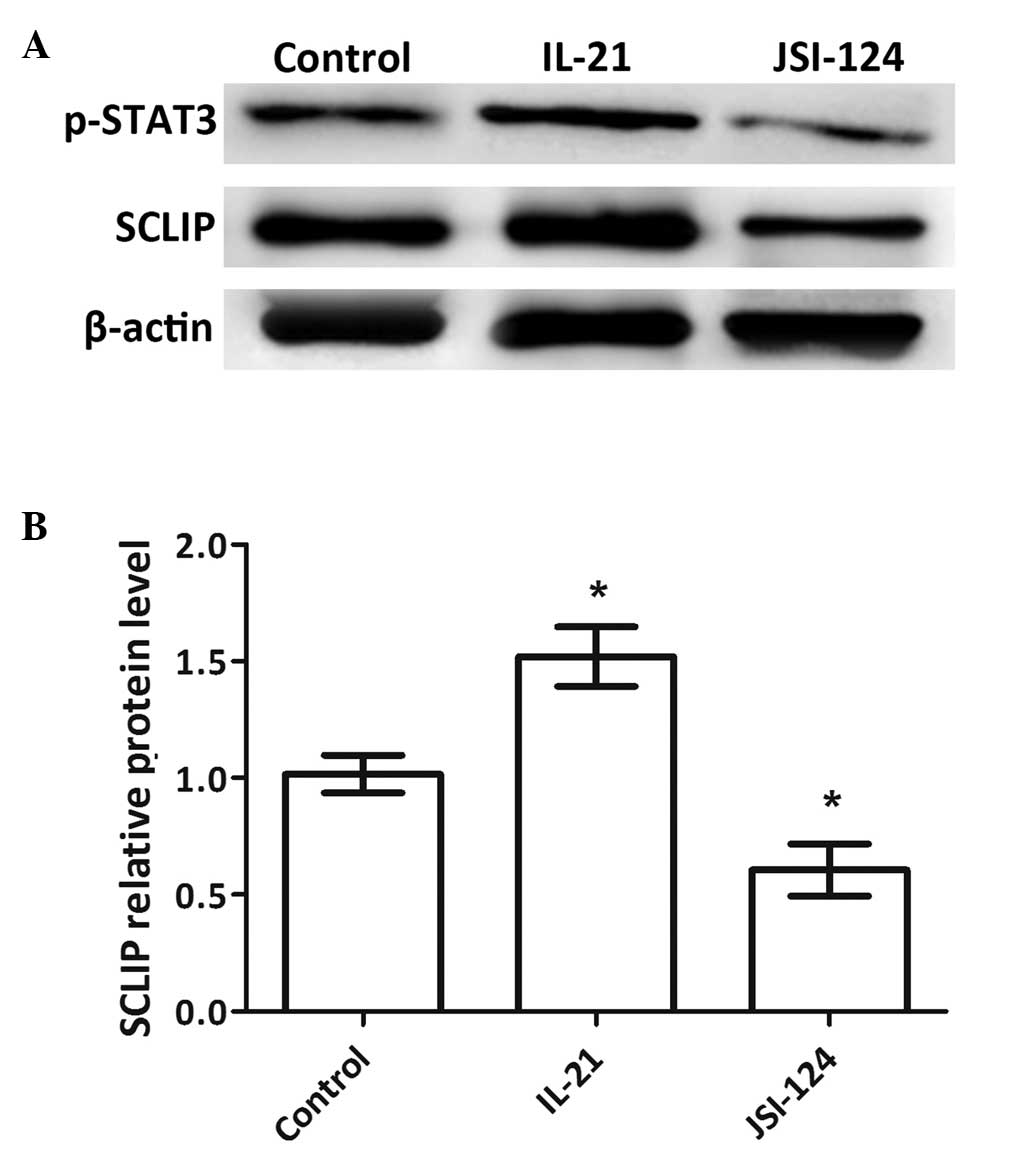
The phosphorylation levels of STAT3 and the protein
expression levels of SCLIP were detected by western blotting. The
results showed a positive correlation between the expression of
phosphorylated STAT3 (p-STAT3) and SCLIP (Fig. 1A and B): When the phosphorylation of
STAT3 was promoted by IL-21, the expression of SCLIP was
upregulated and when the phosphorylation of STAT3 was inhibited by
JSI-124, the expression of SCLIP was downregulated. The differences
in the expression levels of SCLIP in the two treatment groups
compared with those in the control group were significant
(P<0.05). Since the expression of SCLIP could be activated by
p-STAT3, these results imply that SCLIP might be a downstream
molecule of STAT3.
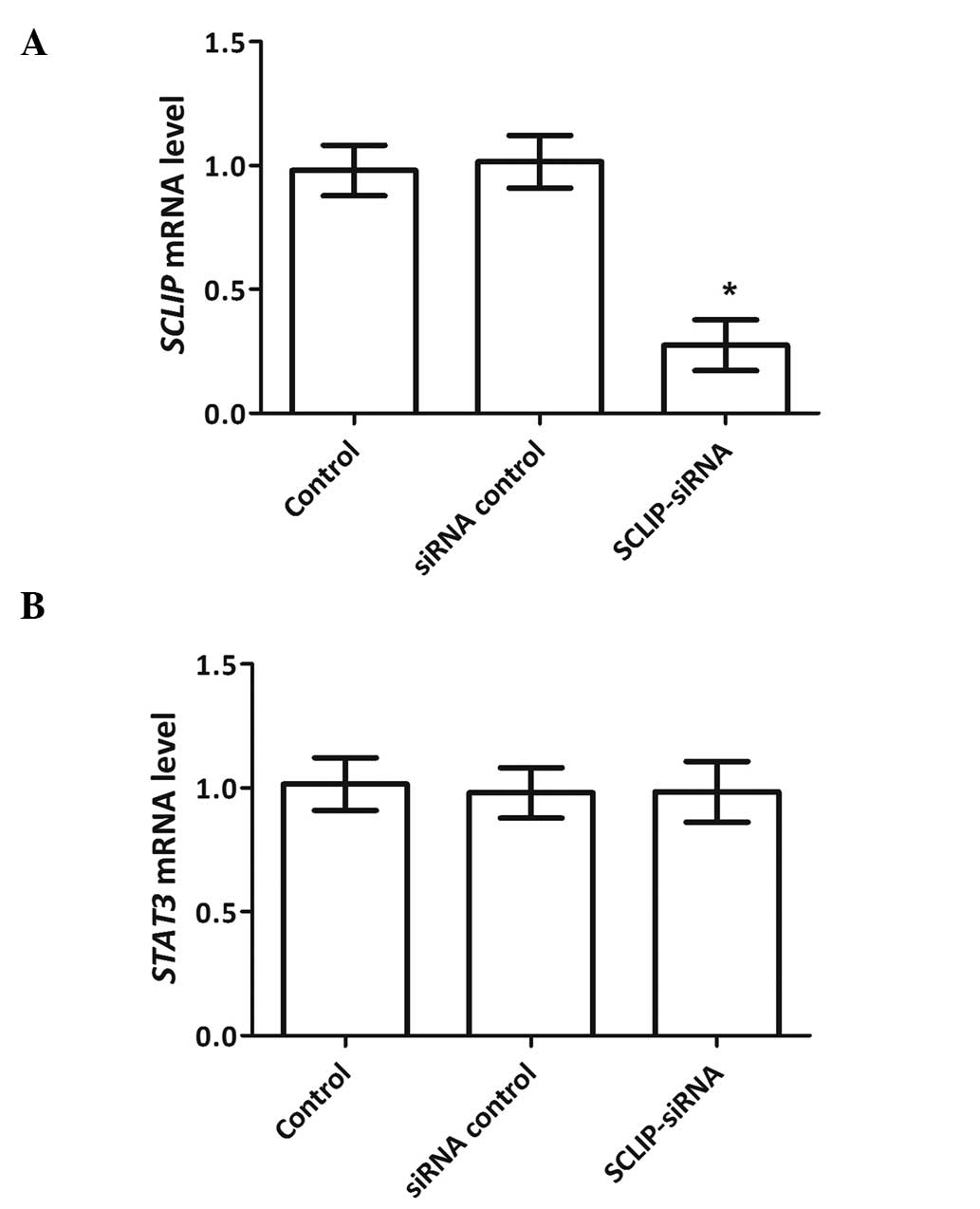
Following the transfection of the cells with the
SCLIP-specific siRNA or negative control siRNA, the mRNA expression
levels of SCLIP and STAT3 in each group were detected
by RT-qPCR. The expression levels of SCLIP and STAT3
mRNA in the siRNA control group were similar to those in the
untransfected control group (Fig.
2). In the SCLIP-siRNA group, the expression of SCLIP
mRNA was significantly downregulated (P<0.05) compared with that
in the control group indicating that effective knockdown of
SCLIP. However, the expression of STAT3 mRNA was
hardly changed as SCLIP was inhibited, which implies that
STAT3 is located upstream of SCLIP. These results together with the
previous western blotting results, indicate that SCLIP may be
upregulated by the phosphorylation of its upstream molecule,
STAT3.
Knockdown of SCLIP inhibits cell
viability and promotes apoptosis
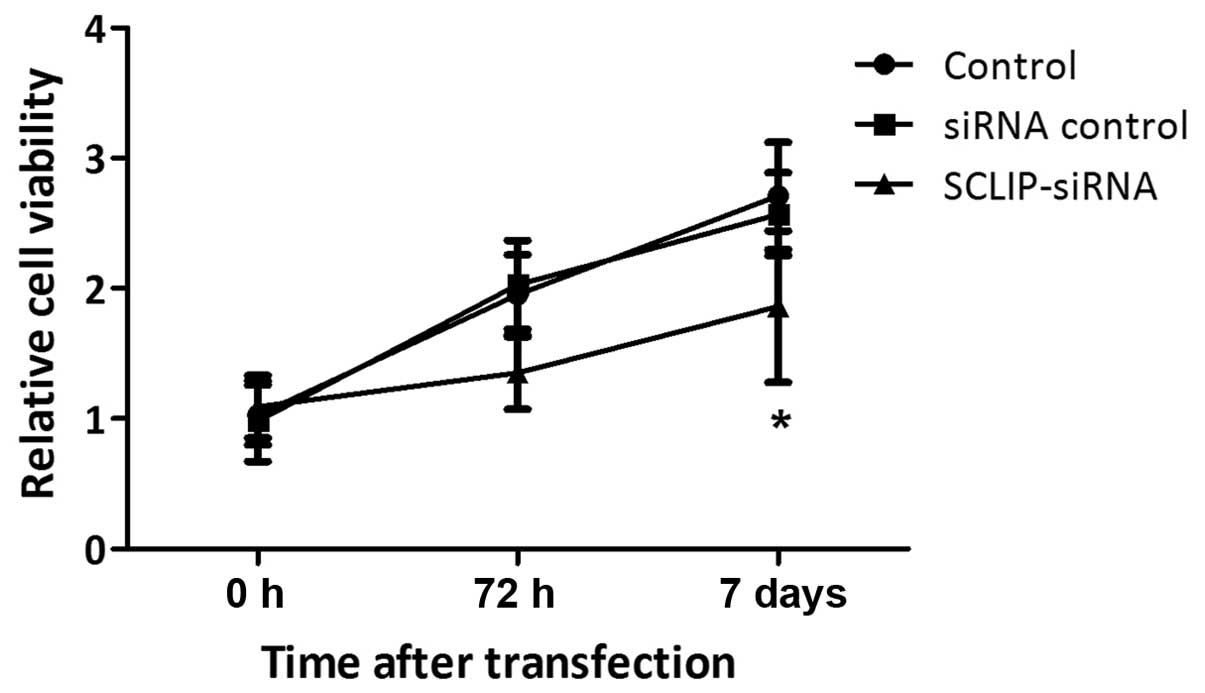
The effects of SCLIP on K562 cell viability and
apoptosis were tested by MTT and cell apoptosis assays,
respectively. The cell viability of the SCLIP-siRNA group was
inhibited and exhibited a significant difference from the control
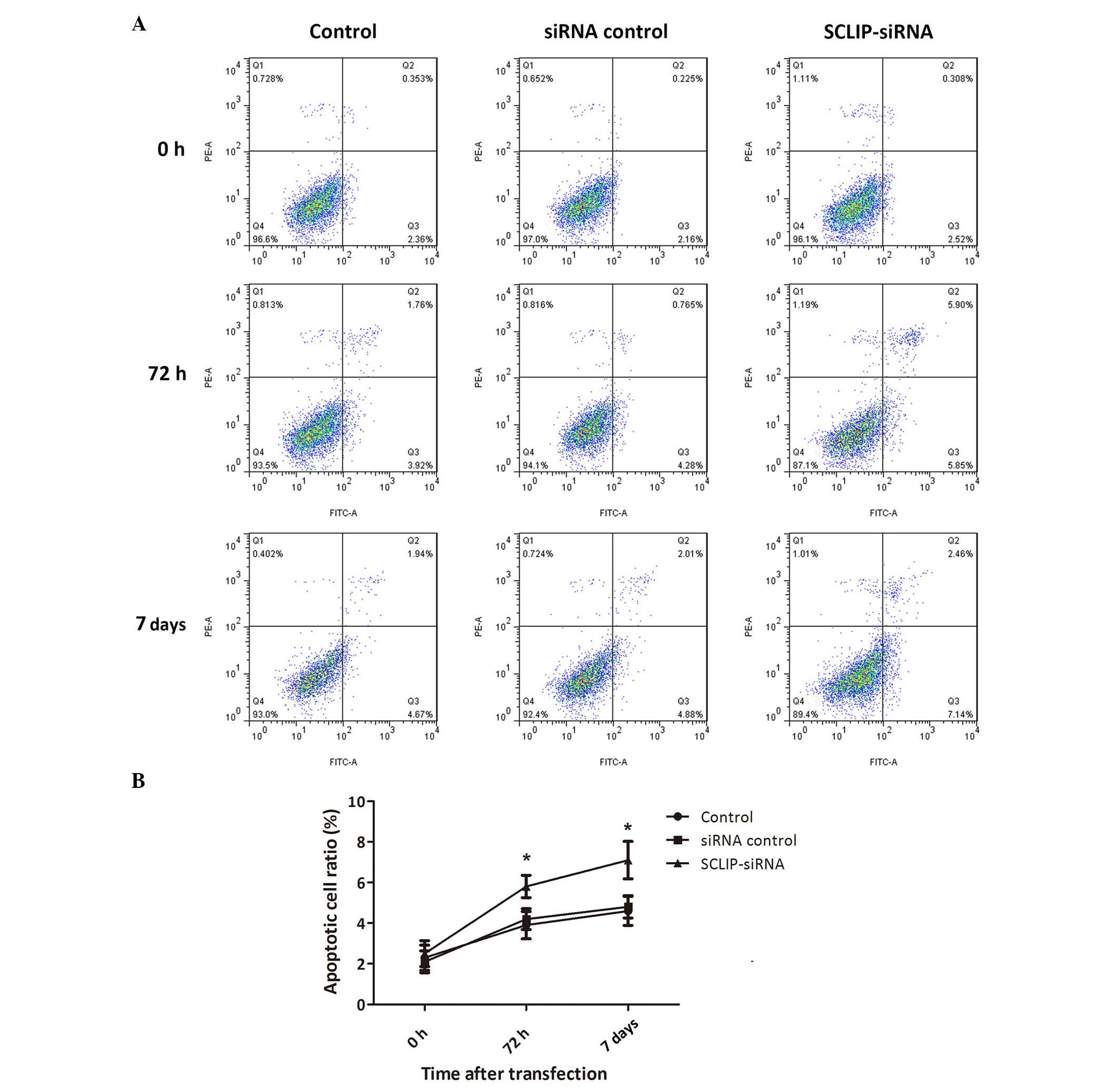
group at 7 days after transfection (P<0.05; Fig. 3). Similarly, the apoptotic cell ratio
in the SCLIP-siRNA group was significantly increased at both 72 h
and 7 days after transfection compared with that in the control
group (P<0.05), increasing from 4.28 to 5.85% and from 4.88 to
7.14%, respectively (Fig. 4). These
data indicate that SCLIP was involved in the promotion of cell
viability and inhibition of cell apoptosis in the K562 cells,
implicating SCLIP as a regulator of the viability and apoptosis of
CML cells.
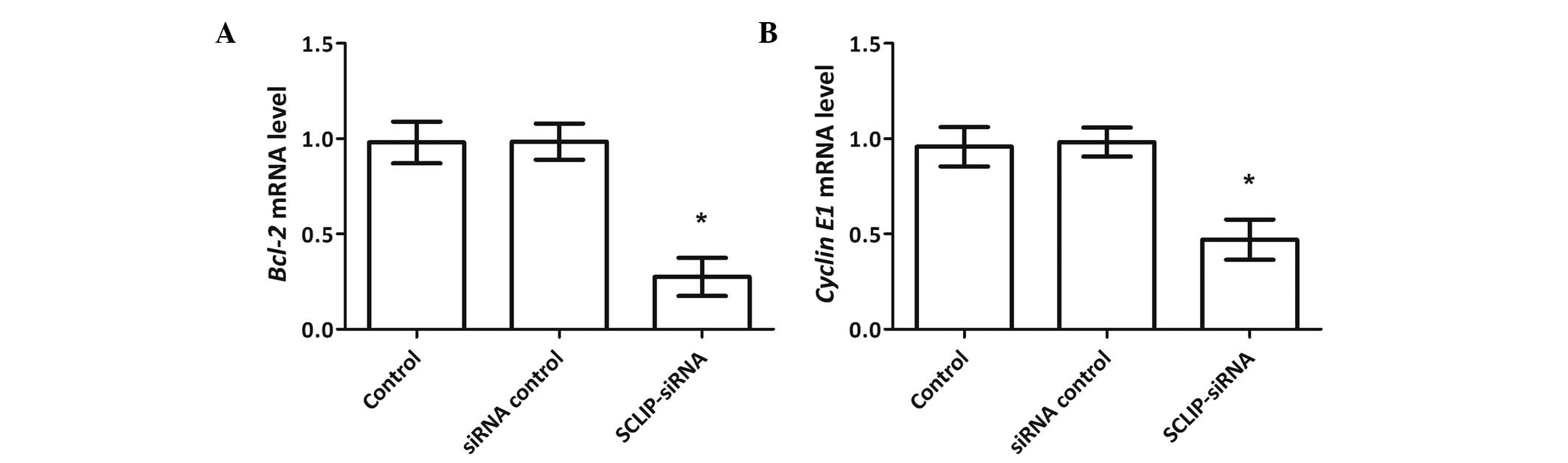
Knockdown of SCLIP regulates the
expression of Bcl-2 and cyclin E1
The possible regulatory mechanisms of SCLIP in CML
were studied by analyzing the changes in the expression of two
anti-apoptosis genes, namely, Bcl-2 and cyclin E1,
following the knockdown of SCLIP. The RT-qPCR results showed that
the expression patterns of the two genes were positively associated
with the expression of SCLIP (Fig.
5). That is, with the knockdown of SCLIP, both Bcl-2 and
cyclin E1 mRNA expression levels were downregulated,
exhibiting significant differences compared with the control group
(P<0.05). Therefore, Bcl-2 and cyclin E1 are two possible
downstream factors of SCLIP. It may be hypothesized that the two
factors, together with STAT3 and SCLIP, constitute a part of the
signaling pathway that regulates the viability and apoptosis of CML
cells.
Discussion
In this study, SCLIP is demonstrated to be a
downstream factor of STAT3, since its expression is activated by
the phosphorylation of STAT3, while the knockdown of SCLIP has no
effect on the expression of STAT3 mRNA. In addition, SCLIP
knockdown is demonstrated to inhibit the viability and promote the
apoptosis of K562 cells. Two anti-apoptosis factors, Bcl-2 and
cyclin E1, were observed to be downregulated when SCLIP was knocked
down, indicating they may be the downstream factors of SCLIP.
Therefore, the data in the present study suggest that the
STAT3-SCLIP pathway consisting of these four factors (SCLIP, STAT3,
Bcl-2 and cyclin E1) is involved in regulating the viability and
apoptosis of CML cells.
This study presents a relatively detailed
delineation of the STAT3-SCLIP pathway involving STAT3, SCLIP,
Bcl-2 and cyclin E1. Since IL-21 can bind to its receptor and
phosphorylate STAT3 to initiate the transcription of regulated
genes (18), while cucurbitacin I
(JSI-124) is able to reduce the phosphorylation level of STAT3
(19), these two factors were used
in the present study to regulate the phosphorylation of STAT3. The
results showed that the expression of SCLIP was regulated by
p-STAT3 levels and the knockdown of SCLIP did not affect the
expression of STAT3. Together with previous studies showing that
STAT3 regulates and even interacts directly with SCLIP (16,20), it
could be inferred that SCLIP is a direct downstream factor of STAT3
in K562 cells. The presence of a direct regulatory association
between SCLIP and Bcl-2 or cyclin E1 is unclear because there was
no explicit evidence ruling out other possible factors between the
two cascades, although microarray analyses have found that STAT3 is
able to regulate the expression of Bcl-2 (21,22).
Therefore, it could be deduced from the existing results that Bcl-2
and cyclin E1 are located downstream of SCLIP. Considering the
aforementioned data together, the basic part of this pathway is
hypothesized to involve direct regulation by STAT3 of its
downstream SCLIP, and two factors further downstream, Bcl-2 and
cyclin E1, that are regulated by SCLIP.
SCLIP was demonstrated to promote the viability and
inhibit the apoptosis of K562 cells in the present study, implying
its role in the regulation of CML. Similar functions of SCLIP have
been demonstrated in other cell lines and diseases. For example,
SCLIP has been shown to facilitate the proliferation, invasion and
migration of non-small cell lung cancer cells (23). It also promotes neurite growth by
pheochromacytoma PC12 cells (24).
Similarly, Bcl-2 and cyclin E1 have been demonstrated to be
anti-apoptosis factors; Bcl-2 knockout or the overexpression of
factors targeting Bcl-2 causes the acceleration of cell apoptosis
(25,26). The abnormal expression of cyclin E1
contributes to tumorigenesis (27)
and its knockdown induces apoptosis in cancer cells (28). In the present study, these two
anti-apoptosis factors were downregulated along with the knockdown
of SCLIP, indicating their consistent roles in the regulation of
the STAT3-SCLIP pathway. Therefore, generally, the activation of
the STAT3-SCLIP pathway, including the upregulation of STAT3,
SCLIP, Bcl-2 or cyclin E1, might result in the promotion of cell
viability and inhibition of cell apoptosis. These key factors in
this pathway could serve as promising molecular targets of CML
treatment.
Since CML is regulated by multiple signaling
pathways (10), there is a great
possibility that the signaling pathway that is partly defined in
the present study is connected to other important factors, thus
constituting a complex regulatory network. As already mentioned,
STAT3 is a member of the JAK/STAT pathway that regulates CML
(12). It also participates in the
phosphatidylinositol 3-kinase/mammalian target of rapamycin/STAT3
pathway to regulate cell viability in breast cancer stem-like cells
(29). Bcl-2 is involved in
pathways, such as the extracellular signaling regulated kinase
pathway and the nuclear factor κB/Bcl-2 pathway, that regulate
cancer cells (30,31). Furthermore, the cyclin E1-Cdk2
complex is the endpoint of several pathways in the growth control
of cancer cells (32). It may be
deduced that CML is regulated by a complex pathway network
involving STAT3, SCLIP, Bcl-2 and cyclin E1. Therefore, a
comprehensive understanding of the regulatory mechanisms is of
great significance.
In summary, this study demonstrates that SCLIP, as a
direct downstream factor of STAT3, is able to regulate the
viability and apoptosis of CML cells. The STAT3-SCLIP pathway, a
basic pathway model for CML regulation is described, including the
direct interaction between STAT3 and SCLIP, and factors further
downstream, Bcl-2 and cyclin E1. These data facilitate future
molecular studies of CML regulation and suggest alternative
molecular targets for CML treatment. A more profound understanding
of the associated signaling pathway network is necessary for
further clinical research.
References
|
1
|
Stein B and Smith BD: Treatment options
for patients with chronic myeloid leukemia who are resistant to or
unable to tolerate imatinib. Clinical Therapeutics. 32:804–820.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chaudhary V, Sachdeva P, Karanth P and
Arora R: Spontaneous hemoperitoneum in chronic myeloid leukemia: An
unusual etiology. J Hematol (Brossard). 2:40–41. 2013.
|
|
3
|
Faderl S, Talpaz M, Estrov Z and
Kantarjian HM: Chronic myelogenous leukemia: Biology and therapy.
Ann Intern Med. 131:207–219. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hehlmann R, Hochhaus A and Baccarani M:
European LeukemiaNet: Chronic myeloid leukaemia. Lancet.
370:342–350. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kabel AM and Elmaaboud MAA: Cancer: Role
of nutrition, pathogenesis, diagnosis and management. World Journal
of Nutrition and Health. 2:48–51. 2014.
|
|
6
|
Shah NP, Guilhot F, Cortes JE, Schiffer
CA, le Coutre P, Brümmendorf TH, Kantarjian HM, Hochhaus A,
Rousselot P, Mohamed H, et al: Long-term outcome with dasatinib
after imatinib failure in chronic-phase chronic myeloid leukemia:
Follow-up of a phase 3 study. Blood. 123:2317–2324. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bixby D and Talpaz M: Seeking the causes
and solutions to imatinib-resistance in chronic myeloid leukemia.
Leukemia. 25:7–22. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weisberg E, Manley PW, Cowan-Jacob SW,
Hochhaus A and Griffin JD: Second generation inhibitors of BCR-ABL
for the treatment of imatinib-resistant chronic myeloid leukaemia.
Nat Rev Cancer. 7:345–356. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pavlů J and Apperley JF: Allogeneic stem
cell transplantation for chronic myeloid leukemia. Curr Hematol
Malig Rep. 8:43–51. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Deininger MW, Goldman JM and Melo JV: The
molecular biology of chronic myeloid leukemia. Blood. 96:3343–3356.
2000.PubMed/NCBI
|
|
11
|
Chai SK, Nichols GL and Rothman P:
Constitutive activation of JAKs and STATs in BCR-Abl-expressing
cell lines and peripheral blood cells derived from leukemic
patients. J Immunol. 159:4720–4728. 1997.PubMed/NCBI
|
|
12
|
Zhu JF, Ll ZI, Zhang GS, Meng K, Kuang WY,
Li J, Zhou XF, Ll RJ, Peng HI, Dai CW, et al: Icaritin shows potent
anti-leukemia activity on chronic myeloid leukemia in vitro and in
vivo by regulating MAPK/ERK/JNK and JAK2/STAT3/AKT signalings. PLoS
One. 6:e237202011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jung JH, Kwon TR, Jeong SJ, Kim EO, Sohn
EJ, Yun M and Kim SH: Apoptosis induced by tanshinone IIA and
cryptotanshinone is mediated by distinct JAK/STAT3/5 and SHP1/2
signaling in chronic myeloid leukemia K562 cells. Evid Based
Complement Alternat Med. 2013:8056392013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kiper HD, Kaymaz B Tezcanli, Gokbulut AA,
Selvi N, Avci CB, Kosova B, Iskender G, Yandim MK, Gunduz C, Sahin
F, et al: STAT pathway in the regulation of zoledronic acid-induced
apoptosis in chronic myeloid leukemia cells. Biomed Pharmacother.
67:527–532. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kaymaz BT, Cetintaş VB, Aktan C and Kosova
B: MicroRNA-520a-5p displays a therapeutic effect upon chronic
myelogenous leukemia cells by targeting STAT3 and enhances the
anticarcinogenic role of capsaicin. Tumour Biol. 35:8733–8742.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ng DC, Lim CP, Lin BH, Zhang T and Cao X:
SCG10-like protein (SCLIP) is a STAT3-interacting protein involved
in maintaining epithelial morphology in MCF-7 breast cancer cells.
Biochem J. 425:95–105. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhu M, Pleasic-Williams S, Lin TH,
Wunderlich DA, Cheng JB and Masferrer JL: pSTAT3: A target
biomarker to study the pharmacology of the anti-IL-21R antibody
ATR-107 in human whole blood. J Transl Med. 11:652013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Qi J, Xia G, Huang CR, Wang JX and Zhang
J: JSI-124 (Cucurbitacin I) inhibits tumor angiogenesis of human
breast cancer through reduction of STAT3 phosphorylation. Am J Chin
Med. 43:337–347. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang Y, Ni S, Huang B, Wang L, Zhang X
and Li X, Wang H, Liu S, Hao A and Li X: Overexpression of SCLIP
promotes growth and motility in glioblastoma cells. Cancer Biol
Ther. 16:97–105. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gritsko T, Williams A, Turkson J, Kaneko
S, Bowman T, Huang M, Nam S, Eweis I, Diaz N, Sullivan D, et al:
Persistent activation of stat3 signaling induces survivin gene
expression and confers resistance to apoptosis in human breast
cancer cells. Clin Cancer Res. 12:11–19. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang D, He D, Xue Y, Wang R, Wu K, Xie H,
Zeng J, Wang X, Zhau HE, Chung LW, et al: PrLZ protects prostate
cancer cells from apoptosis induced by androgen deprivation via the
activation of Stat3/Bcl-2 pathway. Cancer Res. 71:2193–2202. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nair S, Bora-Singhal N, Perumal D and
Chellappan S: Nicotine-mediated invasion and migration of non-small
cell lung carcinoma cells by modulating STMN3 and GSPT1 genes in an
ID1-dependent manner. Mol Cancer. 13:1732014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kang SW, Shin YJ, Shim YJ, Jeong SY, Park
IS and Min BH: Clusterin interacts with SCLIP (SCG10-like protein)
and promotes neurite outgrowth of PC12 cells. Exp Cell Res.
309:305–315. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
van Delft MF, Wei AH, Mason KD, Vandenberg
CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, et
al: The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and
efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized.
Cancer Cell. 10:389–399. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Y, Li M, Zang W, Ma Y, Wang N, Li P,
Wang T and Zhao G: MiR-429 up-regulation induces apoptosis and
suppresses invasion by targeting Bcl-2 and SP-1 in esophageal
carcinoma. Cell Oncol (Dordr). 36:385–394. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Spruck CH, Won KA and Reed SI: Deregulated
cyclin E induces chromosome instability. Nature. 401:297–300. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gurzov EN and Izquierdo M: Cyclin E1
knockdown induces apoptosis in cancer cells. Neurol Res.
28:493–499. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang
Y, Deng J, Margolick JB, Liotta LA, Petricoin E III and Zhang Y:
Activation of the PTEN/mTOR/STAT3 pathway in breast cancer
stem-like cells is required for viability and maintenance. Proc
Natl Acad Sci USA. 104:16158–16163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Galante JM, Mortenson MM, Bowles TL,
Virudachalam S and Bold RJ: ERK/BCL-2 pathway in the resistance of
pancreatic cancer to anoikis. J Surg Res. 152:18–25. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Buchholz TA, Garg AK, Chakravarti N,
Aggarwal BB, Esteva FJ, Kuerer HM, Singletary SE, Hortobagyi GN,
Pusztai L, Cristofanilli M and Sahin AA: The nuclear transcription
factor kappaB/bcl-2 pathway correlates with pathologic complete
response to doxorubicin-based neoadjuvant chemotherapy in human
breast cancer. Clin Cancer Res. 11:8398–8402. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Möröy T and Geisen C: Cyclin E. Int J
Biochem Cell Biol. 36:1424–1439. 2004. View Article : Google Scholar : PubMed/NCBI
|