Introduction
Asthma is characterized by chronic airway
inflammation, airway hyper-responsiveness and reversible airflow
restriction. It is generally considered to be a T helper type 2
(Th2) driven inflammatory disorder (1). However, in addition to the adaptive
immune system, the innate immune system is now considered to serve
a key role in the pathogenesis of asthma (2).
Macrophages are innate immune cells with key roles
in immune defense mechanisms, including tissue homeostasis, primary
responses to pathogens, immune resolution, coordination of adaptive
immune responses, inflammation and tissue repair (3). Depending on the microenvironment,
macrophages can polarize into distinct subsets and the
heterogeneity of circulating macrophage precursors (monocytes) may
predefine the polarization fate that they assume upon entering a
tissue (4,5). Polarized macrophages are broadly
classified into two main groups: Classically activated macrophages
(M1) and alternatively activated macrophages (M2) (6,7). Induced
by interferon (IFN)-γ and tumor necrosis factor (TNF)-α, M1
macrophages are potent cells that typically attack microorganisms
and tumor cells, and are primarily associated with pathological
type 1 inflammation, express inducible nitric oxide synthase (iNOS)
and the majority of Toll-like receptors (TLRs), and also secrete
interleukin (IL)-12, TNF-α, IL-1β, IL-6, chemokine ligand (CCL)-5
and IFN-γ-induced protein 10 (8,9). By
contrast, M2 macrophages are induced by IL-4 and IL-13, and serve
roles in Th1 inflammatory responses and adaptive immunity,
enhancement of Th2 inflammatory responses, angiogenesis and tissue
repair (6–9). M2 macrophages are characterized by high
expression of the mannose receptor [also known as cluster of
differentiation (CD)-206], major histocompatibility class II and a
number of other signature genes, including arginase 1 (Arg1),
transglutaminase 2 (TG2), chitinase-like molecules (Ym1/2 and acid
mammalian chitinase) and resistin-like molecule-α (3,10–12).
However, in the presence of immune complexes, macrophages polarize
into distinct regulatory macrophages (6,8). A key
feature of regulatory macrophages is high-level production of the
anti-inflammatory cytokine IL-10 (13), and regulatory macrophages have been
demonstrated to reduce inflammation and pathological changes in a
mouse model of lethal endotoxemia (14) and a mouse model of experimental
autoimmune encephalomyelitis (EAE) (15).
In the respiratory tract, macrophages are among the
most abundant cells and are grouped into two main populations
depending on their localization: Alveolar macrophages (AMs), which
line the surface of the alveoli, and interstitial macrophages
(IMs), which remain in the space between the alveolar and vascular
endothelia (16). While AMs are well
characterized (17), the phenotype
and in vivo functions of IMs are not fully understood.
Previous clinical and in vivo studies have indicated that M2
macrophages are associated with asthma severity. Based on analyses
of bronchial biopsies, it has been documented that individuals with
asthma exhibit higher percentages of macrophages expressing CD206
(CD206+) and TG2 compared with healthy individuals
(18,19). In addition, Kim et al
(20) demonstrated that individuals
with severe asthma had higher counts of IL-13-positive M2
macrophages in the bronchoalveolar lavage fluid (BALF) compared
with healthy individuals. Levels of chitinase molecules and the
percentage of CD206+ macrophages may also correlate with
asthma severity (19,21). Furthermore, a recent study in three
murine models of house-dust-mite-induced asthma has indicated that
the number of M2 macrophages positively correlates with the
severity of airway inflammation (22). By contrast, lower levels of IL-10
have been observed in lung macrophages isolated from asthmatic
patients compared to those from healthy individuals (23). It has also been observed that
macrophages from severe asthmatic patients produced higher levels
of IL-6 and IL-8, relative to individuals with moderate asthma. In
addition, IL-10 was undetectable in the patients with severe asthma
(24).
Under normal pathological conditions, lung IMs may
suppress airway hyper-responsiveness to harmless allergens,
principally by producing IL-10 and inhibiting the maturation and
migration of dendritic cells from the lymph nodes (25). Macrophages that produce IL-10 are
considered to be regulatory macrophages (15,25).
Therefore, the present study used an ovalbumin (OVA)-induced mouse
model of asthma to determine whether lung IMs undergo a phenotypic
switch from a regulatory IL-10-producing macrophage state under
normal conditions to an M2-polarized state under asthmatic
conditions.
Materials and methods
Mice
A total of 60 wild-type (WT) female BALB/c mice (6–8
weeks old), with a mean weight of 20 g, were obtained from the
Animal Biosafety Level 3 Laboratory of the Center for Animal
Experiments at Wuhan University (Wuhan, China). Mice were housed in
environmentally controlled and specific pathogen-free conditions
(22°C, 12:12 h light/dark cycle; 40–70% humidity) and were given
free access to tap water and pellet food. All animal care and
handling protocols were approved by the Animal Welfare Committee of
Wuhan University.
OVA sensitization and airway
challenge
WT BALB/c mice were sensitized intraperitoneally on
days 0 and 14 of an airway challenge experiment with 20 µg chicken
OVA (grade V; Sigma-Aldrich; Merck KgaA, Darmstadt, Germany)
emulsified in 2 mg aluminum hydroxide (Pierce; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) in 200 µl phosphate-buffered
saline (PBS). Intranasal OVA challenges (100 µg in 50 µl PBS) were
administered on days 25, 26 and 27. Age- and sex-matched control
mice (6 mice per group) from the same batch of wild-type (WT)
female BALB/c mice were simultaneously treated with PBS alone. Mice
were sacrificed 24 h after the final OVA challenge with an
intraperitoneal injection of sodium pentobarbitone (100 µg/kg;
Mintal; Mitsubishi Tanabe Pharma, Osaka, Japan) and their lungs
were harvested for analysis.
Cell isolation and culture
Isolated lungs were perfused with 20 ml PBS through
the right ventricle. Lungs were then cut into pieces and digested
with 1 mg/ml collagenase I (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) and 0.05 mg/ml DNase (Roche Diagnostics,
Basel, Switzerland) in Hanks' Balanced Salt Solution (Beyotime
Institute of Biotechnology, Haimen, China) at 37°C for 1 h. The
lung mononuclear cells (MNCs) were isolated by centrifugation at
800 × g for 20 min at room temperature in a Lymphoprep
gradient (density=1.081 mg/ml; BD Biosciences, Franklin Lakes, NJ,
USA). Erythrocytes were lysed by the addition of 10% volume of Red
Blood Cell Lysis Buffer (Sigma-Aldrich; Merck KgaA, Darmstadt,
Germany) and incubation for 8 min at 25°C followed by two washes
with flow cytometry buffer (00–4222-57; eBioscience Inc., San
Diego, CA, USA). Previous studies have demonstrated that lung IMs
express the macrophage marker F4/80, but do not express the
dendritic cell marker CD11c (25,26).
Thus, lung IMs were sorted as F4/80+CD11c- by flow cytometry. Cell
purity following sorting was ≥99%, determined by flow cytometry
analysis and cytospin analysis using a Rapi-diff Stain set
(Clinical Sciences Diagnostics Cc, Johannesburg, South Africa)
according to the manufacturer's protocol.
Isolated lung IMs from immunized and control mice
were cultured in duplicate at a density of 1×106
cells/ml in RPMI 1640 medium supplemented with 10% fetal calf
serum, 2 mM L-glutamine, 0.1 mM nonessential amino acids, 50 µM
mercaptoethanol, 50 U/ml penicillin and 50 mg/ml streptomycin (all
from Invitrogen; Thermo Fisher Scientific, Inc.) in the presence or
absence of OVA supplemented with lipopolysaccharide (LPS;
Sigma-Aldrich Merck KgaA; OVALPS; 100 µg/ml LPS-free OVA
and 10 ng/ml LPS). After 16 h incubation at 37°C, cell culture
supernatants were collected to measure the secretion levels of the
following cytokines and chemokines by ELISA: IL-10, IL-12, CCL17,
CCL22 and CCL24.
Flow cytometry analysis
A single cell suspension of lung cells obtained from
control and OVA-induced asthmatic mice was resuspended in
fluorescence-activated cell sorting (FACS) buffer (563503; BD
Biosciences, San Jose, CA, USA) at a density of 2×106 cells/ml at
room temperature, according to the manufacturer's protocol.
Subsequent staining reactions were performed at 4°C. To reduce
nonspecific binding, cells were first blocked with anti-CD16/CD32
antibody (553141; clone 2.4G2; BD Biosciences) in a staining buffer
(Hank's buffer containing 2% fetal calf serum and 0.1% sodium
azide) on ice for 15 min, then washed once with FACS buffer for 5
min and labeled with a saturating amount of isotype controls of
antibodies on ice for 45 min in the staining buffer. The antibodies
(all 1:100) were as follows: Fluorescein isothiocyanate
(FITC)-conjugated anti-F4/80 (11–4801-82; eBioscience Inc),
phycoerythrin (PE)-cyano 5 (cy5)-conjugated anti-CD11c (15-0114-82;
eBioscience, Inc.), PE-conjugated anti-CD206 (141705; BioLegend,
Inc., San Diego, CA, USA), PE-conjugated anti-IL-10 (12-7101-41;
eBioscience Inc.), or PE-conjugated anti-iNOS (12-5920-82;
eBioscience Inc.). After washing once with FACS buffer on ice for 5
min, to characterize the co-expression of iNOS or CD206 with
F4/80+CD11c-, FITC-conjugated anti-F4/80 and PE-cy5-conjugated
anti-CD11c labeled cells were fixed in a fixation buffer (2%
paraformaldehyde in PBS) at room temperature for 10 min, then
incubated with PE-conjugated CD206 (141705; 1:100; BioLegend,
Inc.), PE-conjugated anti-IL-10 (12-7101-41; 1:100; eBioscience
Inc.), or PE-conjugated anti-iNOS (12-5920-82; 1:100; eBioscience
Inc.) on ice for 45 min in the staining buffer following membrane
permeabilization with a Leucoperm kit (AbD Serotec, Raleigh, NC,
USA), according to the manufacturer's protocol. Cells were sorted
and analyzed using an EPICS Altra Flow Cytometer (Epics Altra II;
Beckman Coulter, Inc., Brea, CA, USA).
Detection of IL-10, IL-12, CCL17,
CCL22 and CCL24 by ELISA
ELISA was used to quantify the secretion levels of
the cytokines IL-10 and IL-12 and chemokines CCL17, CCL22 and CCL24
in the culture supernatant of lung IMs. IL-10 (catalogue number:
BMS614/2) and IL-12 (catalogue number: BMS616) ELISA kits were
purchased from eBioscience, Inc. and CCL17 (catalogue number:
MCC170), CCL22 (catalogue number: MCC220) and CCL24 (catalogue
number: DY528) ELISA kits were purchased from R&D Systems, Inc.
(Minneapolis, MN, USA). All assays were performed according to the
manufacturers' protocols.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from FACS-sorted lung IMs
using TRIzol reagent (Applied Biosystems; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. Isolated RNA was
then purified using an RNeasy Microprep kit (Qiagen GmbH, Hilden,
Germany). cDNA was synthesized using a SuperScript II reverse
transcriptase (Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. qPCR was performed using
a LightCycler® FastStart DNA MasterPLUS SYBRGreen I reaction mix
(Roche Diagnostics) on a Lightcycler 480II (Roche Diagnostics). The
levels of mRNA expression for iNOS, Arg1, TG, and
IL-10 were measured using primers purchased from Thermo
Fisher Scientific, Inc. The primer sequences were as follows: For
iNOS, forward, 5′-CGGAGCCTTTAGACCTCAACAGA-3′ and reverse,
5′-TAGGACAATCCACAACTCGCTCC-3′, for Arg1, forward,
5′-TGGGAAGACAGCAGAGGAGGTG-3′ and reverse,
5′-GCTTATGGTTACCCTCCCGTTG-3′, for IL-10, forward,
5′-TGGACAACATACTGCTAACCGAC-3′ and reverse
5′-CCTGGGGCATCACTTCTACC-3′, for TG2, forward,
5′-TTCCGGCTGACTCTGTACTTCGAG-3′ and reverse,
5′-ACATTGTCCTGTTGGTCCAGCACT-3′ and for GAPDH, forward,
5′-AGGAGCGAGACCCCACTAACA-3′ and reverse,
5′-AGGGGGGCTAAGCAGTTGGT-3′. The PCR protocol consisted of 60 sec at
95°C, followed by 40 cycles of 15 sec at 95°C, 15 sec at 59°C and
45 sec at 72°C. All data were analyzed using SDS 2.2 software
(Applied Biosystems; Thermo Fisher Scientific, Inc.). Gene
expression was quantified as the average of triplicate samples
after standard curve interpolation (relative standard curve method)
(27) and normalization to GAPDH
gene expression.
Western blot analysis of Arg 1, iNOS,
and IL-10 protein expression
Single cell suspensions of lung IMs isolated from
control and OVA-induced asthmatic mice were prepared and pooled
across mice within each group. Cells were lysed for 20 min at 4°C
in radioimmunoprecipitation assay buffer (10 mM phosphate buffer pH
7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS;
R-2417; AG Scientific Inc., San Diego, CA, USA), according to the
manufacturer's protocol. Lysates were then collected following
centrifugation at 12,000 × g at 4°C for 20 min. Protein
concentration was assayed using a Bicinchoninic Acid (BCA) Protein
Assay kit (Pierce; Thermo Fisher Scientific, Inc.) and whole
lysates were mixed with 4X SDS loading buffer (125 mM Tris-hydrogen
chloride, 4% SDS, 20% glycerol, 100 mM dithiothreitol and 0.2%
bromophenol blue) in a 1:3 ratio. Samples were heated at 100°C for
5 min and separated on SDS-polyacrylamide gels (10×10 cm by 0.75
mm) containing 4 and 12.5% acrylamide in the stacking and
separating gels, respectively. A total of 50 µg protein was loaded
per lane. Separated proteins were then transferred to
polyvinylidene difluoride membranes in a solution of transfer
buffer (25 mM Tris, 192 mM glycine, 0.01% SDS, 20%, v/v, methanol)
at 200 mA for 2 h. After transfer, the membranes were rinsed for 5
min with wash buffer and 0.02% Tween-20. Wash buffer was removed
and the membranes were incubated with 5% blocking buffer (blotting
grade blocker non-fat dry milk; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) for 1 h at room temperature. Membrane blots were
probed with primary antibodies overnight at 4°C, then incubated
with a corresponding horseradish peroxidase (HRP)-conjugated
secondary antibody for 1 h at room temperature (RT). Proteins were
visualized using an enhanced chemiluminescent system (GE Healthcare
Life Sciences, Chalfont, UK) The following primary antibodies were
diluted to 1:500 in TBST buffer (10 mM Tris HCl, 150 mM NaCl, 0.1%
Tween-20) and used: Rabbit anti-mouse anti-Arg1 (610709; BD
Biosciences), -iNOS (14-5920-82; eBioscience, Inc.), -IL-10
(14-71-01; eBioscience, Inc.) and -GAPDH (14-9523-82; eBioscience,
Inc.). The HRP-conjugated secondary antibody was anti-rabbit
(18-4818-82; eBioscience, Inc.). Optical densities (ODs) of bands
were measured using Scion Image software (Scion Image Beta 4.02 for
Windows; Scion Corporation, Frederick, MD, USA). The ODs of Arg1,
iNOS and IL-10 were normalized to that of GAPDH.
iNOS and Arg1 activity assays
FACS-sorted lung IMs isolated from control and
OVA-induced asthmatic mice were plated in triplicate into 96-well
plates at a density of 2×105 cells/well and 2×104 cells/well for
iNOS and Arg1 activity assays, respectively. Isolated cells were
cultured in RPMI 1640 medium supplemented with 10% fetal calf
serum, 2 mM l-glutamine, 1 mM sodium pyruvate, 0.1 mM nonessential
amino acids, 50 µM β-mercaptoethanol, 50 µg/ml streptomycin, and 50
IU/ml penicillin (all Invitrogen; Thermo Fisher Scientific, Inc.)
at 37°C for 48 h. Levels of nitric oxide (NO) in the culture
supernatant were measured based on the Griess reaction using Griess
reagent (G2930; Promega Corporation, Madison, WI, USA) according to
manufacturer's protocol and absorbance was measured at a wavelength
of 570 nm with a microplate reader (MR 710, Dynatech Scientific
Labs, El Paso, TX, USA) to evaluate iNOS activity. Arg1 activity
was determined by measuring the enzymatic conversion of arginine
into urea in the cell culture supernatants using a DARG-200
QuantiChrom™ Arginase Assay kit (BioAssay Systems,
Hayward, CA, USA). Protein concentration was determined using a BCA
Protein Assay kit (Pierce; Thermo Fisher scientific, Inc., Hemel
Hempstead, UK).
Statistical analysis
All data are expressed as the mean ± standard
deviation of at least three independent experiments. Student's
t-test was used to determine significant differences between two
groups. Data were analyzed using GraphPad Prism 5 software
(GraphPad Software Inc., La Jolla, CA, USA) and P<0.05 was
considered to indicate a statistically significant difference.
Results
CD206+F4/80+CD11c− macrophages
accumulate in the lungs of OVA-induced asthmatic mice
Previous studies have demonstrated that lung IMs
express the macrophage marker F4/80 but do not express the
dendritic cell marker CD11c (25,26). To
further characterize IMs in an OVA-induced mouse model of asthma,
whole lungs from BALB/c OVA-induced asthmatic mice and PBS-treated
control mice were digested 24 h after the final OVA challenge (day
27 post-model establishment; 100 µg in 50 µl PBS). Isolated cells
were subsequently stained for F4/80 and CD11c and analyzed by flow
cytometry. It was observed that the percentage of F4/80+CD11c-
macrophages was significantly higher in OVA-induced asthmatic mice
relative to that in control mice (P<0.01; Fig. 1A and B). It was also observed that
lung IMs were polarized towards an M2 macrophage phenotype. This
was demonstrated by a significant increase in the expression of
CD206, as a marker of M2 macrophages, on IMs (F4/80+CD11c- cells)
isolated from OVA-induced asthmatic mice, relative to control mice
(P<0.01; Fig. 1C and D). By
contrast, the number of IL-10+F4/80+CD11c- macrophages, which are
considered to be regulatory macrophages, significantly decreased in
OVA-induced asthmatic mice relative to control mice (P<0.01;
Fig. 1E and F). Furthermore, lung
IMs from control and OVA-induced asthmatic mice expressed markedly
low levels of iNOS (Fig. 1G), which
is a marker of classically activated (M1) macrophages.
Collectively, these results indicate that M2-polarized lung IMs
accumulate in the lungs of an OVA-induced asthma mouse model.
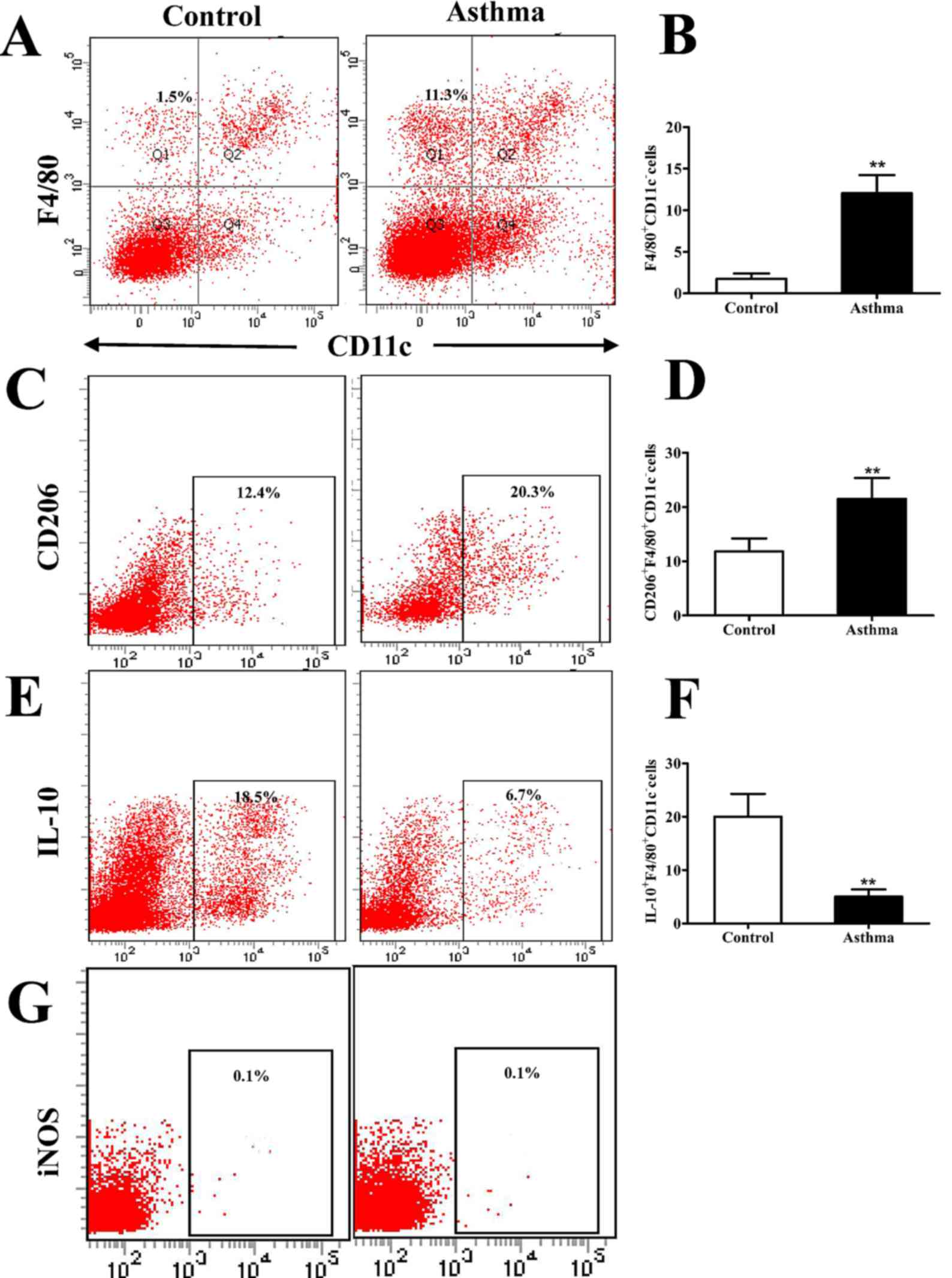 | Figure 1.Flow cytometry analysis of
CD206+F4/80+CD11c− cells in the
lungs of OVA-induced asthmatic mice. (A) Flow cytometry was used to
identify F4/80+ and CD11c− lung IMs isolated
from the lungs of OVA-induced asthmatic mice and PBS-treated
control mice. Boxes indicate the gating region of lung IMs
(F4/80+CD11c−; gate B). (B) Relative
percentages of F4/80+CD11c− cells in control
and OVA-induced asthmatic mice. (C) Detection of lung IMs positive
for the cell surface marker CD206. Boxes indicate the gating region
of CD206+F4/80+CD11c− cells (gate
C). (D) Relative percentages of
CD206+F4/80+CD11c− cells in
control and OVA-induced asthmatic mice. (E) Detection of lung IMs
positive for IL-10. Boxes indicate the gating region of
IL-10+F4/80+CD11c− cells (gate C).
(F) Percentages of lung IMs expressing IL-10 in control and
OVA-asthmatic mice (G) Detection of lung IMs positive for iNOS.
Boxes indicate the gating region of
iNOS+F4/80+CD11c− cells (gate P2).
Percentages on each dot plot represent positively stained cells.
All data are presented as the mean ± standard deviation, n=6.
**P<0.01 vs. control. CD, cluster of differentiation; F4/80,
epidermal growth factor family cell surface antigen; IM,
interstitial macrophages; IL-10, interleukin-10; OVA, ovalbumin;
iNOS, inducible nitric oxide synthase; PBS, phosphate-buffered
saline; PE-cy5, phycoerythrin-cyano 5 dye; FITCH, fluorescein
isothiocyanate. |
Expression of signature genes
associated with M2 F4/80+CD11c− macrophages
is increased in the lung IMs of OVA-induced asthmatic mice
Lung IMs isolated from control and OVA-induced
asthmatic mice were sorted 24 h after the final OVA challenge and
their RNA was isolated. The expression of mRNA associated with
classically and alternatively activated macrophages, along with
IL-10 mRNA, were measured by RT-qPCR. It was observed that levels
of Arg1 and TG2 mRNA, as markers of alternatively activated
macrophages, were significantly increased relative to control mice
(P<0.01; Fig. 2A and B). In
addition, IL-10 mRNA expression, as a primary feature of regulatory
macrophages, was significantly decreased in OVA-induced asthmatic
mice relative to control mice (P<0.05; Fig. 2C). However, levels of iNOS mRNA, as a
marker of classically activated macrophages, did not significantly
differ between OVA-induced asthmatic mice and control mice
(P>0.05; Fig. 2D).
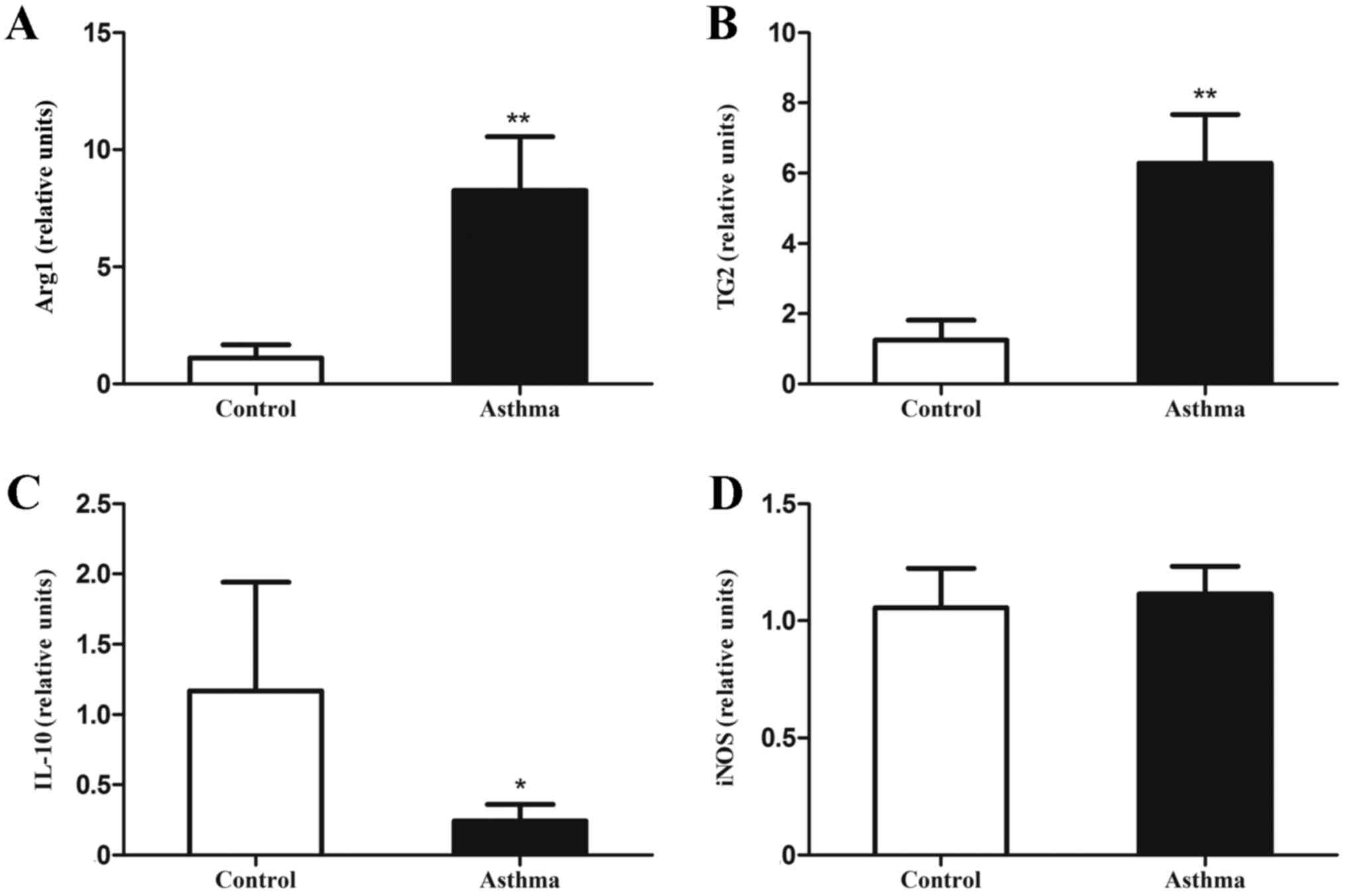 | Figure 2.Expression of genes associated with
alternatively activated macrophages in the lungs of OVA-induced
asthmatic mice. Lung IMs from OVA-induced asthmatic mice and
PBS-treated control mice were sorted 24 h after the final OVA
challenge (day 27 post-model establishment; 100 µg in 50 µl PBS)
and the levels of (A) Arg1, (B) TG2, (C) IL-10 mRNA and (D) iNOS
mRNA, as markers of classically activated macrophages, were
measured by reverse transcription-quantitative polymerase chain
reaction. Expression of Arg1, TG2, iNOS, and IL-10 was normalized
to GAPDH expression. Data are presented as the mean ± standard
deviation, n=6. *P<0.05 and **P<0.01 vs. control. OVA,
ovalbumin; IM, interstitial macrophage; Arg1, arginase 1; TG2,
transglutaminase 2; iNOS, inducible nitric oxide synthase; IL-10,
interleukin-10; PBS, phosphate-buffered saline. |
Expression of signature proteins
associated with M2 F4/80+CD11c− macrophages
is increased in the lung IMs of OVA-induced asthmatic mice
It was observed following western blot analysis
(Fig. 3A) that the levels of Arg1
protein in the lung IMs of OVA-induced asthmatic mice were
significantly increased, relative to control mice (P<0.01;
Fig. 3B). However, levels of iNOS
protein did not significantly differ between the two groups
(P>0.05; Fig. 3C). In addition,
the expression of IL-10 in the lung IMs of OVA-induced asthmatic
mice was significantly decreased, relative to control mice
(P<0.01; Fig. 3D). Collectively,
these data indicate that lung IMs in an OVA-induced asthma mouse
model exhibit increased expression of signature genes and proteins
associated with M2 polarization.
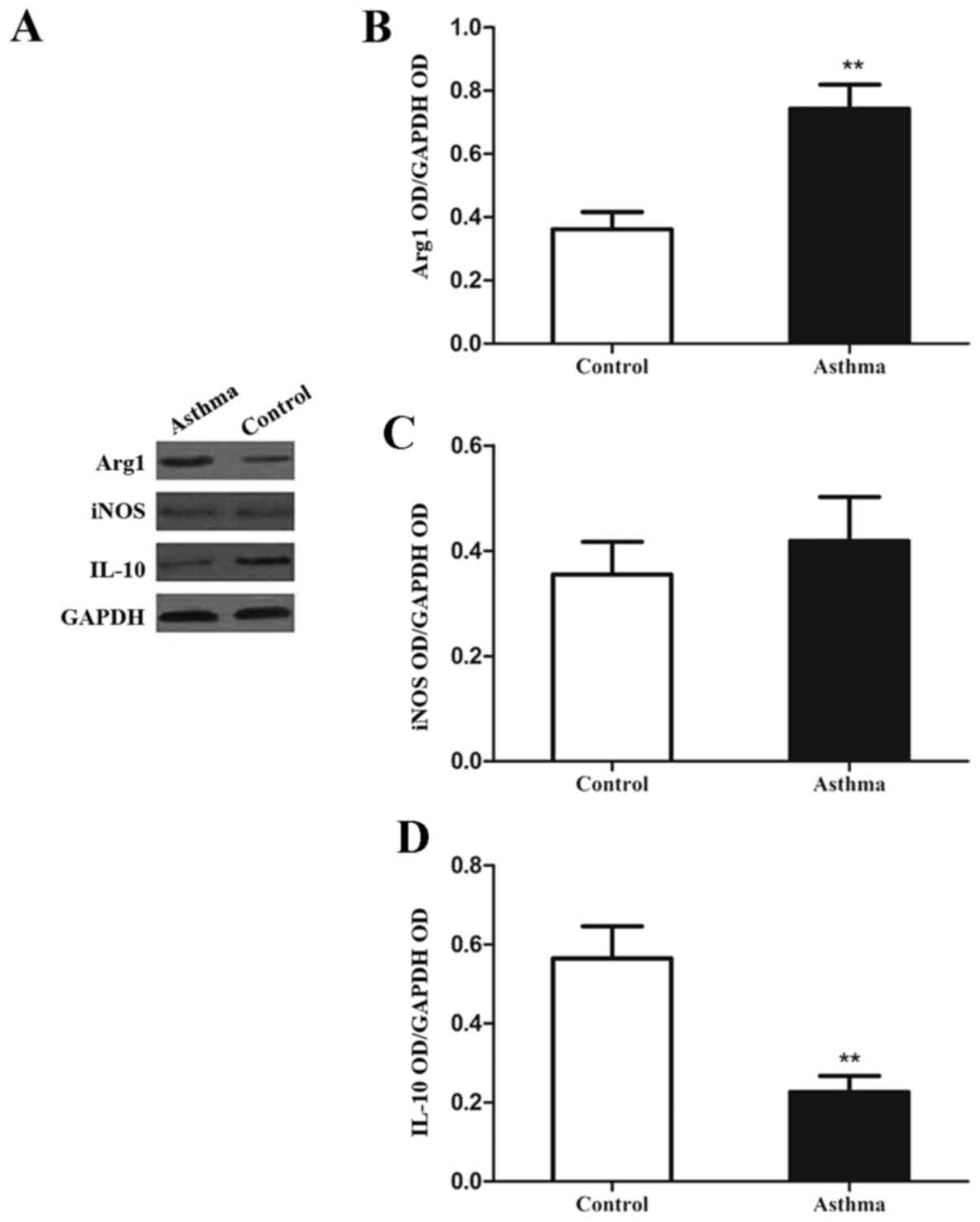 | Figure 3.Expression of Arg1, iNOS and IL-10 in
lung IMs of OVA-induced asthmatic mice. Lung IMs from OVA-induced
asthmatic mice and PBS-treated control mice were sorted 24 h after
the final OVA challenge (day 27 post-model establishment; 100 µg in
50 µl PBS) and (A) western blot analysis was used to measure Arg1,
IL-10 and iNOS protein expression. GAPDH was used as a protein
loading control and images are representative of 6 mice in each
group. ODs of (B) Arg1, (C) iNOS and (D) IL-10 levels normalized to
GAPDH. Data are presented as the mean ± standard deviation, n=6.
**P<0.01 vs. control. Arg1, arginase 1; iNOS, inducible nitric
oxide synthase; IL-10, interleukin-10; OVA, ovalbumin; IM,
interstitial macrophage; PBS, phosphate-buffered saline; OD,
optical density. |
IL-10 is reduced while chemokine
secretion is increased in lung IMs of OVA-induced asthmatic
mice
To determine whether lung IMs produce relevant
inflammatory factors of the different macrophage phenotypes
(cytokines IL-10 and IL-12 and chemokines CCL17, CCL22 and CCL24),
lung IMs isolated from control and OVA-induced asthmatic mice were
cultured in the presence or absence of OVALPS. After 16
h, culture supernatants were collected and analyzed for cytokine
and chemokine expression. It was observed that IL-10 concentration
was significantly lower in the culture supernatant of unstimulated
lung IMs (no OVALPS group) of OVA-induced asthmatic
mice, relative to control mice (no OVALPS group;
P<0.01; Fig. 4A). The
concentration of IL-12, as a pro-inflammatory cytokine released by
classically activated macrophages (28) did not significantly differ in the
culture supernatants of unstimulated lung IMs of OVA-induced
asthmatic mice, relative to control mice (P>0.05; Fig. 4B). The concentrations of CCL17, CCL22
and CCL24, as chemokines typically secreted by alternatively
activated macrophages (23), were
significantly higher in the culture supernatant of unstimulated
lung IMs from OVA-induced asthmatic mice, relative to unstimulated
lung IMs from control mice (P<0.01; Fig. 4C). Stimulation with OVALPS
had no significant effect on the levels of IL-10 produced by the
lung IMs of OVA-induced asthmatic mice, relative to unstimulated
cells in the OVA-asthmatic group (P>0.05; Fig. 4A). By contrast,
OVALPS-stimulated lung IMs from control mice produced
significantly more IL-10 than unstimulated control cells
(P<0.01; Fig. 4A). In addition,
stimulation with OVALPS had no significant effect on the
levels of IL-12 produced by lung IMs of OVA-induced asthmatic or
control mice, relative to unstimulated lung IMs in their respective
groups (both P>0.05; Fig. 4B).
Stimulation with OVALPS also had no significant effect
on the production of CCL17, CCL22 or CCL24 from lung IMs of control
mice (P>0.05; Fig. 4C). However,
OVALPS-stimulated lung IMs produced significantly more
CCL17, CCL22 and CCL24 than unstimulated lung IMs from OVA-induced
asthmatic mice (all P<0.01; Fig.
4C). Collectively, these data suggest that the quiescent
phenotype of lung IMs switches to an M2 polarized state in an
OVA-induced asthma mouse model.
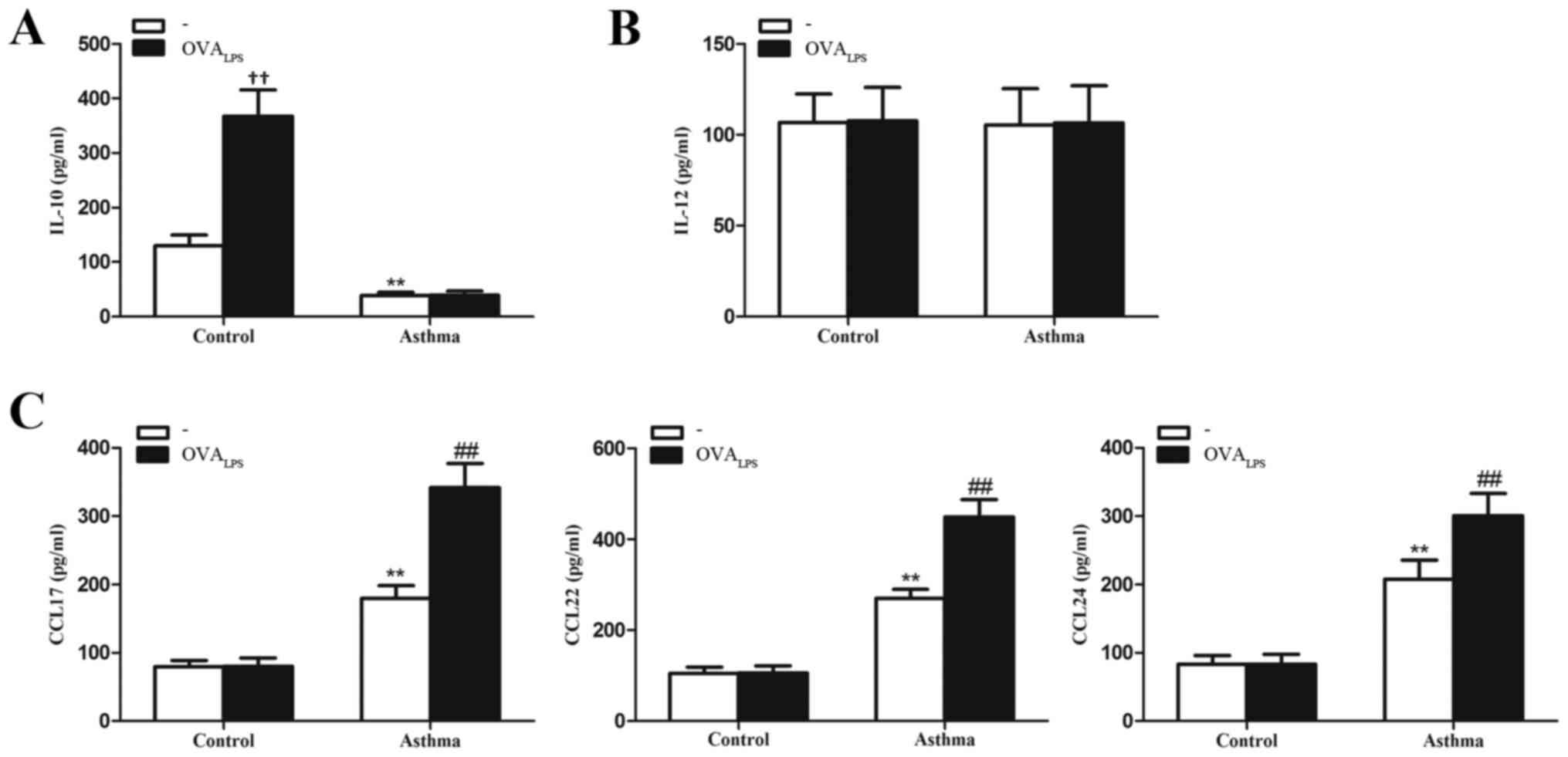 | Figure 4.Expression of cytokines IL-10 and
IL-12 and chemokines CCL17, CCL22 and CCL24 in lung IMs of
OVA-induced asthmatic mice. Lung IMs from OVA-induced asthmatic
mice and PBS-treated control mice were sorted 24 h after the final
OVA challenge (day 27 post-model establishment; 100 µg in 50 µl
PBS) and cultured in the presence or absence of OVALPS.
After 16 h, ELISA was used to measure the levels of the cytokines
(A) IL-10 and (B) IL-12 and (C) the chemokines CCL17, CCL22 and
CCL27 in the culture supernatant. Data are presented as the mean ±
standard deviation, n=6. **P<0.01 vs. non
OVALPS-stimulated control mice; ††P<0.01
vs. non OVALPS-stimulated control mice;
##P<0.01 vs. non OVALPS-stimulated
asthmatic mice. IL, interleukin; CCL, chemokine ligand; OVA,
ovalbumin; OVALPS, lipopolysaccharide supplemented OVA;
IM, interstitial macrophage; PBS, phosphate-buffered saline. |
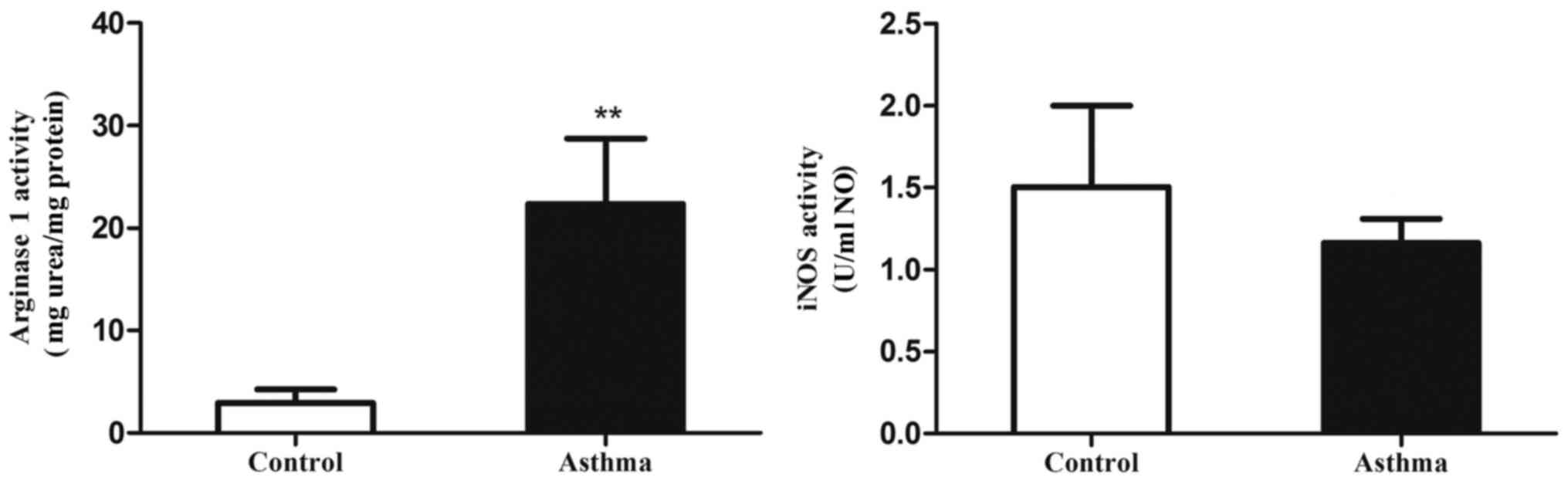
Arg1 activity is in the lung IMs of
OVA-induced asthmatic mice
Increased arginase activity is also a characteristic
of alternatively activated macrophages (29). Therefore, the present study measured
the levels of alternatively activated macrophages in the lungs of
an OVA-induced asthmatic mouse model through ex vivo assays of Arg1
and iNOS catalytic activity (Fig.
5). It was observed that Arg1 activity in the lung IMs of
OVA-induced asthmatic mice was significantly elevated relative to
control mice (P<0.01). In addition, iNOS activity did not
significantly differ between the control and asthmatic groups. This
suggests that the Arg1/iNOS balance in the lung IMs of an
OVA-induced asthma mouse model is skewed towards an M2 polarization
state.
Discussion
The innate immune system is associated with the
induction and progression of airway diseases, including asthma.
Although macrophages are a key part of the innate immune system,
their contribution to the pathogenesis of asthma is currently
unknown. A previous study indicated that alternatively activated
macrophages and the inflammatory molecules they produce are
associated with asthma (30). AMs
and IMs are the two main populations of macrophages in the lungs
and occupy distinct compartments (16). The characteristics of AMs have been
widely studied (17), however the IM
phenotype and its in vivo functions are not fully
understood. Therefore, the present study evaluated the subtypes and
expression profiles of lung IMs in a mouse model of OVA-induced
asthma. It was principally demonstrated that the lung IMs of
OVA-induced asthmatic mice express hallmark genes of macrophage
polarization towards an alternatively activated state (M2), thus
implying a causative role of these cells in the pathogenesis of
asthma.
Macrophage activation is a dynamic process, in which
cells that are initially involved in pro-inflammatory and cytotoxic
reactions subsequently participate in the resolution of
inflammation and wound healing (7,30–33).
Therefore, macrophage differentiation is an adaptable process,
enabling macrophages to modulate their response to changing
microenvironments (7,31,33).
Macrophages are activated by different stimuli and the cells
subsequently express distinct patterns of chemokines, cytokines,
surface markers and metabolic enzymes that ultimately generate the
diversity of macrophage function observed in inflammatory and
noninflammatory settings (6,28). Macrophage activation is defined by
two distinct polarization states: M1 (classically activated
macrophages) and M2 (alternatively activated macrophages) (6,28). Other
macrophage subtypes also exist, including regulatory macrophages
(6,8). Macrophage activation into an M2
polarization state involving tumor-associated macrophages or
parasitic infections has been previously documented in different
tumor settings, and is considered to modulate tissue repair and
prevent excess inflammation (30,34). In
the present study, lung IMs exhibited increased levels of CD206
expression, as a surface marker of alternatively activated
macrophages (31), indicating that
M2-polarized lung IMs accumulate in the lungs of OVA-induced
asthmatic mice.
The balance between arginase and iNOS activity is an
important distinction between M1 and M2 polarization states. Both
arginase and iNOS utilize L-arginine, though only arginase
production is increased in M2-polarized macrophages (35). In addition, arginase blocks iNOS
activity through a number of mechanisms, including outcompeting
iNOS for the arginine substrate, which is required for NO
production (35). Based on direct
measurements of arginase activity and gene expression, including
for Arg1 and TG2, as markers of M2 macrophages, the present study
demonstrated that lung IMs of an OVA-induced asthma mouse model
exhibit increased arginase activity, compared with control mice.
Furthermore, it was observed that chemokines typically produced by
alternatively activated macrophages, namely CCL17, CCL22 and CCL24,
are produced in greater quantities by lung IMs from OVA-induced
asthmatic mice, relative to controls. However, lung IMs from
control and OVA-induced asthmatic mice exhibited similar levels of
iNOS, along with similar levels of IL-12 secretion. In addition,
levels of iNOS activity in the lung IMs of OVA-induced asthmatic
mice were similar to controls. Collectively, these data suggest
that lung IMs in an OVA-induced asthma mouse model are polarized
towards an alternative activation phenotype.
The existence of regulatory macrophages has
previously been indicated (6). A key
feature of regulatory macrophages is high-level production of the
anti-inflammatory cytokine IL-10 (13). In addition, regulatory macrophages
have been demonstrated to reduce inflammation and pathological
changes in a mouse model of lethal endotoxemia (14) and a mouse model of EAE (15). Therefore, in contrast to the M1 and
M2 macrophage phenotypes, regulatory macrophages may be crucial in
modulating excessive immune responses and preventing inflammatory
disease. Interestingly, it has been observed that lung macrophages
from asthmatic subjects produced less IL-10 than those from healthy
individuals (23). Furthermore, in
severe asthma, lung macrophages have been found to produce higher
levels of IL-6, IL-8 and monocyte-derived chemokines, while IL-10
was undetectable in the cells, relative to those from patients with
moderate asthma (24). This suggests
that in cases of severe asthma, the inhibitory functions of lung
macrophages are absent, while their pro-inflammatory functions are
enhanced (24). Results also
indicate that under normal physiological conditions, lung IMs with
regulatory characteristics serve key roles in the maintenance of
respiratory tract immune homeostasis and prevention of airway
allergic responses (25). In the
present study, significant differences in the levels of IM IL-10
expression between asthmatic and control mice indicates that IL-10
is a potential homeostatic regulator under normal conditions.
Therefore, lung IMs may lose their protective regulatory effects in
OVA-induced asthmatic mice.
Macrophages may differentiate into an M2 phenotype
following exposure to IL-4, IL-13 or a combination of both
(6,7). IL-4 and IL-13 are abundant in the lungs
of asthmatic individuals, which may explain why pulmonary IMs were
polarized towards the M2 state in OVA-induced asthmatic mice in the
present study. However, the potential roles of M2-polarized lung
IMs in the pathogenesis of asthma are currently unknown. In a
previous murine model of asthma, it was observed that alternatively
activated macrophages were involved in disease development
(19,36). Previous results also indicate that
asthmatic mice have higher counts of pulmonary M2 macrophages
relative to controls. In addition, it has been observed that
increasing the number of M2 macrophages in the lungs by
intratracheal instillation, prior to the induction of allergic
inflammation, leads to greater airway inflammation. In patients
with severe asthma, increased counts of alternatively activated
macrophages have been detected in the BALF when compared with
healthy controls. It has also been observed that the number of
pulmonary M2 macrophages correlated with peak expiratory flow
variation (20). These data suggest
that alternatively activated macrophages may serve a key regulatory
role in asthma. Similarly, previous results have demonstrated that
adoptive transfer of IL-4 receptor (R)-α+/+ macrophages,
but not IL-4Rα−/− macrophages, by intraperitoneal
injection was sufficient to strengthen Th2-dependent, OVA-induced
allergic airway inflammation in mice (37). Although these results have been
questioned, because it has previously been reported that
IL-4Rα-dependent alternatively activated macrophages do not serve
an important role in the pathology of disease by using
LysMcreIL-4Rα−/lox mice in ovalbumin- and
house dust mite-induced allergic airway diseases (38), collectively the aforementioned
findings suggest that alternatively activated macrophages are key
contributors of Th2-driven airway inflammation, rather than simply
bystander cells that respond to Th2 cytokines. Although all
macrophages may respond to IL-4 and IL-13, it is unknown whether
AMs and/or IMs transform into an alternatively activated macrophage
phenotype within asthmatic individuals. In the current study, lung
IMs were polarized towards an alternative activation phenotype in
murine OVA-induced asthma. Therefore, M2-polarized lung IMs may
contribute to a Th2 response, though this warrants further
investigation.
In conclusion, the present data indicate that lung
IMs undergo a phenotypic switch from a regulatory macrophage
phenotype under normal conditions to an alternative activation
state in OVA-induced asthmatic mice. Further studies are now
warranted to identify which factors trigger the phenotypic switch
of lung IMs in asthmatic individuals, and to determine if targeted
therapies can be developed that modify the characteristics of lung
IMs, particularly at the levels of cytokine, metabolite and enzyme
expression.
Acknowledgements
The current study was supported by staff at the
Center for Medical Research at the Hubei Key Laboratory of Allergy
and Immune-Related Diseases (Wuhan University, Wuhan, China) and
the National Natural Science Foundation of China (grant nos.
81500021 and 81270076).
Glossary
Abbreviations
Abbreviations:
|
IM
|
interstitial macrophage
|
|
AM
|
alveolar macrophage
|
|
OVA
|
ovalbumin
|
|
Th2
|
T helper type 2
|
|
Arg1
|
arginase 1
|
|
TG2
|
transglutaminase 2
|
|
NO
|
nitric oxide
|
|
iNOS
|
inducible nitric oxide synthase
|
|
IL
|
interleukin
|
|
TNF
|
tumor necrosis factor
|
|
CCL
|
chemokine ligand
|
|
IFN
|
interferon
|
|
CD
|
cluster of differentiation
|
|
BALF
|
bronchoalveolar lavage fluid
|
References
|
1
|
Barnes PJ: Immunology of asthma and
chronic obstructive pulmonary disease. Nat Rev Immunol. 8:183–192.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Anderson GP: Endotyping asthma: New
insights into key pathogenic mechanisms in a complex, heterogeneous
disease. Lancet. 372:1107–1119. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Martinez FO, Helming L and Gordon S:
Alternative activation of macrophages: An immunologic functional
perspective. Annu Rev Immunol. 27:451–483. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Strauss-Ayali D, Conrad SM and Mosser DM:
Monocyte subpopulations and their differentiation patterns during
infection. J Leukoc Biol. 82:244–252. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Auffray C, Fogg D, Garfa M, Elain G,
Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G and
Geissmann F: Monitoring of blood vessels and tissues by a
population of monocytes with patrolling behavior. Science.
317:666–670. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gordon S and Taylor PR: Monocyte and
macrophage heterogeneity. Nat Rev Immunol. 5:953–964. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Martinez FO, Sica A, Mantovani A and
Locati M: Macrophage activation and polarization. Front Biosci.
13:453–461. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mosser DM and Edwards JP: Exploring the
full spectrum of macrophage activation. Nat Rev Immunol. 8:958–969.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gorden S and Martinez FO: Alternative
activation of macrophages: Mechanism and functions. Immunity.
32:593–604. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Loke P, Nair MG, Parkinson J, Guiliano D,
Blaxter M and Allen JE: IL-4 dependent alternative activated
macrophages have a distinct in vivo gene expression phenotype. BMC
Immunol. 3:72002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nair MG, Cochrane DW and Allen JE:
Macrophages in chronic type 2 inflammation have a novel phenotype
characterized by the abundant expression of Ym1 and Fizzl that can
be partly replicated in vivo. Immunol Lett. 85:173–180. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sandler NG, Mentink-Kane MM, Cheever AW
and Wynn TA: Global gene expression profiles during acute
pathogen-induced pulmonary inflammation reveal divergent roles for
Th1 and Th2 responses in tissue repair. J Immunol. 171:3655–3667.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sutterwala FS, Noel GJ, Salgame P and
Mosser DM: Reversal of proinflammatory responses by ligating the
macrophage Fcgamma receptor type I. J Exp Med. 188:217–222. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gerber JS and Mosser DM: Reversing
lipopolysaccharide toxicity by ligating the macrophage Fc gamma
receptors. J Immunol. 166:6861–6868. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fleming BD and Mosser DM: Regulatory
macrophages: Setting the threshold for therapy. Eur J Immunol.
41:2498–2502. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schneberger D, Aharonson-Raz K and Singh
B: Monocyte and macrophage heterogeneity and Toll-like receptors in
the lung. Cell Tissue Res. 343:97–106. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Holt PG, Strickland DH, Wikström ME and
Jahnsen FL: Regulation of immunological homeostasis in the
respiratory tract. Nat Rev Immunol. 8:142–152. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Martinez FO, Helming L, Milde R, Varin A,
Melgert BN, Draijer C, Thomas B, Fabbri M, Crawshaw A, Ho LP, et
al: Genetic programs expressed in resting and IL-4 alternatively
activated mouse and human macrophages: Similarities and
differences. Blood. 121:e57–e69. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Melgert BN, ten Hacken NH, Rutgers B,
Timens W, Postma DS and Hylkema MN: More alternative activation of
macrophages in lungs of asthmatic patients. J Allergy Clin Immunol.
127:831–833. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim EY, Battaile JT, Patel AC, You Y,
Agapov E, Grayson MH, Benoit LA, Byers DE, Alevy Y, Tucker J, et
al: Persistent activation of an innate immune response translates
respiratory viral infection into chronic lung disease. Nat Med.
14:633–640. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chupp GL, Lee CG, Jarjour N, Shim YM, Holm
CT, He S, Dziura JD, Reed J, Coyle AJ, Kiener P, et al: A
chitinase-like protein in the lung and circulation of patients with
severe asthma. N Engl J Med. 357:2016–2027. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Draijer C, Robbe P, Boorsma CE, Hylkema MN
and Melgert BN: Characterization of macrophage phenotypes in three
murine models of house-dust-mite-induced asthma. Mediators Inflamm.
2013:6320492013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maneechotesuwan K, Supawita S,
Kasetsinsombat K, Wongkajornsilp A and Barnes PJ: Sputum
indoleamine-2, 3-dioxygenase activity is increased in asthmatic
airways by using inhaled corticosteroids. J Allergy Clin Immunol.
121:43–50. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fitzpatrick AM, Higgins M, Holguin F,
Brown LA and Teague WG: National Institutes of Health/National
Heart, Lung, and Blood Institute's Severe Asthma Research Program:
The molecular phenotype of severe asthma in children. J Allergy
Clin Immunol. 125:851–857.e18. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bedoret D, Wallemacq H, Marichal T, Desmet
C, Calvo F Quesada, Henry E, Closset R, Dewals B, Thielen C, Gustin
P, et al: Lung interstitial macrophages alter dendritic cell
functions to prevent airway allergy in mice. J Clin Invest.
119:3723–3738. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lagranderie M, Nahori MA, Balazuc AM,
Kiefer-Biasizzo H, e Silva JR Lapa, Milon G, Marchal G and
Vargaftig BB: Dendritic cells recruited to the lung shortly after
intranasal delivery of Mycobacterium bovis BCG drive the primary
immune response towards a type 1 cytokine production. Immunology.
108:352–364. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Larionov A, Krause A and Miller W: A
standard curve based method for relative real time PCR data
processing. BMC Bioinformatics. 6:622005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mantovani A, Sica A, Sozzani S, Allavena
P, Vecchi A and Locati M: The chemokine system in diverse forms of
macrophage activation and polarization. Trends Immunol. 25:677–686.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gordon S: Alternative activation of
macrophages. Nat Rev Immunol. 3:23–35. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhu Z, Zheng T, Homer RJ, Kim YK, Chen NY,
Cohn L, Hamid Q and Elias JA: Acidic mammalian chitinase in
asthmatic Th2 inflammation and IL-13 pathway activation. Science.
304:1678–1682. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Porcheray F, Viaud S, Rimaniol AC, Léone
C, Samah B, Dereuddre-Bosquet N, Dormont D and Gras G: Macrophage
activation switching: An asset for the resolution of inflammation.
Clin Exp Immunol. 142:481–489. 2005.PubMed/NCBI
|
|
32
|
Laskin DL, Sunil VR, Gardner CR and Laskin
JD: Macrophages and tissue injury: Agents of defense or
destruction? Annu Rev Pharmacol Toxicol. 51:267–288. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Stout RD and Suttles J: Functional
plasticity of macrophages: Reversible adaptation to changing
microenvironments. J Leukoc Biol. 76:509–513. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mantovani A, Sozzani S, Locati M, Allavena
P and Sica A: Macrophage polarization: Tumor-associated macrophages
as a paradigm for polarized M2 mononuclear phagocytes. Trends
Immunol. 23:549–555. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bronte V and Zanovello P: Regulation of
immune responses by L-arginine metabolism. Nat Rev Immunol.
5:641–654. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Melgert BN, Oriss TB, Qi Z, Dixon-McCarthy
B, Geerlings M, Hylkema MN and Ray A: Macrophages: Regulators of
sex differences in asthma? Am J Respir Cell Mol Biol. 42:595–603.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ford AQ, Dasgupta P, Mikhailenko I, Smith
EM, Noben-Trauth N and Keegan AD: Adoptive transfer of IL-4Rα+
macrophages is sufficient to enhance eosinophilic inflammation in a
mouse model of allergic lung inflammation. BMC Immunol. 13:62012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nieuwenhuizen NE, Kirstein F, Jayakumar J,
Emedi B, Hurdayal R, Horsnell WG, Lopata AL and Brombacher F:
Allergic airway disease is unaffected by the absence of
IL-4Rα-dependent alternatively activated macrophages. J Allergy
Clin Immunol. 130:743–750.e8. 2012. View Article : Google Scholar : PubMed/NCBI
|