Introduction
Thalidomide (THD) was introduced in the 1950s in
Europe due to its antiemetic and sedative effects (1). It is a synthetic derivative of glutamic
acid that contains two imide rings: Glutarimide and phthalimide.
Due to its neurotoxicity, THD caused devastating teratogenic
effects in the 1960s (2,3) and subsequently, the use of this drug
was rapidly forbidden as a result. In the early 1990s, THD was
identified to possess antiangiogenic properties (4). In 1999, it was revealed that THD may be
used in the treatment of multiple myeloma (5). Additionally, in 2006, the Food and Drug
Administration (USA) approved the use of THD for the treatment of
multiple myeloma (4). In the
following years, a number of studies demonstrated that THD has
important antitumor effects in several types of cancer, including
prostate, colorectal, non-small-cell lung and breast cancer and
renal cell carcinoma (6–10). The mechanism of action of THD is
associated with angiogenesis suppression (3,11),
proliferation inhibition and apoptosis induction (12,13). THD
is also classified as an immunomodulatory drug that inhibits the
production of tumor necrosis factor-α and may also affect the
production of interleukin (IL)-1β, IL-2, IL-4, IL-5, IL-6, IL-10
and interferon-γ (3,11). However, the failure of THD to inhibit
tumor growth and angiogenesis has also been observed in a murine
model in vivo (14). The
mechanism underlying the therapeutic failure of THD is
uncertain.
Vascular endothelial growth factor (VEGF)/vascular
endothelial growth factor receptor (VEGFR) signaling pathways are
the key drivers of angiogenesis, and enhance tumor cell survival
and promote tumor aggressiveness (15–17).
VEGFR-1, also known as fms-like tyrosine kinase receptor 1, has an
important role in neovascularization under pathological conditions
(18). However, the expression of
VEGFR1 is not limited to endothelial cells (19). VEGFR1 has been reported to be
expressed by multiple types of cancer cells, including gastric
cancer cells (20), colorectal
carcinoma cells (21), breast cancer
cells (22) and lymphoma cells
(23). VEGFR1-positive hematopoietic
progenitors have been suggested to initiate the formation of a
pre-metastatic niche (24).
Furthermore, VEGFR1 may be presented on a subset of macrophages in
various types of breast cancers, which are significantly enriched
in metastatic sites (25).
Additionally, the overexpression of VEGFR1 in peripheral blood has
been associated with advanced clinical stages of cancer (26) These findings have contributed to the
suggestion that VEGFR1 may be a novel therapeutic target for
anti-angiogenesis strategies and cancer therapy (19,27).
F56, a VEGFR1-specific peptide, has been shown to displace VEGF
from its receptor VEGFR1 and inhibit tumor growth (27,28).
However, the role of VEGFR1 in the response of cancer therapy
requires further investigation.
In the present study, the effect of THD on CT26
tumor cells and human umbilical vein endothelial cells (HUVECs)
in vitro was determined. Specifically, its effect on cell
proliferation, apoptosis and VEGFR1 expression was investigated. In
addition, the effect of THD on reactive oxygen species (ROS), and
the effect of ROS suppression on THD-induced VEGFR1 expression were
evaluated. Furthermore, whether VEGFR1 acts as a stress-associated
protein in response to chemotherapy, radiotherapy and thermotherapy
was examined. The findings of this study may be useful for the
future application of VEGFR1-targeted therapy with current
therapeutics.
Materials and methods
Reagents
THD, cisplatin, doxorubicin, paclitaxel and
5-fluorouracil (5-FU) were all purchased from Selleckchem (Houston,
TX, USA), N-acetyl-L-cysteine (NAC) was obtained from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Cell lines and cell culture
CT26, HUVECs, SW480, SW620 and HCT116 were obtained
from the American Type Culture Collection (ATCC; Manassas, VA,
USA). CT26 and SW480 cells were grown in 100-cm2 cell
culture plates and maintained in RPMI-1640 medium (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
heat-inactivated fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml streptomycin
at 37°C in a humidified atmosphere containing 95% air and 5%
CO2. SW620 and HCT116 cells were maintained in
Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc.) and HUVECs were maintained in DMEM-F12 (Gibco;
Thermo Fisher Scientific, Inc.). DMEM and DMEM-F12 were also
supplemented with 10% heat-inactivated fetal bovine serum (Gibco;
Thermo Fisher Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml
streptomycin at 37°C in a humidified atmosphere containing 5%
CO2. Cells in the exponential growth phase were used for
all experiments.
Cell proliferation assays
CT26 cells and HUVECs were plated into 24-well
plates at a density of 1×105 cells/well. THD dissolved
in dimethyl sulfoxide was incubated with the cells at different
concentrations (0, 12.5, 25, 50 and 100 µM) with three replicates
being performed for each concentration. Following 24, 48 or 72 h
incubation at 37°C, the cell proliferation was determined manually
by cell number counting.
Annexin V/propidium iodide (PI)
assay
CT26 cells and HUVECs were seeded into 6-well plates
at a density of 2×105 cells/well and exposed to THD at
various concentrations (0, 25 and 100 µM) at 37°C. Cells were
harvested following 24-h incubation and washed twice with PBS.
Subsequently, cells were stained using an Annexin V/PI double
staining solution from an Annexin V-FITC apoptosis detection kit
(BD Pharmingen; BD Biosciences; San Jose, CA, USA) at room
temperature, according to the manufacturer's instructions.
Following 15 min of incubation, the Annexin V/PI stained cells were
detected using a FACSCalibur flow cytometer (BD Biosciences) and
analyzed using CellQuest software (version 5.1; BD
Biosciences).
Cell cycle analysis
CT26 cells and HUVECs were seeded into 6-well plates
at a density of 2×105 cells/well and incubated with THD
at various concentrations (0, 25, 50 and 100 µM) for 24 h at 37°C.
Subsequently, cells were harvested and washed three times with cold
PBS. Cells were fixed in 70% ice-cold ethanol overnight, washed
twice with PBS, stained with PI/RNase staining buffer (BD
Pharmingen; BD Biosciences) at room temperature for 15 min and
detected using a flow cytometer. Data were analyzed using Modifit
software (version 4.1; Verity Software House, Topsham, ME,
USA).
Cell treatments and protein
extraction
CT26 cells and HUVECs were seeded into 6-well plates
at a density of 2×105 cells/well. Cells were exposed to
THD (0, 25, 50 and 100 µM), cisplatin (0, 0.5, 0.8 and 1 µg/ml),
paclitaxel (0, 10, 20 and 40 ng/ml), 5-FU (0, 1, 2 and 4 µM) and
doxorubicin (0, 5 and 10 µM), irradiated with 2 Gy X-rays (29,30) or
heated at 47°C for 3 min as previously described (31). To inhibit intracellular reactive
oxygen species (ROS), the antioxidant NAC (2 and 4 mM,
respectively) was added to the culture media, then they were
exposed to 100 µM THD for 24 h at 37°C. In addition, SW480, SW620
and HCT116 cells were seeded at a density of 2×105
cells/well and were exposed to THD at various concentrations (0,
25, 50 and 100 µM). Following 24-h incubation at 37°C, all cells
were harvested by centrifugation (475 × g for 3 min at room
temperature), washed twice using cold PBS, and subsequently lysed
in radioimmunoprecipitation assay lysis buffer (Beyotime Institute
of Biotechnology, Haimen, China) on ice for 15 min for protein
extraction.
Western blot assay
Protein concentrations were determined using a
bicinchoninic acid assay. Sample proteins (40 µg/sample) were
separated on 10% SDS-PAGE and then transferred onto polyvinylidene
difluoride membranes. Membranes were blocked with 5% non-fat milk
in Tris-buffered saline with Tween-20 (TBST) at room temperature
for 1 h. Membranes were subsequently incubated at 4°C overnight
with the primary rabbit monoclonal antibodies against VEGFR-1 (cat.
no. ab32125), cyclin dependent kinase (CDK)6 (cat. no. ab131439),
cyclin D1 (cat. no. ab134175), C-MYC (cat. no. ab39688), B-cell
lymphoma-2 (BCL-2; cat. no. ab59348), BCL-2-associated X protein
(BAX; cat. no. ab182733) and caspase-3 (ab32351, all 1:1,000; all
from Abcam, Cambridge, MA, USA). GAPDH protein was also determined
using a specific antibody (dilution 1:1,000, cat. no. 2118; Cell
Signaling Technology, Inc., Danvers, MA, USA) as a loading control.
The membranes were washed with TBST three times and incubated with
the appropriate horseradish peroxidase-conjugated secondary
antibody (cat. no. ab6721, 1:5,000; Abcam) at room temperature for
1 h. The membranes were washed with TBST three times and visualized
using an enhanced chemiluminescence detection system (Beyotime
Institute of Biotechnology).
ROS analysis
CT26 cells and HUVECs were respectively seeded at
2×105 cells/well in 6-well plates and incubated with THD
at various concentrations (0, 25, 50 and 100 µM) for 6 h at 37°C.
Cells were harvested and washed three times with cold PBS.
Subsequently, cells were resuspended in diluted
2′,7′-dichlorofluorescein diacetate (DCFH-DA) at a cell
concentration of 1×106 to 20×106 cells/ml
according to the manufacturer's instructions and incubated at 37°C
for 20 min. Cells were washed three times with serum-free cell
culture medium to sufficiently remove DCFH-DA that had not entered
the cells (ROS kit, cat. no. s0033; Beyotime Institute of
Biotechnology). Analysis was conducted using a flow cytometer and
CellQuest software (version 5.1).
Animal models
The present study was approved by the Animal
Experimental Ethics Committee of the State Key Laboratory of
Biotherapy, Sichuan University (Sichuan, China). A total of 20
female BALB/c mice (6–8 weeks old, 18–20 g) were obtained from the
Vital River Laboratory Animal Technology (Beijing, China), and
housed in a specific pathogen-free environment (temperature,
21±2°C; humidity, 40–70%; light cycle, 12/12 h; free access to food
and water). A total of 5×105 CT26 tumor cells were
injected into the abdominal cavity of mice. Tumor-bearing mice were
divided into two groups (n=10) and treatment began at 3 days
post-inoculation. Mice in the THD group were administered 100 mg/kg
THD every day by gavage. Mice in the control group received normal
saline. The effect of THD on mice was evaluated at day 7 based on
body weight, ascites volume and tumor weight.
Immunohistochemical staining
Fresh tumor nodules were harvested, flash frozen and
sliced into 4–8 µm sections. Frozen sections were treated with 3%
hydrogen peroxide in methanol for 20 min to quench endogenous
peroxidase activity. Subsequent to washing in PBS (5 min, 3 times;
pH 7.6), sections were blocked with 10% normal goat serum (cat. no.
s8080; Solarbio Science and Technology Co., Ltd., Beijing, China)
for 10 min. Sections were subsequently incubated with rabbit
monoclonal antibody against VEGFR1 (ab32152, 1:250 dilution; Abcam)
overnight at 4°C, and then washed in PBS (5 min, 3 times; pH 7.6).
The washed sections were incubated with biotinylated goat
anti-rabbit IgG (1:100 dilution, cat. no. ZB-2301; OriGene
Technologies, Inc., Beijing, China) for 30 min followed by a
streptavidin-peroxidase conjugate for 30 min at room temperature. A
solution of 0.02% diaminobenzidine hydrochloride containing 0.03%
hydrogen peroxide was used as chromogen to visualize peroxidase
activity at room temperature for 5–8 min. The preparations were
counterstained with hematoxylin at room temperature for 30 sec to 2
min, dried, mounted and examined using light microscopy.
Statistical analysis
All statistical analyses were performed using
GraphPad Prism 6.01 software (GraphPad Software, Inc., La Jolla,
CA, USA). Data analysis was performed using the one-way analysis of
variance and Dunnett's test for multiple groups. The Student's
t-test was used for the analysis of two groups. All data were
presented as the mean ± standard deviation of three independent
experiments. P<0.05 was considered to indicate a statistically
significant difference.
Results
In vitro effects of THD on the growth
of CT26 tumor cells and HUVECs
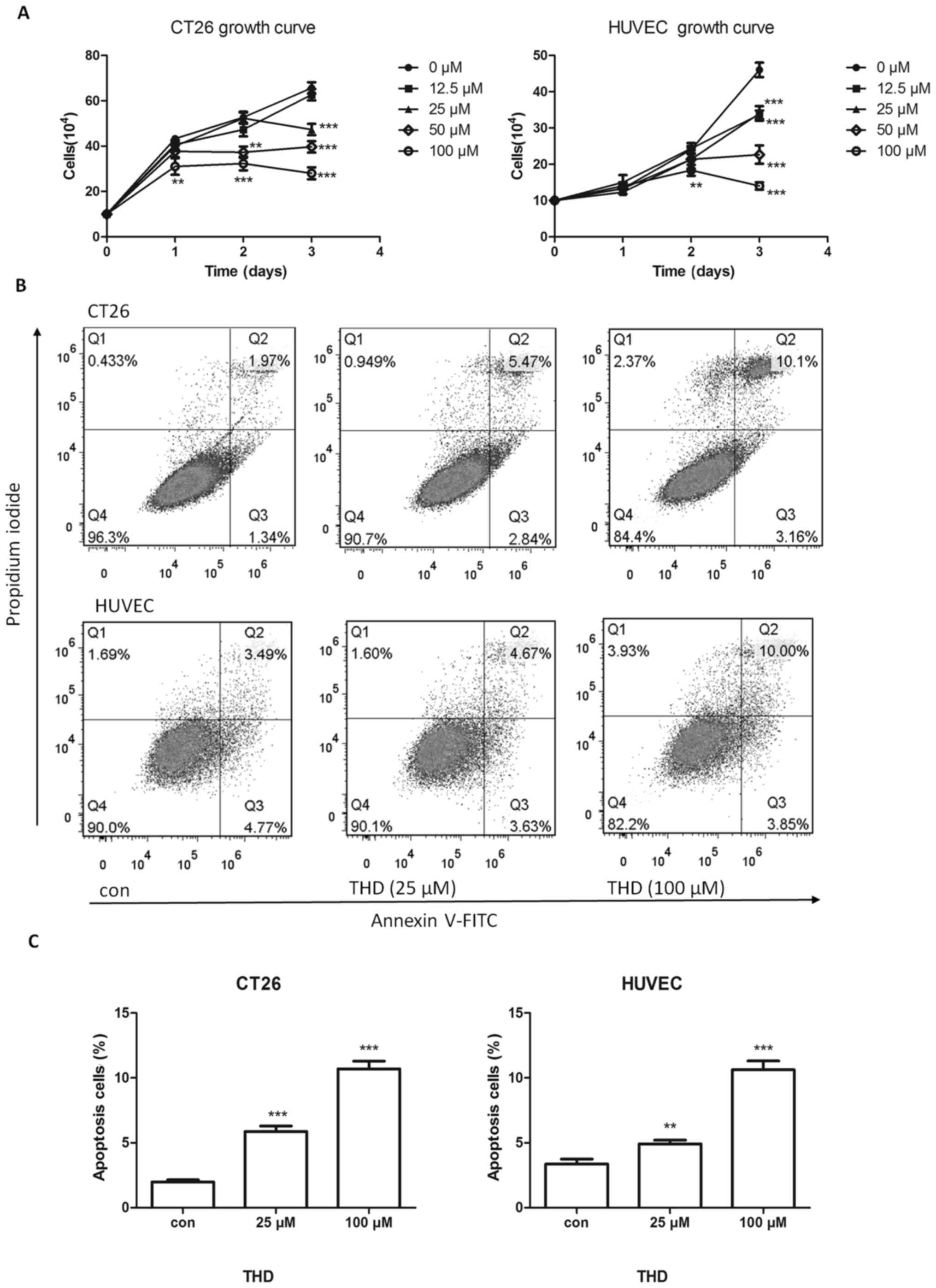
The effect of THD on murine CT26 colorectal tumor
cells in vitro was evaluated. CT26 tumor cells were treated
with various concentrations (0, 12.5, 25, 50 and 100 µM) of THD for
24, 48 and 72 h. The number of surviving tumor cells was
determined. Results indicated (P<0.001; Fig. 1A).
As THD has antiangiogenic properties, and
endothelial cells have a key role in angiogenesis (32,33), the
effect of THD on HUVECs was evaluated. The results revealed that
THD inhibited the proliferation of HUVECs compared with the control
group (Fig. 1A).
The effect of THD on the apoptosis of CT26 cells and
HUVECs was determined using an Annexin V/PI assay. The apoptosis
rate was increased following exposure to THD for 24 h in a
dose-dependent manner (Fig. 1B) and
this difference was statistically significant (P<0.01; Fig. 1C). Collectively, these data revealed
that THD had a growth-arresting and apoptosis-inducing effect on
tumor cells and endothelial cells.
Effect of THD on the cell cycle and
apoptosis-associated proteins
The effect of THD on cell cycle and
apoptosis-associated molecules was investigated. CT26 tumor cells
and HUVECs were exposed to different concentrations of THD for 24
h. Flow cytometry revealed that THD arrested the cell cycle at the
G0/G1 phase in CT26 tumor cells, and at the S phase in HUVECs
(Fig. 2A).
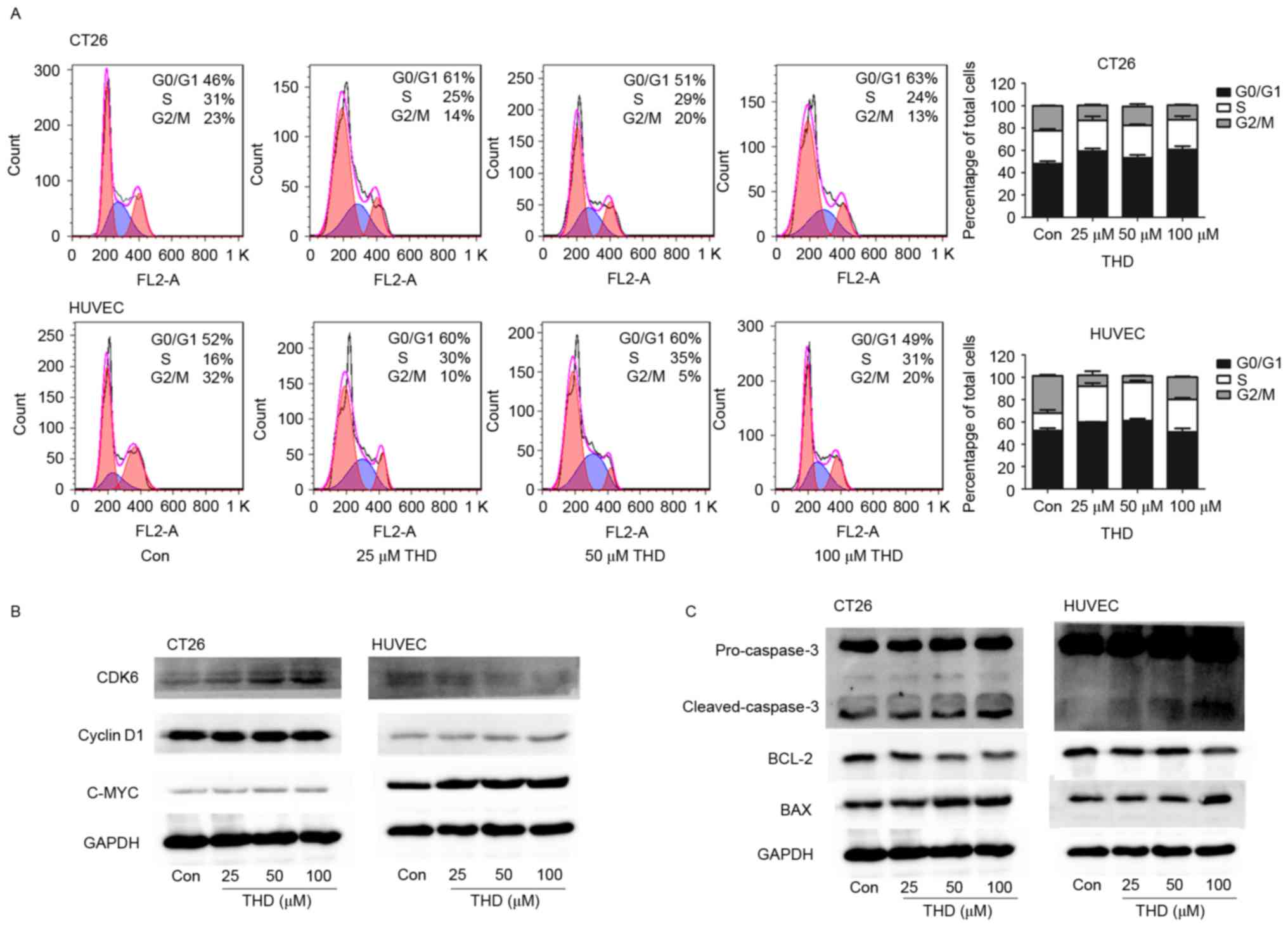 | Figure 2.Effect of THD on cell cycle and
apoptosis-associated proteins. (A) CT26 tumor cells and HUVECs were
incubated with increasing concentrations of THD for 24 h. Cell
cycle analysis was performed using flow cytometry. (B and C)
Expression of the CDK6, cyclin D1, C-MYC, cleaved-caspase-3, BCL-2
and BAX proteins in the CT26 cells and HUVECs was determined by
western blot analysis. Data are presented as the mean ± standard
deviation. THD, thalidomide; Con, control; HUVECs, human umbilical
vein endothelial cells; CDK6, cyclin dependent kinase 6; BCL-2,
B-cell lymphoma-2; BAX, BCL-2-associated X protein. |
The protein expression levels of CDK6, cyclin D1,
C-MYC, BCL-2, BAX and cleaved-caspase 3 were examined by western
blot analysis. CDK6 is a protein kinase that activates cell
proliferation and is associated with restriction in the cell cycle
(34). Cyclin D1 protein required
for progression through the G1 phase of the cell cycle; it is
synthesized rapidly and accumulates in the nucleus during the G1
phase and is degraded as the cell enters the S phase (35). The protein product of the
proto-oncogene C-MYC is a transcription factor that regulates a
range of cellular processes that serve a key role in cell
proliferation, most notably in the regulation of G1 specific cyclin
dependent kinases (36). The results
indicated that in CT26 cells the protein expression levels of CDK6
were markedly upregulated, whereas the protein expression levels of
cyclin D1 exhibited no notable change following THD treatment
compared with the control (Fig. 2B).
However, exposure of HUVECs to THD downregulated CDK6 protein
expression levels as the concentration increased. Conversely, the
protein expression levels of cyclin D1 were only marginally
increased as a result of THD exposure compared with the control
group (Fig. 2B). Furthermore, the
protein expression levels of C-MYC were markedly increased in the
two cell lines, particularly in HUVECs, following THD treatment
(Fig. 2B). BCL-2 is an
anti-apoptotic protein, whereas BAX promotes apoptosis (37). In the present study, the protein
expression levels of anti-apoptotic protein BCL-2 were
downregulated with increased THD concentration, whereas the protein
expression levels of pro-apoptotic protein BAX and
cleaved-caspase-3 were upregulated compared with the control group
(Fig. 2C). These findings suggested
that THD alters the protein expression levels of cell cycle and
apoptosis-associated molecules.
Effect of THD on VEGFR1 expression
levels in vitro
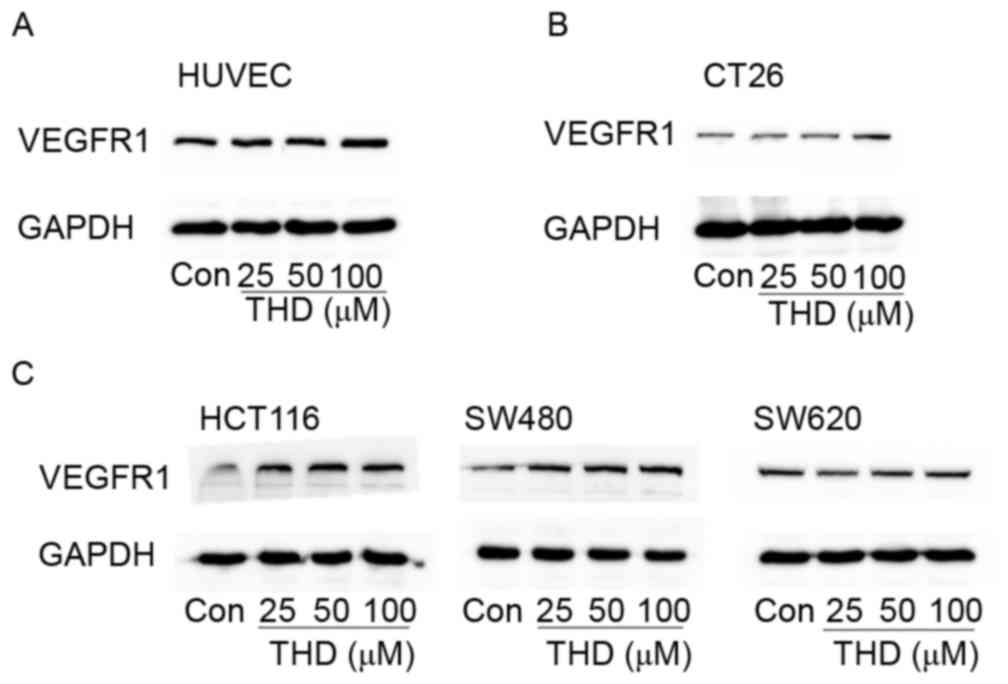
To determine whether THD influenced the protein
expression levels of VEGR1 in endothelial cells, HUVECs were
exposed to different concentrations (0, 25, 50 and 100 µM) of THD
for 24 h. Results indicated that THD at a high concentration (100
µM) markedly upregulated the protein expression levels of VEGFR1
compared with the control group (Fig.
3A). Subsequently, this phenomenon was assessed in tumor cells.
The CT26 tumor cells treated with THD exhibited clearly upregulated
VEGFR1 expression levels compared with the control group (Fig. 3B). Furthermore, similar phenomena
were observed in the human colorectal cell lines SW480 and HCT116.
In the SW620 cell line, no obvious differences in VEGFR1 expression
were observed between different dosages of THD. It may be that
VEGFR1 expression is only affected by higher concentrations of THD
(Fig. 3C). These data suggest that
THD upregulates VEGFR1 protein expression levels in
vitro.
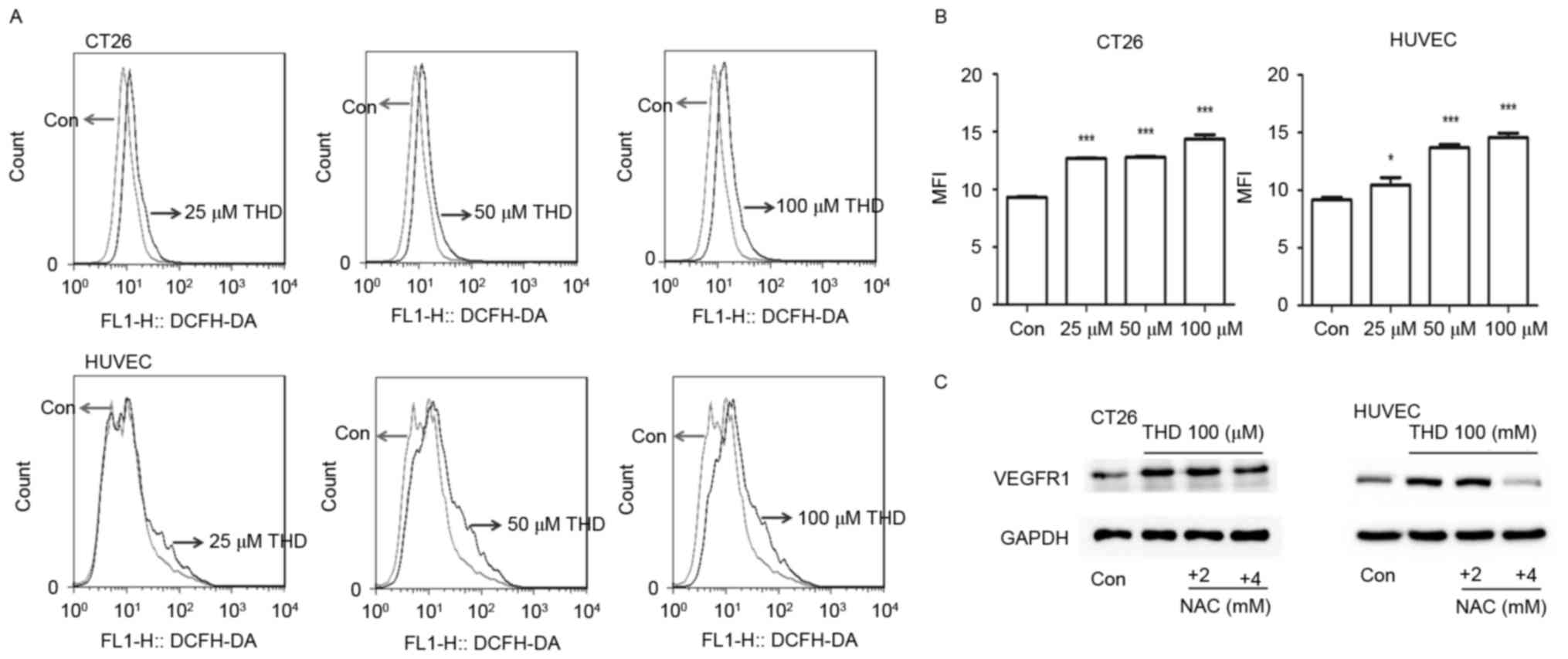
Effect of THD on ROS and its
association with VEGFR1
Considering the cytotoxic effect of THD on CT26
tumor cells and HUVECs, and the upregulation of VEGFR1 observed
in vitro, it was speculated that the upregulation of VEGFR1
may be induced by cellular stress caused by THD. The levels of
intracellular ROS were subsequently analyzed using the probe
DCFH-DA. ROS levels were significantly increased in the two cell
types following exposure to THD (Fig. 4A
and B). Notably, the ROS antagonist NAC reversed VEGFR1
elevation to some extent in CT26 cells and HUVECs (Fig. 4C). These results suggested that the
elevation of VEGFR1 was associated with oxidative cellular
stress.
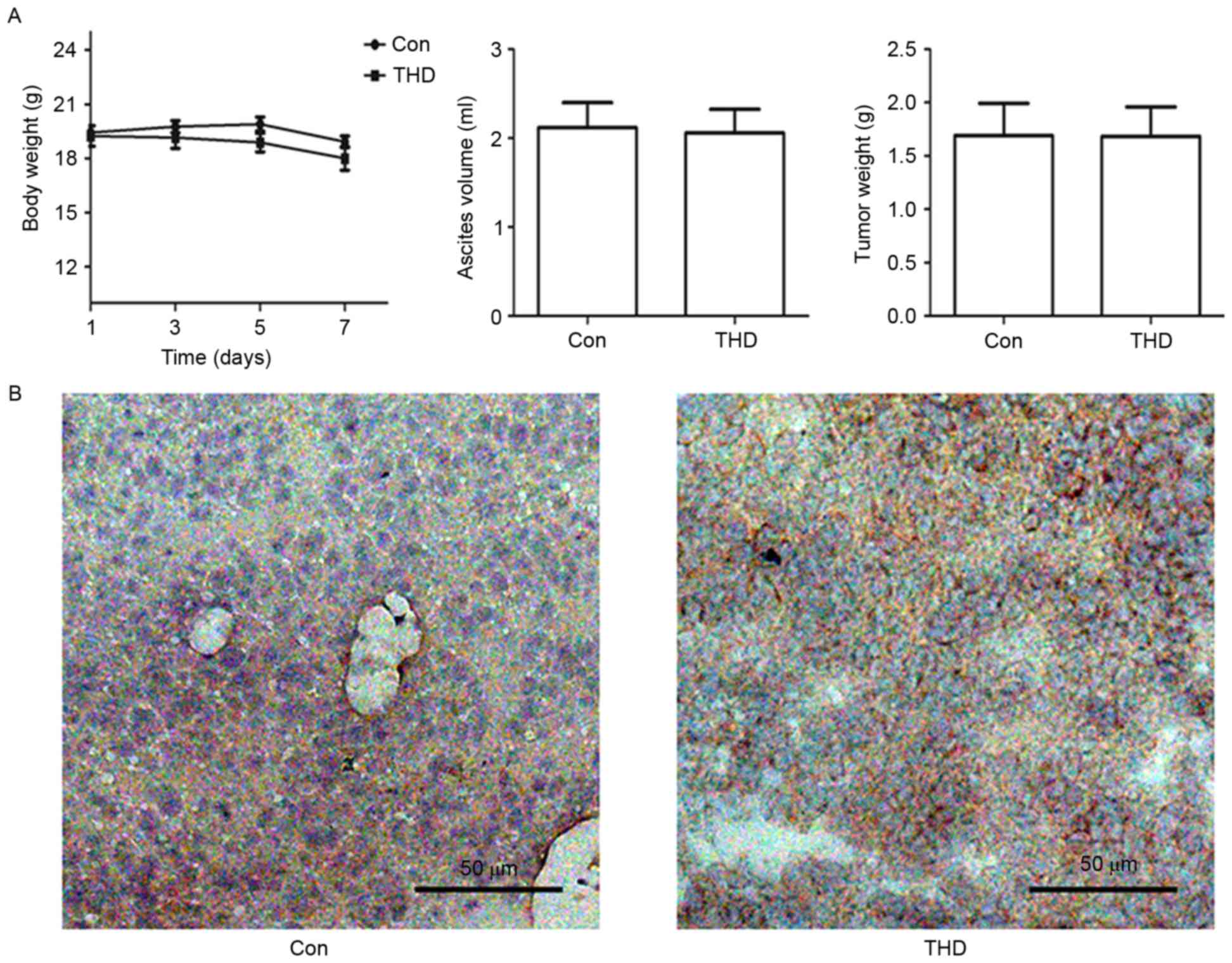
Effect of THD on VEGFR1 expression in
vivo
To determine the effect of THD upregulation on
VEGFR1 protein expression levels in vivo, CT26 cells were
injected into the abdominal cavity of BALB/c mice. The antitumor
effect of THD was evaluated based on body weight, ascites volume
and tumor weight over the course of 7 days. There was no
significant difference in body weight, ascites volume or tumor
weight between the THD group and control group (P>0.05; Fig. 5A). Furthermore, the protein
expression levels of VEGFR1 were increased in the THD-treated group
compared with the control group according to immunohistochemical
staining analysis (Fig. 5B). These
data suggest that THD failed to inhibit CT26 murine tumor growth,
but upregulated VEGFR1 protein expression levels in
vivo.
VEGFR1 is a stress-inducible
molecule
In order to further determine whether VEGFR1 is a
stress-inducible molecule, VEGFR1 expression levels were assessed
in cells subjected to various stress-related situations. CT26 cells
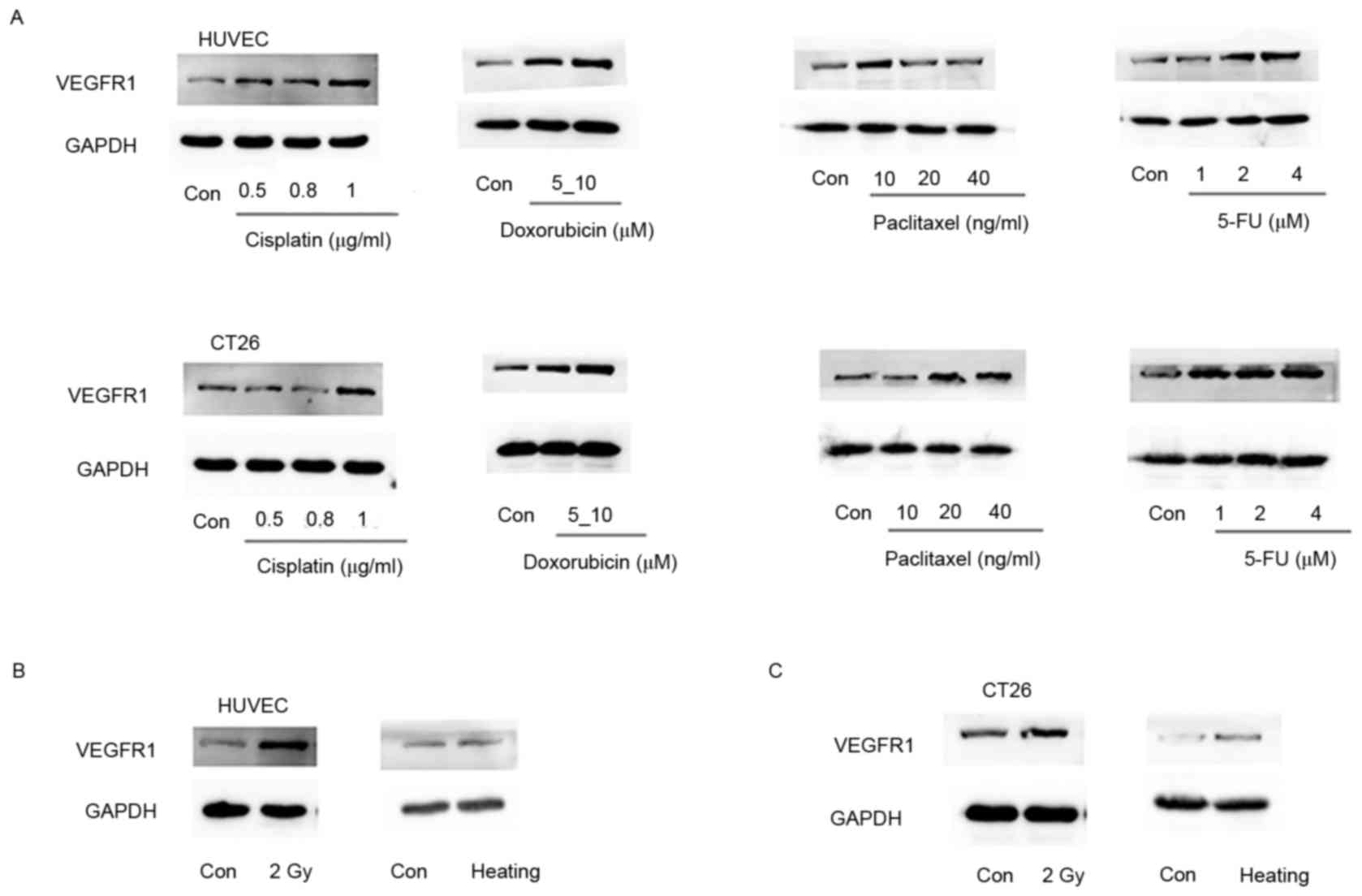
and HUVECs were exposed to chemotherapeutic drugs, including
cisplatin, doxorubicin, paclitaxel and 5-FU, respectively. VEGFR1
expression levels were mostly upregulated following exposure to
these chemotherapeutic agents compared with the respective control
groups. However VEGFR1 in HUVECs for 20 and 40 ng/ml paclitaxel was
lower than for 10 ng/ml. It was hypothesized that an appropriate
dosage may produce a stress response and increase the expression of
VEGFR1, whereas too high a dosage would increase cell apoptosis.
VEGFR1 expression did not increase in HUVECs treated with 20 or 40
ng/ml paclitaxel. (Fig. 6A).
Furthermore, stress was induced in CT26 cells and HUVECs by
irradiation with 2 Gy X-rays. VEGFR1 expression levels were
markedly upregulated following irradiation compared with the
control group (Fig. 6B and C).
Additionally, cellular stress was induced in CT26 tumor cells and
HUVECs by heating the cells at 47°C for 3 min. VEGFR1 protein
expression levels were increased in comparison with those in with
the control group (Fig. 6B and C).
Together, the data suggest VEGFR1 may be a stress-inducible
molecule.
Discussion
In the present study, the growth-arresting and
apoptosis-inducing effects of THD on tumor cells and endothelial
cells were determined. Notably, VEGFR1 protein expression levels
were upregulated in response to high concentrations of THD in
vitro and in vivo. Furthermore, results suggested that
the levels of ROS were increased in response to THD exposure, and
the inhibition of ROS reduced VEGFR1 protein expression levels to
an extent. Thus, we postulated that VEGFR1 may act as a
stress-associated protein, and the results of further experiments
indicated that VEGFR1 protein expression levels were elevated in
response to various stress-associated situations, including
chemotherapy, radiotherapy and thermotherapy.
VEGFR1 has been reported to be expressed on
endothelial cells and in various types of cancer cells, including
gastric cancer cells, colorectal carcinoma cells, breast cancer
cells and lymphoma cells (19–23).
However, the biological function of VEGFR1 remains to be fully
elucidated. The present study assessed the effect of THD on HUVECs
in vitro, and indicated that THD elevated the protein
expression levels of VEGFR1. This upregulation of VEGFR1 protein
expression was also observed on CT26 tumor cells. Similar phenomena
were also observed in the SW480 and HCT116 human colorectal tumor
cells. Furthermore, in the constructed in vivo murine model,
THD failed to inhibit tumor growth and the protein expression
levels of VEGFR1 were elevated by THD treatment. The extent of the
apoptosis induced by THD treatment in CT26 cells and HUVECs
suggests that the upregulation of VEGFR1 could be due to cellular
stress caused by THD. The biosynthesis and accumulation of ROS,
including superoxide anion radicals, hydrogen peroxide, hydroxyl
radicals and oxygen, are central to oxidative stress-associated
metabolism (38). The results of the
present study suggested that exposure to THD increased
intracellular ROS levels. Pretreatment of CT26 tumor cells with the
antioxidant NAC prior to exposure to THD reversed the THD-induced
elevation of VEGFR1 protein expression. Several stress-associated
conditions for cells were explored in the present study, including
chemotherapeutic agents, irradiation and heat shock. Exposure to
these factors upregulated the protein expression levels of VEGFR1
in CT26 cells and HUVECs. These results indicate that the increased
cell surface expression of VEGFR1 may be a cellular stress
response.
Cellular stress responses, which are defense
reactions, are an important part of physiology to either ensure
cell survival or eliminate damaged or unwanted cells (39). Depending on the type of cellular
stress and its severity, there are four primary types of response,
including the heat shock response, the unfolded protein response,
the DNA damage response and the response to oxidative stress
(40). Stress-associated proteins
have protective effects on cell survival (40). Well-known stress proteins are the
heat shock protein (HSP) family. HSPs protect cells against damage
in stressful conditions, which may facilitate tissue homeostasis
and tissue regeneration (40). HSP70
enhances the survival of fibroblasts and promotes recovery from
heat shock (41). HSP70 enhancement
has a critical role in the recovery of striated muscle
post-exercise (42). In a zebrafish
model, treatment with HSP inhibitor I inhibited axonal elongation
or visual function following injury (43). HSPs attenuate cellular apoptosis
(44). HSP27 protects tumor cells
from ultraviolet-induced apoptosis via the Akt and p21 signaling
pathways (45). Increased HSP
expression is associated with chemoresistance (46). High levels of HSP90AA1 have been
demonstrated to increase chemoresistance to the chemotherapeutic
agent cisplatin in SKOV3 cells (46). Furthermore, HSPs are upregulated in
various tumors, including lung cancer (47), prostate cancer (48), breast cancer (49) and gastric cancer (50). They are typically associated with a
poor prognosis (51); thus, they may
provide a potential molecular target in cancer therapy (52). A previous study indicated that
VEGF165 promotes the survival of leukemia cells via the
HSP90-mediated induction of BCL-2 expression and apoptosis
inhibition (53). However, further
investigations are required to determine whether VEGFR1 serves a
similar role, and protects cells from apoptosis.
In conclusion, the present study indicated a novel
pharmacological property of THD and the results suggested that
VEGR1 may be a stress-inducible molecule. The findings provide a
basis for future investigations into the application of
VEGFR1-targeted therapy to enhance the efficacy of current
therapies.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81501609
and 81272523).
References
|
1
|
McBride WG: Thalidomide embryopathy.
Teratology. 16:79–82. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kim JH and Scialli AR: Thalidomide: The
tragedy of birth defects and the effective treatment of disease.
Toxicol Sci. 122:1–6. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Melchert M and List A: The thalidomide
saga. Int J Biochem Cell Biol. 39:1489–1499. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ito T, Ando H and Handa H: Teratogenic
effects of thalidomide: Molecular mechanisms. Cell Mol Life Sci.
68:1569–1579. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Singhal S, Mehta J, Desikan R, Ayers D,
Roberson P, Eddlemon P, Munshi N, Anaissie E, Wilson C, Dhodapkar
M, et al: Antitumor activity of thalidomide in refractory multiple
myeloma. N Engl J Med. 341:1565–1571. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
de Souza CM, Silva Araújo e AC, de Jesus
Ferraciolli C, Moreira GV, Campos LC, dos Reis DC, Lopes MT,
Ferreira MA, Andrade SP and Cassali GD: Combination therapy with
carboplatin and thalidomide suppresses tumor growth and metastasis
in 4T1 murine breast cancer model. Biomed Pharmacother. 68:51–57.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee SM and Hackshaw A: A potential new
enriching trial design for selecting non-small-cell lung cancer
patients with no predictive biomarker for trials based on both
histology and early tumor response: Further analysis of a
thalidomide trial. Cancer Med. 2:360–366. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lv J, Liu N, Liu KW, Ding AP, Wang H and
Qiu WS: A randomised controlled phase II trial of the combination
of XELOX with thalidomide for the first-line treatment of
metastatic colorectal cancer. Cancer Biol Med. 9:111–114.
2012.PubMed/NCBI
|
|
9
|
Rezvani H, Haghighi S, Ghadyani M and
Attarian H: Efficacy of taxotere, thalidomide, and prednisolone in
patients with hormone-resistant metastatic prostate cancer. Urol J.
9:673–677. 2012.PubMed/NCBI
|
|
10
|
Tunio MA, Hashmi A, Qayyum A, Naimatullah
N and Masood R: Low-dose thalidomide in patients with metastatic
renal cell carcinoma. J Pak Med Assoc. 62:876–879. 2012.PubMed/NCBI
|
|
11
|
de Almeida MV, Teixeira FM, de Souza MV,
Amarante GW, Alves CC, Cardoso SH, Mattos AM, Ferreira AP and
Teixeira HC: Thalidomide analogs from diamines: Synthesis and
evaluation as inhibitors of TNF-alpha production. Chem Pharm Bull
(Tokyo). 55:223–226. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dmoszynska A, Podhorecka M, Manko J,
Bojarska-Junak A, Rolinski J and Skomra D: The influence of
thalidomide therapy on cytokine secretion, immunophenotype, BCL-2
expression and microvessel density in patients with resistant or
relapsed multiple myeloma. Neoplasma. 52:175–181. 2005.PubMed/NCBI
|
|
13
|
Marriott JB, Clarke IA, Czajka A, Dredge
K, Childs K, Man HW, Schafer P, Govinda S, Muller GW, Stirling DI
and Dalgleish AG: A novel subclass of thalidomide analogue with
anti-solid tumor activity in which caspase-dependent apoptosis is
associated with altered expression of bcl-2 family proteins. Cancer
Res. 63:593–599. 2003.PubMed/NCBI
|
|
14
|
Gutman M, Szold A, Ravid A, Lazauskas T,
Merimsky O and Klausner JM: Failure of thalidomide to inhibit tumor
growth and angiogenesis in vivo. Anticancer Res. 16:3673–3677.
1996.PubMed/NCBI
|
|
15
|
Fantozzi A, Gruber DC, Pisarsky L, Heck C,
Kunita A, Yilmaz M, Meyer-Schaller N, Cornille K, Hopfer U,
Bentires-Alj M and Christofori G: VEGF-mediated angiogenesis Links
EMT-induced cancer stemness to tumor Initiation. Cancer Res.
74:1566–1575. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Takahashi S: Vascular endothelial growth
factor (VEGF), VEGF receptors and their inhibitors for
antiangiogenic tumor therapy. Biol Pharm Bull. 34:1785–1788. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fiorelli A, Vicidomini G, Di Domenico M,
Napolitano F, Messina G, Morgillo F, Ciardiello F and Santini M:
Vascular endothelial growth factor in pleural fluid for
differential diagnosis of benign and malignant origin and its
clinical applications. Interact Cardiovasc Thorac Surg. 12:420–424.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shibuya M: Structure and dual function of
vascular endothelial growth factor receptor-1 (Flt-1). Int J
Biochem Cell Biol. 33:409–420. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shibuya M: Vascular endothelial growth
factor receptor-1 (VEGFR-1/Flt-1): A dual regulator for
angiogenesis. Angiogenesis. 9:225–231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu H, Zhao C, Liu F, Wang L, Feng J, Zhou
Z, Qu L, Shou C and Yang Z: Radiolabeling and evaluation of
(64)Cu-DOTA-F56 peptide targeting vascular endothelial growth
factor receptor 1 in the molecular imaging of gastric cancer. Am J
Cancer Res. 5:3301–3310. 2015.PubMed/NCBI
|
|
21
|
Lesslie DP, Summy JM, Parikh NU, Fan F,
Trevino JG, Sawyer TK, Metcalf CA, Shakespeare WC, Hicklin DJ,
Ellis LM and Gallick GE: Vascular endothelial growth factor
receptor-1 mediates migration of human colorectal carcinoma cells
by activation of Src family kinases. Br J Cancer. 94:1710–1717.
2006.PubMed/NCBI
|
|
22
|
Ning Q, Liu C, Hou L, Meng M, Zhang X, Luo
M, Shao S, Zuo X and Zhao X: Vascular endothelial growth factor
receptor-1 activation promotes migration and invasion of breast
cancer cells through epithelial-mesenchymal transition. PLoS One.
8:e652172013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang ES, Teruya-Feldstein J, Wu Y, Zhu Z,
Hicklin DJ and Moore MA: Targeting autocrine and paracrine VEGF
receptor pathways inhibits human lymphoma xenografts in vivo.
Blood. 104:2893–2902. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kaplan RN, Riba RD, Zacharoulis S, Bramley
AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, et
al: VEGFR1-positive haematopoietic bone marrow progenitors initiate
the pre-metastatic niche. Nature. 438:820–827. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qian BZ, Zhang H, Li J, He T, Yeo EJ,
Soong DY, Carragher NO, Munro A, Chang A, Bresnick AR, et al: FLT1
signaling in metastasis-associated macrophages activates an
inflammatory signature that promotes breast cancer metastasis. J
Exp Med. 212:1433–1448. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kosaka Y, Mimori K, Fukagawa T, Ishikawa
K, Etoh T, Katai H, Sano T, Watanabe M, Sasako M and Mori M:
Identification of the high-risk group for metastasis of gastric
cancer cases by vascular endothelial growth factor receptor-1
overexpression in peripheral blood. Br J Cancer. 96:1723–1728.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou Z, Zhao C, Wang L, Cao X, Li J, Huang
R, Lao Q, Yu H, Li Y, Du H, et al: A VEGFR1 antagonistic peptide
inhibits tumor growth and metastasis through VEGFR1-PI3K-AKT
signaling pathway inhibition. Am J Cancer Res. 5:3149–3161.
2015.PubMed/NCBI
|
|
28
|
An P, Lei H, Zhang J, Song S, He L, Jin G,
Liu X, Wu J, Meng L, Liu M and Shou C: Suppression of tumor growth
and metastasis by a VEGFR-1 antagonizing peptide identified from a
phage display library. Int J Cancer. 111:165–173. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ye L, Yu G, Wang C, Du B, Sun D, Liu J, Qi
T, Yu X, Wei W, Cheng J and Jiang Y: MicroRNA-128a, BMI1 polycomb
ring finger oncogene and reactive oxygen species inhibit the growth
of U-87 MG glioblastoma cells following exposure to X-ray
radiation. Mol Med Rep. 12:6247–6254. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen H, Ye H, Meng DQ, Cai PC, Chen F, Zhu
LP, Tang Q, Long ZX, Zhou Q, Jin Y, et al: Reactive oxygen species
and X-Ray disrupted spontaneous [Ca2+]I oscillation in
alveolar macrophages. Radiat Res. 179:485–492. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gu ZT, Wang H, Li L, Liu YS, Deng XB, Huo
SF, Yuan FF, Liu ZF, Tong HS and Su L: Heat stress induces
apoptosis through transcription-independent p53-mediated
mitochondrial pathways in human umbilical vein endothelial cell.
Sci Rep. 4:44692014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Komorowski J, Jerczyńska H, Siejka A,
Barańska P, Ławnicka H, Pawłowska Z and Stepień H: Effect of
thalidomide affecting VEGF secretion, cell migration, adhesion and
capillary tube formation of human endothelial EA.hy 926 cells. Life
Sci. 78:2558–2563. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Aydoğan S, Celiker U, Türkçüoğlu P, Ilhan
N and Akpolat N: The effect of thalidomide on vascular endothelial
growth factor and tumor necrosis factor-alpha levels in retinal
ischemia/reperfusion injury. Graefes Arch Clin Exp Ophthalmol.
246:363–368. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kozar K and Sicinski P: Cell cycle
progression without cyclin D-CDK4 and cyclin D-CDK6 complexes. Cell
Cycle. 4:388–391. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Baldin V, Lukas J, Marcote MJ, Pagano M
and Draetta G: Cyclin D1 Is a nuclear-protein required for cell
cycle progression in G1. Gene Dev. 7:812–821. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Robson S, Pelengaris S and Khan M: c-Myc
and downstream targets in the pathogenesis and treatment of cancer.
Recent Pat Anticancer Drug Discov. 1:305–326. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Qiao Z, Yuan J, Shen J, Wang C, He Z, Hu
Y, Zhang M and Xu C: Effect of thalidomide in combination with
gemcitabine on human pancreatic carcinoma SW-1990 cell lines in
vitro and in vivo. Oncol Lett. 9:2353–2360. 2015.PubMed/NCBI
|
|
38
|
Gill SS and Tuteja N: Reactive oxygen
species and antioxidant machinery in abiotic stress tolerance in
crop plants. Plant Physiol Biochem. 48:909–930. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chan CJ, Smyth MJ and Martinet L:
Molecular mechanisms of natural killer cell activation in response
to cellular stress. Cell Death Differ. 21:5–14. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fulda S, Gorman AM, Hori O and Samali A:
Cellular stress responses: Cell survival and cell death. Int J Cell
Biol. 2010:2140742010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu RY, Li X, Li L and Li GC: Expression
of human hsp70 in rat fibroblasts enhances cell survival and
facilitates recovery from translational and transcriptional
inhibition following heat shock. Cancer Res. 52:3667–3673.
1992.PubMed/NCBI
|
|
42
|
Noble EG, Melling CW and Milne KJ: HSP,
Exercise and Skeletal Muscle. Heat Shock Proteins Whole Body
Physiol. 5:285–316. 2010. View Article : Google Scholar
|
|
43
|
Nagashima M, Fujikawa C, Mawatari K, Mori
Y and Kato S: HSP70, the earliest-induced gene in the zebrafish
retina during optic nerve regeneration: Its role in cell survival.
Neurochem Int. 58:888–895. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Beere HM: ‘The stress of dying’: the role
of heat shock proteins in the regulation of apoptosis. J Cell Sci.
117:2641–2651. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kanagasabai R, Karthikeyan K, Vedam K,
Qien W, Zhu Q and Ilangovan G: Hsp27 protects adenocarcinoma cells
from UV-induced apoptosis by Akt and p21-dependent pathways of
survival. Mol Cancer Res. 8:1399–1412. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chu SH, Liu YW, Zhang L, Liu B and Li L,
Shi JZ and Li L: Regulation of survival and chemoresistance by
HSP90AA1 in ovarian cancer SKOV3 cells. Mol Biol Rep. 40:1–6. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wen W, Liu W, Shao Y and Chen L:
VER-155008, a small molecule inhibitor of HSP70 with potent
anti-cancer activity on lung cancer cell lines. Exp Biol Med
(Maywood). 239:638–645. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Azad AA, Zoubeidi A, Gleave ME and Chi KN:
Targeting heat shock proteins in metastatic castration-resistant
prostate cancer. Nat Rev Urol. 12:26–36. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Calderwood SK: Heat shock proteins in
breast cancer progression-a suitable case for treatment? Int J
Hyperthermia. 26:681–685. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Partida-Rodríguez O, Torres J, Flores-Luna
L, Camorlinga M, Nieves-Ramírez M, Lazcano E and Perez-Rodríguez M:
Polymorphisms in TNF and HSP-70 show a significant association with
gastric cancer and duodenal ulcer. Int J Cancer. 126:1861–1868.
2010.PubMed/NCBI
|
|
51
|
Calderwood SK, Khaleque MA, Sawyer DB and
Ciocca DR: Heat shock proteins in cancer: Chaperones of
tumorigenesis. Trends Biochem Sci. 31:164–172. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Staufer K and Stoeltzing O: Implication of
heat shock protein 90 (HSP90) in tumor angiogenesis: A molecular
target for anti-angiogenic therapy? Current Cancer Drug Targets.
10:890–897. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Dias S, Shmelkov SV, Lam G and Rafii S:
VEGF(165) promotes survival of leukemic cells by Hsp90-mediated
induction of Bcl-2 expression and apoptosis inhibition. Blood.
99:2532–2540. 2002. View Article : Google Scholar : PubMed/NCBI
|