Introduction
Hepatic cirrhosis is a common response to chronic
liver injury, during which the normal liver architecture is
distorted by scar tissue and excessive extracellular matrix (ECM)
is deposited (1). Portal
hypertension (PHT) is a severe and frequently occurring
complication of hepatic cirrhosis (2). The interaction of numerous factors in
the environment forms a complex network system that affects the
process of liver cirrhosis and PHT. Increased hepatic vascular
resistance (HVR) to portal blood flow and increased portal
collateral blood flow is the basis of the pathogenesis of PHT
(3). Portal pressure increases in
response to sinusoidal endothelial dysfunction (3). In addition, PHT is caused by vascular
remodeling, which leads to structural and functional alteration of
vessels (4). In vessels, immature
vascular smooth muscle cells (VSMCs) are prone to proliferation,
migration and synthesis of ECM components (5). The apoptotic decrease of portal vein
smooth muscle cells (PVSMCs) in the vein wall is a cause of portal
vein remodeling in PHT (6).
Currently, there are no approved therapeutic options designed to
reverse the progression of PHT. Therefore, it is important to
search for effective methods to inhibit PHT development.
Calcium activated chloride channels (CaCCs) exist in
multiple tissues and serve critical functions in fundamental
physiological processes, including epithelial secretion, cardiac
and neuronal excitation, regulation of smooth muscle contraction,
sensory transduction, nociception and fertilization (7). In 2008, three laboratories used
different approaches to demonstrate that transmembrane protein 16A
(TMEM16A; also known as TAOS2 or ANO1) is a primary component of
CaCCs (8–10). The coding sequence of TMEM16A is
located within the 11q13 amplicon, one of the most frequently
amplified chromosomal regions carrying tumor-related genes with
poor prognosis, such as cyclin D1 (11). In addition to Ca2+, many
factors, including calmodulin, protons, cholesterol,
phosphoinositides and thermal and mechanical stimuli, could
regulate TMEM16A function (7).
TMEM16A dysfunction has been implicated in multiple diseases,
including cancer, hypertension, gastrointestinal motility disorders
and cystic fibrosis (12).
It has been confirmed that TMEM16A is present in
various smooth muscle cells of ear, coronary, aortic and mesenteric
arteries and the portal vein (12).
The level of TMEM16A expression and its activity are significantly
upregulated in hypertension and pulmonary hypertension models
(13,14). To the best of our knowledge, the role
of TMEM16A in portal vein remodeling induced by PHT has not been
studied yet. Numerous investigations have indicated that TMEM16A
mediates tumor progression, including cell proliferation, migration
and invasion (15–17). It has previously been confirmed that
TMEM16A is expressed in PVSMCs (12). Factors that regulate the growth of
VSMCs may contribute to the development of hypertension by
promoting the thickening of vessels (18). Since TMEM16A is regarded as a
regulator of cell proliferation, it is reasonable to assume that
the effect of TMEM16A overexpression on PVSMC proliferation may
influence PHT. Several vasoconstrictors, including urotensin II,
angiotensin II (Ang II) and endothelin, have been indicated to be
involved in the increased HVR in PHT (19). Consequently, an influence of TMEM16A
on these and other pathogenesis pathways should also be considered
and studied. Therefore, the present study aimed to determine
whether TMEM16A could promote proliferation in PVSMCs to aggravate
portal vein remodeling and PHT. The results indicate a functional
role for TMEM16A in portal vein remodeling and PHT.
Materials and methods
Animals
All animal experimental procedures were approved by
the Institutional Animal Care and Use Committee of Wuhan University
(Wuhan, China) and adhered to the ethical guidelines of the
International Association for the Study of Pain (20). A total of 20 male Sprague-Dawley
rats, aged 6–8 weeks old and weighing 180–200 g, were purchased
from the Center for Animal Experiment, Wuhan University (Wuhan,
China) and maintained in specific pathogen-free conditions. The
animals were housed at a constant temperature (20–24°C) and
humidity (45–50%) under a 12-h light/dark cycle, and provided with
food and water ad libitum. Rats were randomly assigned to two
groups: The bile duct ligation (BDL) group and the control group
(10 rats/group). Biliary hepatic cirrhosis was induced by ligation
of the common bile duct. Briefly, rats were anesthetized with 10%
(w/v) chloral hydrate (3.5 ml/kg; Jiangsu Lianshui Pharmaceutical
Co., Ltd., Lianshui, China) through intraperitoneal injection. In
the BDL group, a 1.5 cm midline incision was made and the common
bile duct was located and double ligated with 3.0 mm silk
ligatures. Sham-operated animals in the control group received a
midline incision and manipulation of the common bile duct, without
ligation. All rats in each group survived the procedure. At 8 weeks
following surgery, the animals were anesthetized and sacrificed by
carbon dioxide euthanasia. At the time of death, the BDL was
confirmed to be intact with proximal dilatation of the common bile
duct. Portions of the right and left liver lobes and the portal
vein were fixed in 10% buffered formalin for 24 h at room
temperature and embedded in paraffin for histological
examination.
Histological analysis
Formalin-fixed, paraffin-embedded portions of the
liver lobes and portal vein were cut into 3 mm sections and stained
with hematoxylin and eosin (H&E) as previously described
(21). Briefly, sections were
deparaffinized, hydrated, stained in alum hematoxylin,
differentiated with acid alcohol, washed with tap water, stained
with eosin and dehydrated. Sections were then observed under a
light microscope (BX51; Olympus Corporation, Tokyo, Japan), by a
pathologist who was blinded to the groups. Ten high-power fields
were randomly collected. The thickness of portal vein wall was
quantified using Image J software version 1.45 (National Institutes
of Health, Bethesda, MD, USA).
Immunohistochemistry was performed on sections of
formalin-fixed, paraffin-embedded liver to detect expression of
TMEM16A, extracellular signal-regulated kinase 1 and 2 (ERK1/2),
phosphorylated ERK1/2 (p-ERK1/2). Briefly, the 3-µm-thick sections
were prepared as described above and incubated with 3% hydrogen
peroxide for 10 min at room temperature to block the endogenous
peroxidase activity. The sections were then boiled in sodium
citrate buffer (pH 6.0) to retrieve antigen. The sections were
incubated with goat serum (Wuhan Boster Biological Technology,
Ltd., Wuhan, China) for 15 min at room temperature. TMEM16A
polyclonal antibody (cat. no. sc-69343; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA; 1:500 dilution), ERK1/2 monoclonal antibody
(cat. no. 4695; 1:400 dilution) and p-ERK1/2 monoclonal antibody
(cat. no. 4370; 1:400 dilution) (both, Cell Signaling Technology,
Inc., Danvers, MA, USA) were added and incubated overnight at 4°C.
The following day, the sections were rinsed in PBS three times and
incubated with horseradish-peroxidase conjugated secondary antibody
(cat. no. HAF008; 1:1,000 dilution; R&D Systems, Inc.,
Minneapolis, MN, USA) for 15 min at room temperature. A
3,3′-diaminobenzidine color developing substrate was added and the
sections were examined microscopically for color development for
5–10 min, mounted and visualized under a light microscope (BX51;
Olympus Corporation).
Single PVSMC isolation and
culture
Primary single PVSMCs were isolated as previously
described (22,23). Briefly, the portal vein was dissected
under sterile conditions. The connective tissues surrounding the
outer membrane and endothelial cells of tunica intima were
carefully stripped off under a dissecting microscope. The smooth
muscle layer of the tunica media was separated from the tunica
adventitia with a blunt dissection technique and then cut into
small fragments (1–2 mm3), which were placed in 25
cm3 culture plates (~20/plate). Dulbecco's modified
Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and
1X strength Antibiotic/Antimycotic (all Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) was carefully added to the
culture plates so as not to disturb adhered explants. Culture
plates were placed in a 37°C incubator (5% CO2). Cells
started growing from explants within 1 week and became confluent in
4 weeks. Cells were fixed for 10 min in glacial acetate at room
temperature and immersed in 0.3% H2O2 for 30
min to quench endogenous peroxidase activity at room temperature.
They were then incubated with primary antibodies directed against
smooth muscle α-actin (cat. no. SAB2500963; 1:1,000 dilution;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) at room temperature
for 1 h and secondary antibodies conjugated to FITC (cat. no.
SAB3700002; 1:1,000 dilution; Sigma-Aldrich; Merck KGaA) for 30 min
at room temperature. Morphometry was performed from three random
fields of each slide using the Image Pro Plus software version 6.0
(Media Cybernetics Inc., Rockville, MD, USA). An Olympus-BX53
microscope (Olympus Corporation, Tokyo, Japan) was used at a
magnification of ×200 to observe the slides and calculate the
number of smooth muscle α-actin positive PVSMCs.
Lentivirus infection and plasmid
transfection
Recombinant pEGFP-TMEM16A and pEGFP-N1 GV358
lentivirus plasmids were packaged using Lenti-Easy Packaging mix
(all Shanghai GeneChem Co., Ltd, Shanghai, China) and the virus
titer was determined using methods of fluorescence enumeration, as
previously described (24). Briefly,
PVSMCs were plated on a 6-well plate with DMEM supplemented with
10% fetal bovine serum in a 37°C incubator with 5% CO2
at a density of 1–1.5×105/ml. After 24 h, pEGFP-TMEM16A
or pEGFP-N1 plasmids were transfected into the cells with
Lipofectamine™ 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) in OPTI-MEM I reduced serum medium (Gibco; Thermo
Fisher Scientific, Inc.), according to the manufacturer's protocol.
After 6 h the cells were rinsed with PBS and switched to 5% fetal
bovine serum-containing DMEM. The expression of green fluorescence
protein was observed under an inverted fluorescent microscope
(Olympus-IX71; Olympus Corporation) 3 days later and the
transfection efficiency was calculated. The average rate of
transfection=positive cells number/total number of cells ×100% (one
visual field of the microscope) (25).
Stock solutions (5 mM) of the TMEM16A inhibitor,
T16Ainh-A01 (cat. no. SML0493; Sigma-Aldrich; Merck KGaA) were made
in dimethyl sulfoxide (DMSO; cat. no. D-2650-5; Sigma-Aldrich) and
stored at −20°C. The compound was freshly diluted on the day of the
experiment to a final concentration of 10 µM. DMSO was used at a
final concentration of 1:500 for the vehicle control. Cells were
treated with 10 µM T16Ainh-A01 at 37°C in 5% CO2 for 6
h.
Flow cytometry
Following treatment with plasmid transfection or the
TMEM16A inhibitor, T16Ainh-A01 the cells were detached from the
6-well plates with 0.2% trypsin, harvested, washed twice with PBS
and centrifuged twice at 300 × g for 5 min at 4°C. Then, the
supernatant was discarded and the pellet was resuspended in 400 µl
Annexin V binding buffer (Beyotime Institute of Biotechnology,
Haimen, China) at 20°C for ≥12 h. Cells were subsequently treated
in PBS with RNase A for 30 min at room temperature and stained with
propidium iodide. Flow cytometric analysis was performed using
EPICS XL-MCL™ software (Beckman Coulter, Inc., Brea, CA,
USA) and a FACScan Flow Cytometer (BD Biosciences, Franklin Lakes,
NJ, USA) was used to determine the DNA contents. A concentration of
1–5×106/ml cells were analyzed for each sample, and the
experiment was repeated ≥3 times. The S-phase cell ratio and
proliferation index were calculated at the same time based on the
following equation: S-phase cell
ratio=S/(G0/G1+S+G2/M);
proliferation
index=(S+G2/M)/(G0/G1
+S+G2/M) (26).
Western blotting
Western blotting analysis was performed as
previously described (27). In
brief, Portal vein samples were lysed in RIPA lysis buffer (50
mmol/l Tris-HCl, pH 7.4, 150 mmol/l NaCl, 10 mmol/l
phenylmethylsulfonyl fluoride, 1 mmol/l EDTA, 0.1% SDS, 1% Triton
X-100 and 1% sodium deoxycholate) for 20–30 min on ice. Protein
concentrations were determined using the Lowry protein assay.
Samples were then boiled in loading buffer and separated by 10%
SDS-PAGE, 20 µg of protein was loaded per lane. After
electrophoresis, protein was transferred onto a nitrocellulose
membrane, which was incubated with blocking solution [10% non-fat
dry milk in TBS containing 0.05% Tween-20 (TBST)] for 2 h.
Membranes were immunoblotted with primary antibodies, including
TMEM16A (1:1,000 dilution), ERK 1/2, p-ERK 1/2 (both 1:2,000
dilution) and β-actin (cat. no, ab8229; 1:10,000 dilution; Abcam,
Cambridge, UK) overnight at 4°C. After washing with TBST, the
membranes were incubated with anti-rabbit immunoglobulin conjugated
to horseradish peroxidase secondary antibody (cat. no. sc-362261;
1:1,000 dilution; Santa Cruz Biotechnology, Inc.) for 2 h at room
temperature. The membranes were developed using an enhanced
chemiluminescence western blotting kit (EMD Millipore, Billerica,
MA, USA), then exposed to X-ray film. The bands of interest were
quantified by Image Pro Plus version 6.0 analysis software.
Statistical analysis
Data are presented as the mean ± standard deviation.
SPSS 19.0 software (IBM Corp., Armonk, NY, USA) was used for data
analysis. Statistical analysis was performed using Student's t-test
for the comparison of two groups, or one-way analysis of variance
followed by a Dunnett's test for multiple comparisons. P<0.05
was considered to indicate a statistically significant
difference.
Results
Establishment of the BDL model
First, it was verified that biliary hepatic
cirrhosis and portal vein remodeling rats by BDL had been
established successfully (Fig. 1A).
H&E staining of liver pathological sections indicated that
lymphocyte infiltration and pseudolobuli formation were obvious in
the liver of the BDL group. Normal structures were observed in the
sham-operated group. The portal vein remodeling in PHT was
characterized by thickening of the vein. The thickness of portal
vein was significantly increased in the BDL group compared with the
sham-operated group (P<0.05; Fig.
1A).
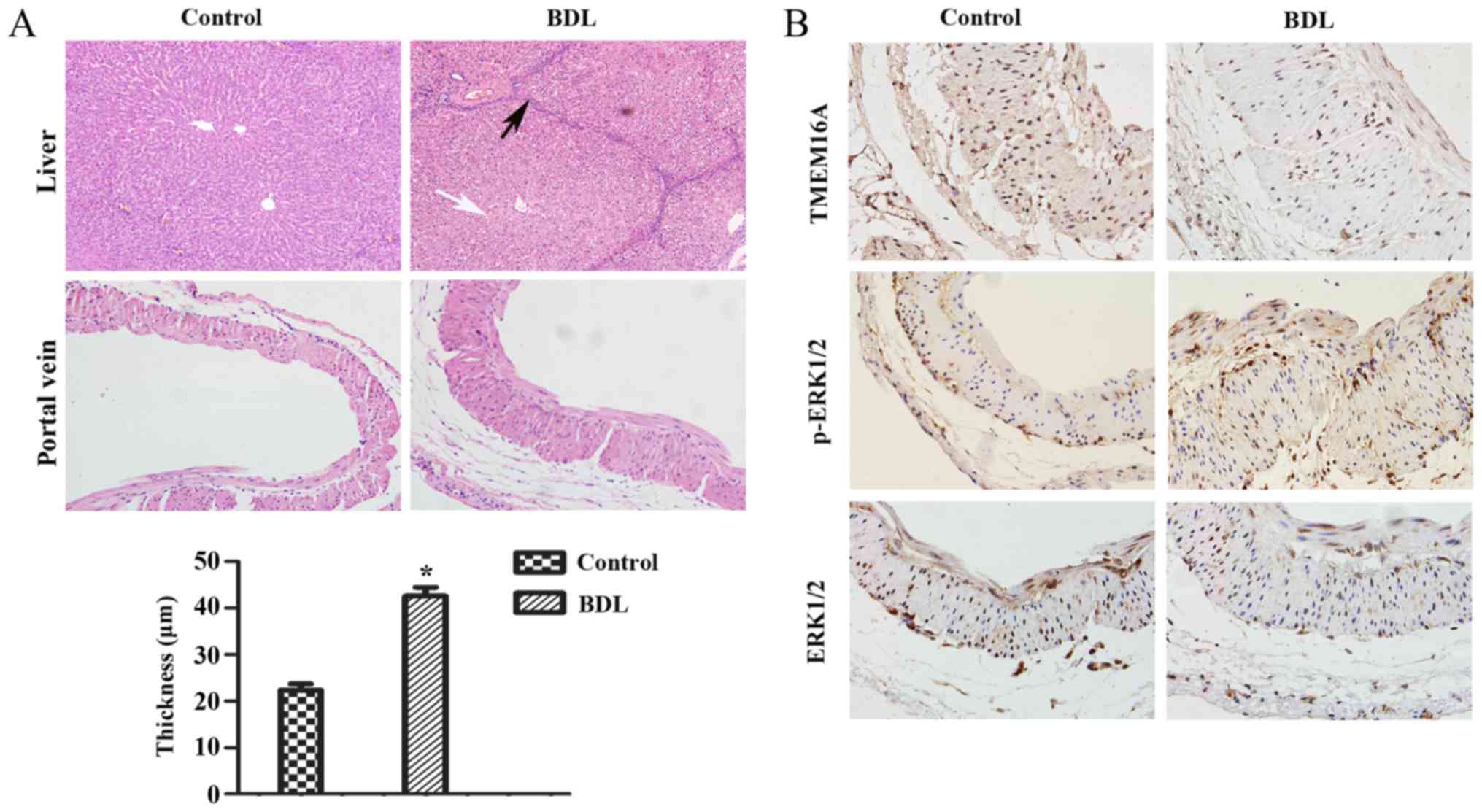 | Figure 1.Pathological alterations and protein
expression levels in the rat cirrhotic liver at 8 weeks after BDL.
(A) The extent of liver fibrosis (magnification, ×200) and the wall
thickness of the portal vein (magnification, ×400) were assessed by
hematoxylin and eosin staining. Data are presented as the mean ±
standard deviation (n=10). Compared with the normal structure of
liver, severe degeneration associated with necrosis were observed
in model group (white arrow), accompanied by inflammatory
infiltration around the portal area, a wide range of hyperplasia in
connective tissues and destruction in lobular structure (black
arrow). Compared with the control group, the thickness of the
portal vein in the model group was increased (21.75±5.56 µm vs.
43.27±9.62 µm). *P<0.05 vs. control group. (B) Protein
expression levels of TMEM16A, p-ERK1/2, ERK1/2 in the portal vein
were visualized by immunohistochemistry. BDL, bile duct ligation;
TMEM16A, transmembrane protein 16A; ERK1/2, extracellular
signal-related kinase 1 and 2; p-ERK1/2, phosphorylated ERK1/2. |
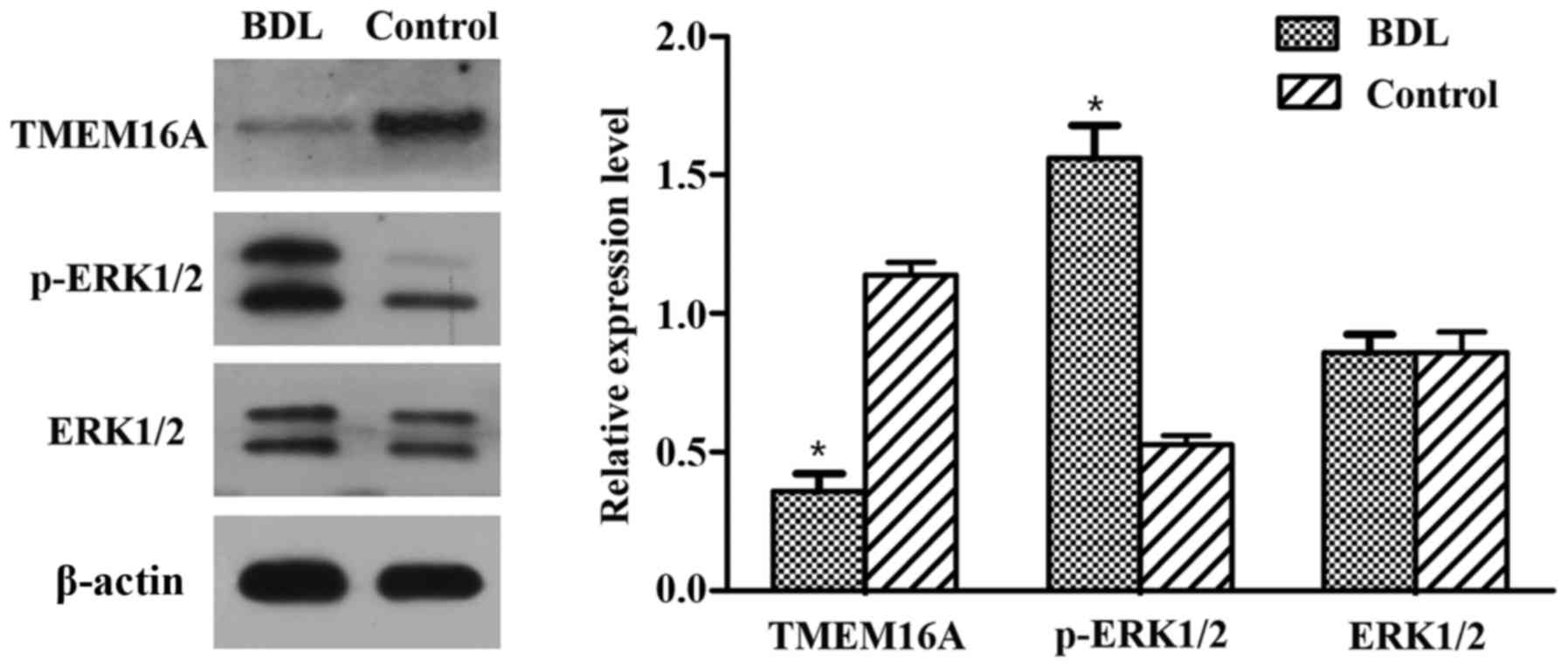
Immunohistochemistry and western blot
analysis of TMEM16A and p-ERK1/2
Immunohistochemistry results indicated that the
TMEM16A protein expression level was decreased in BDL rats compared
with the control (Fig. 1B). Western
blot analysis indicated that the protein expression level of
TMEM16A was significantly decreased in BDL rats (P<0.05;
Fig. 2). p-ERK1/2 expression was
indicated to be increased in BDL rats in the immunohistochemistry
results (Fig. 1B) and significantly
increased in the results of western blot analysis (P<0.05;
Fig. 2). These results suggested
that TMEM16A may have a negative association with PVSMC
proliferation and portal vein remodeling.
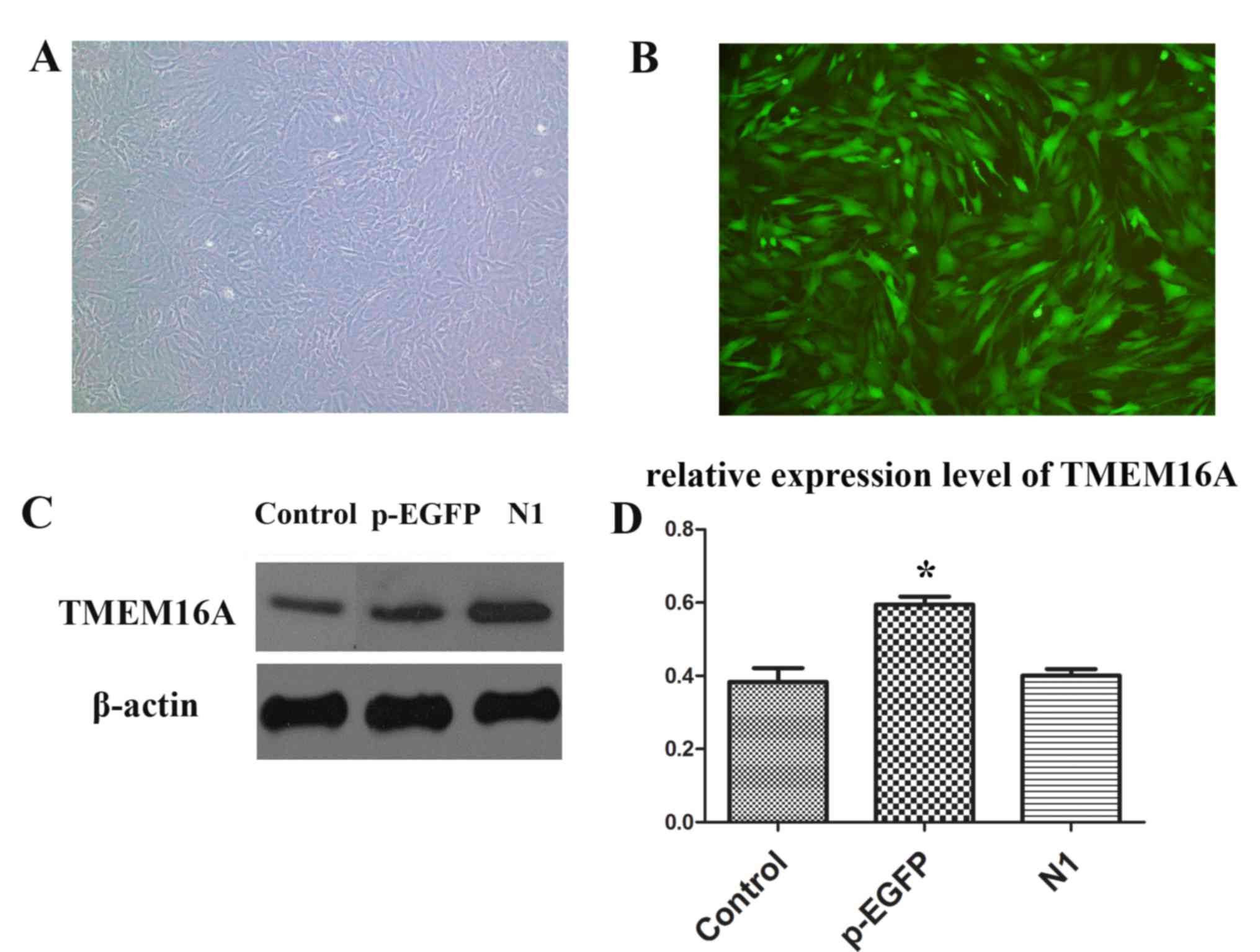
PVSMC isolation and transfection
PVSMCs were successfully isolated and cultured.
Then, pEGFP-TMEM16A and pEGFP-N1 plasmids were used to transfect
PVSMCs (Fig. 3A and B). The
expression of TMEM16A was significantly upregulated in the
pEGFP-TMEM16A transfection plasmids group compared with the control
and pEGFP-N1 plasmids groups (Fig. 3C
and D).
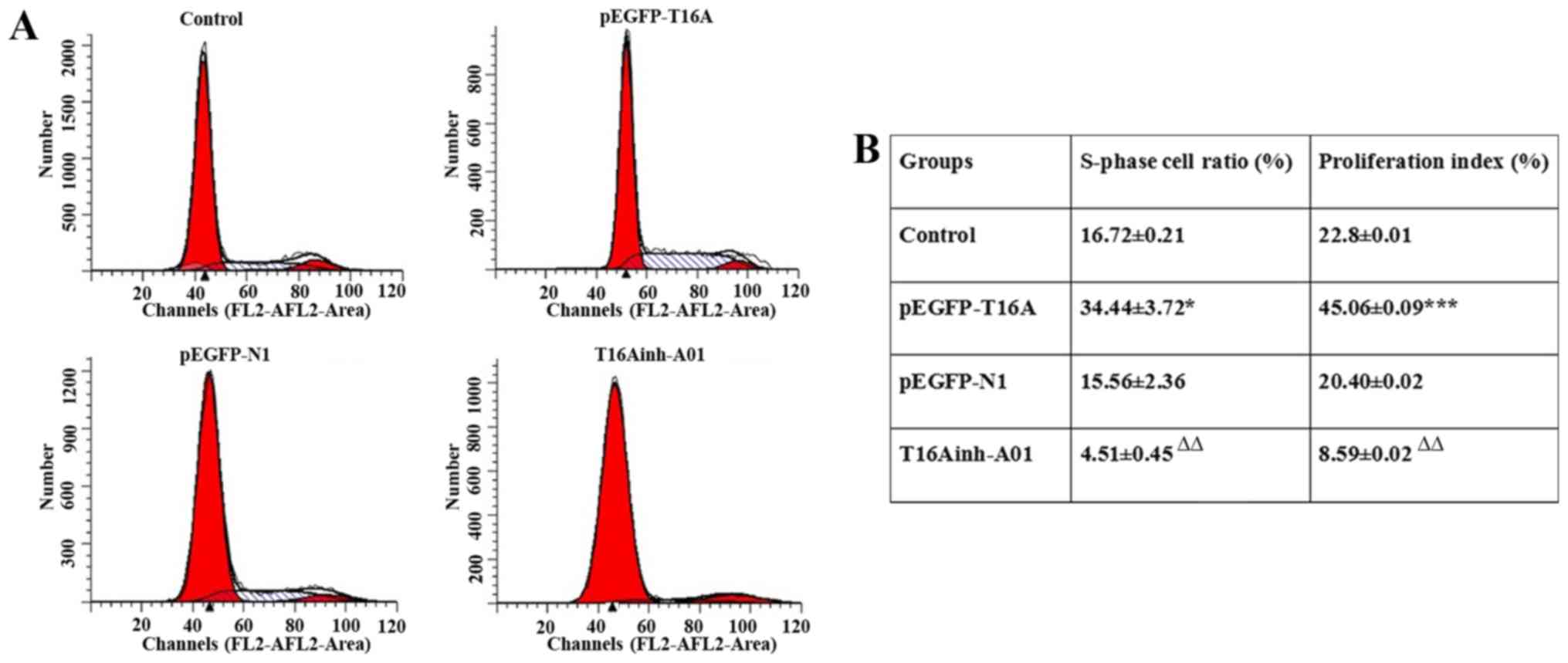
Flow cytometry
The expression of TMEM16A was upregulated by
pEGFP-TMEM16A transfection plasmids as described above and CaCCs
were inhibited by T16Ainh-A01 (10 µM) as described previously
(28). Compared with the negative
control pEGFP-N1 plasmid group, the S rate (34.44±3.72 vs.
15.56±2.36; P<0.05) and proliferation index (45.06±0.09 vs.
20.40±0.02; P<0.001) of pEGFP-TMEM16A plasmid-transfected cells
were significantly increased (Fig.
4). The S rate (4.51±0.45 vs. 16.72±0.21) and proliferation
index (8.59±0.02 vs. 22.8±0.01) of T16Ainh-A01 treated cells were
significantly reduced compared with the control group (P<0.01;
Fig. 4). It was identified that
overexpression of TMEM16A facilitated PVSMC proliferation, while
inhibition of TMEM16A reduced PVSMC proliferation. TMEM16A
contributed to PVSMC proliferation in vitro, but in
vivo, it may be a negative regulator of cell proliferation. The
contradictory results in vivo and in vitro require
further research.
Discussion
The primary focus of research into TMEM16A has been
tumorigenesis and cancer progression (16). The fundamental role of TMEM16A in the
generation of slow waves in interstitial cells of Cajal has also
been demonstrated (29).
Furthermore, TMEM16A is expressed in the renal collecting duct
principal cells and may be involved in the physiological regulation
of NaCl transport in vivo (30). TMEM16A can affect Ca2+
entry and VSMC contraction by regulating membrane potentials by
depolarization (31). However, the
association between TMEM16A and PVSMCs in PHT remains unclear.
To the best of our knowledge, the present study is
the first to indicate that TMEM16A is attenuated in the PVSMCs of a
widely used BDL rat model. This suggests that TMEM16A may be
negatively associated with PVSMC proliferation and portal vein
remodeling in PHT. This is consistent with a previous study, which
reported that downregulation of TMEM16A is a major contributing
factor in the remodeling of the wall of cerebral arteries in a
two-kidney, two clip hypertensive rat model (32). However, in the present in
vitro results, it was observed that upregulation of TMEM16A
promoted PVSMC proliferation, while inhibition of TMEM16A reduced
PVSMC proliferation. These results seem to contradict the in
vivo findings.
Experimental and human studies have consistently
indicated that Ang II is involved in the development of cardiac
hypertrophy and pulmonary hypertension (33–36). Ang
II contributes to the proliferation of hepatic stellate cells and
the progression of liver fibrosis (37). Decreased vasodilatory substances,
including nitric oxide and/or excessive production of
vasoconstrictors, including endothelin and Ang II, was reported to
increase HVR (3). Therefore, the
renin Ang II system is involved in the regulation of intrahepatic
vascular resistance in cirrhosis.
Wang et al (32) reported that Ang II suppresses TMEM16A
expression. The study also identified that knockdown of TMEM16A
facilitates and overexpression of TMEM16A inhibits Ang II-induced
cell cycle transition and cell proliferation. Ang II has been
reported to increase proliferation in VSMCs (38) and inhibit TMEM16A expression via
Krüppel-like factor 5 in cultured VSMCs (39). A previous report that the expression
of Ang II is increased in the portal vein and splenic vein in
patients with liver cirrhosis (40)
suggests that the expression of TMEM16A in the portal vein may be
suppressed by increased Ang II in BDL rats.
Recent studies have revealed that TMEM16A promotes
cancer cell proliferation and tumor growth through the
mitogen-activated protein kinase (MAPK)-ERK signal pathway
(16,41). The present results indicated that the
expression of p-ERK1/2 was increased while TMEM16A was decreased,
which is in opposition to the aforementioned TMEM16A-MAPK-ERK
signal pathway. An alternative possibility is that the positive
effect of Ang II on p-ERK1/2 obscured the negative effect of
downregulation of TMEM16A. This is supported by previous evidence
that Ang II induces a substantial and rapid increase in p-ERK1/2
activity (42). In BDL rats, Ang II
may serve a key function in controlling the expression of TMEM16A
and the MAPK-ERK signal pathway. However, the effect of Ang II may
be context-dependent. One group reported that Ang II significantly
enhances TMEM16A expression in VSMCs via the Ang II type 1
receptor-phosphoinositide 3-kinase-Akt pathway (15). Nonetheless, further experiments are
required to confirm whether Ang II is the negative regulator of
TMEM16A expression in portal vein reconstruction.
A future focus for research will be to examine
TMEM16A expression levels in the portal vein of PHT patients to
provide insight into the clinical significance of TMEM16A in PHT
pathogenesis. The present study identified that a CaCC channel,
TMEM16A, is downregulated in the portal vein of PHT rats in
vivo. However, TMEM16A was also observed to promote PVSMC
proliferation in vitro. The regulatory peptide Ang II may be
involved in regulating these opposite situations. This study
indicates a potential mechanism of PHT and provides a basis for
further research into the function of TMEM16A in PVSMC
proliferation.
References
|
1
|
Intengan HD and Schiffrin EL: Vascular
remodeling in hypertension: Roles of apoptosis, inflammation, and
fibrosis. Hypertension. 38:581–587. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
García-Pagán JC, Gracia-Sancho J and Bosch
J: Functional aspects on the pathophysiology of portal hypertension
in cirrhosis. J Hepatol. 57:458–461. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kang SH, Kim MY and Baik SK: Novelties in
the pathophysiology and management of portal hypertension: New
treatments on the horizon. Hepatol Int. Jul 11–2017.(Epub ahead of
print). View Article : Google Scholar
|
|
4
|
Kapoor D and Sarin S: Pathophysiology of
portal hypertension. J Gastroenterol Hepatol. 17 Suppl:S482–S487.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Halka AT, Turner NJ, Carter A, Ghosh J,
Murphy MO, Kirton JP, Kielty CM and Walker MG: The effects of
stretch on vascular smooth muscle cell phenotype in vitro.
Cardiovasc Pathol. 17:98–102. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kun L, Ying L, Lei W, Jianhua Z, Yongbo X,
Tao W, Jinyuan T and Haibo C: Dysregulated apoptosis of the venous
wall in chronic venous disease and portal hypertension. Phlebology.
31:729–736. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ma K, Wang H, Yu J, Wei M and Xiao Q: New
insights on the regulation of Ca2+ -activated chloride channel
TMEM16A. J Cell Physiol. 232:707–716. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schroeder BC, Cheng T, Jan YN and Jan LY:
Expression cloning of TMEM16A as a calcium-activated chloride
channel subunit. Cell. 134:1019–1029. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Caputo A, Caci E, Ferrera L, Pedemonte N,
Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O and
Galietta LJ: TMEM16A, a membrane protein associated with
calcium-dependent chloride channel activity. Science. 322:590–594.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang YD, Cho H, Koo JY, Tak MH, Cho Y,
Shim WS, Park SP, Lee J, Lee B, Kim BM, et al: TMEM16A confers
receptor-activated calcium-dependent chloride conductance. Nature.
455:1210–1215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ruiz C, Martins JR, Rudin F, Schneider S,
Dietsche T, Fischer CA, Tornillo L, Terracciano LM, Schreiber R,
Bubendorf L and Kunzelmann K: Enhanced expression of ANO1 in head
and neck squamous cell carcinoma causes cell migration and
correlates with poor prognosis. Plos One. 7:e432652012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Oh U and Jung J: Cellular functions of
TMEM16/anoctamin. Pflugers Arch. 468:443–453. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Forrest AS, Joyce TC, Huebner ML, Ayon RJ,
Wiwchar M, Joyce J, Freitas N, Davis AJ, Ye L, Duan DD, et al:
Increased TMEM16A-encoded calcium-activated chloride channel
activity is associated with pulmonary hypertension. Am J Physiol
Cell Physiol. 303:C1229–C1243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang B, Li C, Huai R and Qu Z:
Overexpression of ANO1/TMEM16A, an arterial Ca2+-activated
Cl-channel, contributes to spontaneous hypertension. J Mol Cell
Cardiol. 82:22–32. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Duvvuri U, Shiwarski DJ, Xiao D, Bertrand
C, Huang X, Edinger RS, Rock JR, Harfe BD, Henson BJ, Kunzelmann K,
et al: TMEM16A induces MAPK and contributes directly to
tumorigenesis and cancer progression. Cancer Res. 72:3270–4281.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qu Z, Yao W, Yao R, Liu X, Yu K and
Hartzell C: The Ca(2+)-activated Cl(−) channel, ANO1 (TMEM16A), is
a double-edged sword in cell proliferation and tumorigenesis.
Cancer Med. 3:453–461. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shiwarski DJ, Shao C, Bill A, Kim J, Xiao
D, Bertrand CA, Seethala RS, Sano D, Myers JN, Ha P, et al: To
‘grow’ or ‘go’: TMEM16A expression as a switch between tumor growth
and metastasis in SCCHN. Clin Cancer Res. 20:4673–4688. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Su EJ, Lombardi DM, Siegal J and Schwartz
SM: Angiotensin II induces vascular smooth muscle cell replication
independent of blood pressure. Hypertension. 31:1331–1337. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu D, Chen J, Wang J, Zhang Z, Ma X, Jia
J and Wang Y: Increased expression of urotensin II and GPR14 in
patients with cirrhosis and portal hypertension. Int J Mol Med.
25:845–851. 2010.PubMed/NCBI
|
|
20
|
Zimmermann M: Ethical considerations in
relation to pain in animal experimentation. Acta Physiol Scand
Suppl. 554:221–233. 1986.PubMed/NCBI
|
|
21
|
Xu C and Dong W: Role of hypoxia-inducible
factor-1α in pathogenesis and disease evaluation of ulcerative
colitis. Exp Ther Med. 11:1330–1334. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Leik CE, Willey A, Graham MF and Walsh SW:
Isolation and culture of arterial smooth muscle cells from human
placenta. Hypertension. 43:837–840. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Adhikari N, Shekar KC, Staggs R, Win Z,
Steucke K, Lin YW, Wei LN, Alford P and Hall JL; International
Society of Cardiovascular Translational Research, : Guidelines for
the isolation and characterization of murine vascular smooth muscle
cells. A report from the international society of cardiovascular
translational research. J Cardiovasc Transl Res. 8:158–163. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang L, Liu HJ, Li TJ, Yang Y, Guo XL, Wu
MC, Rui YC and Wei LX: Lentiviral vector-mediated siRNA knockdown
of SR-PSOX inhibits foam cell formation in vitro. Acta Pharmacol
Sin. 29:847–852. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Holt JR, Johns DC, Wang S, Chen ZY, Dunn
RJ, Marban E and Corey DP: Functional expression of exogenous
proteins in mammalian sensory hair cells infected with adenoviral
vectors. J Neurophysiol. 81:1881–1888. 1999.PubMed/NCBI
|
|
26
|
Tacev T, Zaloudik J, Janáková L and
Vagunda V: Early changes in flow cytometric DNA profiles induced by
californium-252 neutron brachytherapy in squamocellular carcinomas
of the uterine cervix. Neoplasma. 45:96–101. 1998.PubMed/NCBI
|
|
27
|
Jin C, Wang A, Chen J, Liu X and Wang G:
Relationship between expression and prognostic ability of PTEN,
STAT3 and VEGF-C in colorectal cancer. Exp Ther Med. 4:633–639.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mazzone A, Eisenman ST, Strege PR, Yao Z,
Ordog T, Gibbons SJ and Farrugia G: Inhibition of cell
proliferation by a selective inhibitor of the Ca(2+)-activated
Cl(−) channel, Ano1. Biochem Biophys Res Commun. 427:248–253. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hwang SJ, Blair PJ, Britton FC, O'Driscoll
KE, Hennig G, Bayguinov YR, Rock JR, Harfe BD, Sanders KM and Ward
SM: Expression of anoctamin 1/TMEM16A by interstitial cells of
Cajal is fundamental for slow wave activity in gastrointestinal
muscles. J Physiol. 587:4887–4904. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Svenningsen P, Nielsen MR, Marcussen N,
Walter S and Jensen BL: TMEM16A is a Ca(2+)-activated Cl(−) channel
expressed in the renal collecting duct. Acta Physiol (Oxf).
212:166–174. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qu Z, Wang B, Zhang Z, Ma L, Li D, Zhuang
L, Chi J and Liu J: Functions of ANO1/TMEM16A, Ca2+-activated
Cl-channels in regulation of blood pressure and vascular
remodeling. J Cardiol Ther. 3:543–548. 2016. View Article : Google Scholar
|
|
32
|
Wang M, Yang H, Zheng LY, Zhang Z, Tang
YB, Wang GL, Du YH, Lv XF, Liu J, Zhou JG and Guan YY:
Downregulation of TMEM16A calcium-activated chloride channel
contributes to cerebrovascular remodeling during hypertension by
promoting basilar smooth muscle cell proliferation. Circulation.
125:697–707. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Galán M, Varona S, Guadall A, Orriols M,
Navas M, Aguiló S, de Diego A, Navarro MA, García-Dorado D,
Rodríguez-Sinovas A, et al: Lysyl oxidase overexpression
accelerates cardiac remodeling and aggravates angiotensin
II-induced hypertrophy. FASEB J. 31:3787–3799. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li J, Li Y, Zhang Y, Hu D, Gao Y, Shang H
and Xing Y: The inhibitory effect of WenxinKeli on H9C2
cardiomyocytes hypertrophy induced by angiotensin II through
regulating autophagy activity. Oxid Med Cell Longev.
2017:70428722017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen L, Zhao L, Samanta A, Mahmoudi SM,
Buehler T, Cantilena A, Vincent RJ, Girgis M, Breeden J, Asante S,
et al: STAT3 balances myocyte hypertrophy vis-à-vis autophagy in
response to Angiotensin II by modulating the AMPKα/mTOR axis. PLoS
One. 12:e01798352017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lu Y, Guo H, Sun Y, Pan X, Dong J, Gao D,
Chen W, Xu Y and Xu D: Valsartan attenuates pulmonary hypertension
via suppression of mitogen activated protein kinase signaling and
matrix metalloproteinase expression in rodents. Mol Med Rep.
16:1360–1368. 2017.PubMed/NCBI
|
|
37
|
Bataller R, Ginès P, Nicolás JM, Görbig
MN, Garcia-Ramallo E, Gasull X, Bosch J, Arroyo V and Rodés J:
Angiotensin II induces contraction and proliferation of human
hepatic stellate cells. Gastroenterology. 118:1149–1156. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Daemen MJ, Lombardi DM, Bosman FT and
Schwartz SM: Angiotensin II induces smooth muscle cell
proliferation in the normal and injured rat arterial wall. Circ
Res. 68:450–456. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang XH, Zheng B, Yang Z, He M, Yue LY,
Zhang RN, Zhang M, Zhang W, Zhang X and Wen JK: TMEM16A and
myocardin form a positive feedback loop that is disrupted by KLF5
during Ang II-induced vascular remodeling. Hypertension.
66:412–421. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang L, Yang Z, Shi BM, Li DP, Fang CY
and Qiu FZ: Expression of local renin and angiotensinogen mRNA in
cirrhotic portal hypertensive patient. World J Gastroenterol.
9:1584–1588. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Deng L, Yang J, Chen H, Ma B, Pan K, Su C,
Xu F and Zhang J: Knockdown of TMEM16A suppressed MAPK and
inhibited cell proliferation and migration in hepatocellular
carcinoma. Onco Targets Ther. 14:325–333. 2016.
|
|
42
|
Matrougui K, Eskildsen-Helmond YE,
Fiebeler A, Henrion D, Levy BI, Tedgui A and Mulvany MJ:
Angiotensin II stimulates extracellular signal-regulated kinase
activity in intact pressurized rat mesenteric resistance arteries.
Hypertension. 36:617–621. 2000. View Article : Google Scholar : PubMed/NCBI
|