Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune
inflammatory disease in which patient experience clinical symptoms
of articular pain, cartilage degradation, narrow joint space and
loss of joint flexibility (1–3).
Osteoclasts, which are the cells primarily responsible for bone
dissolution and resorption, serve a critical role in joint
destruction by upregulating inflammation and directly destroying
the adjacent bone (4). Osteoclast
precursors and mature osteoclasts, which also serve an important
role in the inflammatory response are localized at arthritic bone
erosions (5). Among the Chinese
population, the incidence rate of RA is 0.24–0.5% and RA is more
prevalent in women than men (6).
Synovial fibroblasts serve a key role in the
pathogenesis of RA and their tumor-like proliferation leads to the
development of synovial hyperplasia (7). During RA, activated synovial
fibroblasts accumulate in the hyperplastic synovium of patients
with RA. Higher levels of inflammatory cytokines, chemotactic
factors and matrix metalloproteinase mediate inflammation and
degrade cartilage, which eventually leads to the destruction of
joints (8). Several studies have
been conducted to explore the association between synovial
fibroblasts and the regulation of inflammatory pathways in the RA
synovium (9–11); however, the precise mechanism of
action by which this occurs remains unknown (12).
MicroRNAs (miRNAs) are a class of non-coding RNAs
that regulate gene expression by binding to the 3′untranslated
region (UTR) of their target mRNAs. They serve critical roles in
numerous diseases, including different types of cancer,
inflammatory and neurological diseases (13–15).
Studies have suggested that miRNAs are involved in the pathogenesis
of many diseases, such as RA and may therefore be developed as
potential therapeutic targets in the treatment of RA (16–18).
miR-143-3p is a type of miRNA that has been implicated in numerous
diseases, including age-related defective muscle regeneration,
gastric cancer and esophageal squamous cell carcinoma (19). However, to the best of our knowledge,
the key roles and regulatory mechanisms of miR-143-3pin RA remain
unknown.
In the present study, the expression of miR-143-3p
in RA synovial tissues was measured. The regulation of insulin-like
growth factor 1 receptor (IGF1R) and insulin-like growth
factor-binding protein 5 (IGFBP5) by miR-143-3p is considered to be
a key mechanism that mediates the age-related changes in satellite
cell function (20). Furthermore, it
has been demonstrated that the p38 mitogen-activated protein kinase
(MAPK) pathway serves a crucial role in the induction and
maintenance of chronic inflammation in RA (21). Therefore, the present study
investigated the association between miR-143-3p, and IGF1R and
IGFBP5, as well between miR-143-3p and the Ras/p38 MAPK signaling
pathway, to elucidate the potential regulatory mechanism of
miR-143-3p in RA. The MH7A cell line was used as an in vitro
model system.
Materials and methods
Patients
Synovia were obtained from 5 patients with RA and 5
patients with osteoarthritides (OA) that fulfilled the American
College of Rheumatology Criteria for RA or OA (22), as well as one patient undergoing
reconstruction of the anterior cruciate ligament (as a control).
Synovial tissues were collected from 3 patients with RA as they
underwent total knee arthroplasty (TKA) and 2 patients with RA as
they underwent synovectomy. All synovial tissue samples taken from
patients with OA were collected during TKA. The mean age of
patients with RA and OA were 62.9±10.9 (39–78) and 71.6±4.9 (72–81)
years, respectively. As the mean age of patients in the two groups
differed, a multivariate logistic regression model with adjustment
for age was used to evaluate miR-143-3p expression in the synovium
of patients with RA as well as those with OA. Among the patients
with RA, there were 2 males and 3 females; however, all patients
with OA were female. A further patient (male, 45 years old)
undergoing leg amputation but with a normal knee joint was included
as the control. The synovial tissues were harvested from the
control patient during the amputation surgery. The present study
was approved by the Ethics Committee of Shandong Academy of Medical
Science (Jinan, China) and written informed consent was obtained
from all patients that participated in the study.
Cell culture
The human RA synovial cell line MH7A was procured
from the American Type Culture Collection (Manassas, VI, USA). MH7A
cells were stained positively for intercellular adhesion molecule-1
(ICAM-1), interleukin (IL)-1R, cluster of differentiation (CD)16,
CD40, CD80 and CD95 (23); and have
been previously used to investigate the molecular mechanisms
underlying RA (23). Cells were
maintained in RPMI 1640 medium (Hyclone; GE Healthcare, Logan, UT,
USA), supplemented with 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) and 1%
penicillin/streptomycin (1:100, Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany), at 37°C in a relative humidity of 95% and in
5% CO2 (24).
Cell transfection
To assess the effects of miR-143-3p abnormal
expression on MH7A cells, the cells were transfected with
miR-143-3p mimic, miR-143-3p inhibitor or their controls (mimic NC
or inhibitor NC). Briefly, MH7A cells
(1×105-2×105) were transfected with 90 pmol
hsa-miR-143-3p mimic (Takara, Okinawa, Japan) and the mimic control
(mimic NC; 90 pmol) (Takara) three times over 7 days. Cells were
also transfected with mirVana™ hsa-miR-143-3p inhibitor
(Thermo Fisher Scientific, Inc.) (150 pmol), inhibitor NC (150
pmol), small interfering (si)RNA against IGF1R (si-IGFR; 100 pmol)
or IGFBP5 (si-IGFBP5; 100 pmol) or the si-NC control (100 pmol)
(Thermo Fisher Scientific, Inc.) for 72 h. All transfections were
performed using Lipofectamine® RNAiMAX Transfection
Reagent (Thermo Fisher Scientific, Inc.) following the
manufacturer's protocol. The cells in the control group received no
additional treatment. The sequences of the miRs and siRNA used were
as follows: miR-143-3p mimic sense 5′-GGUGCAGUGCUGCAUCUCUGGU-3′ and
anti-sense, 5′-CAGAGAUGCAGCACUGCACCUU-3′; mimic NC sense
5′-UCACAACCUCCUAGAAAGAGUAGA-3′; mimic NC antisense
5′-UCUACUCUUUCUAGGAGGUUGUGA-3′; miR-143-3p inhibitor
5′-ACCAGAGAUGCAGCACUGCACC-3′; inhibitor NC,
5′-UCUACUCUUUCUAGGAGGUUGUG-3′. si-IGFR 5′-GCGGAGAGAUGUCAUGCAAGU-3′;
si-IGFBP5 5′-GGAUCUGUCUCCUCCUCUAGC-3′. si-NC:
5′-UUCUUCGAAGGUGUCACGUTT-3′.
Cell counting kit 8 (CCK-8) assay
MH7A cells were seeded in 96-well plates at 5,000
cells/well for 72 h transfected with miR-143-3p inhibitor,
si-IGF1R, si-IGFBP5 or corresponding controls (inhibitor NC or
si-NC) for 24 h. Subsequently, cells were treated with tumor
necrosis factor (TNF)-α (10 ng/ml; Sigma-Aldrich; Merck KGaA) at
room temperature for 24 h to construct a model of RA. The cells in
the control group were treated with normal RPMI 1640 medium.
Subsequently, CCK-8 solution (Invitrogen; Thermo Fisher Scientific,
Inc.) was added to each well and plates were incubated at 37°C for
2 h. The absorbance of each well was measured at 450 nm using a
microplate reader (25).
Flow cytometric analysis of
apoptosis
Cells or corresponding controls were harvested
following transfection with miR-143-3p inhibitor, si-IGF1R or
si-IGFBP5 for 24 h. Cells were treated with TNF-α, washed twice
with pre-chilled PBS and resuspended in 100 µl binding buffer at a
concentration of 1×106 cells/ml. Annexin V and propidium
iodide (PI) double-staining was performed using an Annexin
V-fluorescein isothiocyanate Apoptosis Detection kit (BD
Biosciences, San Jose, CA, USA) according to the manufacturer's
protocol. Apoptosis analysis was performed using the BD LSRII Flow
Cytometer System with FACSDiva Software version 6.0 (BD
Biosciences) within 1 h (25).
Vector construction, target prediction
and luciferase reporter assay
The potential target genes of miR-143-3p were
analyzed by TargetScan Human 7.0 (targetscan.org/vert_71/). A cut-off value of 0.7 was
applied to predict the target genes. To develop a luciferase
reporter construct, the 3′-UTR fragments of IGF1R or IGFBP5, which
contained putative binding sites for miR-143-3p, were inserted
downstream of firefly luciferase in the pGL3 vector (Promega
Corporation). Mutant 3′-UTR, which carried the mutated sequence in
the complementary site for miR-143-3p, was generated using the
polymerase chain reaction (PCR) method (26) and inserted downstream of firefly
luciferase in a separate pGL3 vector. 293A cells (1×105;
American Type Culture Collection) were cultured in a 48-well plate
were co-transfected with miR-143-3p with a luciferase reporter
comprising wild-type or mutant 3′-UTR of the target gene. The
luciferase assay was performed as previously reported (26). 293A cells were co-transfected with
miRs and wild-type 3′-UTR or mutant 3′-UTR luciferase reporter,
using pRL-TK (Promega Corporation) as a control vector. Following
48 h transfection, luciferase activity was measured using the Dual
Luciferase Assay kit (Promega Corporation) with a β-counter
luminometer. Relative luciferase activity was calculated as the
ratio of the raw firefly luciferase activity and Renilla
luciferase activity (27).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from MH7A cells using the
commercial total RNA miniprep kit (Axygen; Corning Inc., Corning,
NY, USA) according to the manufacturer's protocol. Each sample was
reverse transcribed using a cDNA synthesis kit (Takara). qPCR was
performed using SYBR Green PCR Premix Ex Taq II reagents (Takara)
on a Light Cycler 480 II real-time system (Roche Diagnostics,
Indianapolis, IN, USA). The house-keeping gene GAPDH was used for
normalization. The mRNA expression was quantified by the
comparative 2−(∆∆Cq) method (28). The primers used for target
amplification were as follows: MiR-143 forward,
5′-TGAGATGAAGCACTGTAGCTC-3′ and reverse, 5′-TGGTGTCGTGGAGTCG-3′; U6
forward, 5′-CTCGCTTCGGCAGCACA-3′ and reverse,
5′-AACGCTTCACGAATTTGCGT-3′; B-cell lymphoma (Bcl)-2 forward,
5′-GATGCAGTGCCGGCCTAAG-3′ and reverse, 5′-TTCTCTTGTACGCACGAGCT-3′;
Bax forward, 5′-CTGAGCTGACCTTGGAGC-3′ and reverse,
5′-GACTCCAGCCACAAAGATG-3′; Caspase-3 forward,
5′-ACCGATGTCGATGCAGCTAA-3′ and reverse, 5′-AGGTCCGTTCGTTCCAAAAA3′.
GAPDH forward, 5′-CCACCCATGGCAAATTCCATGGCA-3′ and reverse,
5′-TCTAGACGGCAGGTCAGGTCCACC-3′; IGF1R forward,
5′-GCGAGCTCTCTGGGATAGAAATGTTTAGGTGTA-3′ and reverse,
5′-GCAAGCTTCAGGTGCTGAGAAAGGTGAGATGT-3′; IGFBP5 forward,
5′-ACGCGTCGACATGGGCTCCTTCGTGCAC3′ and reverse,
5′-CGCGGATCCATCACTCAACGTTGCTGCTG-3′. The thermocycling conditions
were as follows: 95°C for 1 min followed by 35 cycles of 95°C for
10 sec and 58°C for 40 sec, with a final extension step of 10 min
at 68°C.
Enzyme-linked immunosorbent assay
(ELISA)
(29). The culture
medium was collected and the concentration of the cytokines was
determined using commercial ELISA kits for IL-1β (ab100704), IL-6
(ab46042), IL-8 (ab46032), MMP-1 (ab100603) and MMP-13 (ab100605)
(all Abcam, Cambridge, UK) following the manufacturer's protocol.
Values were calculated on the basis of a standard curve constructed
for each assay.
Western blot analysis
Cells were solubilized with radioimmunoprecipitation
assay lysis buffer (Thermo Fisher Scientific, Inc.) and protein
concentration was measured using a BCA kit (Thermo Fisher
Scientific, Inc.). Approximately 50 µg of protein from each sample
was separated by 12% SDS-PAGE and transferred to a polyvinylidene
fluoride membrane (EMD Millipore, Burlington, MA, USA). Following
blocking with 5% bovine serum albumin (1:100; Sigma-Aldrich; Merck
KGaA) in Tris-buffered saline with Tween for 2 h at room
temperature, membranes were incubated with Bcl-2 (ab178529), Bax
(ab53154), pro-caspase-3 (ab32150) and active-caspase-3 (ab181418),
phosphorylated (p)-p38 (ab4822), p38 (ab31828) and Ras (ab52939)
primary antibodies at a dilution of 1:1,000 overnight at 4°C (all
Abcam, Cambridge, UK), followed by incubation with goat anti-rabbit
horseradish peroxidase conjugated secondary antibodies (at a
dilution of 1:2,000; ab191866; Abcam) for 1 h at room temperature.
GAPDH (ab8245; dilution 1:2,000; Abcam) was used as an internal
control. Protein blots were visualized and analyzed using a
chemiluminescence system (Bio-Rad Laboratories, Inc., Hercules, CA,
USA) and autoradiography films (Kodak Image Station 440; Kodak,
Rochester, NY, USA).
Statistical analysis
Data are presented as the mean ± standard deviation
from at least three independent experiments. All statistical
analyses were performed using SPSS 16.0 (SPSS, Inc., Chicago, IL,
USA). Student's t test was performed to assess whether differences
between two groups was significant, whereas one-way analysis of
variance followed by a post-hoc Tukey's test was performed to
evaluate whether differences among three or more groups were
significant. P<0.05 was considered to indicate a statistically
significant difference.
Results
Analysis of miR-143-3p expression in
RA tissues and cells
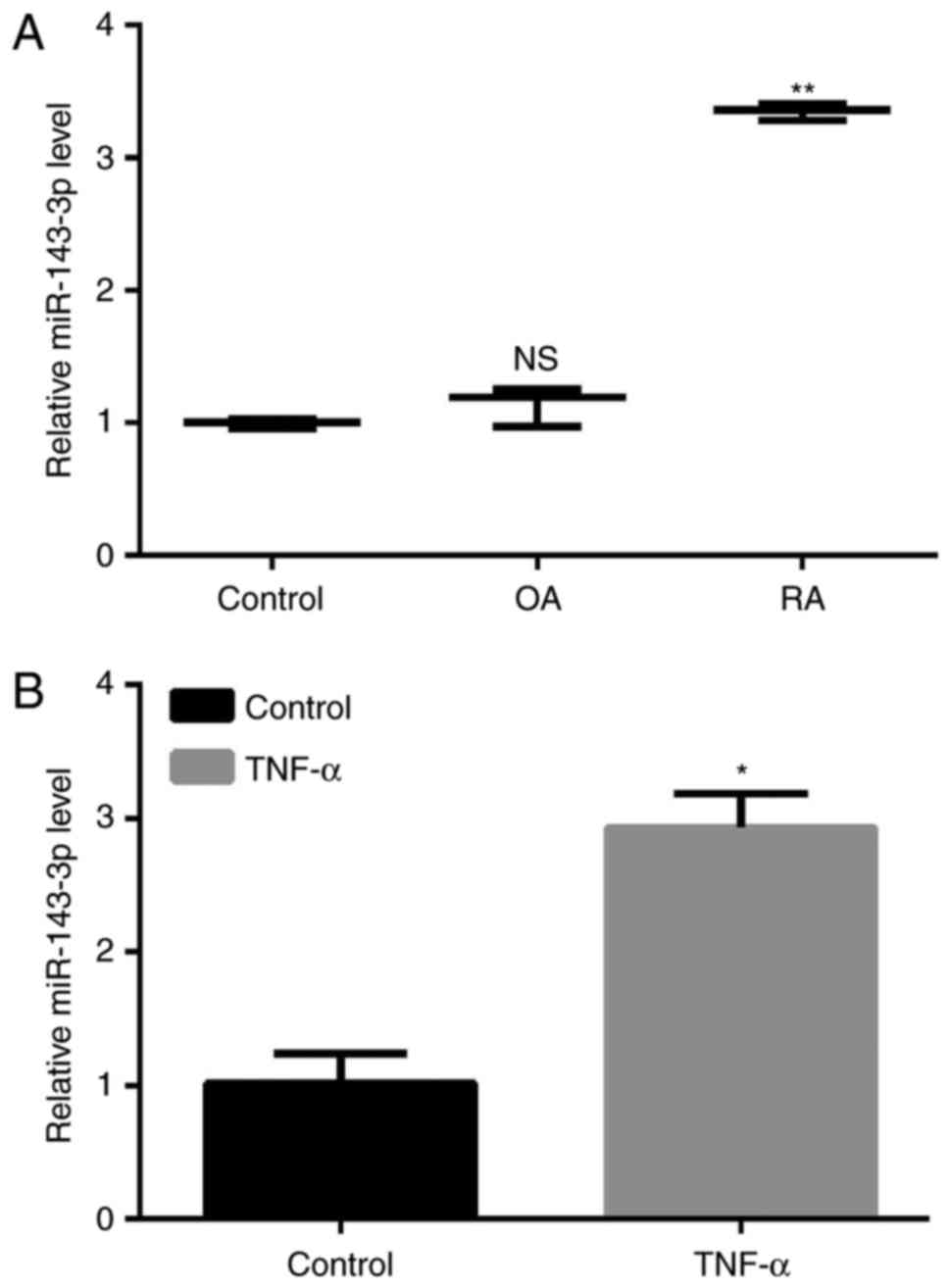
The expression of miR-143-3p in RA was significantly
higher than in patients with OA and the control (P<0.01;
Fig. 1A). MH7A cells were treated
with 10 ng/ml TNF-α to construct a model of RA and the results
demonstrated that the expression of miR-143-3p was significantly
increased in TNF-α-induced RA cells compared with the untreated
control cells (P<0.05, Fig.
1B).
miR-143-3p inhibitor reduces cell
proliferation and promotes apoptosis
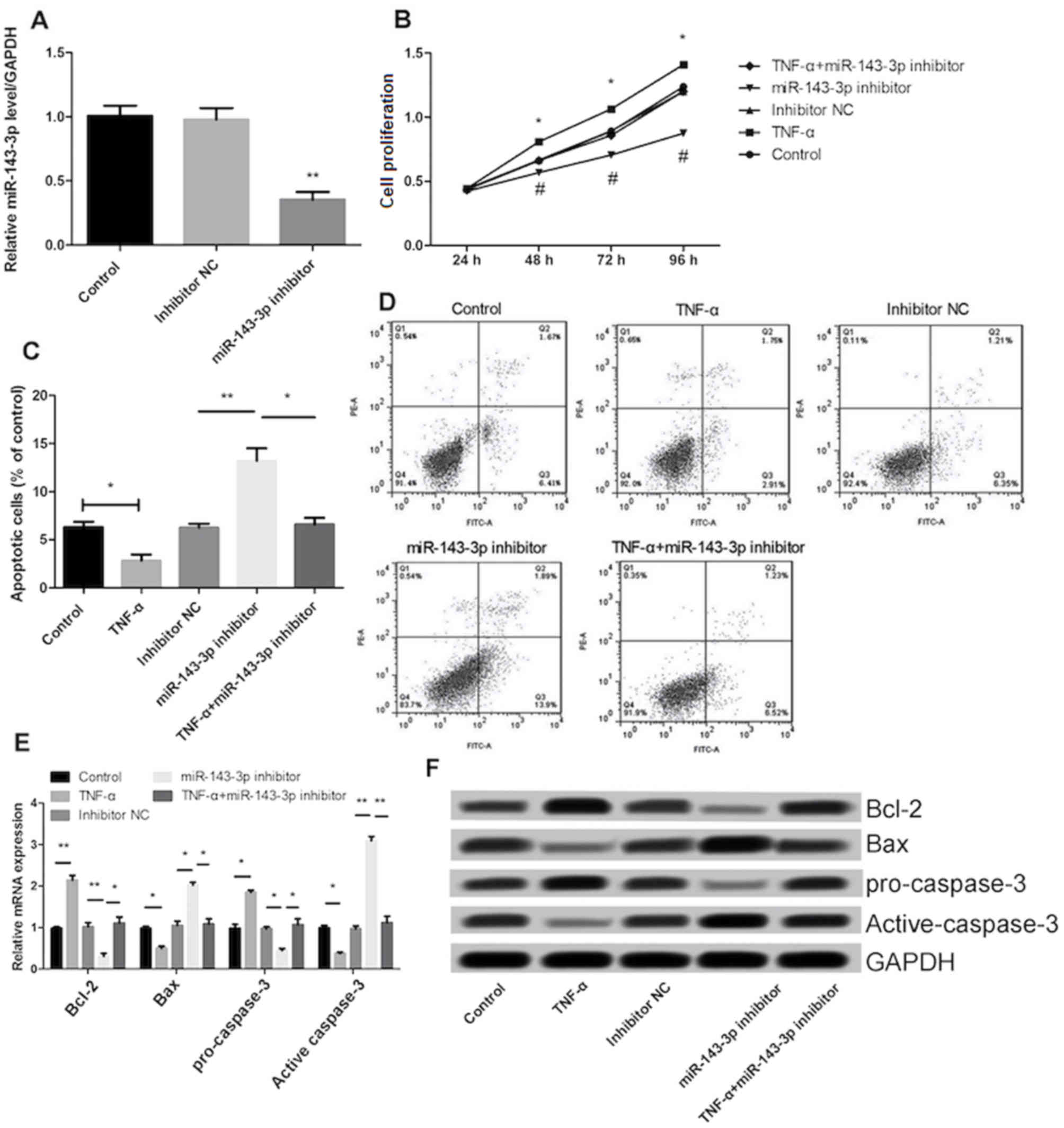
Transfection of the miR-143-3p inhibitor in MH7A
cells significantly decreased miR-143-3p expression (P<0.01),
indicating that transfection was successful (Fig. 2A). Treatment with TNF-α significantly
increased the proliferation and inhibited the apoptosis of MH7A
cells (P<0.05; Fig. 2B and C).
The effect of TNF-α on cell proliferation and apoptosis was
significantly reversed following inhibition of miR-143-3p
(P<0.05; Fig. 2B-D). In addition,
the results demonstrated that transfection with the miR-143-3p
inhibitor significantly promoted the expression of Bax and
inhibited the expression of Bcl-2 (P<0.01; Fig. 2E and F). The expression of cell
apoptosis-associated proteins, including Bcl-2 and pro-caspase-3
was significantly increased in the TNF-α group (P<0.01) compared
with the control group, whereas the expression of Bax and
active-caspase-3 were significantly decreased (P<0.05; Fig. 2E and F). Furthermore, following
transfection with miR-143-3p inhibitor, the expression of
pro-caspase-3 was significantly inhibited and the expression of
active-caspase-3 was significantly promoted.
miR-143-3p inhibitor inhibits levels
of inflammatory factors
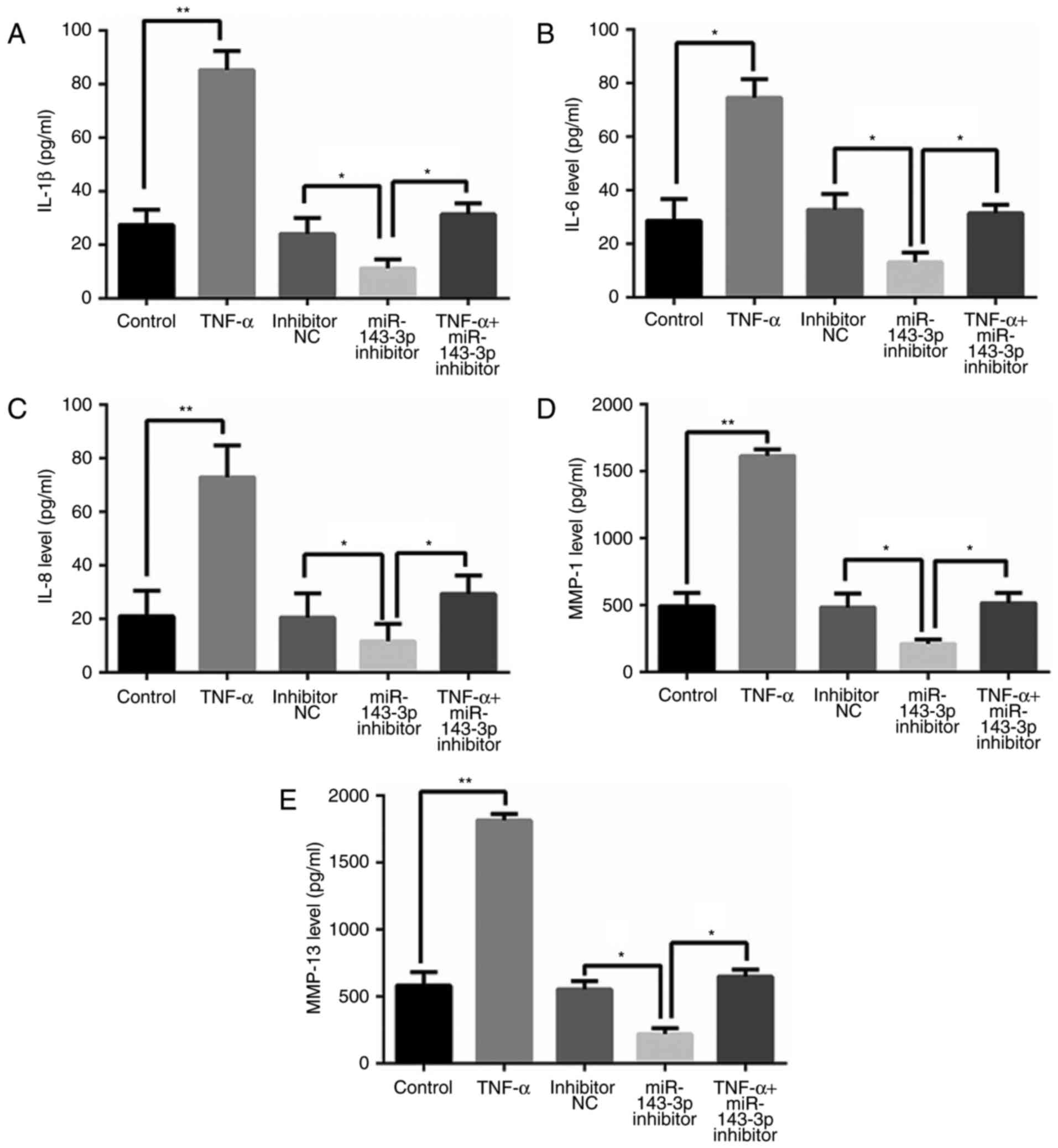
As presented in Fig.
3, TNF-α treatment significantly increased levels of
inflammatory factors, including interleukin (IL)-1β, IL-6, IL-8,
matrix metalloproteinase (MMP)-1 and MMP-13 (P<0.05). However,
cells transfected with miR-143-3p inhibitor exhibited significant
decreases in levels of these inflammatory factors, compared with
control cells (P<0.05; Fig.
3).
Prediction and examination of the
targeting effects of miR-143-3p
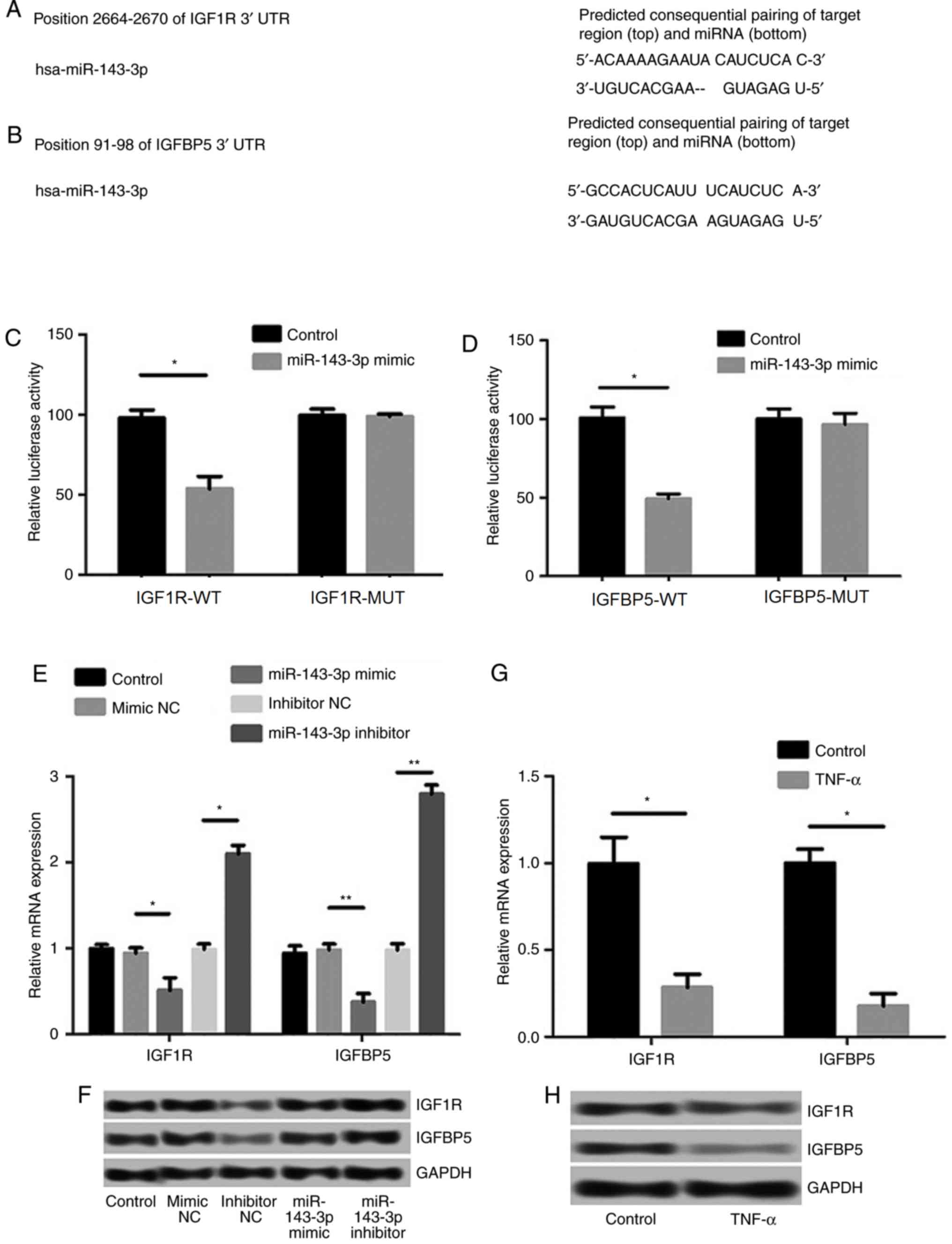
The TargetScan Human 7.0 (targetscan.org/vert_71/) was used to analyze the
potential target gene of miR-143-3p, and the sequence analysis data
indicated that IGF1R and IGFBP5 were targets of miR-143-3p
(Fig. 4A and B). The results of the
luciferase reporter assay confirmed that the 3′-UTR regions of
IGF1R and IGFBP5 possess hsa-miR-143-3p binding sites (P<0.05;
Fig. 4C and D). Moreover, the
expression levels of IGF1R and IGFBP5 were significantly inhibited
following transfection of miR-143-3p mimics (P<0.05), whereas
the transfection of miR-143-3p inhibitors significantly increased
the levels of IGF1R and IGFBP5 (P<0.01; Fig. 4E and F). Reduced levels of IGF1R and
IGFBP5 mRNA and protein expression were observed in RA models
following treatment with TNF-α (P<0.05; Fig. 4G and H).
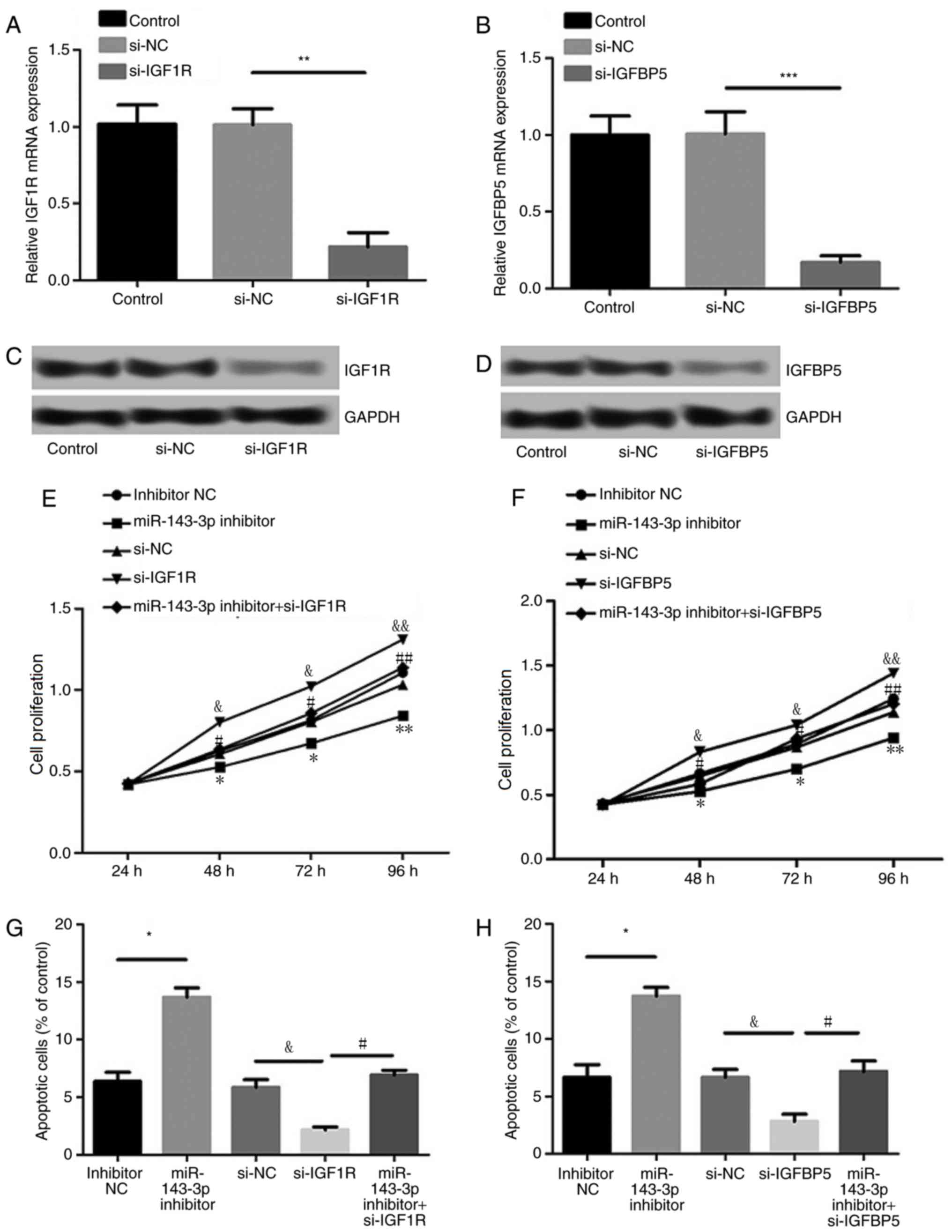
Effects of IGF1R and IGFBP5
suppression on cell proliferation and apoptosis
As presented in Fig.
5A-D, levels of IGF1R and IGFBP5 expression were significantly
suppressed following transfection with siRNAs against IGF1R and
IGFBP5, respectively (P<0.01), indicating efficient
transfection. Furthermore, suppression of IGF1R or IGFBP5
expression significantly reversed the effect of miR-143-3p
inhibition on MH7A cell proliferation and apoptosis (P<0.05;
Fig. 5E-H). These results indicate
that miR-143-3p regulates MH7A cell proliferation and apoptosis by
targeting IGF1R or IGFBP5.
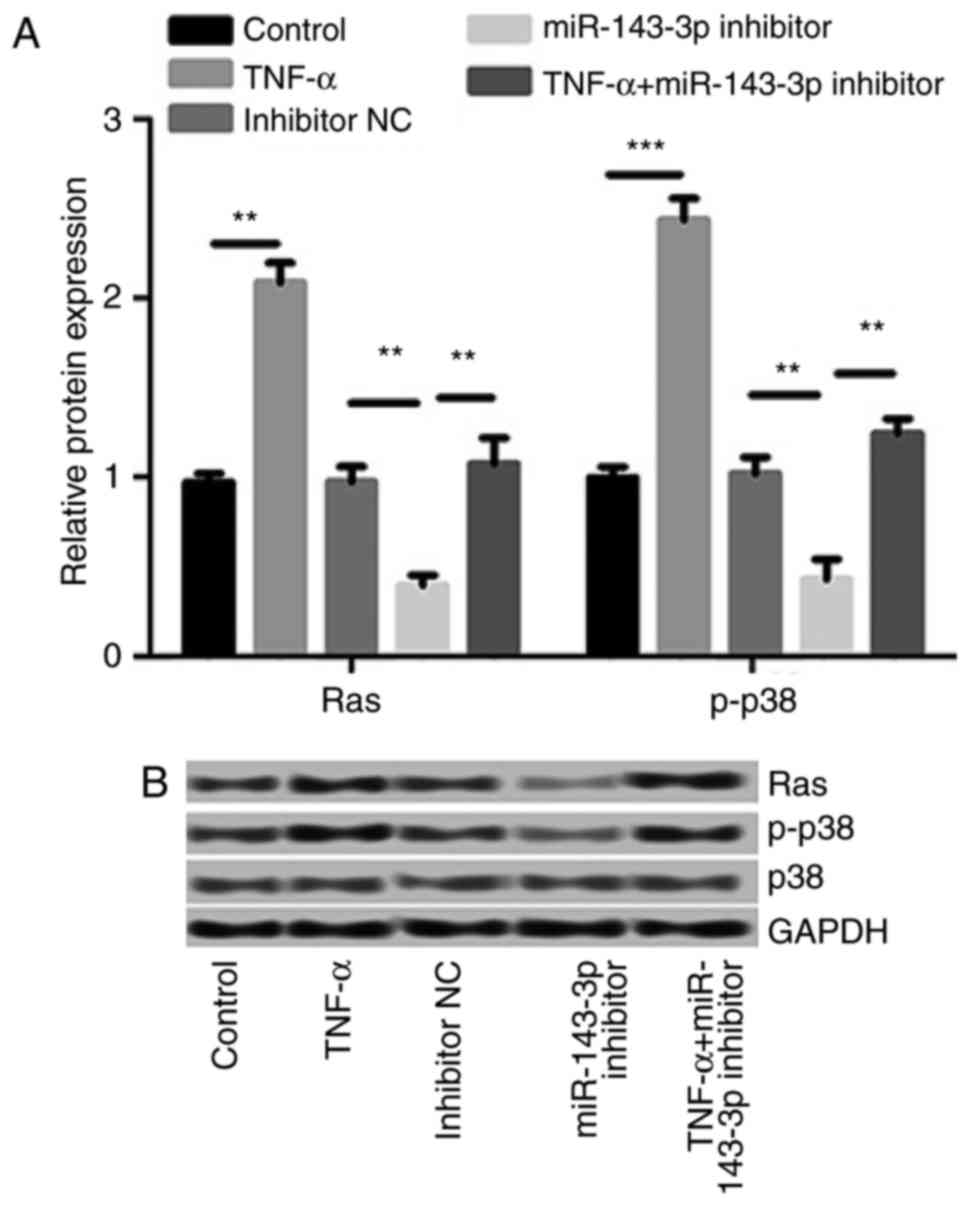
miR-143-3p inhibitor suppressed the
expression of Ras and p-p38/p38
The effect of TNF-α treatment and the miR-143-3p
inhibitor on the Ras/p38 signaling pathway was investigated. The
results demonstrated that TNF-α treatment significantly increased
the expression of Ras (P<0.01) and p-p38/p38 (P<0.001;
Fig. 6). These increases were
significantly reversed by the miR-143-3p inhibitor, indicating that
the miR-143-3p inhibitor inhibits the Ras/p38 MAPK signaling
pathway in a TNF-α-induced RA model.
Discussion
The present study revealed that miR-143-3p was
highly expressed in RA tissue. The inhibition of miR-143-3p
suppressed the proliferation of MH7A cells and promoted their
apoptosis. It was confirmed that IGF1R and IGFBP5 are targets of
miR-143-3p and that miR-143-3p regulated the proliferation and
apoptosis of MH7A cells by targeting IGF1R and IGFBP5. Ultimately,
the miR-143-3p inhibitor suppressed the Ras/p38 MAPK signaling
pathway in TNF-α-induced RA models. These results suggest that
miR-143-3p serves a key role in the development of RA.
It has been reported that miRNAs are able to target
proto-oncogenes, including Bcl-2, Ras or c-Myc (30). Bcl-2 and Bax, which are members of
the anti-apoptotic Bcl-2 family, serve a critical role in
regulating apoptosis and are able to inhibit the activation of
caspase-3 (31). Zhang and Li
(32) indicated that miR-143-3p
regulates the proliferation of cytokine-induced killer cells. The
results of the present study indicated that cells transfected with
the miR-143-3p inhibitor exhibited enhanced expression of Bax, but
decreased expression of Bcl-2, that miR-143-3p promotes cell
apoptosis. In addition, RA is a self-maintaining inflammatory
disease, in which the destruction of the adjacent bone is induced
by inflammation (33). Studies have
demonstrated that inflammatory cytokines, including IL-1β, IL-6,
IL-8, MMP-1 and MMP-13, serve a key role in the pathogenesis of RA
(34,35). The results of the present study
indicated that inhibition of miR-143-3p significantly inhibited the
production of these inflammatory cytokines.
In addition, the present study indicated that IGF1R
and IGFBP5 are target genes of miR-143-3p. It has been reported
that IGF-1R signaling contributes to T cell-dependent inflammation
in RA (35). Furthermore, it has
been suggested that MMP-13 and IGFBP5 are important factors that
mediate the development of OA (36).
In the present study, it was demonstrated that miR-143-3p regulates
the proliferation and apoptosis of MH7A cells by targeting IGF1R or
IGFBP5, implying that miR-143-3p is associated with IGF1R and
IGFBP5 in RA.
The MAPK signaling pathway, which involves p38 and
c-Jun N terminal kinase (JNK)1/2, serves an important role in
various cellular processes, including inflammation, proliferation,
migration and adhesion (37–39). MAPKs participate in
mechanotransduction in bone cells (40). It has been demonstrated that p38α
MAPK modulates JNK-mediated cell proliferation in myogenesis
(41). In addition, the involvement
of scavenger receptor class B type 1 in the p38 MAPK signaling
pathway is a key mechanism of mediating serum amyloid A-induced
angiogenesis in RA (42). Chemokine
C-X-C ligand 16 can upregulate receptor activator of nuclear factor
κ-Β ligand expression to mediate RA development via the p38/MAPK
signaling pathway (43). Wang et
al (44) demonstrated that
miR-451 suppresses the secretion of inflammatory cytokines and the
proliferation of synovial fibroblasts in RA via the p38/MAPK
signaling pathway. The results of the current study demonstrated
that TNF-α stimulates the Ras/p38 MAPK signaling pathway. By
contrast, the inhibition of miR-143-3p suppressed the Ras/p38 MAPK
signaling pathway. Given the key role that the Ras/p38 MAPK
signaling pathway serves in RA, miR-143-3p may contribute to RA by
mediating the Ras/p38 MAPK signaling pathway.
In conclusion, the results of the present study
demonstrate that the increased expression of miR-143-3p may
contribute to the progression of RA by promoting cell proliferation
and inflammatory cytokine secretion, inhibiting the initiation of
apoptosis by targeting IGF1R and IGFBP5 and regulating the Ras/p38
MAPK signaling pathway. These results indicate that miR-143-3p may
be developed as a potential target for RA therapy. Further studies
are required to validate the results of the current study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mertsch S, Schmitz N, Jeibmann A, Geng JG,
Paulus W and Senner V: Slit2 involvement in glioma cell migration
is mediated by Robo1 receptor. J Neurooncol. 87:1–7. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Challal S, Minichiello E, Boissier MC and
Semerano L: Cachexia and adiposity in rheumatoid arthritis.
Relevance for disease management and clinical outcomes. Joint Bone
Spine. 83:127–133. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ji L, Geng Y, Zhou W and Zhang Z: A study
on relationship among apoptosis rates, number of peripheral T cell
subtypes and disease activity in rheumatoid arthritis. Int J Rheum
Dis. 19:167–171. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Goldring MB: Osteoarthritis and cartilage:
The role of cytokines. Curr Rheumatol Rep. 2:459–465. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Okada Y, Naka K, Kawamura K, Matsumoto T,
Nakanishi I, Fujimoto N, Sato H and Seiki M: Localization of matrix
metalloproteinase 9 (92-kilodalton gelatinase/type IV
collagenase=gelatinase B) in osteoclasts: Implications for bone
resorption. Lab Invest. 72:311–322. 1995.PubMed/NCBI
|
|
6
|
Symmons DP: Epidemiology of rheumatoid
arthritis: Determinants of onset, persistence and outcome. Best
Pract Res Clin Rheumatol. 16:707–722. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Muller-Ladner U, Kriegsmann J, Franklin
BN, Matsumoto S, Geiler T, Gay RE and Gay S: Synovial fibroblasts
of patients with rheumatoid arthritis attach to and invade normal
human cartilage when engrafted into SCID mice. Am J Pathol.
149:1607–1615. 1996.PubMed/NCBI
|
|
8
|
Huber LC, Distler O, Tarner I, Gay RE, Gay
S and Pap T: Synovial fibroblasts: Key players in rheumatoid
arthritis. Rheumatology (Oxford). 45:669–675. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lowin T and Straub RH: Synovial
fibroblasts integrate inflammatory and neuroendocrine stimuli to
drive rheumatoid arthritis. Expert Rev Clin Immunol. 11:1069–1071.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McInnes IB and Schett G: Pathogenetic
insights from the treatment of rheumatoid arthritis. Lancet.
389:2328–2337. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Edhayan G, Ohara RA, Stinson WA, Amin MA,
Isozaki T, Ha CM, Haines GK III, Morgan R, Campbell PL, Arbab AS,
et al: Inflammatory properties of inhibitor of DNA binding 1
secreted by synovial fibroblasts in rheumatoid arthritis. Arthritis
Res Ther. 18:872016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Turner JD and Filer A: The role of the
synovial fibroblast in rheumatoid arthritis pathogenesis. Curr Opin
Rheumatol. 27:175–182. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gregory RI, Yan KP, Amuthan G, Chendrimada
T, Doratotaj B, Cooch N and Shiekhattar R: The Microprocessor
complex mediates the genesis of microRNAs. Nature. 432:235–240.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Farh KK, Grimson A, Jan C, Lewis BP,
Johnston WK, Lim LP, Burge CB and Bartel DP: The widespread impact
of mammalian MicroRNAs on mRNA repression and evolution. Science.
310:1817–1821. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ambros V: MicroRNAs and developmental
timing. Curr Opin Genet Dev. 21:511–517. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ammari M, Jorgensen C and Apparailly F:
Impact of microRNAs on the understanding and treatment of
rheumatoid arthritis. Curr Opin Rheumatol. 25:225–233. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Olivieri F, Rippo MR, Procopio AD and
Fazioli F: Circulating inflamma-miRs in aging and age-related
diseases. Front Genet. 4:1212013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sugatani T and Hruska KA: Impaired
micro-RNA pathways diminish osteoclast differentiation and
function. J Biol Chem. 284:4667–4678. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
He Z, Yi J, Liu X, Chen J, Han S, Jin L,
Chen L and Song H: MiR-143-3p functions as a tumor suppressor by
regulating cell proliferation, invasion and epithelial-mesenchymal
transition by targeting QKI-5 in esophageal squamous cell
carcinoma. Mol Cancer. 15:512016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Soriano-Arroquia A, McCormick R, Molloy
AP, McArdle A and Goljanek-Whysall K: Age-related changes in
miR-143-3p: Igfbp5 interactions affect muscle regeneration. Aging
Cell. 15:361–369. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schett G, Zwerina J and Firestein G: The
p38 mitogen-activated protein kinase (MAPK) pathway in rheumatoid
arthritis. Ann Rheum Dis. 67:909–916. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kawasaki T, Inoue K, Ushiyama T and Fukuda
S: Assessment of the American College of Rheumatology criteria for
the classification and reporting of osteoarthritis of the knee.
Ryumachi. 38:2–5. 1998.(In Japanese). PubMed/NCBI
|
|
23
|
Miyazawa K, Mori A and Okudaira H:
Establishment and characterization of a novel human rheumatoid
fibroblast-like synoviocyte line, MH7A, immortalized with SV40 T
antigen. J Biochem. 124:1153–1162. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jia Q, Cheng W, Yue Y, Hu Y, Zhang J, Pan
X, Xu Z and Zhang P: Cucurbitacin E inhibits TNF-α-induced
inflammatory cytokine production in human synoviocyte MH7A cells
via suppression of PI3K/Akt/NF-κB pathways. Int Immunopharmacol.
29:884–890. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen H, Lin YW, Mao YQ, Wu J, Liu YF,
Zheng XY and Xie LP: MicroRNA-449a acts as a tumor suppressor in
human bladder cancer through the regulation of pocket proteins.
Cancer Lett. 320:40–47. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao X, Liu D, Gong W, Zhao G, Liu L, Yang
L and Hou Y: The toll-like receptor 3 ligand, poly(I:C), improves
immunosuppressive function and therapeutic effect of mesenchymal
stem cells on sepsis via inhibiting MiR-143. Stem Cells.
32:521–533. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Song T, Zhang X, Wang C, Wu Y, Cai W, Gao
J and Hong B: MiR-138 suppresses expression of hypoxia-inducible
factor 1alpha (HIF-1α) in clear cell renal cell carcinoma 786-O
cells. Asian Pac J Cancer Prev. 12:1307–1311. 2011.PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ruedel A, Dietrich P, Schubert T,
Hofmeister S, Hellerbrand C and Bosserhoff AK: Expression and
function of microRNA-188-5p in activated rheumatoid arthritis
synovial fibroblasts. Int J Clin Exp Pathol. 8:4953–4962.
2015.PubMed/NCBI
|
|
30
|
Le XF, Merchant O, Bast RC and Calin GA:
The roles of MicroRNAs in the cancer invasion-metastasis cascade.
Cancer Microenviron. 3:137–147. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qin H, Tan W, Zhang Z, Bao L, Shen H, Wang
F, Xu F and Wang Z: 15d-prostaglandin J2 protects cortical neurons
against oxygen-glucose deprivation/reoxygenation injury:
Involvement of inhibiting autophagy through upregulation of Bcl-2.
Cell Mol Neurobiol. 35:303–312. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang H and Li W: Dysregulation of
micro-143-3p and BALBP1 contributes to the pathogenesis of the
development of ovarian carcinoma. Oncol Rep. 36:3605–3610. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wegner N, Lundberg K, Kinloch A, Fisher B,
Malmström V, Feldmann M and Venables PJ: Autoimmunity to specific
citrullinated proteins gives the first clues to the etiology of
rheumatoid arthritis. Immunol Rev. 233:34–54. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hwang SY, Kim JY, Kim KW, Park MK, Moon Y,
Kim WU and Kim HY: IL-17 induces production of IL-6 and IL-8 in
rheumatoid arthritis synovial fibroblasts via NF-kappaB- and
PI3-kinase/Akt-dependent pathways. Arthritis Res Ther. 6:R120–R128.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Erlandsson MC, Töyrä Silfverswärd S,
Nadali M, Turkkila M, Svensson MND, Jonsson IM, Andersson KME and
Bokarewa MI: IGF-1R signalling contributes to IL-6 production and T
cell dependent inflammation in rheumatoid arthritis. Biochim
Biophys Acta. 1863:2158–2170. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tardif G, Hum D, Pelletier JP, Duval N and
Martel-Pelletier J: Regulation of the IGFBP-5 and MMP-13 genes by
the microRNAs miR-140 and miR-27a in human osteoarthritic
chondrocytes. BMC Musculoskelet Disord. 10:1482009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhu C, Qi X, Chen Y, Sun B, Dai Y and Gu
Y: PI3K/Akt and MAPK/ERK1/2 signaling pathways are involved in
IGF-1-induced VEGF-C upregulation in breast cancer. J Cancer Res
Clin Oncol. 137:1587–1594. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li Z, Li C, Du L, Zhou Y and Wu W: Human
chorionic gonadotropin beta induces migration and invasion via
activating ERK1/2 and MMP-2 in human prostate cancer DU145 cells.
PLoS One. 8:e545922013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang W and Liu HT: MAPK signal pathways
in the regulation of cell proliferation in mammalian cells. Cell
Res. 12:9–18. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liedert A, Kaspar D, Blakytny R, Claes L
and Ignatius A: Signal transduction pathways involved in
mechanotransduction in bone cells. Biochem Biophys Res Commun.
349:1–5. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Perdiguero E, Ruiz-Bonilla V, Gresh L, Hui
L, Ballestar E, Sousa-Victor P, Baeza-Raja B, Jardí M, Bosch-Comas
A, Esteller M, et al: Genetic analysis of p38 MAP kinases in
myogenesis: Fundamental role of p38alpha in abrogating myoblast
proliferation. EMBO J. 26:1245–1256. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hong C, Shen C, Ding H, Huang S, Mu Y, Su
H, Wei W, Ma J and Zheng F: An involvement of SR-B1 mediated p38
MAPK signaling pathway in serum amyloid A-induced angiogenesis in
rheumatoid arthritis. Mol Immunol. 66:340–345. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li CH, Xu LL, Zhao JX, Sun L, Yao ZQ, Deng
XL, Liu R, Yang L, Xing R and Liu XY: CXCL16 upregulates RANKL
expression in rheumatoid arthritis synovial fibroblasts through the
JAK2/STAT3 and p38/MAPK signaling pathway. Inflamm Res. 65:193–202.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang ZC, Lu H, Zhou Q, Yu SM, Mao YL,
Zhang HJ, Zhang PC and Yan WJ: MiR-451 inhibits synovial
fibroblasts proliferation and inflammatory cytokines secretion in
rheumatoid arthritis through mediating p38MAPK signaling pathway.
Int J Clin Exp Pathol. 8:14562–14567. 2015.PubMed/NCBI
|