Introduction
Hepatic stellate cells (HSCs) play a crucial role in
the development of liver fibrosis (1,2). During
various types of liver injury, such as that induced by viral
infection, immune stimulation and alcohol, HSCs, which are the
primary source of extracellular matrix (ECM) and overexpression of
α-smooth muscle actin (α-SMA), undergo activation and
proliferation, leading to the development of fibrosis due to the
abnormal deposition of collagen in the liver (3). Prior research has demonstrated that the
reduction of HSC numbers observed during reversion of liver
fibrosis is due to the transformation of HSCs from their activated
to a stationary state (4). It has
also been demonstrated that apoptosis of HSCs is the main mechanism
involved in the restoration stage of liver fibrosis, as this
inhibits the self-activation of HSCs and decreases the production
of ECM (5).
Heme oxygenase-1 (HO-1), also referred to as heat
shock protein 32 (Hsp32), is a microsomal rate-limiting enzyme that
catalyzes the degradation of heme into biliverdin, iron atoms, and
carbon monoxide (CO) (6). HO-1 and
its breakdown products possess biological protective properties,
including anti-oxidant, anti-inflammatory and immunoregulatory
properties (7). HO-1 is closely
associated with cell apoptosis, and most studies suggest that HO-1
may regulate apoptosis by affecting the expression of key proteins
in the apoptotic signaling pathway (8). In addition, HO-1 appears to exert
protective effects on liver cells under adverse conditions, such as
in acute liver injury, alcoholic liver disease, liver
transplantation and ischemia/reperfusion injury (9–11). In
our previous study using rats with cirrhosis induced by carbon
tetrachloride (CCl4) as a research model in vivo,
we demonstrated that HO-1 upregulation was able to alleviate α-SMA
expression, collagen synthesis and liver injury and limit the
extent of fibrosis (12).
Furthermore, we used activated HSC-T6 as a research model in
vitro to demonstrate that the induction of HO-1 can inhibit HSC
proliferation and collagen metabolism. However, during the research
mentioned above, we noticed that the upregulation of HO-1 led to
morphological changes in HSC-T6 cells, such as shrinkage and
nuclear condensation (karyopyknosis). Thus, we aimed to investigate
whether HO-1 is involved in liver protection by regulating the
apoptosis of HSCs, which has not yet been fully elucidated.
This study was undertaken in order to evaluate the
role of HO-1 in the apoptosis of activated HSCs-T6 in vitro.
Our hypothesis was that HO-1 may induce HSC apoptosis while
inhibiting the proliferation of HSCs, which would thus implicate
HO-1 as a viable therapeutic target in preventing or reversing
liver fibrosis.
Materials and methods
Materials and reagent preparation
Dulbecco's modified Eagle's medium (DMEM) and fetal
bovine serum (FBS) were purchased from Invitrogen (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Hemin [ferriprotoporphyrin IX,
HO-1 inducer (13)] and zinc
protoporyphyrin IX [ZnPP-IX, HO-1 inhibitor (14)] were purchased from Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany), and dissolved in 0.2 mol/l NaOH,
titrated to pH 7.4 with 1 mol/l HCl and diluted in 0.9% NaCl
(15). The dose and preparation of
hemin and ZnPP solution were based on the references (16,17). The
final concentrations were 1 mg/ml hemin and 0.5 mg/ml ZnPP-IX.
Polyclonal antibodies against HO-1, α-SMA, nuclear factor (NF)-κB
and caspase-3 were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Monoclonal antibodies against B-cell lymphoma
(Bcl)-2 and anti-β-actin polyclonal antibody were purchased from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). A lactate
dehydrogenase (LDH) release assay kit was obtained from
Sigma-Aldrich; Merck KGaA. An Annexin V-FITC/PI apoptosis detection
kit was purchased from BD Biosciences (Franklin Lakes, NJ, USA). A
terminal deoxynucleotidyl transferase dUTP nick-end labeling assay
(TUNEL) apoptosis detection kit was purchased from Nanjing Keygen
Biotechnology (Nanjing, China).
Cell culture and treatment
Immortalized rat HSC-T6 cells, which were provided
by Professor Zhongfu Zhao (Changzhi Medical College, Changzhi,
China), were cultured at 37°C in a humidified incubator under a 5%
CO2 atmosphere. The cells were passaged every 2 or 3
days and were used for the experiment within 4 to 6 passages.
HSC-T6 cells were incubated with hemin at concentrations of 6.5, 13
and 26 µg/ml, and with Znpp-IX at concentrations of 3, 6 and 12
µg/ml for 12, 24 and 48 h.
Cell viability assay
An MTT assay was used to assess HSC-T6 cell
viability under different concentrations of hemin and Znpp-IX at
12, 24 and 48 h. HSC-T6 cells were plated at a density of
1×104 cells/well in 96-well plates and cultured at 37°C
for 24 h. The medium was then replaced with fresh medium containing
hemin or Znpp-IX at different concentrations, as mentioned above.
Following incubation for 12, 24 and 48 h, the supernatants were
removed and cells were treated with 20 µl MTT solution (5 mg/ml)
for 4 h at 37°C. The formazan precipitates were dissolved in 150 µl
dimethyl sulfoxide, and optical density (OD) was measured at 490 nm
using an enzyme-linked immunometric meter: Cell survival
rate=(ODexperimental group-ODblank
group)/(ODcontrol group-ODblank group)
×100%.
LDH release assay
In order to determine whether the decrease in HSC
proliferative activity was due to apoptosis or necrosis caused by
drugs, the cytotoxic effect of hemin or ZnPP-IX on HSC-T6 cells was
evaluated using the LDH release assay. Preconfluent HSCs were
treated with hemin or ZnPP-IX at the indicated concentrations and
for the indicated times. LDH released by necrotic cells was present
in the supernatant and determined as LDHn, whereas LDH in
precipitates and adherent cell lysates was determined as LDHc. The
results are shown as follows: LDHn%=LDHn/(LDHn + LDHc) ×100. Then,
treatments with hemin at 13 µg/ml and Znpp-IX at 3 µg/ml for 24 h
were applied for subsequent experiments, as these were selected as
the concentrations most prominently affecting the proliferation of
HSC-T6 cells, without observable toxic cell damage.
Apoptosis quantification by flow
cytometry analysis
HSC-T6 cell apoptosis was measured by both flow
cytometry Annexin V/FITC and propidium iodide (PI) double staining
according to the manufacturer's instructions. HSCs were classified
into the control, hemin-treated, Znpp-IX-treated and hemin and
Znpp-IX co-treatment groups. After 24 h of treatment, cells were
detached with ethylenediaminetetraacetic acid (EDTA)-free trypsin
and washed twice with cooled phosphate-buffered saline (PBS), then
re-suspended in 500 µl 1X loading buffer with 5 µl Annexin V and 5
µl PI for 15 min at room temperature in the dark. Binding buffer
was then added and the cells were analyzed by flow cytometry on a
FACSCalibur analyzer. The percentage of Annexin V/FITC+
cells reflected the apoptosis rate.
Qualitative measurement of apoptosis
by TUNEL assay
To further investigate the apoptosis of HSC-T6
cells, a TUNEL assay was used. Preconfluent HSCs cultured in plates
were placed on sterile coverslips and treated with hemin or
Znpp-IX, grouped as mentioned above, for 24 h. Three replicates
were made for each group. The cells were washed with cold PBS three
times before fixation. Apoptotic HSCs were detected with the TUNEL
Apoptosis Assay kit, according to the manufacturer's protocol. The
results were expressed as apoptosis index: Apoptosis index=(number
of apoptotic cells/total number of cells) ×100%.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis of apoptosis-related genes
The effects of HO-1 on the mRNA level of NF-κB,
Bcl-2 and caspase-3 were measured by RT-qPCR. RNA was isolated
using TRI Reagent (Sigma-Aldrich; Merck KGaA), according to the
manufacturer's instructions. RT was performed on total RNA using
random monomers in a final volume of 50 µl. RT was performed in
three steps: 10 min at 25°C, 1 h at 37°C and 5 min at 95°C.
Quantitative detection was performed on the ABI PRISM 7700 (PE
Applied Biosystems, Nieuwerkerk a/d Ijssel, The Netherlands)
initialized by 10 min at 95°C, followed by 40 cycles (15 sec at
95°C, and 1 min at 60°C). Each sample was analyzed in duplicate.
The relative number of mRNA transcripts was normalized against
β-actin using the 2−ΔΔCq method. The specific primer
sequences are listed in Table I.
 | Table I.Primers for reverse
transcription-quantitative polymerase chain reaction analysis. |
Table I.
Primers for reverse
transcription-quantitative polymerase chain reaction analysis.
| Target gene | Primer sequence
(5′-3′) | Size (bp) |
|---|
| β-actin | F:
GTCAGGTCATCACTATCGGCAAT | 147 |
|
| R:
AGAGGTCTTTACGGATGTCAACGT |
|
| HO-1 | F:
CACGCATATACCCGCTACCT | 227 |
|
| R:
AAGGCGGTCTTAGCCTCTTC |
|
| α-SMA | F:
TGTGCTGGACTCTGGAGATG | 222 |
|
| R:
GAAGGAATAGCCACGCTCAG |
|
| NF-κB | F:
AACACTGCCGAGCTCAAGAT | 163 |
|
| R:
CATCGGCTTGAGAAAAGGAG |
|
|
Bcl-2[14] | F:
GGATGACTTCTCTCGTCGCTAC | 100 |
|
| R:
TGACATCTCCCTGTTGACGCT |
|
|
Caspase-3[15] | F:
GAGACAGACAGTGGAACTGACGATG | 147 |
|
| R:
GGCGCAAAGTGACTGGATGA |
|
Western blot analysis of
apoptosis-related proteins
The effect of HO-1 on the protein levels of NF-κB,
Bcl-2 and caspase-3 was measured by western blotting. Approximately
2×107 HSCs-T6 from each group were collected. The
proteins were extracted using radioimmunoprecipitation assay buffer
and loaded on SDS-PAGE gels, then transferred onto PVDF membranes,
which were blocked with 5% bovine serum albumin in TBST buffer
overnight at 4°C. Subsequently, the membranes were incubated with
the following primary antibodies: anti-HO-1 polyclonal antibody at
1:1,000 dilution, anti-NF-κB p65 polyclonal antibody at 1:800
dilution, anti-α-SMA polyclonal antibody at 1:1,000 dilution,
anti-Bcl-2 monoclonal antibody at 1:1,000 dilution, anti-caspase-3
polyclonal antibody at 1:800 dilution and anti-β-actin polyclonal
antibody at 1:4,000 dilution overnight at 4°C. The membranes were
then treated with a horseradish peroxidase-conjugated secondary
antibody. The blots were visualized using a Super ECL detection kit
(Amersham Pharmacia Biotech, Piscataway, NJ, USA), according to the
manufacturer's instructions. Protein bands were detected using a
Kodak Digital Science Imaging System (Kodak, Rochester, NY,
USA).
ELISA analysis for inflammatory
cytokines
To measure the secretion of cytokines from HSCs,
1×105/ml cells/well were cultured for 24 h in a 6-well
plate, then changed to serum-free medium to starve for 12 h. The
cells were further incubated with hemin or Znpp-IX, as mentioned
above, for 24 h. The levels of secreted inflammatory cytokines in
the supernatants were determined by ELISA (Quantikine Cytokine
kits; R&D Systems, Inc., Minneapolis, MN, USA) according to the
manufacturer's instructions. Each treatment was performed in
triplicate. The OD of each well was determined using a microplate
reader at 450 nm. The cytokine levels were derived from standard
curves using the curve-fitting program SOFTmax.
Statistical analysis
The quantitative data were expressed as mean ±
standard deviation. Levene was used in test for homogeneity of
variances. Comparisons between groups were tested by one-way ANOVA
and a post hoc LSD test. P<0.05 was considered to indicate a
statistically significant difference.
Results
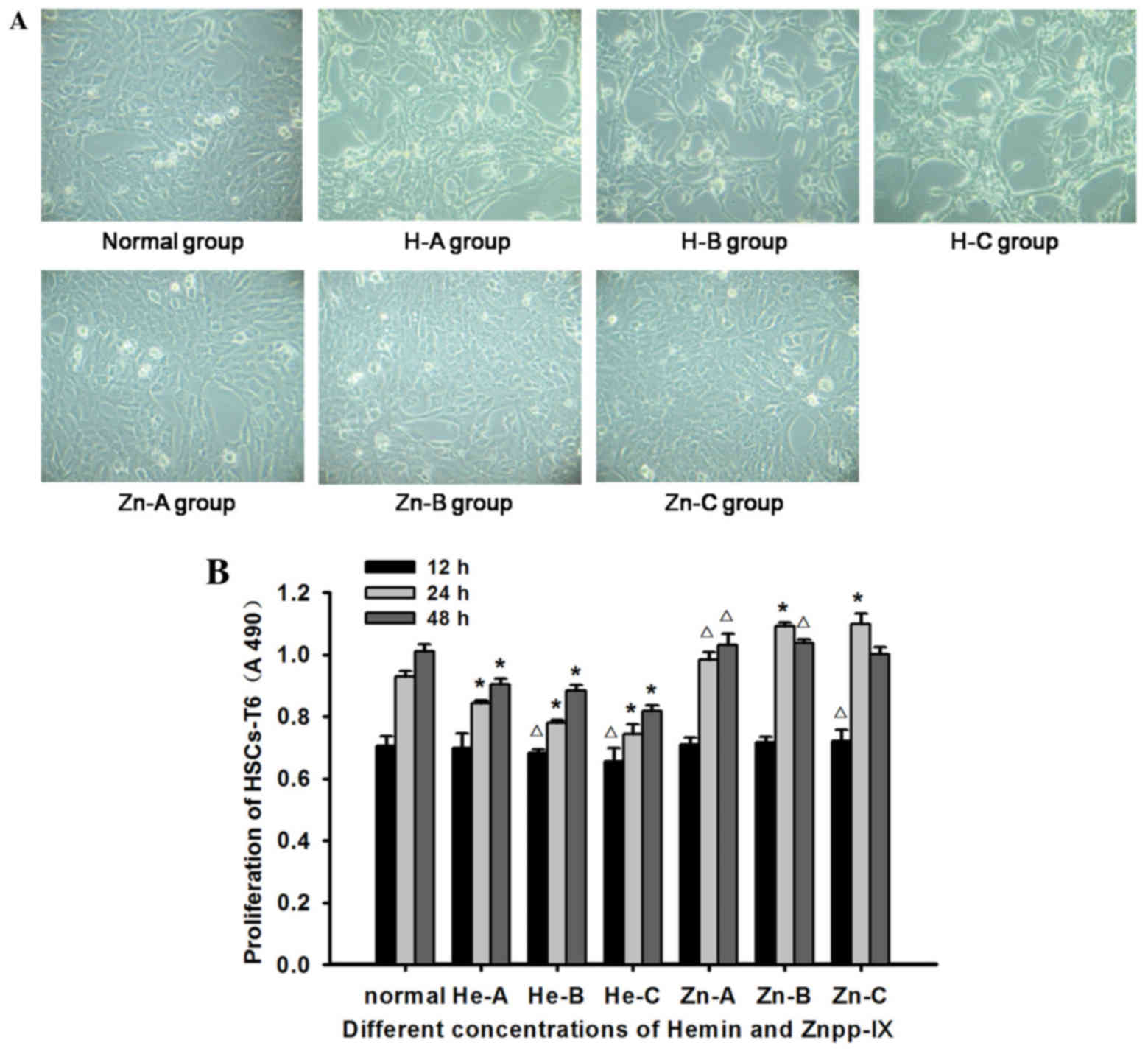
Observation of HSC growth
The morphology of HSCs-T6 in each concentration of
hemin or Znpp-IX at 12 h exhibited no significant changes compared
with that of the normal controls. However, at 24 h, the number of
HSCs was reduced with increasing concentrations of hemin. The cells
were shrunk and rounded, had started to aggregate into clusters,
and the transparency of the cytoplasm was decreased. These changes
were more obvious at 48 h, particularly in the 13- and 26-µg/ml
groups, where cell fragments floating in the supernatant were also
observed. Znpp-IX treatment at all concentrations for 24 h markedly
improved HSC growth and proliferation, and the cells almost
completely covered the bottom of culture flask (Fig. 1A). However, at 48 h in the 6- and
12-µg/ml groups, the cells had become slender, smaller in size, and
larger in pectin cavity, with cell fragments floating in the
supernatant.
HO-1-induced inhibition of HSC
proliferation
The proliferation of HSCs following hemin and
Znpp-IX treatment was determined by MTT assay. As shown in Fig. 1B, compared with normal control, hemin
caused a dose- and time-dependent reduction in cell proliferation.
In particular, HSC proliferation had decreased significantly at 24
and 48 h for all concentrations of hemin (P<0.01). In the
Znpp-IX treated group, HSC proliferation increased with increasing
dosage and treatment time. HSC survival at 12 h in the 12 µg/ml
group and at 24 h in all concentrations was significantly different
from normal control (P<0.05). However, at 48 h, HSC
proliferation exhibited a small decrease in the 12 µg/ml group. The
toxic effect of hemin and Znpp-IX on HSCs was carefully studied
with LDH release assays. As shown in Table II, cells treated with hemin 26 µg/ml
at 24 h and all concentrations at 48 h, and with Znpp-IX 12 µg/ml
at 24 and 48 h and 6 µg/ml at 48 h, exhibited a significant
difference in LDH release compared with the normal control.
Therefore, based on these observations, hemin at 13 µg/ml and
Znpp-IX at 3 µg/ml for 24 h were selected for the apoptosis
analysis.
| Table II.Lactate dehydrogenase release in
cultured HSC-T6 treated with Hemin or Znpp-IX. |
Table II.
Lactate dehydrogenase release in
cultured HSC-T6 treated with Hemin or Znpp-IX.
|
|
| Lactate
dehydrogenase (U/l) |
|---|
|
|
|
|
|---|
| Group | Concentration
(µg/ml) | 12 h | 24 h | 48 h |
|---|
| Normal | – | 22.12±0.65 | 22.83±1.21 | 26.57±0.75 |
| Hemin | 6.5 | 23.81±1.43 | 23.83±0.39 |
31.62±1.36b |
|
| 13 | 22.40±0.31 | 25.21±0.93 |
42.34±2.95a |
|
| 26 | 23.63±0.59 |
31.33±0.75a |
40.82±1.57a |
| Znpp-IX | 3 | 24.04±0.38 | 24.40±1.40 | 29.73±1.32 |
|
| 6 | 24.53±1.39 | 25.24±0.36 |
47.80±1.67a |
|
| 12 |
26.43±0.84a |
28.43±1.03a |
60.23±1.68a |
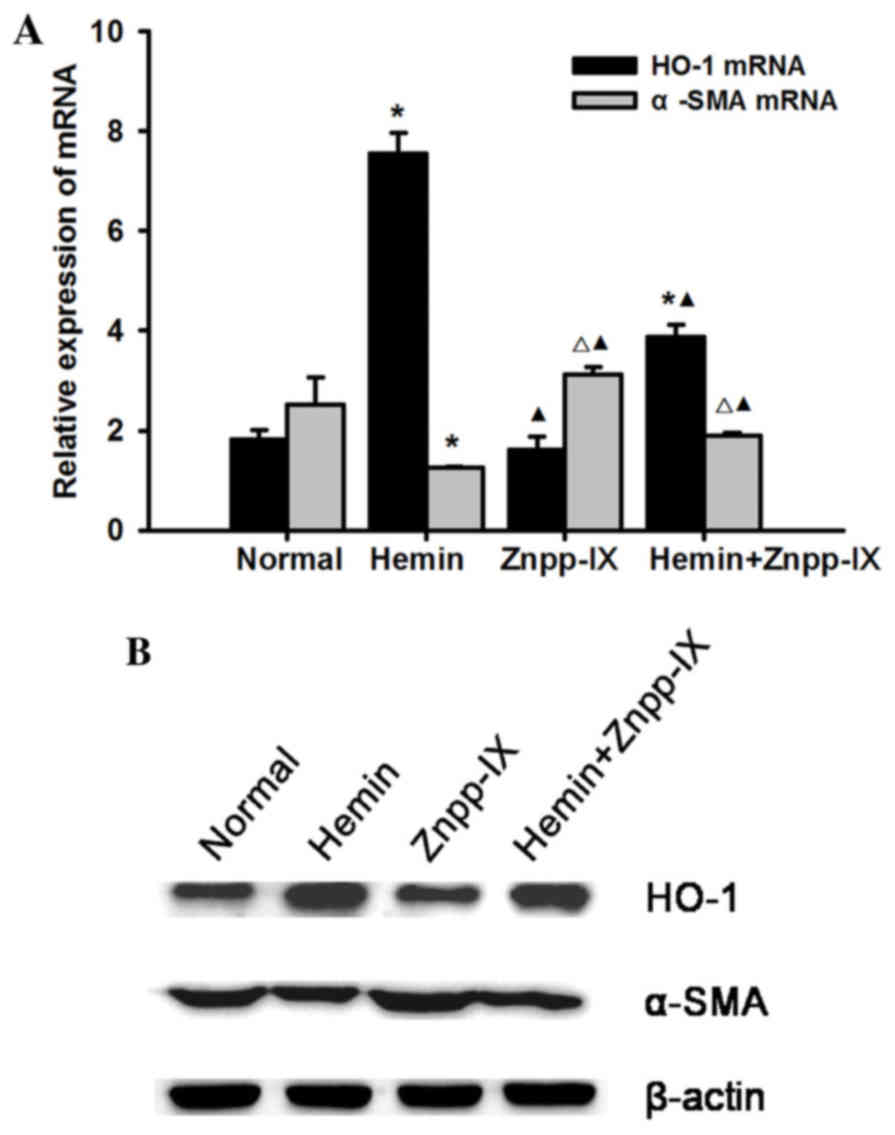
Effects of HO-1 on the expression of
α-SMA in HSC-T6 cells
Under treatment with hemin and Znpp-IX, the HO-1
mRNA and protein expression levels were detected as follows: HO-1
expression increased significantly following 13 µg/ml hemin
treatment compared with the normal control (P<0.01); it was the
lowest in the Znpp-IX group (P<0.05), while it was increased in
the hemin + Znpp-IX group compared with the Znpp-IX group
(P<0.01), but remained lower by 48.7% compared with that in the
hemin group (P<0.01). α-SMA is an important marker of HSC
activation and proliferation. The expression of α-SMA mRNA in the
HO-1 overexpression group decreased significantly compared with the
normal control (P<0.01), but the strongest expression was
observed in the Znpp-IX group (P<0.05). The results of the
protein expression were consistent with the mRNA levels, suggesting
that HO-1 induction in HSCs may decrease the expression of α-SMA,
with a decrease in HSC proliferative activity (Fig. 2).
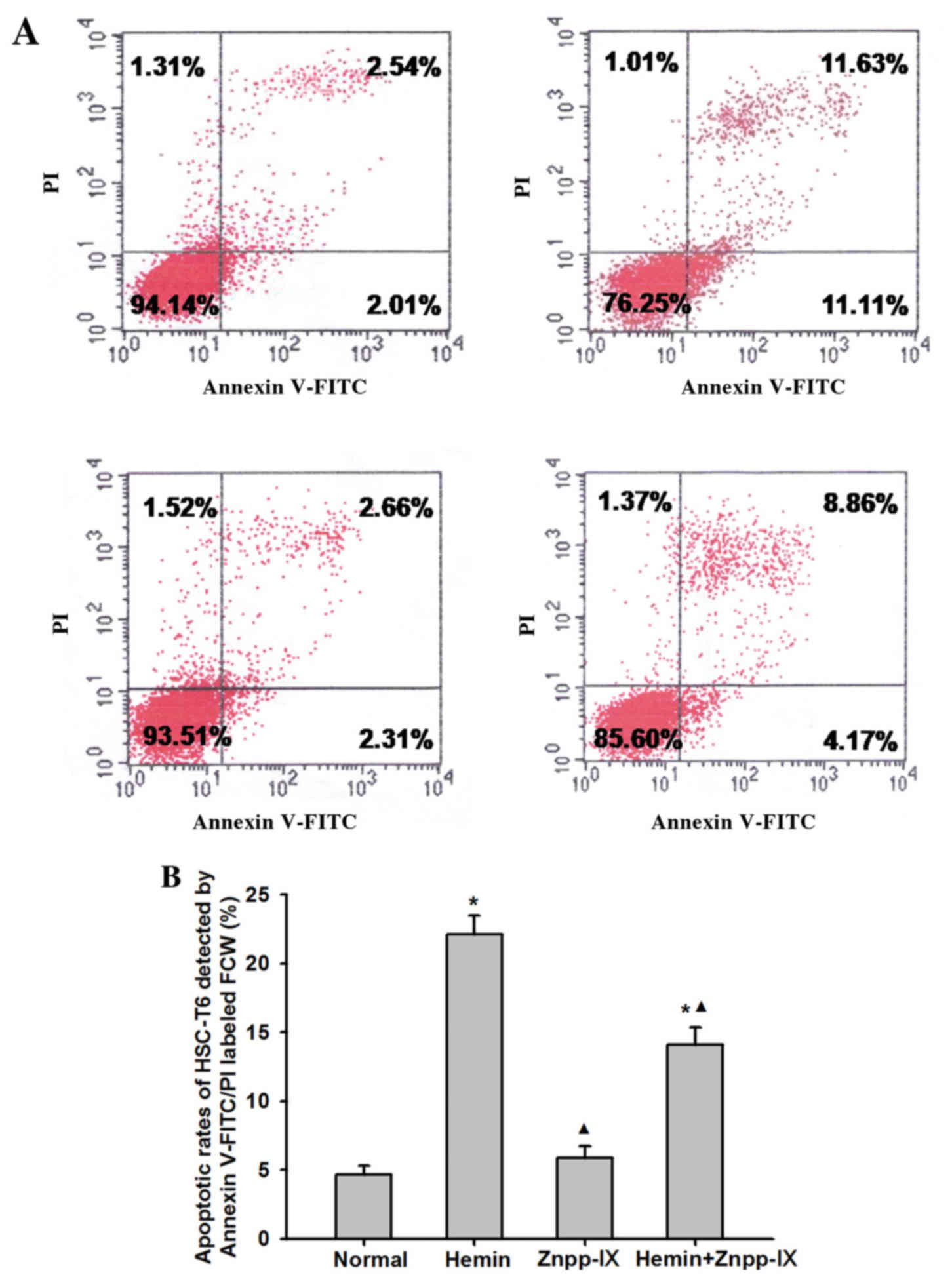
Effects of HO-1 on apoptosis of
activated HSCs
To determine whether the decrease in HSC
proliferation observed in hemin-treated groups was consistent with
the induction of apoptosis, we examined the rate of HSC apoptosis
using Annexin V-FITC/PI labeling. As shown in Fig. 3, the apoptosis rate of HSCs in the
normal control group treated with saline for 24 h was 4.67±0.63%,
whereas in the hemin group it was 22.11±1.38%, which was 3.73 times
higher (P<0.01). In the hemin + Znpp-IX group the apoptosis rate
was 14.07±1.28%, which was 2.01 times higher compared with the
normal control (P<0.01), but lower compared with that in the
hemin group (P<0.01). The apoptosis rate in the Znpp-IX group
did not differ significantly from that in the normal control group
(P=0.435).
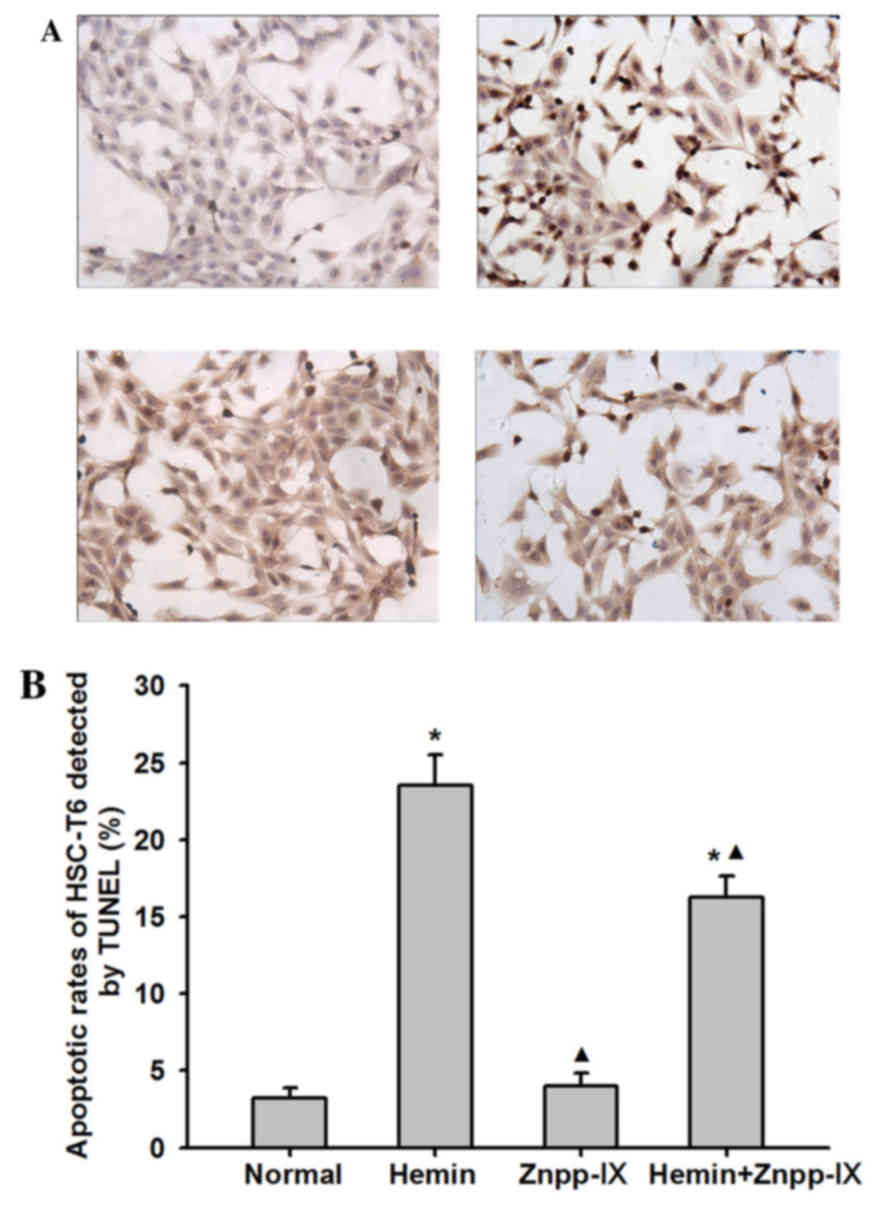
A TUNEL assay was performed to further evaluate the
role of HO-1 in HSC apoptosis. As shown in Fig. 4, the apoptotic index of HSCs in the
hemin group was 23.5±2.02%, which was significantly higher compared
with that in the normal control group (P<0.01), whereas in the
Znpp-IX group it was 4±0.82% (P<0.01). In the hemin + Znpp-IX
group, the apoptosis index was 16.25±1.38%, which was still lower
compared with that in the hemin group (P<0.01). These results
indicated that the inhibitory effect of HO-1 on HSC proliferation
was accompanied by promotion of HSC apoptosis.
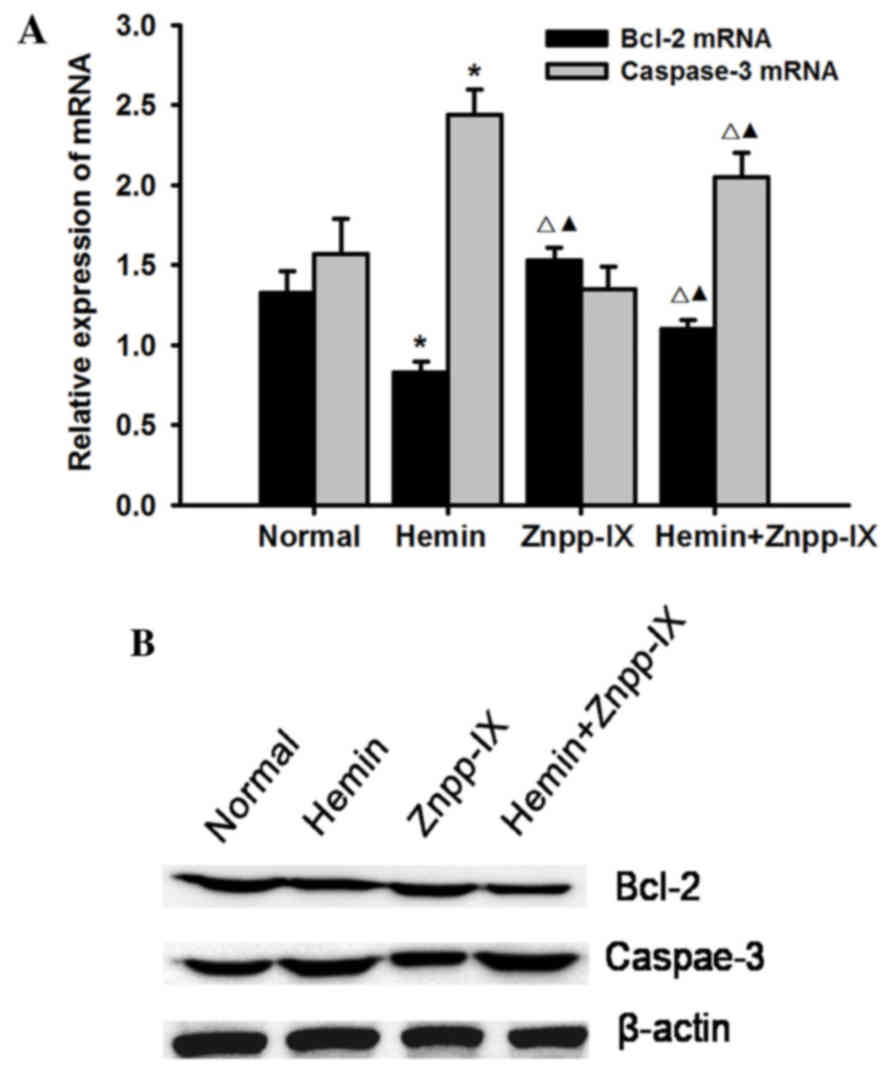
Effects of HO-1 on the expression of
Bcl-2 and caspase-3
To further investigate the involvement of hemin in
cell apoptosis, the expression of Bcl-2 and caspase-3 was measured.
Bcl-2 is an antiapoptotic protein, and its expression at the mRNA
and protein levels was detected as shown in Fig. 5: Bcl-2 expression in the hemin group
was significantly decreased compared with that in the normal
control group (P<0.05), while in the hemin + Znpp-IX group it
was higher compared with that in the hemin group (P<0.05). Bcl-2
expression in the Znpp-IX group was the highest (P<0.05)
compared with other groups.
Caspase-3 plays a key role in the execution phase of
cell apoptosis. HO-1 overexpression significantly increased the
mRNA and protein expression of caspase-3 (P<0.01) in the hemin
group, while it was significantly decreased in the hemin + Znpp-IX
group (P<0.05) and was the lowest in the Znpp-IX group. Taken
together, these results indicated that the regulation of HSC
apoptosis by HO-1 overexpression is mediated by inhibition of Bcl-2
and induction of caspase-3.
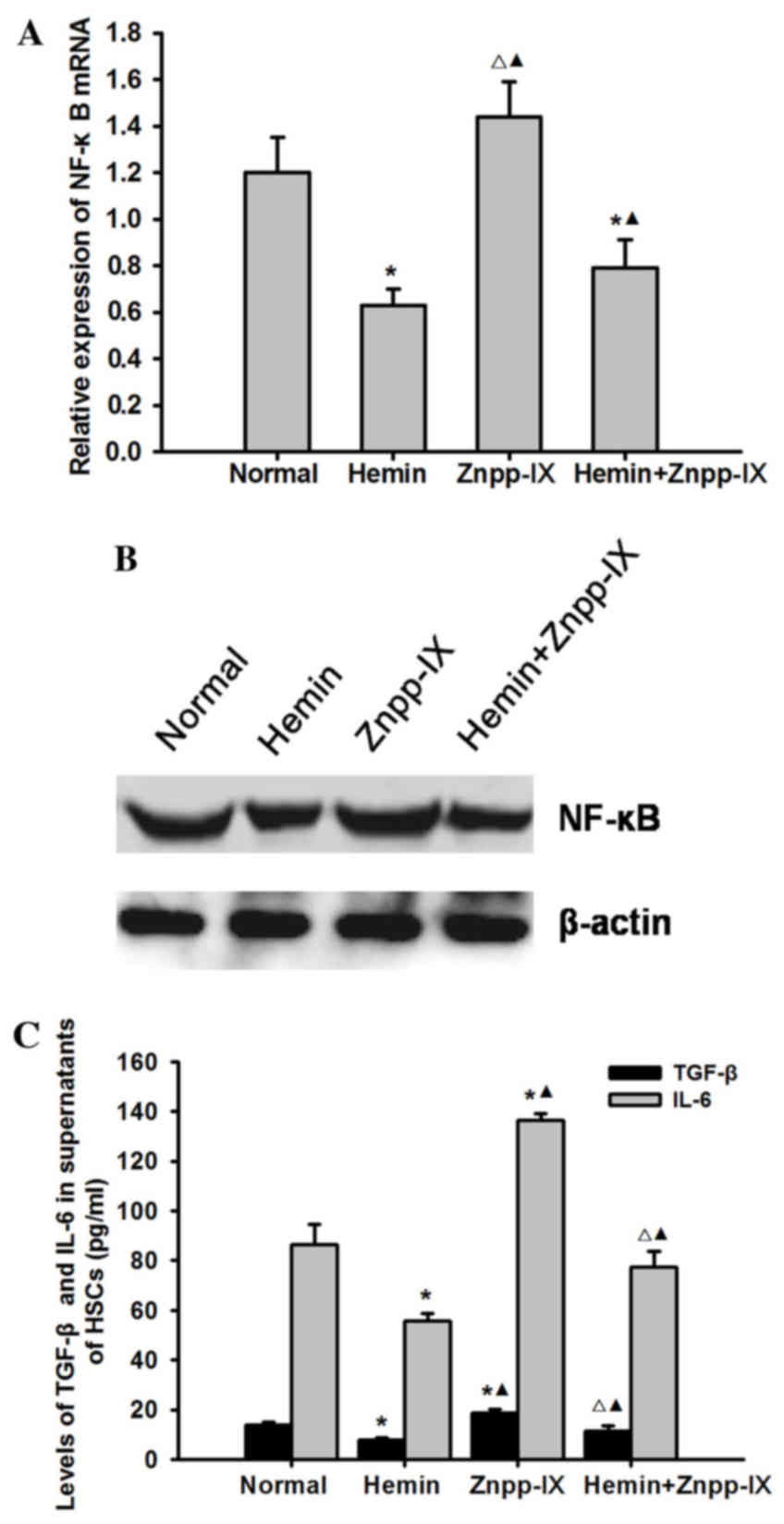
Effects of HO-1 on the expression of
NF-κB p65 and its downstream inflammatory factors TGF-β and
interleukin (IL)-6
To elucidate the possible mechanism underlying the
effect of HO-1 on HSC apoptosis, we examined the mRNA and protein
expression of NF-κB p65 and its downstream inflammatory factors
TGF-β and IL-6 in each experimental group. As shown in Fig. 6, the mRNA and protein expression of
NF-κB p65 in the hemin group was significantly decreased
(P<0.05), while in the hemin + Znpp-IX co-treatment group it was
significantly increased (P<0.05) compared with the hemin group,
but was highest in the Znpp-IX group (P<0.05). The levels of
TGF-β and IL-6 in the HSC supernatants were consistent with NF-κB
p65, also decreasing after hemin treatment (P<0.05) while
increasing after ZnPP-IX treatment.
Discussion
The present study demonstrated that regulation of
HSC activity is key to delaying or reversing hepatic fibrosis.
During the development of hepatic fibrosis, the numbers of HSCs
increase significantly, due to the proliferation of activated cells
and relative inhibition of apoptosis (18). HSC apoptosis, as a potential
regulatory mechanism, significantly reduces the level of specific
tissue inhibitors of matrix metalloproteinases synthesized by
activated HSCs, and increases the degradation of ECM components, so
as to prevent the progression of hepatic fibrosis, or even reverse
this process (19). Therefore,
inducing apoptosis of activated HSCs is one of the major strategies
in the study of prevention of hepatic fibrosis.
HO-1 has anti-inflammatory and anti-oxidant
properties, and is involved in the regulation of apoptosis. HO-1
was shown to markedly affect the cell cycle in renal epithelial
cells by upregulating the expression of p21, leading to alterations
in cell growth pattern and resistance to apoptosis (20). Induction of HO-1 was found to protect
endothelial cells from undergoing apoptosis, mediated through heme
catabolism into CO by the p38 mitogen-activated protein kinase
signal transduction pathway (21).
HO-1 also plays a protective role in preventing nutritional
steatohepatitis through suppressing hepatocyte apoptosis in mice by
upregulating the expression of anti-apoptosis genes and
downregulating pro-apoptosis genes (22). Interestingly, previous findings
suggested that HO-1 may also induce apoptosis. Aung et al
suggested that high-dose DEHP exposure induces caspase-3-dependent
apoptosis in Neuro-2a cells, which was mediated by the upregulation
of the HO-1 gene (23). The study of
Liu et al on the biological role of HO-1 in vascular smooth
muscle cells (SMCs) demonstrated that infection of SMCs with AdHO-1
inhibited serum-stimulated SMC proliferation and stimulated SMC
apoptosis in a dose-dependent manner, as demonstrated by DNA
fragmentation and caspase-3 activation (24); they also found that HO-1-mediated
apoptosis was associated with a marked increase in the expression
of the pro-apoptotic protein p53. In addition, HO-1 has been
reported to induce apoptosis and suppress proliferation and
invasion in breast cancer cells (25,26).
HO-1 was also found to be the key protein involved in determining
the selective effect on cancer cell apoptosis (27). It was first reported that
piperlongumine induced breast cancer cell apoptosis through the
upregulation of HO-1 expression, while protecting normal cells from
piperlongumine-induced apoptosis.
Our previous study demonstrated that HO-1
upregulation decreased α-SMA expression, collagen synthesis, liver
injury and the development of fibrosis in a rat model of
CCl4-induced liver fibrosis (12), which was correlated with the
expression of PPAR and NF-κB in liver tissues. In the present
study, we further demonstrated that the induction of HO-1 reduced
the proliferation and activation of HSC-T6 cells in vitro
and decreased the expression of α-SMA. It was observed that hemin
(a selective HO-1 inducer) inhibited the proliferation and
activation of HSCs in a time- and dose-dependent manner; in
addition, morphological changes reflecting a decrease in the growth
of HSCs were observed under a light microscope, such as group
clustering, volume reduction and karyopyknosis. Similarly, the
group treated with ZnPP-IX (an inhibitor of HO-1) exhibited
increased HSC proliferation with increasing treatment time and
higher concentration. The growth of HSCs under the microscope was
vigorous and folded together on the bottom of the flask. Based on
these results, we hypothesized that HO-1 may be involved in the
regulation of HSC apoptosis. To confirm this hypothesis, the TUNEL
assay demonstrated that HO-1 induction promoted HSC apoptosis with
volume reduction, spindled or crescent-shaped cells, karyopyknosis
and clustered accumulation. Additionally, Annexin V/FITC and PI
double-label flow cytometry was used to detect HSC apoptosis and
the results were consistent with those of the TUNEL assay.
Bcl-2 is an anti-apoptotic protein that acts by
reducing the permeability of the mitochondrial membrane and
inhibiting mitochondrial depolarization and cytochrome C release
(28). Our study demonstrated that
Bcl-2 expression decreased in HSCs in the HO-1 upregulation group
by hemin, recovered in the hemin + Znpp-IX co-treatment group, and
increased in the Znpp-IX group, suggesting that the anti-apoptosis
protein Bcl-2 was involved in the regulatory effect of HO-1 on HSC
apoptosis. Caspase-3 is considered to be the major protein in the
apoptotic cascade reaction, which is closely associated with the
apoptosis of HSCs. Various drugs induce HSC apoptosis by activation
of caspase-3 (29). We observed that
the expression of caspase-3 increased with the upregulation of HO-1
in HSCs, but decreased in the Znpp-IX group. These results indicate
that caspase-3 was also involved in the regulation of HSC apoptosis
by HO-1.
NF-κB, a gene pleiotropism transcription factor, can
activate gene transcription in a number of cell processes,
including the inflammatory response, cell proliferation,
differentiation and apoptosis (30).
Numerous studies have demonstrated that the activity of NF-κB
increases in HSCs during liver fibrosis, resulting in increased
expression of inflammatory factors, such as TNF-α, IL-6, ICAM-1 and
TGF-β, amplifying the inflammatory reaction of the liver (29). Increased activity of NF-κB can also
upregulate its downstream target genes, such as COX-2, cyclin D1
and Bcl-2, while inhibiting the activation of caspase-3 to inhibit
apoptosis (31,32). Accumulating data indicate that the
promoter region of HO-1 has binding sites for NF-κB, and that the
action of HO-1 is directly associated with NF-κB (33). Zhang et al reported that the
protective effect of HO-1 against intestinal barrier dysfunction in
cholestatic liver injury was associated with NF-κB inhibition
(34). Yeh et al suggested
that HO-1 activation may decrease myocardial ischemia-reperfusion
injury with cardioplegia during cardiopulmonary bypass via
inhibition of NF-κB and AP-1 translocation (35). So et al reported that
flunarizine attenuates cisplatin-induced pro-inflammatory cytokine
secretion and cyototoxicity through the downregulation of NF-κB by
Nrf2/HO-1 activation (36). In our
previous research, we also found that HO-1 upregulation decreased
the inflammation and pathological damage of liver tissues in rats
with hepatic fibrosis, while the expression of NF-κB in the liver
was markedly reduced. Consistent with these findings, in order to
elucidate the possible mechanisms underlying the regulation of HSC
apoptosis by HO-1, we examined the expression of NF-κB p65 and its
downstream inflammatory factors TGF-β and IL-6 in each experimental
group. Our results clearly demonstrated that HO-1 upregulation was
able to reduce the expression of NF-κB p65 and the release of TGF-β
and IL-6, whereas co-treatment with Znpp-IX alleviated these
inhibitory effects. By contrast, treatment of Znpp-IX alone exerted
the opposite effects.
The findings of the present study suggested that the
anti-fibrosis effects of HO-1 on activated HSCs were mediated by
inhibiting the proliferation of HSCs, and also through promoting
the apoptosis of HSCs by regulating the expression of
apoptosis-related proteins, such as Bcl-2 and caspase-3. This was
inconsistent with the majority of previous studies that suggested
HO-1 and its metabolites play a major inhibitory role in the
regulation of apoptosis. In combination with our previous animal
experiments, we further confirmed that HO-1 promoted HSC apoptosis
by attenuating the expression of NF-κB and its downstream signaling
molecules in HSCs. This indirect regulation may be advantageous to
the direct regulation of HO-1 itself and its metabolites on the
apoptosis-related pathway and protein expression in HSCs. Thus, the
protective role of HO-1 may be considered as a potential candidate
in the prevention of liver fibrosis, although further experiments
are required to clearly determine its role and potential
applications in liver diseases.
Acknowledgements
The authors would like to thank Dr Adelina Hung
(Yale University School of Medicine) for the English language
editing of this manuscript.
Funding
This study was supported by the International
Technology Cooperation Project of Shanxi Province (grant no.
2014081053-2) and the Foundation of Health and Family Planning
Commission of Shanxi Province (grant no. 201601030).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HY, LoZ and ZZ designed the study. LiZ, JC, XiaoqZ
and XiaohZ performed the experiments. BC and YZ analysed the data.
HY wrote the manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
She H, Xiong S, Hazra S and Tsukamoto H:
Adipogenic transcriptional regulation of hepatic stellate cells. J
Biol Chem. 280:4959–4967. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee SH, Seo GS, Park YN and Sohn DH:
Nephroblastoma overexpressed gene (NOV) expression in rat hepatic
stellate cells. Biochem Pharmacol. 68:1391–1400. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Melhem A, Muhanna N, Bishara A, Alvarez
CE, Ilan Y, Bishara T, Horani A, Nassar M, Friedman SL and Safadi
R: Anti-fibrotic activity of NK cells in experimental liver injury
through killing of activated HSC. J Hepatol. 45:60–71. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Burt AD: C. L. Oakley Lecture (1993).
Cellular and molecular aspects of hepatic fibrosis. J Pathol.
170:105–114. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Di Sario A, Bendia E, Baroni Svegliati G,
Ridolfi F, Casini A, Ceni E, Saccomanno S, Marzioni M, Trozzi L,
Sterpetti P, et al: Effect of pirfenidone on rat hepatic stellate
cell proliferation and collagen production. J Hepatol. 37:584–591.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Araujo JA, Zhang M and Yin F: Heme
oxygenase-1, oxidation, inflammation, and atherosclerosis. Front
Pharmacol. 3:1192012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gozzelino R, Jeney V and Soares MP:
Mechanisms of cell protection by heme oxygenase-1. Annu Rev
Pharmacol Toxicol. 50:323–354. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim KM, Im AR, Lee S and Chae S: Dual
protective effects of flavonoids from petasites japonicus against
UVB-induced apoptosis mediated via HSF-1 activated heat shock
proteins and Nrf2-activated heme oxygenase-1 pathways. Biol Pharm
Bull. 40:765–773. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lijie Z, Ranran F, Xiuying L, Yutang H, Bo
W and Tao M: Soyasaponin Bb protects rat hepatocytes from
alcohol-induced oxidative stress by inducing heme oxygenase-1.
Pharmacogn Mag. 12:302–306. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Park SW, Kang JW and Lee SM: The role of
heme oxygenase-1 in drug metabolizing dysfunction in the alcoholic
fatty liver exposed to ischemic injury. Toxicol Appl Pharmacol.
292:30–39. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu B, Song HL, Yang Y, Yin ML, Zhang BY,
Cao Y, Dong C and Shen ZY: Improvement of liver transplantation
outcome by heme oxygenase-1-transduced bone marrow mesenchymal stem
cells in rats. Stem Cells Int. 2016:92350732016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang H, Zhao LF, Zhao ZF, Wang Y, Zhao JJ
and Zhang L: Heme oxygenase-1 prevents liver fibrosis in rats by
regulating the expression of PPARγ and NF-κB. World J
Gastroenterol. 18:1680–1688. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Supinski GS and Callahan LA: Hemin
prevents cardiac and diaphragm mitochondrial dysfunction in sepsis.
Free Radic Biol Med. 40:127–137. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu H, Song D and Lee SS: Role of heme
oxygenase-carbon monoxide pathway in pathogenesis of cirrhotic
cardiomyopathy in the rat. Am J Physiol Gastrointest Liver Physiol.
280:G68–G74. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Martasek P, Schwartzman ML, Goodman AI,
Solangi KB, Levere RD and Abraham NG: Hemin and L-arginine
regulation of blood pressure in spontaneously hypertensive rats. J
Am Soc Nephrol. 2:1078–1084. 1991.PubMed/NCBI
|
|
16
|
Hualin C, Wenli X, Dapeng L, Xijing L,
Xiuhua P and Qingfeng P: The anti-inflammatory mechanism of heme
oxygenase-1 induced by hemin in primary rat alveolar macrophages.
Inflammation. 35:1087–1093. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Choi KM, Gibbons SJ, Nguyen TV, Stoltz GJ,
Lurken MS, Ordog T, Szurszewski JH and Farrugia G: Heme oxygenase-1
protects interstitial cells of Cajal from oxidative stress and
reverses diabetic gastroparesis. Gastroenterology. 135:2055–2064.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li X, Wang Y, Wang H, Huang C, Huang Y and
Li J: Endoplasmic reticulum stress is the crossroads of autophagy,
inflammation, and apoptosis signaling pathways and participates in
liver fibrosis. Inflamm Res. 64:1–7. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gressner AM: The up-and-down of hepatic
stellate cells in tissue injury: Apoptosis restores cellular
homeostasis. Gastroenterology. 120:1285–1288. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Inguaggiato P, Gonzalez-Michaca L, Croatt
AJ, Haggard JJ, Alam J and Nath KA: Cellular overexpression of heme
oxygenase-1 up-regulates p21 and confers resistance to apoptosis.
Kidney Int. 60:2181–2191. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Soares MP, Usheva A, Brouard S, Berberat
PO, Gunther L, Tobiasch E and Bach FH: Modulation of endothelial
cell apoptosis by heme oxygenase-1-derived carbon monoxide. Anti
oxid Redox Signal. 4:321–329. 2002. View Article : Google Scholar
|
|
22
|
Nan Y, Wang R, Zhao S, Han F, Wu WJ, Kong
L, Fu N, Kong L and Yu J: Heme oxygenase-1 prevents non-alcoholic
steatohepatitis through suppressing hepatocyte apoptosis in mice.
Lipids Health Dis. 9:1242010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Aung KH, Win-Shwe TT, Kanaya M, Takano H
and Tsukahara S: Involvement of hemeoxygenase-1 in di(2-ethylhexyl)
phthalate (DEHP)-induced apoptosis of Neuro-2a cells. J Toxicol
Sci. 39:217–229. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu XM, Chapman GB, Wang H and Durante W:
Adenovirus-mediated heme oxygenase-1 gene expression stimulates
apoptosis in vascular smooth muscle cells. Circulation. 105:79–84.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lin CW, Shen SC, Hou WC, Yang LY and Chen
YC: Heme oxygenase-1 inhibits breast cancer invasion via
suppressing the expression of matrix metalloproteinase-9. Mol
Cancer Ther. 7:1195–1206. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee WY, Chen YC, Shih CM, Lin CM, Cheng
CH, Chen KC and Lin CW: The induction of heme oxygenase-1
suppresses heat shock protein 90 and the proliferation of human
breast cancer cells through its byproduct carbon monoxide. Toxicol
Appl Pharmacol. 274:55–62. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee HN, Jin HO, Park JA, Kim JH, Kim JY,
Kim B, Kim W, Hong SE, Lee YH, Chang YH, et al: Heme oxygenase-1
determines the differential response of breast cancer and normal
cells to piperlongumine. Mol Cells. 38:327–335. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mòdol T, Brice N, de Galarreta Ruiz M,
Garzón García A, Iraburu MJ, Martínez-Irujo JJ and López-Zabalza
MJ: Fibronectin peptides as potential regulators of hepatic
fibrosis through apoptosis of hepatic stellate cells. J Cell
Physiol. 230:546–553. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Elsharkawy AM and Mann DA: Nuclear
factor-kappaB and the hepatic inflammation-fibrosis-cancer axis.
Hepatology. 46:590–597. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Uwagawa T and Yanaga K: Effect of NF-κB
inhibition on chemoresistance in biliary-pancreatic cancer. Surg
Today. 45:1481–1488. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kucharczak J, Simmons MJ, Fan Y and
Gélinas C: To be, or not to be: NF-kappaB is the answer-role of
Rel/NF-kappaB in the regulation of apoptosis. Oncogene.
22:8961–8982. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Son G, Iimuro Y, Seki E, Hirano T, Kaneda
Y and Fujimoto J: Selective inactivation of NF-kappaB in the liver
using NF-kappaB decoy suppresses CCl4-induced liver injury and
fibrosis. Am J Physiol Gastrointest Liver Physiol. 293:G631–G639.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin CC, Chiang LL, Lin CH, Shih CH, Liao
YT, Hsu MJ and Chen BC: Transforming growth factor-beta1 stimulates
heme oxygenase-1 expression via the PI3K/Akt and NF-kappaB pathways
in human lung epithelial cells. Eur J Pharmacol. 560:101–109. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang L, Zhang Z, Liu B, Jin Y, Tian Y,
Xin Y and Duan Z: The protective effect of heme oxygenase-1 against
intestinal barrier dysfunction in cholestatic liver injury is
associated with NF-κB inhibition. Mol Med. 23:2017.(Epub ahead of
print). View Article : Google Scholar
|
|
35
|
Yeh CH, Chen TP, Wang YC, Lin YM and Lin
PJ: HO-1 activation can attenuate cardiomyocytic apoptosis via
inhibition of NF-kappaB and AP-1 translocation following cardiac
global ischemia and reperfusion. J Surg Res. 155:147–156. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
So H, Kim H, Kim Y, Kim E, Pae HO, Chung
HT, Kim HJ, Kwon KB, Lee KM, Lee HY, et al: Evidence that
cisplatin-induced auditory damage is attenuated by downregulation
of pro-inflammatory cytokines via Nrf2/HO-1. J Assoc Res
Otolaryngol. 9:290–306. 2008. View Article : Google Scholar : PubMed/NCBI
|