Introduction
Atherosclerosis (AS) is a common chronic vascular
disease that may lead to cardiovascular and cerebrovascular
disease, affecting the health of the individual (1). A number of epidemiological studies have
revealed that AS occurrence is closely associated with infection,
including infection with human cytomegalovirus (HCMV) or
Chlamydophila pneumonia (2,3).
Endothelial cells (ECs) expressing the 36 kDa annexin II receptor
on their surface are potential hosts of HCMV (4). Bentz et al (5) revealed that HCMV infection of human
microvascular ECs could result in a decrease in the cellular
barrier function and an increase in permeability. This effect was
associated with cytoskeletal rearrangements, including actin stress
fiber depolymerization, and decreased expression of tight junction
proteins, containing occludin and vascular endothelial-cadherin
(6). Vascular endothelial impairment
and barrier function dysfunction are important factors in the
initiation of AS lesions; they facilitate the movement of monocytes
accompanied by peroxidized lipids across the vascular endothelium,
which are then deposited in the intima where monocytes absorb
lipids, resulting in the formation of foam cells, which accumulate
into atherosclerotic plaques (7).
Therefore, elucidating the mechanism by which HCMV infection leads
to the reduction of EC barrier function and promotes increased
permeability through the rearrangement of the cytoskeleton may
improve understanding of the process of AS formation.
Ena/vasodilator-stimulated phosphoprotein (VASP)
homology (EVH) proteins are actin-associated proteins involved in a
range of dynamic processes that are dependent on cytoskeletal
remodeling and cellular polarity, including axon guidance and
formation, lamellipodial and filopodial dynamics, platelet
activation and cell migration (8).
Additionally, as a main member of the EVH family, VASP was also
revealed to serve a crucial role in establishing and maintaining
the barrier functions of endothelial and epithelial cells, which
are closely associated with tight junction protein ZO-1 (ZO-1) at
tight junctions (9,10). ZO-1, which is located near the
tightly connected EC envelope, has a molecular weight of 225 kDa
(11,12) and contains an SH3 domain (10). VASP consists of three functional
regions: EVH1, EVH2 and proline-rich regions (PRR), of which PRR
can bind to an SH3 domain. In a previous study utilizing human
umbilical vein endothelial cells (HUVECs), VASP was phosphorylated
by protein kinase A and distributed to the cell-cell junction,
while the binding between phosphorylated VASP and ZO-1 was
significantly enhanced; also, the polymerization of tight junctions
was increased and EC permeability was significantly reduced
(9). These data demonstrated that
VASP and ZO-1 could jointly regulate EC barrier function. However,
further studies are required.
The Rho family of GTPases contains >20 members,
of which transforming protein RhoA (RhoA), Ras-related C3 botulinum
toxin substrate 1 (Rac1) and cell division control protein 42
homolog (Cdc42) are the main players involved in the regulation of
cell-cell connections and potential actomyosin networks (13,14).
RhoA- and Rac1-mediated signaling pathways can respectively disrupt
or maintain cell barrier function by coordinating actomyosin
contractions and barrier alterations in various cell types
(15,16). Furthermore, Rac1 and Cdc42 activities
are required to maintain barrier integrity (17,18) by
mediating the formation of actin filaments that associate with
proteins from junctional complexes, including ZO-1 and α-catenin at
the cell periphery (19). In
addition, Rac1 regulates the alterations of endothelial
permeability by mediating skeletal protein remodeling (20). It has been demonstrated that VASP is
a downstream effector of Rac1 in osteosarcoma cells (21). Therefore, Rac1-mediated VASP
activation may be involved in maintaining the barrier function of
ECs.
In the preent study, HCMV-induced EC barrier
dysfunction was used to investigate the role of Rac1-mediated VASP
activation in regulating vascular permeability, which may
contribute to elucidating the molecular mechanism underlying the
development of AS following HCMV infection.
Materials and methods
Plasmids, small interfering (si)RNAs
and antibodies
To generate green fluorescent protein (GFP)-VASP
overexpression plasmids, the VASP cDNA sequence was cloned into the
pEGFP-C1 (304 mg/ml; Clontech Laboratories, Inc., Mountainview, CA,
USA) multicloning site between the EcoRI and BamHI
restriction sites. To generate flag tag protein (Flag)-Rac1
overexpression plasmids, the Rac1 DNA sequence was cloned into the
pflag-CMV (285 mg/ml; Clontech Laboratories, Inc.) multicloning
site between the EcoRI and BamHI restriction sites.
The 21-nucleotide RNA oligos corresponding to human Rac1 (Genbank
accession number, 006908) were synthesized by Shanghai GenePharma
Co., Ltd. (Shanghai, China) as follows: siRNA1 targeted nucleotides
439-459 (sense, 5′-GGAGATTGGTGCTGTAAAA-3 and anti-sense,
5-UUUUACAGCACCAAUCUCC-3), siRNA2 targeted nucleotides 199-217
(sense, 5-CUACUGUCUUUGACAAUUA-3 and anti-sense,
5-UAAUUGUCAAAGACAGUAG-3) and siRNA3 targeted nucleotides 328-346
(sense, 5-CAUCCUAGUGGGAACUAAA-3′ and anti-sense,
5-UUUAGUUCCCACUAGGAUG3), with dTdT overhangs at each 3 terminus.
The following RNA oligos containing 21 nucleotides corresponding to
human VASP (Genbank accession number, 003370) were also synthesized
by Shanghai GenePharma Co., Ltd.: siRNA1 targeted nucleotides
942-960 (sense, 5-CAACCUUGCCAAGGAUGAATT-3′ and anti-sense,
5-UUCAUCCUUGGCAAGGUUGTG-3), siRNA2 targeted nucleotides 1,002-1,020
(sense, 5-CGCCCAGCUCCAGUGAUUATT-3′ and anti-sense,
5-UAAUCACUGGAGCUGGGCGTG-3) and siRNA3 targeted nucleotides
1,024-1,042 (sense, 5-GGACCUACAGAGGGUGAAATT-3 and anti-sense,
5′-UUUCACCCUCUGUAGGUCCGA-3). A negative control siRNA
(5′-AGUUCAACGACCAGUAGUCdTdT-3′) was also used in the experiment
(Shanghai GenePharma Co., Ltd). The anti-Rac1 (cat. no. 610651;
dilution 1:1,000; BD Biosciences, Franklin Lakes, NJ, USA),
anti-VASP (cat. no. 0010-10; dilution, 1:1,000; ImmunoGlobe GmbH,
Himmelstadt, Germany), anti-GAPDH (cat. no. SC-69778; dilution,
1:5,000) and anti-tubulin (cat. no. SC-73242; dilution, 1:5,000)
(both Santa Cruz Biotechnology, Inc., Dallas, TX, USA) antibodies
were used in the study.
Cell culture and transfection
Two endothelial cell lines, the primary HUVECs and
HUVEC-CRL1730, were obtained from the Key State Laboratory of
Virology of Wuhan University (Wuhan, China). The cells were
cultured in Dulbecco's modified Eagle's medium (DMEM, Hyclone; GE
Healthcare, Logan, UT, USA) supplemented with 10% fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA), 100 U/ml penicillin G and 100 U/ml streptomycin sulfate at
37°C in a humidified incubator supplemented with 5% CO2.
Prior to transfection, the HUVEC-CRL1530 cells were seeded at a
density of 2×105 cells/well in 6-well plates and grown
overnight in DMEM (without FBS or antibiotics) until they reached
60-70% confluence. Transfection of siRNAs or plasmids was carried
out using Lipofectamine® 2000 reagent (Invitrogen;
Thermo Fisher Scientific, Inc.). Lipofectamine and siRNAs or
plasmids were each diluted in DMEM. Diluted Lipofectamine was then
mixed with diluted siRNAs or plasmids, and this mixture was
incubated for 20 min at room temperature for complex formation.
Following the addition of DMEM (2 ml/well) to the cells, the
Lipofectamine/siRNA or Lipofectamine/plasmid mixture was added to
each well, resulting in a final concentration of 100 pmol siRNAs or
4 µg plasmids. At 48 h after transfection, the cells were collected
for western blotting or incubated with TRIzol (Invitrogen; Thermo
Fisher Scientific, Inc.) for reverse transcription-quantitative
(RT-q) PCR analysis.
HCMV titration assay and observation
of cell morphology
The HCMV AD169 He was obtained from the Perinatal
Laboratory of Tongji Medical College, Huazhong University of
Science and Technology (Wuhan, China). The HUVEC-CRL1730 cells were
seeded at 5×103 cells/well in 96-well plates and
cultured in DMEM (supplemented with 2% FBS and antibiotics) at 37°C
in 5% CO2. To determine the CCID50/ml, serial 10-fold dilutions
followed by serial 8-fold dilutions of the HCMV viral sample were
made in DMEM. The following day the cells were inoculated a 96-well
plate with the same dilution of virus, 6 replicate wells were set
up for each dilution each well was inoculated with 0.1 ml diluted
virus solution in a 37°C, 5% CO2 incubator. Following HCMV
infection, the cells become larger and rounder and specific
‘hawk-eye’ changes appeared, which were observed under a light
microscope (Olympus IX 73 DP80, Olympus Corporation, Tokyo, Japan).
Following inoculation, depending on the speed of the HUVEC-CRL1530
cell lesions, observations were continued for several days until
the cytopathies did not progress. Cytopathic effect (CPE) is a
specific lesion that occurs when cells are infected with a virus.
The virus dilution corresponding to 50% CPE is the titer of the
virus. The dilution of half of the lesions was taken as the 50%
infection unit; CCID50 (where CCID50 represents the 50% cell
culture infectious dose in HUWEC-CRL1530 cells).
Endothelial monolayer cell
permeability assay
In vitro cell permeability analysis was
performed with fluorescein isothiocyanate (FITC)-dextran
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) as follows.
Following treatment with a time gradient HCMV (10−5
dosage) infection at 0, 4, 8, 12 and 24 h or plasmid and siRNA
transfection, the HUVEC-CRL-1730 cells were seeded into the upper
chambers of a Costar Transwell 24-well plate at a density of
1×103 cells/cm2 and initially plated with 1%
gelatin (membrane diameter, 6.5 µm; pore size, 0.4 µm). Following
adherence, the cells were cultured in serum-free DMEM for 24 h. The
medium in the upper layer was subsequently replaced with medium
containing 100 µg/ml FITC-dextran (100 µl) and the lower chamber
was filled with normal medium. After incubation for 45 min, the
fluorescence intensity of the sample was measured in a black
96-well plate with 100 µl sample from the upper and lower chambers.
An excitation wavelength of 490 nm and an emission wavelength of
520 nm were used to measure the fluorescence in each well using a
microplate reader (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) and the volume of liquid in the lower chamber was measured.
The permeability of the EC monolayer to FITC-labeled dextran was
expressed as ‘Pa’ and calculated as follows: Pa=[A]/t × 1/A +
V/[L]. In the formula, ‘t’ was the time in seconds; ‘[A]’ was the
FITC-labeled dextran concentration in the upper layer (expressed in
terms of fluorescence intensity); ‘V’ was the volume of liquid in
the lower chamber in ml; ‘[L]’ was the FITC-labeled dextran
concentration in the lower chamber (expressed in terms of
fluorescence intensity); and ‘A’ was the filter area in
cm2.
Isolation of RNA and RT-qPCR
analysis
The HUVEC-CRL-1730 cells were seeded into the 6-well
plate with HCMV infection and/or plasmid and siRNA transfection and
the mRNA expression was measured at 0, 4, 8, 12, 16, 24 and 48 h.
Total RNA was extracted from cells using TRIzol reagent, and 2 mg
total RNA was used for first-strand cDNA synthesis with a
RevertAid™ First-Strand cDNA Synthesis kit (Fermentas; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
The semi-quantitative PCR was performed using the CFX96 Real Time
PCR Detection system (Bio-Rad Laboratories, Inc., Hercules, CA,
USA) in the presence of SYBR Green (Thermo Fisher Scientific,
Inc.). All qPCRs were run in triplicate and repeated at least three
times. Relative mRNA expression was calculated using the
2−ΔΔCq method (22) using
GAPDH as the internal reference. The primers used were as follows:
VASP, sense 5-GGAAAGTCAGCAAGCAGG-3 and antisense
5′-TGTGCGGAAAGGAGAAGC-3; Rac1, sense 5-AGACGGAGCTGTAGGTAAAA-3 and
antisense 5-GCAGGACTCACAAGGGA-3; GAPDH, sense
5-AGGTCCACCACTGACACGTT-3′ and antisense 5′-GCCTCAAGATCATCAGCAAT-3′.
Rac1 reaction parameters were 94°C for 40 sec, 54°C for 30 sec and
72°C for 30 sec for 40 cycles. VASP reaction parameters were 94°C
for 40 sec, 60°C for 30 sec and 72°C for 30 sec for 40 cycles.
Western blotting
The HUVEC-CRL-1730 cells were seeded into the 6-well
plate treatment with HCMV infection and/or plasmid and siRNA
transfection at the protein expression was measured at 0, 4, 8, 12,
16, 24 and 48 h. Following that the cells were washed with PBS and
lysed in 200 µl modified RIPA buffer (Thermo Fisher Scientific,
Inc.). Protein concentration was measured with a Pierce BCA Protein
Assay kit (Thermo Fisher Scientific, Inc.). Equal amounts of total
protein (10 µg/lane) were loaded, electrophoresed on 10% SDS-PAGE
gels and transferred to polyvinylidene difluoride membranes (EMD
Millipore, Billerica, MA, USA). The membranes were blocked with
Tris-buffered saline containing 5% nonfat milk for 2 h at room
temperature, then probed with antibodies against VASP (1:1,000
dilution), Rac1 (1:1,000 dilution) and GAPDH at 4°C overnight. The
membranes were then incubated with horseradish peroxidase-linked
goat anti-rabbit IgG (cat. no. 16473-1-AP; 1:1,000 dilution) and
anti-mouse IgG (cat. no. 10285-1-AP; 1:1,000 dilution) (both
ProteinTech Group, Inc., Chicago, IL, USA) secondary antibodies for
2 h at room temperature. The bound antibodies were detected by
Pierce™ ECL Western Blotting substrate (Thermo Fisher Scientific,
Inc.). Quantification of band densities was performed using ImageJ
software (version 1.5.0.26; National Institutes of Health,
Bethesda, MD, USA). The experiments were repeated three times.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. Statistical significance between groups was tested using
one-way analysis of variance followed by a Bonferroni's post-hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
HCMV infection induces increased EC
permeability
The normal primary HUVECs appeared closely linked
and flat with typical spindle shapes (Fig. 1A). After 6 passages, the morphology
of primary HUVECs had changed when viewed under an inverted
microscope, with an increased number of irregular protrusions
decreased density and more cellular debris in the cytoplasm
(Fig. 1B). After 7 days of HCMV
infection, the morphology of the primary HUVEC cells was altered,
with evident nuclear enlargement and cell swelling, and gradual
rounding of the cells (Fig. 1C).
Typical dense basophilic inclusion bodies surrounded by a halo in
the nucleus and ‘owl eye’-like alterations of the nuclei were
observed. The normal HUVEC-CRL-1730 cells appeared closely linked
and flat with typical spindle shapes (Fig. 1D). After 10 days of HCMV infection,
the HUVEC-CRL-1730 cell boundaries became blurred and numerous
cells merged to form giant cells, and a number of the cells died
(Fig. 1E).
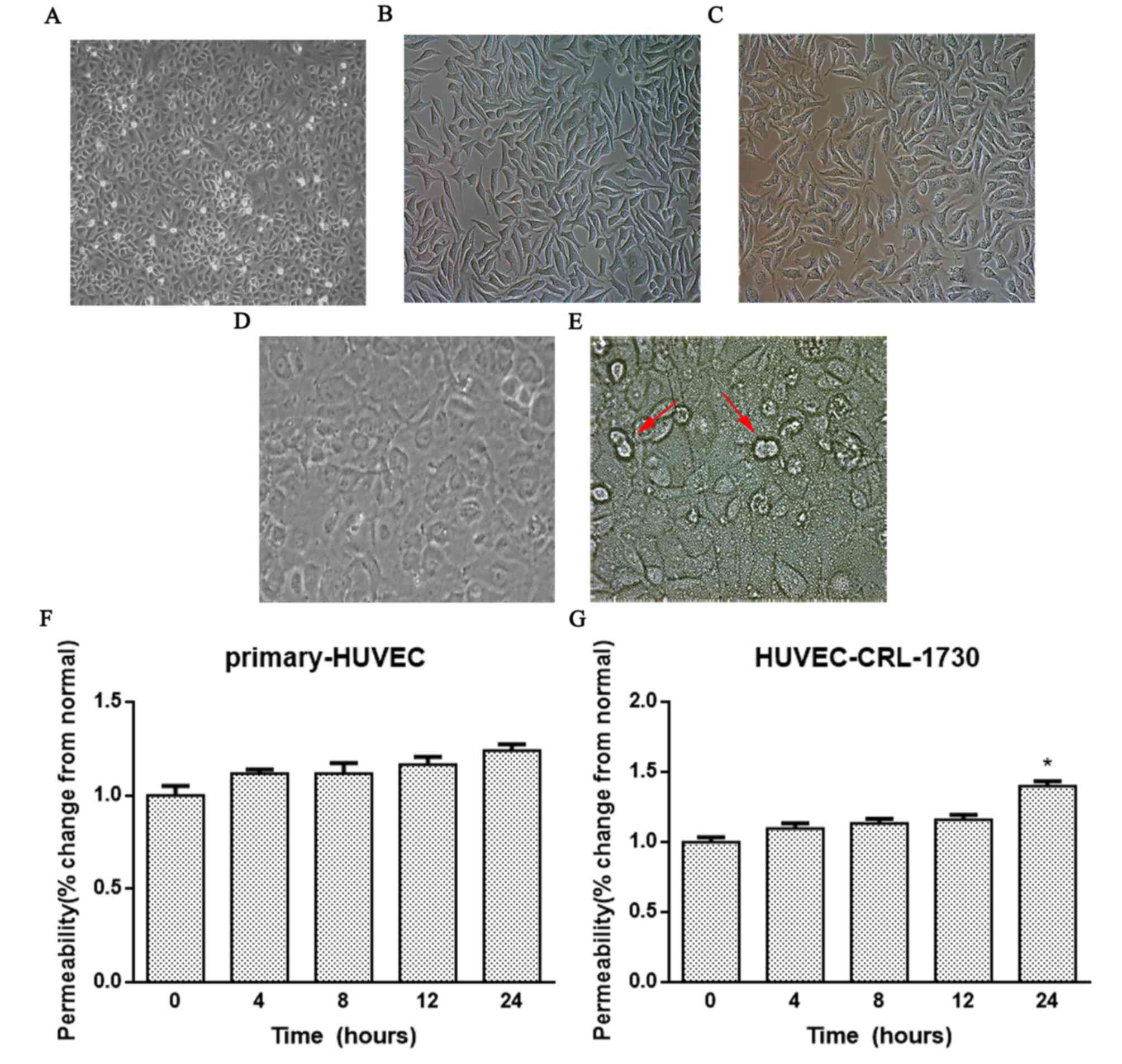 | Figure 1.HCMV infection induces increased EC
permeability. Cell morphology was observed in cultures of (A)
normal primary HUVECs (magnification, ×100), (B) primary HUVECs
following 6 passages (magnification, ×400) and (C) primary HUVECs 7
days after HCMV infection (magnification, ×400). Cell morphology
was observed in cultures of (D) normal HUVEC-CRL-1730 cells
(magnification, ×400). (E) HUVEC-CRL-1730 10 days after HCMV
infection (magnification, ×400), the red arrows indicate the ‘owl
eye’. (F) Primary HUVECs and (G) HUVEC-CRL-1730 monolayer cells
were infected with tissue culture infective
dose50 (10−5) HCMV AD169 for 0, 4, 8, 12
and 24 h. A Transwell assay was then performed to detect
endothelial permeability using fluorescein isothiocyanate-labeled
dextran. Data are expressed as the mean ± standard error of the
mean (n=3/group). *P<0.05 vs. the 0 h group. HUVEC, human
umbilical vein endothelial cell; HCMV, human cytomegalovirus; EC,
endothelial cell. |
Next, the effect of HCMV infection on the
permeability of the two EC cell lines was studied. First, the 50%
cell culture infective dose (CCID50) of HCMV was
determined to be 10−5 by a virus titration assay. The
monolayer cells were infected with CCID50 HCMV AD169 for
4, 8, 12 and 24 h. A Transwell assay was subsequently used to
analyze the intercellular permeability of FITC-labeled dextran. As
revealed in Fig. 1F and G, the
permeability of HUVEC-CRL-1730 cells was significantly increased
after HCMV infection compared with the negative control group
(P<0.05), while no significant difference was identified in
primary HUVECs. These results indicated that HCMV infection
increased the permeability of monolayer HUVEC-CRL-1730 cells,
leading to a decline in cell barrier function.
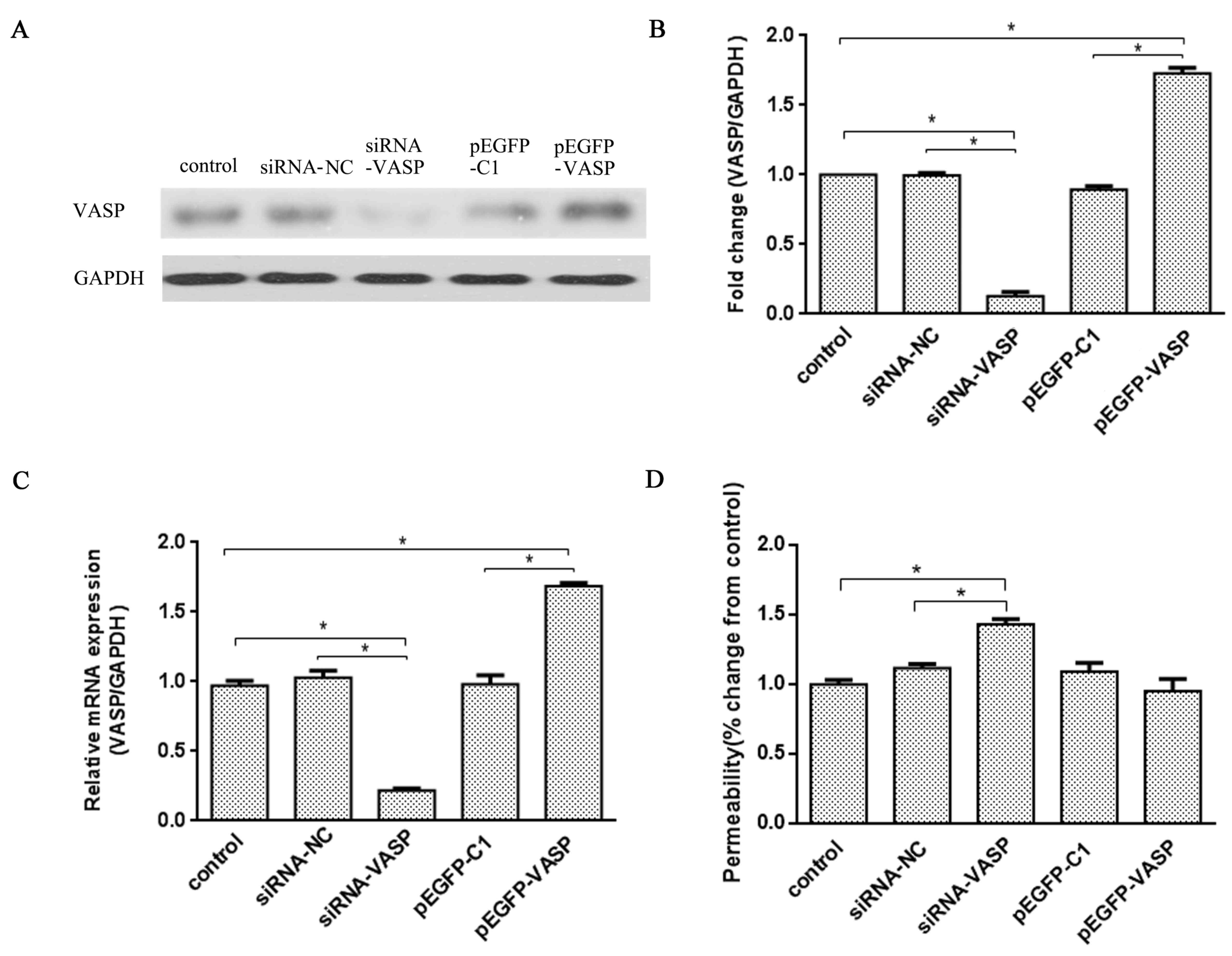
VASP is involved in maintaining EC
permeability
VASP is involved in maintaining EC permeability in
HUVEC-CRL-1730 cells via binding to ZO-1 at the cell junction
(9). In order to verify whether the
expression level of VASP affects the permeability of ECs, a
loss-of-function assay was utilized by transfecting HUVEC-CRL-1730
cells with siRNA-VASP or the overexpression plasmid pEGFP-VASP.
siRNA-VASP significantly inhibited VASP gene transcription in the
cells, which resulted in the protein expression level being
decreased to 85% (P<0.05; Fig. 2A and
B) and the mRNA expression level of VASP being reduced to 79%
(P<0.05; Fig. 2C) compared with
the siRNA-NC group. On the contrary, compared with the pEGFP-C1
group, transfection with pEGFP-VASP upregulated VASP protein and
mRNA expression by 72 and 68%, respectively (both P<0.05;
Fig. 2B and C).
In addition, a Transwell assay was used to detect
permeability 24 h after transfection. VASP knockdown revealed a
40±2% increase in endothelial permeability (both P<0.05;
Fig. 2D) compared with the control
or siRNA-NC groups, whereas the overexpression plasmid resulted in
no significant difference in endothelial permeability.
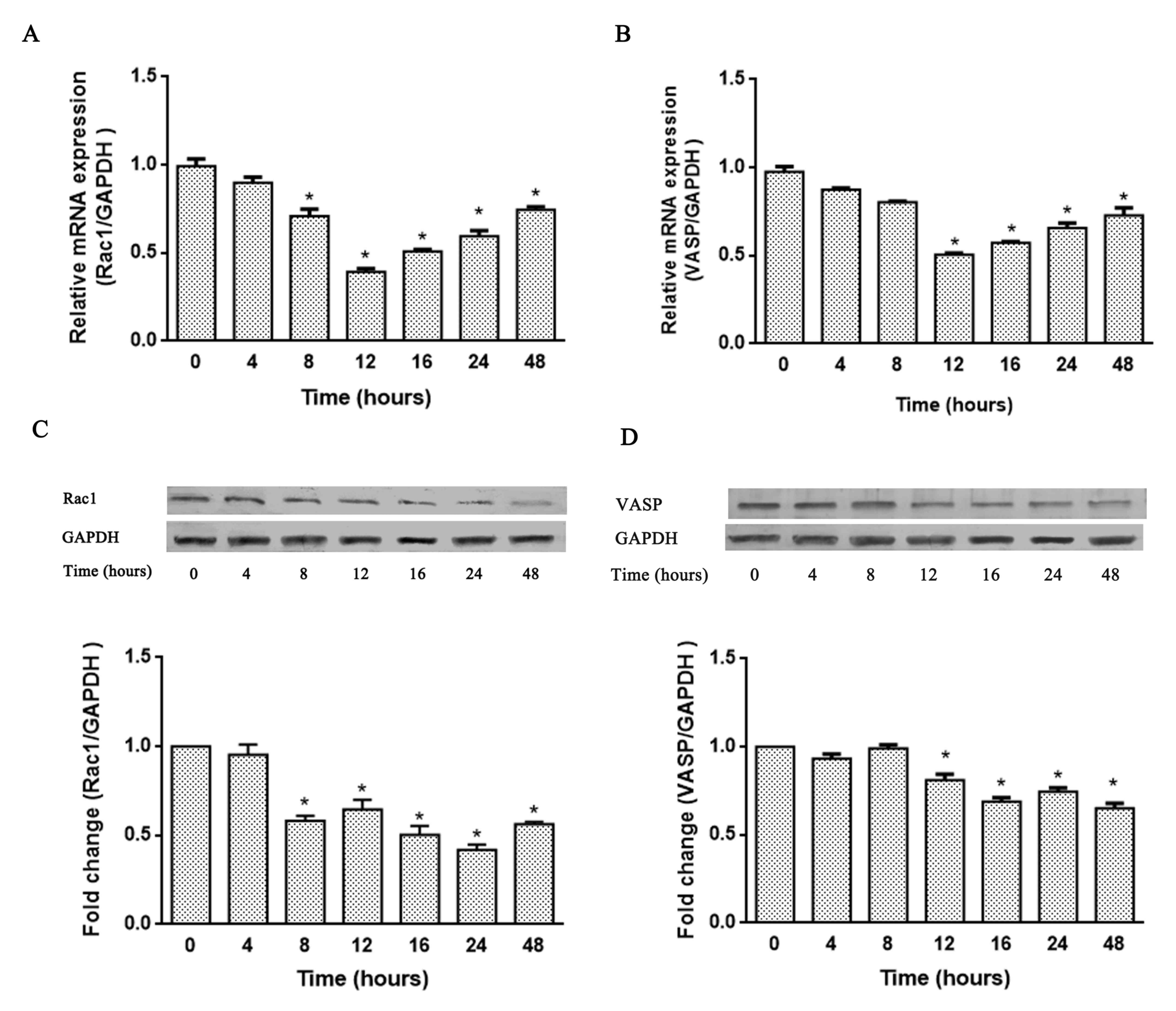
HCMV infection reduces the expression
of Rac1 and VASP in HUVEC-CRL-1730 cells
The Rac1/VASP signaling pathway is involved in the
regulation of endothelial permeability (23) and HCMV infection increased the
permeability of the monolayer HUVEC-CRL-1730 cells. To investigate
the potential signaling pathway involved in HCMV infection-induced
hyperpermeability in ECs, the expression of Rac1 and VASP at the
mRNA and protein levels were examined in HCMV-pretreated cells over
a range of times. The results demonstrated that HCMV infection
significantly inhibited Rac1 and VASP mRNA expression starting at 8
and 12 h, respectively, compared to the 0 h group (all P<0.05;
Fig. 3A and B); maximum inhibition
was achieved at 12 h. HCMV infection also significantly inhibited
Rac1 and VASP protein expression starting at 8 and 12 h,
respectively, compared with the 0 h group (all P<0.05; Fig. 3A). Maximum Rac1 and VASP protein
inhibition was achieved at 24 and 48 h, respectively. Therefore,
the data demonstrated that Rac1 and VASP expression levels can be
affected by HCMV infection, which also suggested that VASP
expression was positively associated with Rac1. These data suggest
that HCMV infection-induced increases in EC permeability may be
achieved by altering the expression levels of VASP and/or Rac1.
HCMV infection induces
hyperpermeability through the Rac1/VASP signaling pathway in
HUVEC-CRL-1730 cells
HCMV infection inhibited the expression of Rac1 and
VASP, and, therefore, to investigate the association between Rac1
and VASP, silencing or over-expression of Rac1 was carried out
using siRNA-Rac1 or a pflag-Rac1 plasmid, respectively. At the mRNA
and protein level, siRNA-Rac1 oligonucleotides demonstrated a
significant inhibitory effect on Rac1 and VASP expression, and the
pflag-Rac1 plasmid significantly increased Rac1 and VASP expression
compared with the control group and their respective negative
controls (all P<0.05; Fig. 4A-C).
These data indicated that VASP may act as a downstream effector of
Rac1 in HUVEC-CRL-1730 cells.
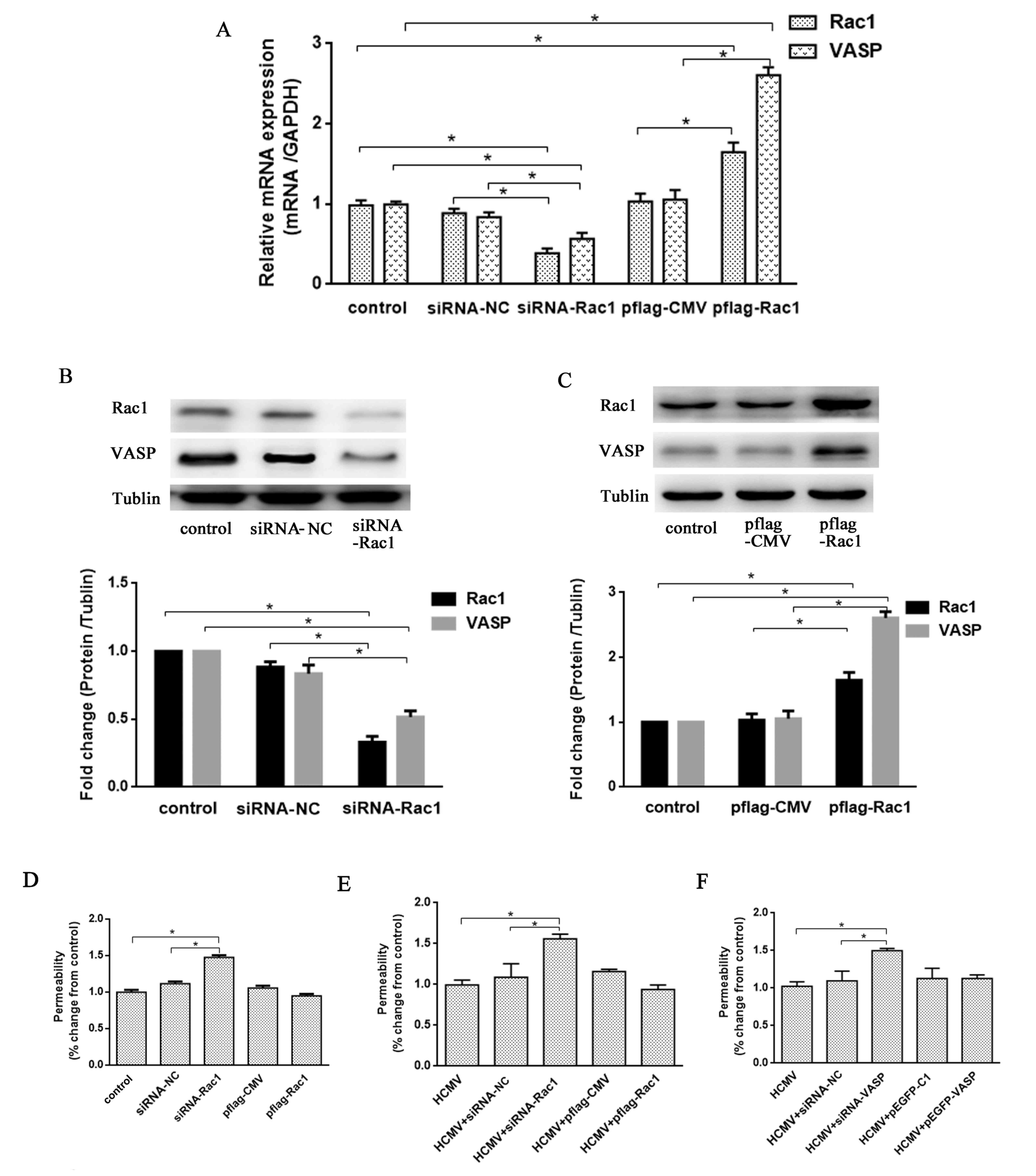 | Figure 4.HCMV infection induces
hyperpermeability through the Rac1/VASP signaling pathway in
HUVEC-CRL-1730 cells. (A) Rac1 and VASP mRNA quantification,
relative to GAPDH mRNA, was assessed by reverse
transcription-polymerase chain reaction, in cells treated with
siRNA-NC, siRNA-Rac1 or pflag-Rac1 for 48 h. Rac1 and VASP protein
expression was determined and quantified by western blot analysis
in cells treated with (B) siRNA-NC or siRNA-Rac1 and (C) pflag-CMV
or pflag-Rac1 for 48 h. (D) The permeability of HUVEC-CRL-1730
cells, which were treated with siRNA-NC, siRNA-Rac1, pflag-CMV or
the overexpression plasmid pflag-Rac1 for 24 h, was assessed using
FITC-labeled dextran in a Transwell assay. After 24 h of HCMV
infection, the permeability of HUVEC-CRL-1730 cells, which were
treated with siRNA-NC and (E) siRNA-Rac1, pflag-CMV or the
overexpression plasmid pflag-Rac1, or (F) siRNA-VASP, pEGFP-C1 or
the overexpression plasmid pEGFP-VASP for 24 h, was assessed using
FITC-labeled dextran in a Transwell assay. Data are expressed as
the mean ± standard error of the mean (n=3 per group). *P<0.05
vs. the control group. si, small interfering; HUVEC, human
umbilical vein endothelial cell; VASP, vasodilator-stimulated
phosphoprotein; flag, tag protein; GFP, green fluorescent protein;
NC, negative control; Rac1, Ras-related C3 botulinum toxin
substrate 1; FITC, fluorescein isothiocyanate. |
Following transfected with siRNA-Rac1 or the
pflag-Rac1 plasmid in HUVEC-CRL-1730 cells, a Transwell assay was
used to detect endothelial permeability at 24 h (Fig. 4D). Rac1 knockdown revealed
significant increase in endothelial permeability compared with the
control and siRNA-NC groups (P<0.05), which is a 45±2% increase
compared with the control group, whereas the Rac1 overexpression
plasmid resulted in no significant difference in endothelial
permeability. Furthermore, the potential role of the Rac1/VASP
signaling pathway in the regulation of HCMV infection-induced
hyperpermeability in the cells was analyzed.
After HCMV infection for 24 h, the cells were
transfected with siRNA-Rac1, the pflag-Rac1 overexpression plasmid,
siRNA-VASP, the pEGFP-VASP overexpression plasmid or their
respective controls using Lipofectamine 2000. After 48 h,
HUVEC-CRL-1730 cell permeability was assessed using a Transwell
assay. The endothelial permeability of Rac1 knockdown cells
significantly increased compared with the control and siRNA-NC
groups (both P<0.05), which was an increase of 58±3% compared to
the control cells (Fig. 4E). The
endothelial permeability of the Rac1 overexpressing cells revealed
no significant alteration. As demonstrated in Fig. 4F, the endothelial permeability of
VASP knockdown cells was also significantly increased compared with
the control and siRNA-NC groups (both P<0.05), an increase of
50±3% compared to the control cells. The endothelial permeability
of the VASP overexpressing cells revealed no significant
alteration. These data suggested that HCMV infection induces
hyperpermeability via the Rac1/VASP signaling pathway.
Discussion
AS is a common chronic vascular pathological process
and the most common cause of cardiovascular and cerebrovascular
diseases (24). The pathogenesis of
AS is complex; the high etiological risk factors associated with
the development of AS include genetic polymorphisms and
susceptibility, long-term exposure to various chemicals, smoking,
hypertension and hyperlipidemia (25). Furthermore, numerous epidemiological
studies and clinical retrospective analyses have demonstrated that
viral or bacterial infections, including HCMV (26) and Chlamydophila pneumonia
(27), may affect the formation of
AS.
EC injury is the first step in AS development and is
frequently caused by specific chemicals, high fat diets, genetic
factors, trauma and pathogen infection (3). Yi et al (28) found HCMV DNA, antigens, and
immediate-early (IE) genes and proteins in the internal carotid
arteries of patients suffering ischemic stroke. Blum et al
(29) found a high-titer of
immunoglobulin G anti-CMV antibodies in the sera of patients with
high-risk atherosclerotic cardiovascular disease. Additionally, a
clinical cohort study revealed that CMV infection is an independent
risk factor for AS and coronary heart disease (30). These studies suggest that the viral
infection is closely associated with the development and
progression of AS.
ECs are potential hosts of HCMV, and the entry of
HCMV into cells is mediated by cellular annexin II (4), a 36 kDa HCMV-binding protein that
exists on the cellular membrane. In the present study, in order to
demonstrate that endothelial damage can be caused by viral
invasion, the effect of HCMV infection on the morphology of ECs was
observed. Under normal physiological conditions, HUVECs were
closely connected, flat, long and spindle-shaped, and exhibited the
typical ‘paving stone’ morphology. After the cells were infected
with HCMV for 7-10 days, the nuclei had increased in size and some
of the cells had become rounded. In addition, the boundaries
between cells became blurred and numerous cells merged to form
giant cells, while a number of cells died. The data from the
current study are consistent with that of a previous report, which
demonstrated that, following HCMV infection, the cells from the two
HUVEC cell lines became rounded and intercellular fissure formation
occurred (27). This alteration
promoted the attraction of a variety of inflammatory cells and
factors through ECs, which began to accumulate peroxidized lipids
from the blood vessels, and gradually evolved into foam cells,
which are involved in atherosclerotic plaque formation (31). These phenomena suggest that the
reduction of barrier function, which is closely associated with the
morphological alterations of ECs, is crucial to the development of
AS.
In the present study, an increase in Rac1 expression
was demonstrated to induce hyperpermeability, which disrupted EC
barrier function. Baumer et al (32) demonstrated that Rac1 activation
likely contributed to the barrier-stabilizing effects of cyclic
adenosine monophosphate in the microvascular endothelium. These
effects may in part, be mediated by the Epac/Rap1 signaling pathway
(33). Cytoskeletal rearrangements
are the structural basis for the modification of EC morphology and
associated barrier function (34).
VASP, an actin-associated protein, belongs to a family of Ena/VASP
proteins, which localize to the intercellular junction, focal
adhesion terminals of stress fibers and highly dynamic areas of
change at the cellular membrane (35). VASP has been revealed to serve an
important role in the maintenance of endothelial barrier function
(36,37), and paracellular permeability
(38). Luyer et al (39) demonstrated that the expression of the
tight junction protein, ZO-l, was abolished and levels of
phosphorylated VASP increased under low intestinal blood flow
conditions in a rat model hemorrhagic shock and, at the same time,
the permeability of the intestinal mucosa and bacteria
translocation also increased. Bogatcheva et al (40) reported that the bacterial endotoxin
lipopolysaccharide secreted by human pulmonary microvascular ECs
can cause the redistribution of VASP- and ZO-1-associated proteins.
In VASP-depleted pulmonary microvascular ECs, the cell barrier
function was reduced, and the mechanism involved may be associated
with the cell-cell tight junction damage caused by the disorder of
the ZO-1-F-actin arrangement (41).
In the current study, after HCMV infection,
endothelial permeability and the expression of Rac1 and VASP were
analyzed at the mRNA and protein levels in two HUVEC cell lines.
The mRNA level of Rac1 and VASP reached the minimum level at 12 h.
The reason for the increase after 12 h may be associated with HCMV
infection-induced activation of cytokine-associated signaling
pathways. For example, following HCMV infection, the cellular
factor, nuclear factor-κB p105 subunit, was activated and its
expression increased, which was important for the efficient
transcription of IE genes (42);
also, hypoxia-inducible factor (HIF)-1α transcription was clearly
stimulated (43). The aforementioned
results also demonstrated that HCMV-induced hyperpermeability was
accomplished by decreasing Rac1 and VASP expression in
HUVEC-CRL-1730 cells.
In addition, a further decrease of VASP expression
using siRNA-VASP in HCMV-infected cells aggravated the permeability
increase. These results indicated that HCMV-induced endothelium
hyperpermeability may be mediated by Rac1 and VASP-associated
cell-cell junction remodeling. In addition, as VASP is a downstream
effector of Rac1 in osteosarcoma cells, interference of Rac1 in the
cells led to a significant reduction in VASP expression (21). Additionally, consistent with previous
reports, the EC data from the current study also revealed that
interfering with the expression of Rac1 using siRNA-Rac1 could
inhibit the expression of VASP. Therefore, HCMV-induced endothelial
cytoskeleton remodeling may be controlled by the Rac1/VASP
signaling pathway. In conclusion, the current study demonstrated
that HCMV infection could induce impairment of the barrier function
in monolayer HUVEC-CRL-1730 cells via downregulation of
Rac1-mediated VASP expression. The present study identified that
the presence of pathogens, which cause a breakdown in the barrier
function of endothelial cells had a high correlation with the
occurrence and development of atherosclerosis. These results
indicate that an effective strategy for the prevention of
atherosclerosis would be to strengthen the protection of
endothelial cells.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no.
81572943).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YT, LW and YH designed the study and wrote the
manuscript. XX and YM analyzed the data. LZ, JZ and LX performed
the cell experiments.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Tarp JB, Jensen AS, Engstrom T,
Holstein-Rathlou NH and Sondergaard L: Cyanotic congenital heart
disease and atherosclerosis. Heart. 103:897–900. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhou YF, Leon MB, Waclawiw MA, Popma JJ,
Yu ZX, Finkel T and Epstein SE: Association between prior
cytomegalovirus infection and the risk of restenosis after coronary
atherectomy. N Engl J Med. 335:624–630. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Danesh J, Whincup P, Walker M, Lennon L,
Thomson A, Appleby P, Gallimore JR and Pepys MB: Low grade
inflammation and coronary heart disease: Prospective study and
updated meta-analyses. BMJ. 321:199–204. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pietropaolo RL and Compton T: Direct
interaction between human cytomegalovirus glycoprotein B and
cellular annexin II. J Virol. 71:9803–9807. 1997.PubMed/NCBI
|
|
5
|
Bentz GL, Jarquin-Pardo M, Chan G, Smith
MS, Sinzger C and Yurochko AD: Human cytomegalovirus (HCMV)
infection of endothelial cells promotes naive monocyte
extravasation and transfer of productive virus to enhance
hematogenous dissemination of HCMV. J Virol. 80:11539–11555. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Conforti G, Zanetti A, Colella S, Abbadini
M, Marchisio PC, Pytela R, Giancotti F, Tarone G, Languino LR and
Dejana E: Interaction of fibronectin with cultured human
endothelial cells: Characterization of the specific receptor.
Blood. 73:1576–1585. 1989.PubMed/NCBI
|
|
7
|
Etingin OR, Silverstein RL and Hajjar DP:
von Willebrand factor mediates platelet adhesion to virally
infected endothelial cells. Proc Natl Acad Sci USA. 90:5153–5156.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Loureiro JJ, Rubinson DA, Bear JE, Baltus
GA, Kwiatkowski AV and Gertler FB: Critical roles of
phosphorylation and actin binding motifs, but not the central
proline-rich region, for Ena/vasodilator-stimulated phosphoprotein
(VASP) function during cell migration. Mol Biol Cell. 13:2533–2546.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Comerford KM, Lawrence DW, Synnestvedt K,
Levi BP and Colgan SP: Role of vasodilator-stimulated
phosphoprotein in PKA-induced changes in endothelial junctional
permeability. FASEB J. 16:583–585. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lawrence DW, Comerford KM and Colgan SP:
Role of VASP in reestablishment of epithelial tight junction
assembly after Ca2+ switch. Am J Physiol Cell Physiol.
282:C1235–C1245. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ross R: Atherosclerosis-an inflammatory
disease. N Engl J Med. 340:115–126. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lum H and Malik AB: Mechanisms of
increased endothelial permeability. Can J Physiol Pharmacol.
74:787–800. 1996. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hall A: Rho family GTPases. Biochem Soc
Trans. 40:1378–1382. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rojas AM, Fuentes G, Rausell A and
Valencia A: The Ras protein superfamily: Evolutionary tree and role
of conserved amino acids. J Cell Biol. 196:189–201. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Abbasi T and Garcia JG: Sphingolipids in
lung endothelial biology and regulation of vascular integrity.
Handb Exp Pharmacol. 201–226. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
van Nieuw Amerongen GP, Beckers CM,
Achekar ID, Zeeman S, Musters RJ and van Hinsbergh VW: Involvement
of Rho kinase in endothelial barrier maintenance. Arterioscler
Thromb Vasc Biol. 27:2332–2339. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Samarin S and Nusrat A: Regulation of
epithelial apical junctional complex by Rho family GTPases. Front
Biosci (Landmark Ed). 14:1129–1142. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kouklis P, Konstantoulaki M, Vogel S,
Broman M and Malik AB: Cdc42 regulates the restoration of
endothelial barrier function. Circ Res. 94:159–166. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ridley AJ, Schwartz MA, Burridge K, Firtel
RA, Ginsberg MH, Borisy G, Parsons JT and Horwitz AR: Cell
migration: Integrating signals from front to back. Science.
302:1704–1709. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Winge MCG and Marinkovich MP: Epidermal
activation of the small GTPase Rac1 in psoriasis pathogenesis.
Small GTPases. 5:1–6. 2017. View Article : Google Scholar
|
|
21
|
Wu G, Wei L, Yu A, Zhang M, Qi B, Su K, Hu
X and Wang J: Vasodilator-stimulated phosphoprotein regulates
osteosarcoma cell migration. Oncol Rep. 26:1609–1615.
2011.PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schlegel N and Waschke J: VASP is involved
in cAMP-mediated Rac 1 activation in microvascular endothelial
cells. Am J Physiol Cell Physiol. 296:C453–C462. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Writing Group M, Mozaffarian D, Benjamin
EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S,
Despres JP, et al: Heart disease and stroke statistics-2016 update:
A report from the american heart association. Circulation.
133:e38–e360. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ross R: The pathogenesis of
atherosclerosis-an update. N Engl J Med. 314:488–500. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Z, Cai J, Zhang M, Wang X, Chi H,
Feng H and Yang X: Positive expression of human cytomegalovirus
phosphoprotein 65 in atherosclerosis. Biomed Res Int.
2016:40676852016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kreutmayer S, Csordas A, Kern J, Maass V,
Almanzar G, Offterdinger M, Ollinger R, Maass M and Wick G:
Chlamydia pneumoniae infection acts as an endothelial stressor with
the potential to initiate the earliest heat shock protein
60-dependent inflammatory stage of atherosclerosis. Cell Stress
Chaperones. 18:259–268. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yi L, Lin JY, Gao Y, Feng ZJ and Wang DX:
Detection of human cytomegalovirus in the atherosclerotic cerebral
arteries in han population in china. Acta Virol. 52:99–106.
2008.PubMed/NCBI
|
|
29
|
Blum A, Peleg A and Weinberg M:
Anti-cytomegalovirus (CMV) IgG antibody titer in patients with risk
factors to atherosclerosis. Clin Exp Med. 3:157–160. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Adam E, Melnick JL, Probtsfield JL, Petrie
BL, Burek J, Bailey KR, McCollum CH and DeBakey ME: High levels of
cytomegalovirus antibody in patients requiring vascular surgery for
atherosclerosis. Lancet. 2:291–293. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Badimon L and Vilahur G: Thrombosis
formation on atherosclerotic lesions and plaque rupture. J Intern
Med. 276:618–632. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Baumer Y, Drenckhahn D and Waschke J: cAMP
induced Rac 1-mediated cytoskeletal reorganization in microvascular
endothelium. Histochem Cell Biol. 129:765–778. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Birukova AA, Zagranichnaya T, Fu P,
Alekseeva E, Chen W, Jacobson JR and Birukov KG: Prostaglandins
PGE(2) and PGI(2) promote endothelial barrier enhancement via PKA-
and Epac1/Rap1-dependent Rac activation. Exp Cell Res.
313:2504–2520. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li YS, Haga JH and Chien S: Molecular
basis of the effects of shear stress on vascular endothelial cells.
J Biomech. 38:1949–1971. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schmit MA, Mirakaj V, Stangassinger M,
Konig K, Kohler D and Rosenberger P: Vasodilator phosphostimulated
protein (VASP) protects endothelial barrier function during
hypoxia. Inflammation. 35:566–573. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rentsendorj O, Mirzapoiazova T, Adyshev D,
Servinsky LE, Renne T, Verin AD and Pearse DB: Role of
vasodilator-stimulated phosphoprotein in cGMP-mediated protection
of human pulmonary artery endothelial barrier function. Am J
Physiol Lung Cell Mol Physiol. 294:L686–L697. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schlegel N, Burger S, Golenhofen N, Walter
U, Drenckhahn D and Waschke J: The role of VASP in regulation of
cAMP- and Rac 1-mediated endothelial barrier stabilization. Am J
Physiol Cell Physiol. 294:C178–C188. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Profirovic J, Han J, Andreeva AV, Neamu
RF, Pavlovic S, Vogel SM, Walter U and Voyno-Yasenetskaya TA:
Vasodilator-stimulated phosphoprotein deficiency potentiates
PAR-1-induced increase in endothelial permeability in mouse lungs.
J Cell Physiol. 226:1255–1264. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Luyer MD, Buurman WA, Hadfoune M, Jacobs
JA, Konstantinov SR, Dejong CH and Greve JW: Pretreatment with
high-fat enteral nutrition reduces endotoxin and tumor necrosis
factor-alpha and preserves gut barrier function early after
hemorrhagic shock. Shock. 21:65–71. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bogatcheva NV, Zemskova MA, Kovalenkov Y,
Poirier C and Verin AD: Molecular mechanisms mediating protective
effect of cAMP on lipopolysaccharide (LPS)-induced human lung
microvascular endothelial cells (HLMVEC) hyperpermeability. J Cell
Physiol. 221:750–759. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ikari A, Ito M, Okude C, Sawada H, Harada
H, Degawa M, Sakai H, Takahashi T, Sugatani J and Miwa M:
Claudin-16 is directly phosphorylated by protein kinase A
independently of a vasodilator-stimulated phosphoprotein-mediated
pathway. J Cell Physiol. 214:221–229. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Caposio P, Dreano M, Garotta G, Gribaudo G
and Landolfo S: Human cytomegalovirus stimulates cellular IKK2
activity and requires the enzyme for productive replication. J
Virol. 78:3190–3195. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Frede S, Stockmann C, Freitag P and
Fandrey J: Bacterial lipopolysaccharide induces HIF-1 activation in
human monocytes via p44/42 MAPK and NF-kappaB. Biochem J.
396:517–527. 2006. View Article : Google Scholar : PubMed/NCBI
|