Introduction
Mounting evidence suggests that intestinal smooth
muscle cells (SMCs) may be involved in different inflammatory
diseases that affect the bowel, leading to altered morphology,
contractility and augmented production of various inflammatory
cytokines (1,2). Studies conducted using different animal
models of gastrointestinal diseases have demonstrated that growth
and contractile properties of SMCs are substantially altered during
mucosal inflammation in the gastrointestinal tract due to increased
expression of different cytokines (3,4).
Patients who suffer from IBD experience symptoms associated with
abnormal intestinal motility, resulting from abnormal proliferation
and contractility of intestinal SMCs (3). Numerous studies have demonstrated that
intestinal SMCs may produce different inflammatory mediators,
including interleukin (IL)-6 and tumor necrosis factor-α (TNF-α)
during various pathological conditions (2,5). Shi and
Sarna (6) demonstrated that TNF-α
binds to cognate receptors expressed on human CSMCs, resulting in
activation of nuclear factor (NF)-κB and induction of expression of
different cytokines and chemokines, including monocyte chemotactic
protein (MCP)-1, IL-8 and intercellular adhesion molecule-1.
Furthermore, exposing human colonic (C)SMCs to different
inflammatory stimuli led to enhanced expression of IL-1α, IL-6,
IL-8, cyclooxygenase-2 and regulated on activation, normal T cell
expressed and secreted (RANTES) (7).
Ulcerative colitis and Crohn's disease are two
distinct forms of IBD, affecting the colon and small intestine,
respectively, and are characterized by chronic and relapsing
intestinal inflammation (8). IBD may
result from dysregulation of the mucosal immune response triggered
by a combination of genetic, environmental and immunological
factors, resulting in mucosal and submucosal inflammation (8). Furthermore, IBD is usually associated
with other co-morbid diseases, including rheumatoid arthritis,
multiple sclerosis, systemic lupus, psoriasis, hypothyroidism and
diabetes mellitus (9). Diabetes
mellitus is one of the major conditions associated with IBD,
resulting in significant clinical and therapeutic consequences
(10). The incidence and prevalence
of IBD and associated comorbidities are increasing worldwide,
resulting in significant healthcare costs and impaired quality of
life for patients (10,11). Despite advances in understanding IBD
pathophysiology and its biological therapies, IBD remains a
non-curable condition highlighting the need to develop novel
treatment approaches (11).
Metformin is a biguanide derivative used in type 2
diabetes treatments as a first-line therapy and is one of the major
prescribed oral hypoglycemic drugs (12). Metformin increases peripheral uptake
of glucose, decreases hepatic glucose production and increases
insulin sensitivity in liver and skeletal muscle (12). Metformin may mediate its hypoglycemic
effect through several molecular mechanisms, including activation
of the adenosine 5′-monophosphate kinase (AMPK) signaling pathway,
which regulates different physiological processes, including
cellular growth and proliferation, mitochondrial function and
biogenesis, insulin sensitivity, inflammation, oxidative stress and
autophagy (12).
Notably, clinical and experimental research has
demonstrated an array of potential benefits of metformin beyond its
hypoglycemic effect in an AMPK-dependent and independent manner
(13). Metformin may exert
anti-inflammatory, anticancer, cardioprotective and
antiatherosclerotic effects, and it may decrease macrovascular
complications of diabetes (14–17). It
has been demonstrated that metformin may inhibit NF-κB activation
and inflammatory marker expression through the AMPK signaling
pathway by reducing signal transduction and activator of
transcription (STAT)3 activity and tumor suppressor
phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase and
dual-specificity protein phosphatase through inhibition of NF-κB
and downstream inflammatory genes in multiple cell types (15).
In an experimental IBD model, metformin reduced
disease activity index scores, decreased colonic histopathological
score, reduced expression of inflammatory mediators and preserved
colon length (18). Additionally,
treatment with metformin upregulated phosphorylated (p)-AMPK levels
and simultaneously inhibited expression of IL-17, p-STAT3 and
p-mammalian target of rapamycin (18). Furthermore, metformin decreased
expression of inflammatory cytokines in a dose-dependent manner in
inflamed human intestinal epithelial HT-29 cells (18). Metformin also decreased
phosphorylation and activation of pro-inflammatory proteins,
including protein kinase B, p38, extracellular signal regulated
kinase and protein kinase C in vascular endothelial cells under
hyperglycemic conditions (19).
Mouse CSMCs have previously been identified as being
capable of expressing multiple cytokines and chemokines including
TNF-α, IL-1α, macrophage colony stimulating factor (M-CSF), T cell
activation gene-3 (TCA-3) and stromal cell-derived factor-1
(SDF-1), when exposed to inflammatory stimuli including
lipopolysaccharides (LPSs) (20).
Although multiple studies have demonstrated that metformin
suppresses NF-κB activation and cytokine production in various cell
types, little information is available on the effect of metformin
on CSMC expression and secretion of pro-inflammatory cytokines and
chemokines (14,15). Therefore, the current study
hypothesized that metformin regulates NF-κB signaling in CSMCs, by
influencing cytokine and chemokine expression, and may provide a
novel adjunct therapy to treat IBD particularly in patients with
diabetes.
Materials and methods
Solutions, drugs and chemicals
A smooth muscle buffer (SMB) was prepared in-house
and contained the following: 120 mM NaCl, 4 mM KCl, 2.6 mM
KH2PO4, 2 mM CaCl2, 25 mM HEPES
(N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid), 14 mM
glucose and 2.1% essential amino acid mixture; the pH was adjusted
to 7.4. Tissue digestion solution contained 0.5 mg collagenase and
0.33 mg soybean trypsin inhibitor per ml of SMB. All of these
chemicals were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt,
Germany). Cell culture media used during incubation processes was
prepared by adding 10% fetal bovine serum (FBS; GE Healthcare Life
Sciences, Little Chalfont, UK), 100 U/ml penicillin, 100 µg/ml
streptomycin and 2.5 µg/ml amphotericin B to Dulbecco's modified
Eagle medium (DMEM) with L-glutamine (Capricorn Scientific GmbH,
Ebsdorfergrund, Germany). Remaining reagents were purchased from
EuroClone S.p.A. (Pero, Italy). LPS was purchased from
Sigma-Aldrich (Merck KGaA). Metformin was purchased from Merck
KGaA. Specific ELISA kits for mouse TNF-α (cat. no. RAB0477), M-CSF
(cat. no. RAB0099), IL-1α (cat. no. RAB0271), TCA-3 (cat. no.
RAB0041) and SDF-1 (cat. no. RAB0125) were purchased from
Sigma-Aldrich (Merck KGaA). Nuclear protein extraction kit (cat.
no. ab113474) and an NF-κB p65 (pS536) ELISA kit (cat. no.
ab176663) were purchased from Abcam (Cambridge, UK). A 500-µm Nitex
mesh was purchased from Sigma-Aldrich (Merck KGaA).
Animals
A total of 20 young mature male BALB/c mice (~12
weeks of age, 26.5–30 g) were provided by the animal house of
Jordan University of Science and Technology (Irbid, Jordan). Mice
were housed in the animal facility at the Jordan University of
Science and Technology under 12-h light/dark cycles in polyethylene
cages at −22°C and 50% humidity. Mice were fed standard chow rodent
diet and water available ad libitum. Mice (age, ~14 weeks)
were euthanized by inhalation of CO2 and the colon was
excised. The colon was cut into pieces (2–3 cm in length) and
placed in cold SMB. All procedures were approved and performed
according to the guidelines of the Animal Care and Use Committee at
Jordan University of Science and Technology.
Preparation of dispersed CSMCs
CSMCs were isolated from the circular and
longitudinal smooth muscle layers of the colons of BALB/c mice
(sacrificed at 14 weeks of age) by enzymatic digestion of muscle
strips, followed by filtration, and centrifugation as previously
described (20). Mucosa was scraped
off murine colon tissue with fine scissors; tissues were cut into
thin slices (2 mm long; 2 mm thin) and incubated for 20 min in SMB
containing 0.5 mg/ml collagenase and 0.33 mg/ml soybean trypsin
inhibitor in an agitated water bath at 31°C. Tissues were
continuously supplied with 100% oxygen during the isolation
procedure. Partly digested tissue was washed twice with 50 ml
collagenase-free SMB and muscle cells were allowed to disperse
spontaneously for 10 min in collagenase-free medium. Cells were
harvested by filtration through 500-µm Nitex mesh and centrifuged
twice at 350 × g for 10 min at 4°C to eliminate broken cells and
organelles. The process was repeated 4–5 times. Cells were counted
in a hemocytometer and viability was assessed using a trypan blue
exclusion assay. Cell suspensions (100 µl) were mixed with 100 µl
of 0.4% trypan blue dye (Thermo Fisher Scientific, Inc., Waltham,
MA, USA) and stained at room temperature (23°C) for 1 min. Cells
were then immediately loaded into hemocytometer (Thermo Fisher
Scientific, Inc.) and examined under an inverted microscope (Nikon
Corporation, Tokyo, Japan; magnification, ×40). In this assay,
trypan blue dye permeates unviable cells; while viable cells
exclude this dye because they possess intact plasma membranes.
Therefore blue-stained cells were counted and considered unviable
cells. It was identified that >95% of cells were viable and
therefore were suitable for further experimentation.
Identification and characterization of
CSMCs
Isolated mouse CSMCs were viewed at a ×20
magnification using an inverted Nikon TMS-f microscope (Nikon
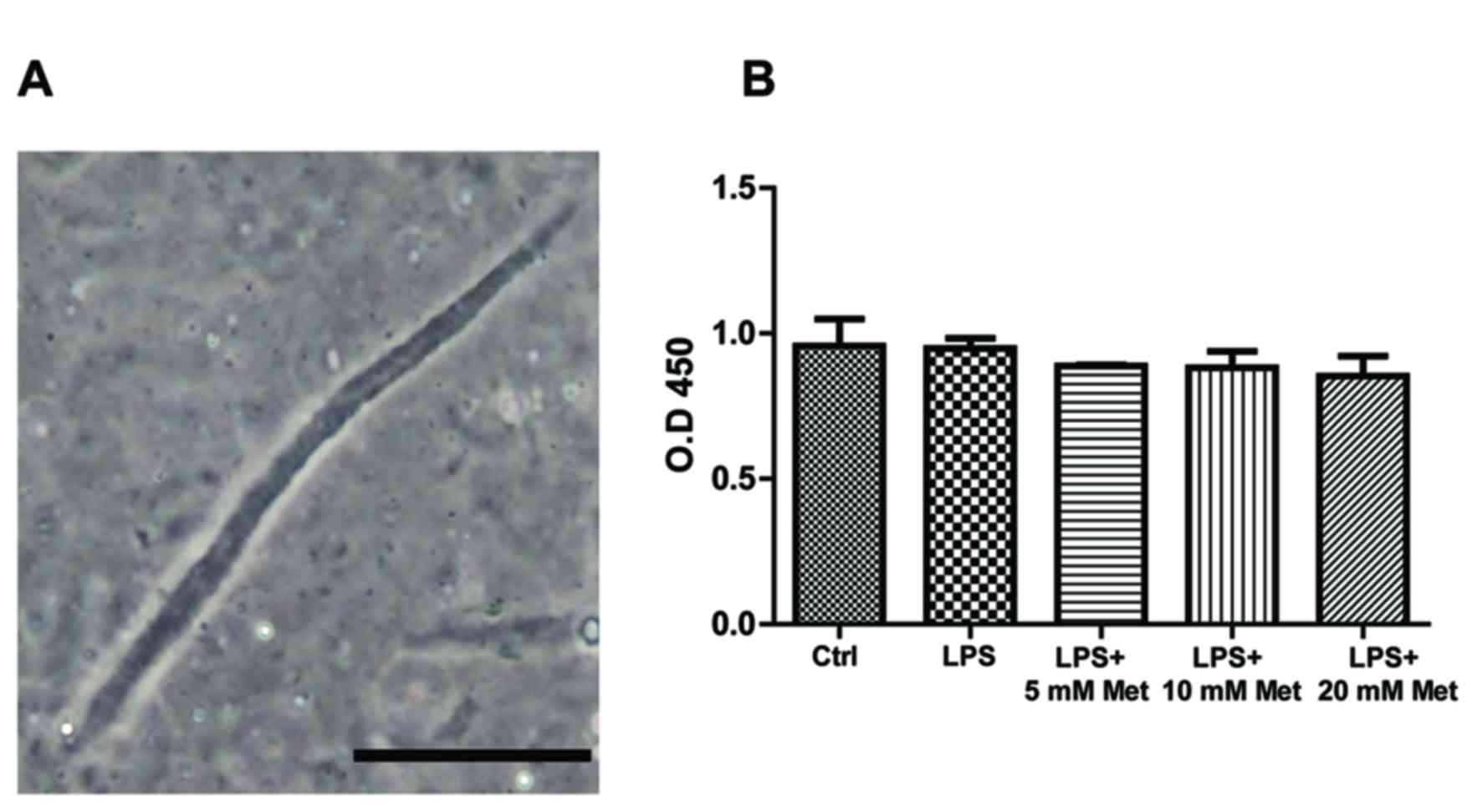
Corporation). Fig. 1A presents CSMCs
as spindle-shaped cells with an average length of ~140 µM.
CSMC viability assay
Isolated CSMCs were cultured in 96-well plates at
1×104 cells/well (n=3 per group). Cells were
serum-deprived for 24 h prior to treatment with LPS (1 µg/ml) and
varying amounts of metformin (0, 5, 10 and 20 mM) at 37°C. Cell
viability was assessed following 24 h using MTT assays (Thermo
Fisher Scientific, Inc.), according to the manufacturer's protocol.
CSMCs were incubated with MTT reagent for 4 h at 37°C. The MTT
reagent was converted to an insoluble formazan. Formazan was then
solubilized with a solubilizing reagent provided in the kit, and
the concentration determined by optical density at 570 nm.
Quantification of protein expression
by ELISA
A total of 10,000 dispersed mouse CSMCs (n=3 per
group) were seeded per well in a 6-well plate containing DMEM with
10% FBS, 100 U/ml penicillin, 100 µg/ml streptomycin and 2.5 µg/ml
amphotericin B. Cells were incubated in a humidified incubator with
95% air and 5% CO2 at 37°C for 24 h with LPS (1 µg/ml)
and varying amounts of metformin (0, 5, 10 and 20 mM). Following
the incubation period treated samples were centrifuged at 350 × g
for 5 min at 4°C. Conditioned media was stored at −20°C for further
analysis and cells pellets were lysed immediately. Cell lysates
were prepared using BashingBeads Lysis tubes from Zymo Research
Corp. (Irvine, CA, USA) and cell lysis buffer containing protease
inhibitor cocktail provided the a whole cell extraction kit (Abcam;
cat. no. ab113475), according to the manufacturer's protocol. A
nuclear protein extraction kit (Abcam; cat. no. ab113474) was used
to extract total nuclear proteins from another set of control and
treated samples. Lysates were centrifuged for 10 min at 10,000 × g
at 4°C and supernatants were collected for further analysis. Total
protein concentration of supernatants was measured using the DC
protein assay kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Protein concentration was adjusted to 100 µg/ml in all samples.
Protein levels of specific cytokines were evaluated by ELISA assay.
Specific ELISA kits for TNF-α, IL-1α, M-CSF, TCA-3, SDF-1 and
nuclear NF-κB p65 (pS536) were used to measure cytokine levels in
lysates and conditioned media for control and treated samples
according to the manufacturer's protocols.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). One-way
analysis of variance followed by Fisher's post-hoc analysis was
used to examine significant differences between groups. All data
are presented as mean ± standard error of the mean. Values
presented are representative of three independent experiments
performed in triplicate. P<0.05 was considered to indicate a
statistically significant difference.
Results
CSMC viability assay
To ensure that neither LPS nor metformin affected
CSMCs viability, an MTT assay was performed on the treated samples.
The results indicated that cell viability was not significantly
affected by LPS or metformin treatment following a 24 h period in
all treatment groups, suggesting that growth of CSMCs remained
unchanged during the treatment period (Fig. 1B).
Evaluation of cytokine and chemokine
expression and secretion
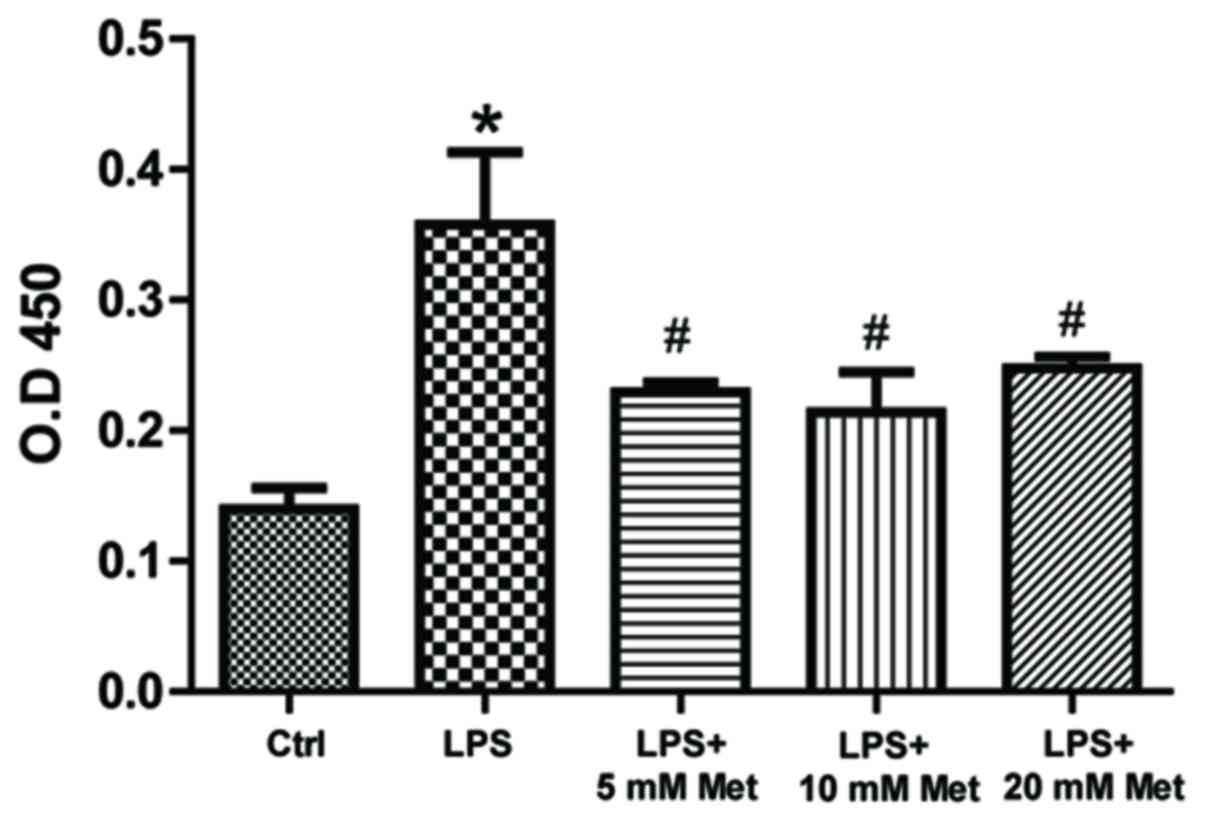
To measure cytokine and chemokine levels in the
control and treated samples, specific ELISA assays were used. The
data demonstrated that LPS treatment resulted in a significant
increase (~1.5–2 fold) in TNF-α, IL-1α, M-CSF, TCA-3 and SDF-1
expression (P<0.05; Fig. 2). On
the other hand, co-treatment with metformin (5 or 10 mM)
significantly reduced expression by ~20-40% compared with LPS alone
(P<0.05). Furthermore, evaluation of cytokine and chemokine
secretion by CSMCs into the media was assessed by ELISA. The data
demonstrated that TNF-α, IL-1α, M-CSF and TCA-3 levels were
significantly elevated following LPS treatment, while co-treatment
with metformin (5 and 10 mM) significantly reduced secretion into
the media compared with LPS alone (P<0.05; Fig. 3). In addition, LPS treatment
upregulated nuclear NF-κB p65 (pS536) protein levels, while
co-treatment with metformin significantly reduced levels compared
with LPS alone (P<0.05), suggesting that metformin may suppress
inflammatory cytokine and chemokine expression and secretion by
interfering with NF-κB signaling pathway activation (Fig. 4).
 | Figure 2.Effect of metformin treatment on
expression of inflammatory cytokines by mouse CSMCs, evaluated
using ELISAs. Levels of (A) TNF-α, (B) IL-1α, (C) M-CSF, (D) TCA-3
and (E) SDF-1 in CSMCs treated with LPS (1 µg/ml) and varying
amounts of metformin (0, 5, 10 and 20 mM). *P<0.01 vs. the Ctrl
group; #P<0.01 vs. the LPS group; §P<0.05 vs. the LPS group.
LPS, lipopolysaccharide; CSMC, colonic smooth muscle cell; Met,
metformin; TNF-α, tumor necrosis factor-α; IL, interleukin; M-CSF,
macrophage-colony stimulating factor; TCA-3, T cell activation
gene-3; SDF-1, stromal cell-derived factor-1; Ctrl, control. |
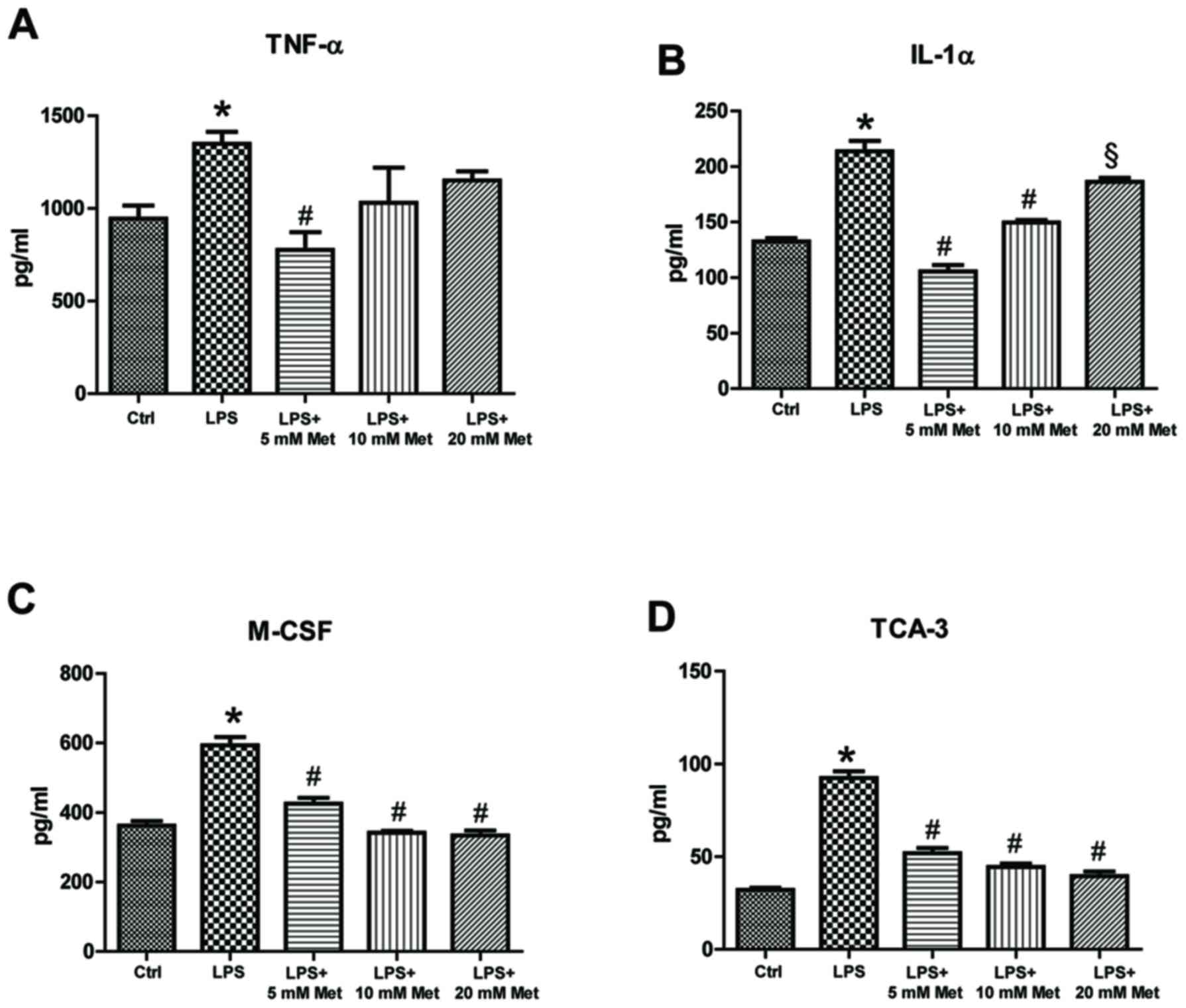 | Figure 3.Effect of metformin treatment on
secretion of inflammatory cytokines by mouse CSMCs into the
conditioned media, evaluated using ELISA. Levels of (A) TNF-α, (B)
IL-1α, (C) M-CSF and (D) TCA-3 in CSMCs following treatment with
LPS (1 µg/ml) and varying amounts of metformin (0, 5, 10 and 20
mM). *P<0.01 vs. the Ctrl group; #P<0.01 vs. the LPS group;
§P<0.05 vs. the LPS group. LPS, lipopolysaccharide; CSMC,
colonic smooth muscle cell; Met, metformin; TNF-α, tumor necrosis
factor-α; IL, interleukin; M-CSF, macrophage-colony stimulating
factor; TCA-3, T cell activation gene-3; Ctrl, control. |
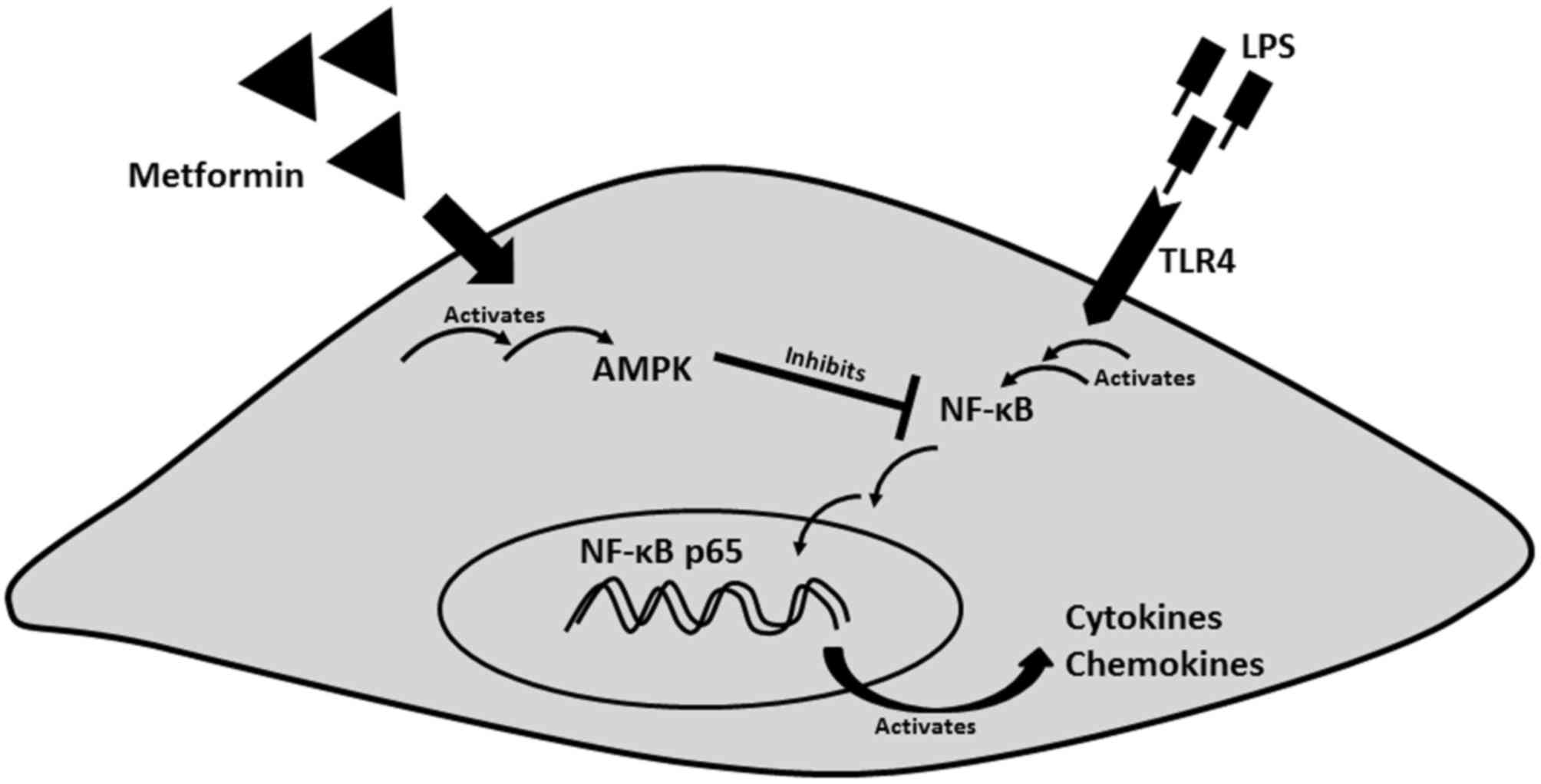
Collectively, these results suggest that metformin
may attenuate the expression and secretion of several cytokines and
chemokines from mouse CSMCs in the presence of inflammatory
stimulus. Fig. 5 represents an
integrative hypothetical model for how metformin may exert its
anti-inflammatory effect in CSMCs. It was hypothesized that
metformin activates the AMPK pathway, thereby inhibiting downstream
inflammatory gene expression in CSMCs.
Discussion
Most inflammatory conditions of the bowel result in
activation and recruitment of different inflammatory cells that
alter the surrounding environment, leading to activation of a
complex integrated inflammatory cascade (21). These events result in major hallmarks
of intestinal inflammation and loss of epithelial tight junctions
(21). Furthermore, it has been
reported that inflammatory conditions affecting the bowel may lead
to significant functional and morphological changes in the
intestinal SMCs. In the present study, it was demonstrated that
metformin may exert significant anti-inflammatory effects on
expression and secretion of different inflammatory mediators from
mouse CSMCs under LPS-induced inflammation in vitro.
It was previously identified that mouse CSMCs are
capable of expressing different cytokines and chemokines, including
TNF-α, IL-1α, M-CSF, TCA-3 and SDF-1, when stimulated with LPS
(20). Therefore, the
anti-inflammatory effects of metformin on expression and secretion
of these inflammatory mediators were investigated. The current
study demonstrated that low doses of metformin (5 or 10 mM) rather
than a high dose (20 mM) caused a significant reduction in
expression and secretion of these inflammatory mediators in CSMCs.
Additionally, metformin significantly reduced LPS-induced nuclear
NF-kB p65 (S635) levels in CSMCs. These results indicated that
metformin may be effective in suppressing expression and secretion
of TNF-α, IL1-α, MCSF, TCA-3 and SDF-1 from CSMCs through
inhibition of NF-kB p65 phosphorylation and nuclear
translocation.
TNF-α is potent pro-inflammatory cytokine produced
as a transmembrane protein that is released into medium through
proteolytic cleavage via metalloproteinase TNF-converting enzyme
(22). TNF-α is primarily secreted
from activated macrophages, as well as a variety of other cell
types (22). Abnormal production of
TNF-α has been associated with different ailments and it has been
reported that expression of both membrane-bound and soluble TNF-α
by submucosal inflammatory cells is increased during active stages
of IBD (22). TNF-α expression in
IBD stimulates multiple pro-inflammatory actions, including
initiation of systemic inflammatory response, increased
angiogenesis, Paneth cell death, production of matrix
metalloproteinases and damage of intestinal epithelial cells
(23). Anti-TNF-α antibodies,
including infliximab and adalimumab, are commonly used in the
management of IBD with high success rates, demonstrating an
essential role of TNF-α in the pathogenesis of IBD (24,25). In
the current study, co-treatment with metformin attenuated
LPS-induced expression and secretion of TNF-α, suggesting an
important role of TNF-α derived from CSMCs in the development of
colonic inflammation.
IL-1α is a key inflammatory mediator that serves an
important role in early phases of IBD. Increased expression of
IL-1α has been reported in experimental models of colitis and colon
biopsies from IBD patients (26,27). It
serves an important role in recruitment of neutrophils, stimulation
of IL-6 production by macrophages and promotion of intestinal
tumorigenesis (28). Cominelli and
Pizarro (29) reported that blockage
of IL-1α receptors inhibited inflammatory responses associated with
immune complex-induced colitis, suggesting a central role for IL-1α
in IBD pathogenesis. Expression of IL-1α is enhanced during
different inflammatory conditions where it initiates a cascade of
events to increase production of several inflammatory mediators.
Enhanced production of both IL-1α and TNF-α together aggravates the
inflammatory process by inducing a cascade of other
pro-inflammatory events (23).
M-CSF is a cytokine produced by different cell types
in the body, where it mediates expression of various inflammatory
genes that control survival, proliferation, adhesion, migration and
differentiation of macrophages (30,31).
Effects of M-CSF are mediated through a tyrosine kinase receptor
(M-CSFR) that is expressed primarily on mononuclear phagocytic
cells and it has been demonstrated that M-CSF, M-CSFR and
macrophages are increased in the gastrointestinal tract in the
presence of IBD (32). It is
postulated that M-CSF may serve an essential role in pathogenesis
of IBD (31). Studies on animal
models of experimental colitis demonstrated that blockage of M-CSF
with a neutralizing antibody reduced symptoms of disease and
cytokine expression by immune cells (31). The present study has demonstrated
that metformin inhibited M-CSF expression and secretion by CSMCs in
the presence of an inflammatory stimulus.
Chemotaxis is an important inflammatory process that
serves an essential role in activation and recruitment of
inflammatory cells during IBD. High levels of different chemokines,
including IL-8 and RANTES, have been detected in colonic biopsies
from patients with IBD during the active phase of colitis (33,34).
Furthermore, mRNA levels of MCP-1 were demonstrated to increase in
endothelial cells and intestinal SMCs during the active phase of
the disease (35). In the present
study, expression of TCA-3 and SDF-1 was increased following
treatment with LPS, while co-treatment with metformin significantly
reversed expression of these chemokines, suggesting that metformin
may inhibit attraction and recruitment of inflammatory cells by
reducing chemokine production from CSMCs.
Activated T lymphocytes are a major source of TCA-3,
which plays an important role as a chemotactic factor for
macrophages and leukocytes. TCA-3 is a β-chemokine that affects
expression of other chemokines, including myoinhibitory-like
protein-1α and MCP-1, thereby affecting leukocyte recruitment to
inflammatory lesions (36).
Chronically inflamed colons of IL-10 knockout mice displayed
elevated levels of TCA-3 and other chemokines when compared with
wild-type mice. Interestingly, remission of colitis in this animal
model was associated with decreased expression of TCA-3 (37).
SDF-1 is a chemokine secreted by different
inflammatory cells. SDF-1 expression is increased in intestinal
epithelial cells in IBD, therefore it has been linked to IBD
pathogenesis (33). In a mouse model
of colitis, blockage of SDF-1 receptor reduced signs of colonic
inflammation and reduced release of other pro-inflammatory
mediators (38).
Despite the important role of CSMCs in IBD
pathogenesis, their inflammatory and secretory functions are rarely
investigated as the majority of studies focus on morphological
alterations and abnormal contractility that occurs during
gastrointestinal inflammation. Synthetic corticosteroids are
commonly used drugs to treat IBD during different stages of the
disease, to inhibit inflammatory cell function, cytokine production
and to suppress disease activity (10). However, a large number of patients
develop steroid resistance and become susceptible to relapse
following treatment termination. Additionally, corticosteroid
therapy is associated with serious side effects, including
increased vulnerability to infection, fluid retention,
hypertension, muscle wasting, hyperglycemia, insulin resistance and
diabetes mellitus (10).
Corticosteroid-induced diabetes is a major systemic manifestation
in IBD and represents a challenge for treatment approaches
(10).
Previous preclinical and clinical studies have
demonstrated that oral hypoglycemic drugs like metformin may
improve chronic inflammatory conditions in diabetic patients
through reduction of hyperglycemia-induced oxidative stress,
improvement of insulin sensitivity, dyslipidemia and its direct
anti-inflammatory effects on different tissues (16,39).
Several studies have demonstrated that metformin may downregulate
the inflammatory process by inhibition of NF-κB activity through
AMPK-dependent and independent signaling pathways, therefore it may
be potentially used as an adjunct therapy of different inflammatory
conditions, including atherosclerosis, diabetic nephropathy,
nonalcoholic steatohepatitis, neurodegenerative disorders and
asthma (15,39). Furthermore, metformin has been
demonstrated to reduce cardiovascular events in diabetic patients
through reductions in systemic inflammatory markers (40).
Metformin exerts its various functions through
activation of AMPK, which is a serine/threonine protein kinase that
functions as a cellular metabolic sensor and insulin sensitizer
(12,13). It has been demonstrated that agents
that activate AMPK may provide multiple anti-inflammatory effects
through inhibition of other inflammatory cell functions and
cytokine production (41,42). The mechanism by which metformin
reduced expression of cytokines and chemokines in CSMCs was beyond
the scope of the present study. It may be speculated that increased
intracellular AMPK activity results in attenuation of NF-κB
phosphorylation and translocation to the nucleus. NF-κB is an
important transcription factor that controls different inflammatory
functions and has been demonstrated to be suppressed by AMPK
(41,43). In addition, several experimental
studies have demonstrated inhibitory activities of metformin on
NF-κB (13,15,19).
Cytokines and chemokines serve an important role in
driving intestinal inflammation, local complications (including
colon cancer) and systemic manifestations in patients with IBD,
therefore agents that inhibit their production are considered
extremely beneficial in treating IBD (23,44).
However, therapies that target single cytokines, including
infliximab, may only benefit certain subgroups of patients due to
the nature of the cytokine network, where blockage of a specific
cytokine may lead to development of a compensatory increase in
other cytokines. Therefore, agents that inhibit multiple cytokines
including metformin may provide great benefits for patients with
IBD (23,45).
In conclusion, metformin, an AMPK activator,
successfully attenuated expression and secretion of several
cytokines and chemokines in mouse CSMCs following LPS challenge.
Fig. 5 represents an integrative
hypothetical model for how metformin may reduce inflammatory marker
expression in CSMCs. Metformin may confer a great advantage in
preventing chronic inflammatory diseases and colon cancer, beyond
its ability to normalize blood glucose levels, and thus it may be
possible to utilize it as an adjunct therapy to treat IBD. The
present study has certain limitations since it was performed in
vitro, therefore future work should evaluate anti-inflammatory
effects of metformin on CSMCs in animal models of IBD in
vivo, and potentially evaluate inflammatory gene expression in
colonic tissues from IBD patients treated with metformin.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Deanship of
Research, Jordan University of Science and Technology, Irbid,
Jordan (grant nos. 20160138 and 20150147).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
AA: Conception and design of study, acquisition of
data, analysis and interpretation of data, drafting the manuscript
and revising the manuscript critically for important intellectual
content. MoA, OA and MaA: Analysis and interpretation of data and
revising the manuscript critically for important intellectual
content. DA: Acquisition of data, analysis and interpretation of
data, drafting and revising the manuscript critically for important
intellectual content.
Ethics approval and consent to
participate
All procedures were approved and performed according
to the guidelines of the Animal Care and Use Committee at Jordan
University of Science and Technology, Irbid, Jordan.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Snape WJ Jr, Williams R and Hyman PE:
Defect in colonic smooth muscle contraction in patients with
ulcerative colitis. Am J Physiol. 261:G987–G991. 1991.PubMed/NCBI
|
|
2
|
Severi C, Sferra R, Scirocco A, Vetuschi
A, Pallotta N, Pronio A, Caronna R, Di Rocco G, Gaudio E,
Corazziari E and Onori P: Contribution of intestinal smooth muscle
to Crohn's disease fibrogenesis. Eur J Histochem. 58:24572014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vermillion DL, Huizinga JD, Riddell RH and
Collins SM: Altered small intestinal smooth muscle function in
Crohn's disease. Gastroenterology. 104:1692–1699. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vrees MD, Pricolo VE, Potenti FM and Cao
W: Abnormal motility in patients with ulcerative colitis: The role
of inflammatory cytokines. Arch Surg. 137:439–446. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shea-Donohue T, Notari L, Sun R and Zhao
A: Mechanisms of smooth muscle responses to inflammation.
Neurogastroenterol Motil. 24:802–811. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shi XZ and Sarna SK: Transcriptional
regulation of inflammatory mediators secreted by human colonic
circular smooth muscle cells. Am J Physiol Gastrointest Liver
Physiol. 289:G274–G284. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Salinthone S, Singer CA and Gerthoffer WT:
Inflammatory gene expression by human colonic smooth muscle cells.
Am J Physiol Gastrointest Liver Physiol. 287:G627–G637. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Abraham C and Cho JH: Inflammatory bowel
disease. N Engl J Med. 361:2066–2078. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bähler C, Schoepfer AM, Vavricka SR,
Brüngger B and Reich O: Chronic comorbidities associated with
inflammatory bowel disease: Prevalence and impact on healthcare
costs in Switzerland. Eur J Gastroenterol Hepatol. 29:916–925.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tamez-Pérez HE, Quintanilla-Flores DL,
Rodríguez-Gutiérrez R, González-González JG and Tamez-Peña AL:
Steroid hyperglycemia: Prevalence, early detection and therapeutic
recommendations: A narrative review. World J Diabetes. 6:1073–1081.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fakhoury M, Negrulj R, Mooranian A and
Al-Salami H: Inflammatory bowel disease: Clinical aspects and
treatments. J Inflamm Res. 7:113–120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pernicova I and Korbonits M:
Metformin-mode of action and clinical implications for diabetes and
cancer. Nat Rev Endocrinol. 10:143–156. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cameron AR, Morrison VL, Levin D, Mohan M,
Forteath C, Beall C, McNeilly AD, Balfour DJ, Savinko T, Wong AK,
et al: Anti-inflammatory effects of metformin irrespective of
diabetes status. Circ Res. 119:652–665. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hirsch HA, Iliopoulos D and Struhl K:
Metformin inhibits the inflammatory response associated with
cellular transformation and cancer stem cell growth. Proc Natl Acad
Sci USA. 110:972–977. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim SA and Choi HC: Metformin inhibits
inflammatory response via AMPK-PTEN pathway in vascular smooth
muscle cells. Biochem Biophys Res Commun. 425:866–872. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kita Y, Takamura T, Misu H, Ota T, Kurita
S, Takeshita Y, Uno M, Matsuzawa-Nagata N, Kato K, Ando H, et al:
Metformin prevents and reverses inflammation in a non-diabetic
mouse model of nonalcoholic steatohepatitis. PLoS One.
7:e430562012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cao X, Li H, Tao H, Wu N, Yu L, Zhang D,
Lu X, Zhu J, Lu Z and Zhu Q: Metformin inhibits vascular
calcification in female rat aortic smooth muscle cells via the
AMPK-eNOS-NO pathway. Endocrinology. 154:3680–3689. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee SY, Lee SH, Yang EJ, Kim EK, Kim JK,
Shin DY and Cho ML: Metformin ameliorates inflammatory bowel
disease by suppression of the STAT3 signaling pathway and
regulation of the between Th17/Treg balance. PLoS One.
10:e01358582015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Isoda K, Young JL, Zirlik A, MacFarlane
LA, Tsuboi N, Gerdes N, Schönbeck U and Libby P: Metformin inhibits
proinflammatory responses and nuclear factor-kappaB in human
vascular wall cells. Arterioscler Thromb Vasc Biol. 26:611–617.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Al-Dwairi A, Alqudah TE, Al-Shboul O,
Alqudah M, Mustafa AG and Alfaqih MA: Glucagon-like peptide-1
exerts anti-inflammatory effects on mouse colon smooth muscle cells
through the cyclic adenosine monophosphate/nuclear factor-κB
pathway in vitro. J Inflamm Res. 11:95–109. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Berkes J, Viswanathan VK, Savkovic SD and
Hecht G: Intestinal epithelial responses to enteric pathogens:
Effects on the tight junction barrier, ion transport, and
inflammation. Gut. 52:439–451. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brynskov J, Foegh P, Pedersen G, Ellervik
C, Kirkegaard T, Bingham A and Saermark T: Tumour necrosis factor
alpha converting enzyme (TACE) activity in the colonic mucosa of
patients with inflammatory bowel disease. Gut. 51:37–43. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Neurath MF: Cytokines in inflammatory
bowel disease. Nat Rev Immunol. 14:329–342. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rizzo G, Pugliese D, Armuzzi A and Coco C:
Anti-TNF alpha in the treatment of ulcerative colitis: A valid
approach for organ-sparing or an expensive option to delay surgery?
World J Gastroenterol. 20:4839–4845. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lv R, Qiao W, Wu Z, Wang Y, Dai S, Liu Q
and Zheng X: Tumor necrosis factor alpha blocking agents as
treatment for ulcerative colitis intolerant or refractory to
conventional medical therapy: A meta-analysis. PLoS One.
9:e866922014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Scarpa M, Kessler S, Sadler T, West G,
Homer C, McDonald C, de la Motte C, Fiocchi C and Stylianou E: The
epithelial danger signal IL-1α is a potent activator of fibroblasts
and reactivator of intestinal inflammation. Am J Pathol.
185:1624–1637. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Papadakis KA and Targan SR: Role of
cytokines in the pathogenesis of inflammatory bowel disease. Annu
Rev Med. 51:289–298. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dionne S, D'Agata DD, Hiscott J, Vanounou
T and Seidman EG: Colonic explant production of IL-1 and its
receptor antagonist is imbalanced in inflammatory bowel disease
(IBD). Clin Exp Immunol. 112:435–442. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cominelli F and Pizarro TT: Interleukin-1
and interleukin-1 receptor antagonist in inflammatory bowel
disease. Aliment Pharmacol Ther. 10:49–53. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ghia JE, Galeazzi F, Ford DC, Hogaboam CM,
Vallance BA and Collins S: Role of M-CSF-dependent macrophages in
colitis is driven by the nature of the inflammatory stimulus. Am J
Physiol Gastrointest Liver Physiol. 294:G770–G777. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Marshall D, Cameron J, Lightwood D and
Lawson AD: Blockade of colony stimulating factor-1 (CSF-I) leads to
inhibition of DSS-induced colitis. Inflamm Bowel Dis. 13:219–224.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Franzè E, Marafini I, de Simone V,
Monteleone I, Caprioli F, Colantoni A, Ortenzi A, Crescenzi F, Izzo
R, Sica G, et al: Interleukin-34 induces Cc-chemokine ligand 20 in
gut epithelial cells. J Crohns Colitis. 10:87–94. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Werner L, Guzner-Gur H and Dotan I:
Involvement of CXCR4/CXCR7/CXCL12 interactions in inflammatory
bowel disease. Theranostics. 3:40–46. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Danese S and Gasbarrini A: Chemokines in
inflammatory bowel disease. J Clin Pathol. 58:1025–1027. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mazzucchelli L, Hauser C, Zgraggen K,
Zgraggen K, Wagner HE, Hess MW, Laissue JA and Mueller C:
Differential in situ expression of the genes encoding the
chemokines MCP-1 and RANTES in human inflammatory bowel disease. J
Pathol. 178:201–206. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Luo Y and Dorf ME: Beta-chemokine TCA3
binds to mesangial cells and induces adhesion, chemotaxis, and
proliferation. J Immunol. 156:742–748. 1996.PubMed/NCBI
|
|
37
|
Scheerens H, Hessel E, de Waal-Malefyt R,
Leach MW and Rennick D: Characterization of chemokines and
chemokine receptors in two murine models of inflammatory bowel
disease: IL-10-/-mice and Rag-2-/-mice reconstituted with CD4+
CD45RBhigh T cells. Eur J Immunol. 31:1465–1474. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xia XM, Wang FY, Zhou J, Hu KF, Li SW and
Zou BB: CXCR4 antagonist AMD3100 modulates claudin expression and
intestinal barrier function in experimental colitis. PLoS One.
6:e272822011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Koh SJ, Kim JM, Kim IK, Ko SH and Kim JS:
Anti-inflammatory mechanism of metformin and its effects in
intestinal inflammation and colitis-associated colon cancer. J
Gastroenterol Hepatol. 29:502–510. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kothari V, Galdo JA and Mathews ST:
Hypoglycemic agents and potential anti-inflammatory activity. J
Inflamm Res. 9:27–38. 2016.PubMed/NCBI
|
|
41
|
Salminen A, Hyttinen JM and Kaarniranta K:
AMP-activated protein kinase inhibits NF-κB signaling and
inflammation: Impact on healthspan and lifespan. J Mol Med (Berl).
89:667–676. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
He C, Li H, Viollet B, Zou MH and Xie Z:
AMPK suppresses vascular inflammation in vivo by inhibiting signal
transducer and activator of transcription-1. Diabetes.
64:4285–4297. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Atreya I, Atreya R and Neurath MF:
NF-kappaB in inflammatory bowel disease. J Intern Med. 263:591–596.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nair DG, Miller KG, Lourenssen SR and
Blennerhassett MG: Inflammatory cytokines promote growth of
intestinal smooth muscle cells by induced expression of PDGF-Rβ. J
Cell Mol Med. 18:444–454. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Guan Q and Zhang J: Recent advances: The
imbalance of cytokines in the pathogenesis of inflammatory bowel
disease. Mediators Inflamm. 2017:48102582017. View Article : Google Scholar : PubMed/NCBI
|