Introduction
Hepatic fibrosis (HF) is a chronic liver disease,
and a major cause of morbidity and mortality worldwide (1). HF can be induced by viral hepatitis,
alcohol/nonalcoholic steatohepatitis and several other etiologies,
which then cause persistent damage and stimulation (2). In the development of HF, an enhanced
fibrotic matrix is identified, which is due to the disturbance of
extracellular matrix (ECM) synthesis and degradation (3). With the progression of HF, it may
further result in liver cirrhosis and hepatocellular carcinoma,
inducing a major public health concern (4). Hence, it is particularly important to
explore an effective therapy method for patients with HF.
Hepatic fibrosis is a pathological process in which
a large number of activated fibroblasts are produced in the liver,
resulting in excessive precipitation of diffuse extracellular
matrix in the liver (5). In recent
years, it has been identified that hepatocytes and bile duct
epithelial cells can be transformed into fibroblasts through the
Epithelial-Mesenchymal Transition (EMT) process and participate in
the development of liver fibrosis (6). Lim et al (7) reported that the hepatic stellate cell
(HSC) may be an epithelial cell in resting state. After HSC was
activated, E-cadherin was transformed into N-cadherin, a marker of
interstitial cells (8). These
results suggest that HSC can form fibroblasts through EMT
activation and participate in the occurrence of liver fibrosis
(8). Hence, the present study aimed
to examine the changes of EMT markers that are closely associated
with liver fibrosis.
Transforming growth factor (TGF)-β is a key
regulator in chronic liver disease where it promotes the process of
fibrogenesis through inflammation (9). Within the liver, TGF-β is secreted by
platelets and Kupffer cells upon stimulation of the inflammatory
response (10). A significant
enhancement in TGF-β expression is identified in the activated
hepatic stellate cells, which is accompanied by the accelerated
accumulation of ECM (11). In
general, TGF-β exerts its biological function via activating
downstream regulators, mothers against decapentaplegic homolog
(SMAD) 2 and 3 (12). Subsequently,
SMADs interact with other signaling pathways, including the
mitogen-activated protein kinase (MAPK) and nuclear factor (NF)-κB
signaling pathways, thereby driving the process of hepatic fibrosis
(12).
Currently, increasing evidence has indicated that
microRNAs (miRs) are widely involved in TGF-β-mediated liver
fibrosis (13,14). For instance, miR-130a-3p is
demonstrated to be negatively regulating HSC activation and
proliferation in the development of nonalcoholic steatohepatitis by
suppressing the expression TGFBR1 and TGFBR2 via the TGF-β/SMAD
signaling pathway (15).
Additionally, miR-9-5p was shown to inhibit TGF-β1-induced HSC
activation via targeting TGFBR1 and TGFBR2 (16). Abnormal expression of miR-291-3p has
been widely identified in different tumors, including malignant
melanoma and colorectal carcinoma (17,18). A
previous study has indicated that miR-291-3p induces liver cell
apoptosis via targeting RNA binding protein HuR (19). Additionally, increased miR-291-3p
expression levels led to abnormal glucose and lipid metabolism in
liver cells (20,21). These data suggested that abnormal
miR-291-3p level in liver cells could result in liver cell damage.
However, whether miR-291-3p is involved in the progression of HF
has never been explored. In the present study, the expression and
functional role of miR-291-3p in the development of liver fibrosis
was evaluated for the first time, to the best of our knowledge.
Patients and methods
Human specimens
Peripheral blood or liver tissue were collected from
a total of 40 patients diagnosed with posthepatitic cirrhosis and
admitted into First People's Hospital of Kunshan Affiliated with
Jiangsu University from February to October in 2017. These included
22 males and 18 females with an average age of 52.5±13.4 years. Two
of them were diagnosed at stage I, three were at stage II, 10 were
at stage III, and 25 were at stage IV. Another 40 healthy
volunteers from the physical examination center of the hospital
were enrolled as the normal control group, including 24 males and
16 females with an average age of 43.6±14.5 years. The normal liver
samples were obtained from residual liver tissues because of
technical reasons (the fibrotic tissues were harvested from an
improper location). Normal liver tissues were determined using
immunohistochemical analysis. Liver cirrhosis was diagnosed by
liver biopsy and/or a typical appearance of the liver on abdominal
ultrasound and/or computed tomography scan. Informed consent for
use of liver samples was obtained from all participants. Peripheral
blood (10 ml) was collected from 40 healthy control and 40 patients
with liver cirrhosis. The present study was approved by the Ethics
Committee of the First People's Hospital of Kunshan Affiliated with
Jiangsu University (KSAJSU-20160819) and was performed in
compliance with the Declaration of Helsinki. All the patients have
provided written informed consent for this study. The details for
the control and patients with liver cirrhosis were included in
Table I.
 | Table I.Clinical characteristics of healthy
control and patients with liver cirrhosis. |
Table I.
Clinical characteristics of healthy
control and patients with liver cirrhosis.
| Characteristic | Control (n=40) | Liver cirrhosis
(n=40) |
|---|
| Sex
(male/female) | 24/16 | 22/18 |
| Age (years) | 43.6±14.5 | 52.5±13.4 |
| Tumor stage
(n) |
|
|
| Stage
I |
| 2 |
| Stage
II |
| 3 |
| Stage
III |
| 10 |
| Stage
IV |
| 25 |
| TBIL (µB) | 11.8±3.8 |
106.7±157.9a |
| ALT (U/l) | 21.3±5.6 |
122.4±235.1a |
| AST (U/l) | 20.3±4.8 |
116.4±182.5a |
| ALB (g/l) | 46.8±2.7 |
32.5±8.1a |
| GGT (U/l) | 23.5±8.2 |
116.4±138.9a |
| AKP (U/l) | 56.2±8.4 |
163.2±96.5a |
| PT time (sec) | 12.1±0.5 |
18.3±13.6a |
| HBV DNA level |
|
6.7±15.9a |
Isolation and culture of rat hepatic
stellate cells (HSCs)
Adult male Sprague-Dawley rats (body weight, 400–500
g; age, 6–8 weeks) were purchased from Shanghai Silaike
Experimental Animal Limited Liability Company (Shanghai, China) and
were used for HSC isolation, as previously described (22). Rats were maintained in the animal
experimental center of Wenzhou Medical University and four animals
were housed per cage with a 12 h light/dark cycle. Room temperature
was maintained at 23±1°C, humidity was maintained at ~60% and all
rats had free access to food and water. The present study was
approved by the Animal Ethics Committee of the First People's
Hospital of Kunshan Affiliated with Jiangsu University
(KSAJSU-20160819). Cells were cultured in Dulbecco's modified
Eagle's medium (DMEM; Hyclone; GE Healthcare Life Sciences, Logan,
UT, USA) supplemented with 10% fetal bovine serum (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA), 100 U/ml
penicillin and 100 U/ml streptomycin. The harvested primary HSCs
were studied at day 0 after isolation throughout all the studies.
Primary HSCs at a density of 106 cells/well were
transfected with miR-291-3p mimics/inhibitors or the negative
control for miR-291-3p mimic (NC)/miR-291-3p inhibitors (NCi; both
Shanghai GeneChem, Co., Ltd., Shanghai, China) at 37°C for 48 h
using Lipofectamine RNAiMAX (Invitrogen, Thermo Fisher Scientific,
Inc. Waltham, MA, USA) at a final concentration of 10 nM. Following
48 h of transfection, cells were collected for subsequent
experimentation.
Transfection of siRNA targeting
Smad2
Primary HSCs at the density 106
cells/well were transfected with siRNA targeting Smad2 or an NC
(both Shanghai GeneChem Co., Ltd.) at 37°C for 48 h using
Lipofectamine RNAiMAX (Invitrogen; Thermo Fisher Scientific, Inc.)
at a final concentration of 10 nM. Following 48 h of transfection,
cells were collected for subsequent experimentation.
Cell culture
293T cells were purchased from the Chinese Academy
of Medical Sciences (Beijing, China). Cells were cultured in
DMEM/F12 (GE Healthcare Life Sciences) supplemented with 10% fetal
bovine serum, 100 U/ml penicillin and 100 U/ml streptomycin in 25
cm3 culture flasks at 37°C in a humidified atmosphere
containing 5% CO2.
Transwell migration assay
Cells were placed in the top chamber of Transwell
migration chambers (8 µm; Millipore, Billerica, MA, USA) at a
density of 105 cells/well in 1 ml DMEM culture. After 48
h, cells which had not migrated to the lower chamber were removed
from the upper surface of the Transwell membrane with a cotton
swab. Migrating cells on the surface of the lower membrane were
fixed with methanol for 15 min at room temperature. Methanol was
then discarded and washed using PBS. The cells were further stained
with 0.5% crystal violet for 20 min at room temperature. Cells were
subsequently imaged and counted using a light microscope (XDS-500D;
Shanghai Caikon Optical Instrument Co., Ltd., Shanghai, China) with
100× magnification. Experiments were assayed in triplicates, and ≥5
fields were counted in each experiment.
Proliferation assay
Cell proliferation was evaluated using the cell
counting kit-8 (CCK-8; Dojindo, Kumamoto, Japan), according to the
manufacturer's protocol. Cells were seeded in 96-well plates at a
density of 1×103 cells per well and cultured for 24 h at 37°C.
Subsequently, cells were transfected with miR-291-3p mimics. After
48 h, CCK-8 solution was added to each well in the 96-well plates.
Then, cells were incubated for an additional 2 h at 37°C.
Absorbance was determined at 450 nm on a microplate reader
(Molecular Devices, Sunnyvale, CA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RNA was isolated from liver tissues, peripheral
blood or primary HSCs using RNA TRIzol reagent (Life Sciences;
Thermo Fisher Scientific, Inc.) following the manufacturer's
protocol. Total RNA was subsequently reverse transcribed to cDNA
using SuperScript™ III Reverse Transcriptase
(Invitrogen; Thermo Fisher Scientific, Inc.) following the
manufacturer's protocol. qPCR was performed using SYBR-Green PCR
Master Mix (Roche Diagnostics, Basel, Switzerland) on an Applied
Biosystems ViiA 7 real-time PCR system (Thermo Fisher Scientific,
Inc.). The final reaction volume was 10 µl and contained 5 µl
SYBR-Green PCR Master Mix (2X), 0.5 µl forward and 0.5 µl reverse
primers (10 mM), 2 ml cDNA and 2 µl double-distilled water. The
thermocycling conditions of qPCR were: Denaturation at 95°C for 10
min, followed by 40 cycles of 95°C for 10 sec and 60°C for 60 sec.
The results were normalized to U6 expression to obtain ∆Cq values
and fold changes in expression were calculated using the
2−∆∆Cq method (23).
The primers used in the current study were listed as
follows: Snai1 forward, 5′-GCAGAGTTGTCTACCGACCT-3′ and reverse,
5′-AGGTGAACTCCACACACGC-3′; VE-cadherin forward,
5′-CCAGAATTTGCCCAGCCCTA-3′ and reverse 5′-GTCCTCGTTCTTCAGGGCAA-3′;
Vimentin forward, 5′-TCCTTCGAAGCCATGTCCAC-3′ and reverse,
5′-GTGGTCACATAGCTCCGGTT-3′; TGF-β1 forward,
5′-GACTCTCCACCTGCAAGACC-3′ and reverse, 5′-GGACTGGCGAGCCTTAGTTT-3′;
glial fibrillary acidic protein (GFAP) forward,
5′-TTGACCTGCGACCTTGAGTC-3′ and reverse,
5′-GAGTGCCTCCTGGTAACTCG-3′.
Dual luciferase reporter assay
MiRNAs targets were predicted using the TargetScan
website (www.targetscan.org). Wild-type (WT)
and mutant seed regions of miR-291-3p in the 3′-untranslated region
(UTR) of the Smad2 gene were chemically synthesized in vitro,
XhoI and NdeI restriction sites were added, and then
cloned into pmirGLO luciferase reporter vector (Promega
Corporation, Madison, WI, USA).
Prior to conducting the dual reporter assay,
5×104 293T cells/well were seeded in 24-well plates with
500 µl DMEM and cultured for 18 h at 37°C. The cells were
transfected with the modified firefly luciferase reporter vector
(500 ng/µl) mixed with Vigofect transfection reagent (Vigorous,
Beijing, China), according to the manufacturer's protocol. After
continuous exposure of miR-291-3p/pmirGLO-Smad2-3′UTR or NC/pmirGLO
blank vector for 48 h, the luciferase activities of firefly and
Renilla were measured with the Dual-Luciferase® Reporter
Assay System (Promega Corporation), according to the manufacturer's
protocol. Firefly luciferase activity was normalized to Renilla
luciferase activity.
Western blotting
Liver tissues or primary HSCs were treated with
radioimmunoprecipitation assay buffer containing 1% (v/v)
phenylmethylsulfonyl fluoride (both Beijing Solarbio Science and
Technology Co., Ltd., Beijing, China), 0.3% (v/v) protease
inhibitor (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and 0.1%
(v/v) phosphorylated proteinase inhibitor (Sigma-Aldrich; Merck
KGaA). A bicinchoninic protein assay kit (Pierce; Thermo Fisher
Scientific, Inc.) was used to determine the protein concentration.
Subsequently, supernatants were extracted from the lysates
following centrifugation at 11,000 × g at 4°C for 15 min. Equal
amounts of protein (30 µg/lane) were separated using 10% SDS-PAGE
at 300 mA for 2 h and transferred onto a polyvinylidene fluoride
membrane, as previously reported (10). The following primary antibodies were
used: β-Actin (cat. no. 4970), Snai1 (cat. no. 3879), vascular
endothelial cadherin (VE-cadherin; cat. no. 2500), Vimentin (cat.
no. 5741), TGF-β1 (cat. no. 3711), GFAP (cat. no. 80788; all
1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA). HSCs
are the main contributors of hepatic fibrosis (24). Glial Fibrillary Acidic Protein
(GFAP), first identified in astroglial cells, belongs to
intermediate filaments, which preserves cell's mechanical strength
and structure (25). TGF-β-mediated
activation of GFAP has been extensively reported (26,27). A
previous study has demonstrated that GFAP could be a more useful
marker of early Hepatic Stellate Cells (HSC) activation than α-SMA
(24). It is suggested that GFAP may
act as a mesenchymal marker of early HSCs activation in liver
fibrosis (28). Hence, the present
study evaluated the expression of GFAP. Following several washes
with TBS with Tween (TBST), the membranes were incubated with
horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG
(1:5,000; cat. no. ZB-2306, Zhongshan Gold Bridge Biological
Technology Co., Beijing, China) for 2 h at room temperature and
then washed. The proteins were detected using enhanced
chemiluminescence, according to the manufacturer's protocol (Merck
KGaA). ImageJ 1.8.0 (National Institutes of Health, Bethesda, MD,
USA) was used to quantify the relative protein levels. β-Actin was
used as an internal control.
Statistical analysis
The data are represented as the mean ± standard
deviation (SD). Two-tailed unpaired Student's t-tests were used for
comparisons of two groups. Comparisons of means among multiple
groups were determined using one-way analysis of variance followed
by a Turkey's post-hoc test. Receiver operating characteristic
(ROC) curves were used to assess miR-291-3p as a biomarker and the
area under the curve was reported (version 20.0; SPSS, Inc.,
Chicago, Illinois). P<0.05 was considered to indicate a
statistically significant difference.
Results
Decreased miR-291-3p in the peripheral
blood and liver tissues of patients with HF
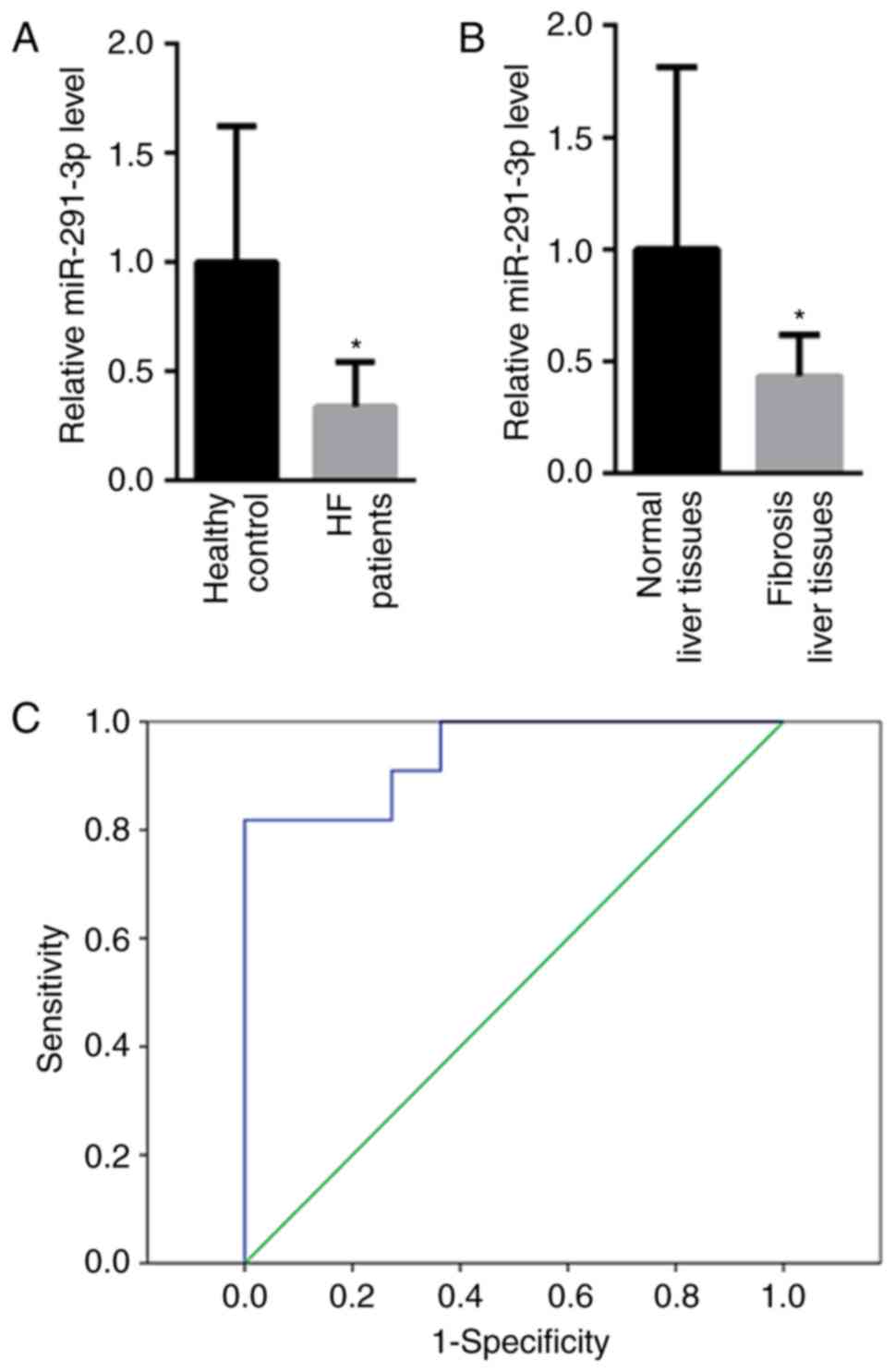
First, the levels of miR-291-3p in the peripheral
blood and liver tissues of patients with HF were detected. Compared
with healthy controls (1±0.69), the levels of miR-291-3p in the
peripheral blood were decreased in patients with HF (0.34±0.32;
Fig. 1A). Furthermore, the levels of
miR-291-3p in fibrosis liver tissues and the normal liver tissues
were tested. As shown in Fig. 1B, a
much lower miR-291-3p level (0.42±0.28) was identified in fibrotic
liver tissues than that of the normal liver tissues (1±0.83). In
addition, the present study also evaluated whether miR-291-3p
present in peripheral blood may be a useful biomarker to determine
possible HF in patients. Receiver operating characteristic (ROC)
curve analysis showed that peripheral blood miR-291-3p may
differentiate patients with HF from healthy controls, with a ROC
curve area of 0.942 (95% confidence interval: 0.858–1.000;
P<0.001; Fig. 1C). Meanwhile, the
sensitivity and specificity for peripheral blood miR-291-3p were
85.4 and 93.2%, respectively, at the cut-off of 0.208.
miR-291-3p suppresses TGF-β
signaling
TGF-β signaling pathway has become a novel
therapeutic in the prevention and treatment of HF (29). Thus, the effect of miR-291-3p on the
TGF-β signaling pathway was evaluated. RT-qPCR analysis showed that
transfection of miR-291-3p mimic significantly enhanced the level
of miR-291-3p (36.8±4.5) than that of NC (1±0.12), but suppressed
the mRNA levels of Snai1, VE-cadherin, Vimentin, TGF-β1, and GFAP
(Fig. 2A). In comparison,
transfection with miR-291-3p inhibitor decreased miR-291-3p, but
increased the mRNA levels of Snai1, VE-cadherin, Vimentin, TGF-β1
and GFAP (Fig. 2B). Additionally,
the protein expression levels of Snai1, VE-cadherin, Vimentin,
TGF-β1 and GFAP were decreased in HSCs transfected with miR-291-3p
mimics (Fig. 2C). Furthermore, the
protein expression of Snai1, VE-cadherin, Vimentin, TGF-β1, and
GFAP was also observed to be enhanced in HSCs transfected with
miR-291-3p inhibitors compared with that of negative controls
(Fig. 2D).
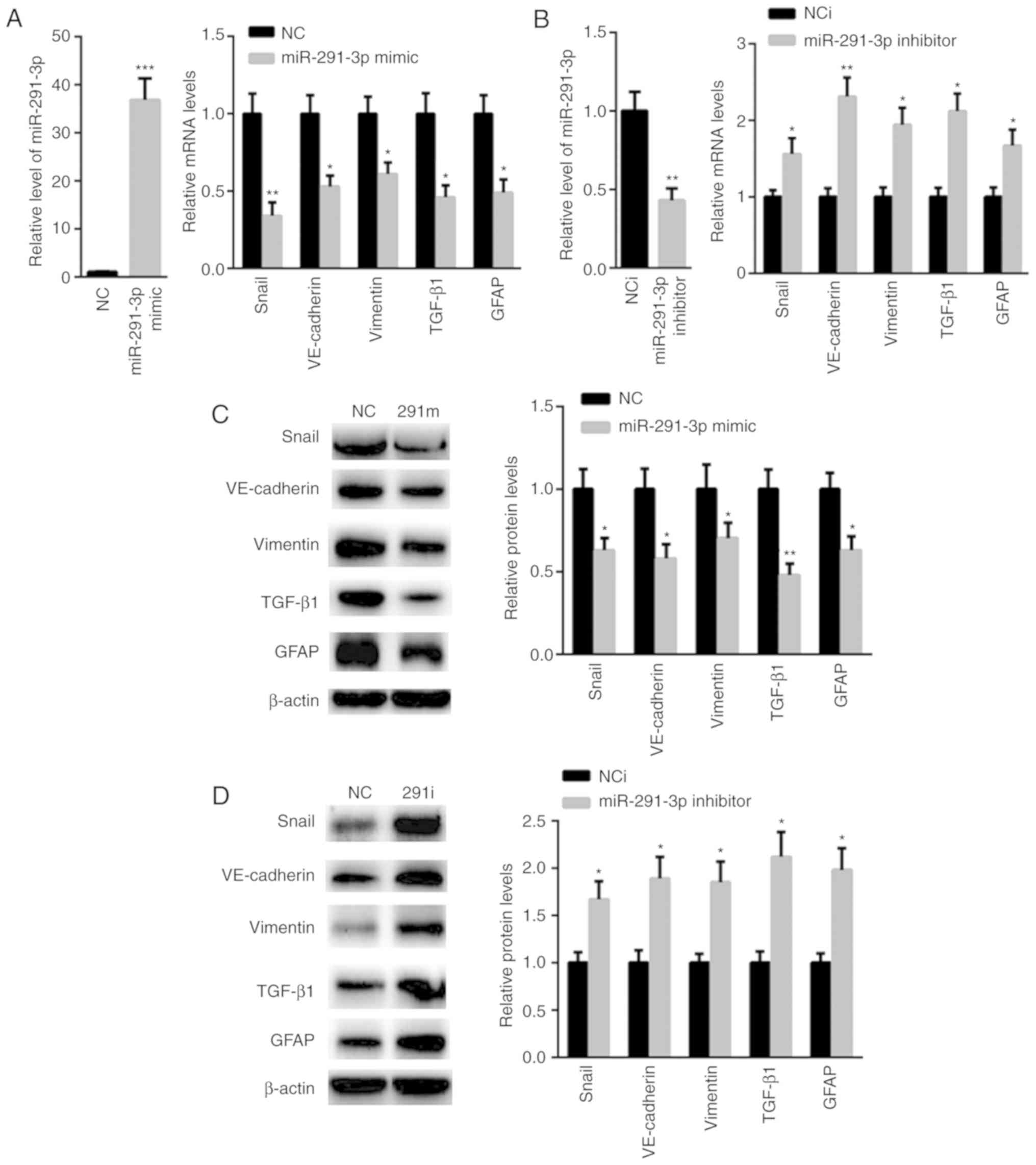 | Figure 2.miR-291-3p suppressed TGF-β
signaling. (A) Reverse transcription-quantitative polymerase chain
reaction showed that transfection of the miR-291-3p mimic
significantly enhanced the level of miR-291-3p than that of NC
(left panel), but significantly suppressed the mRNA levels of
Snai1, VE-cadherin, Vimentin, TGF-β1, and GFAP (right panel). (B)
Transfection of miR-291-3p inhibitor decreased miR-291-3p (left
panel), but increased the mRNA levels of Snai1, VE-cadherin,
Vimentin, TGF-β1, and GFAP. (C) The protein levels of Snai1,
VE-cadherin, Vimentin, TGF-β1, and GFAP were decreased in HSC
transfected with miR-291-3p mimics (right panel). (D) The
expression of Snai1, VE-cadherin, Vimentin, TGF-β1, and GFAP was
also found to be enhanced in HSC transfected with miR-291-3p
inhibitors compared to that of negative controls. *P<0.05,
**P<0.01 and ***P<0.001 vs. negative control (NC). GFAP,
glial fibrillary acidic protein; HSC, hepatic stellate cells; miR,
microRNA; TGF, transforming growth factor; VE-cadherin, vascular
endothelial cadherin. |
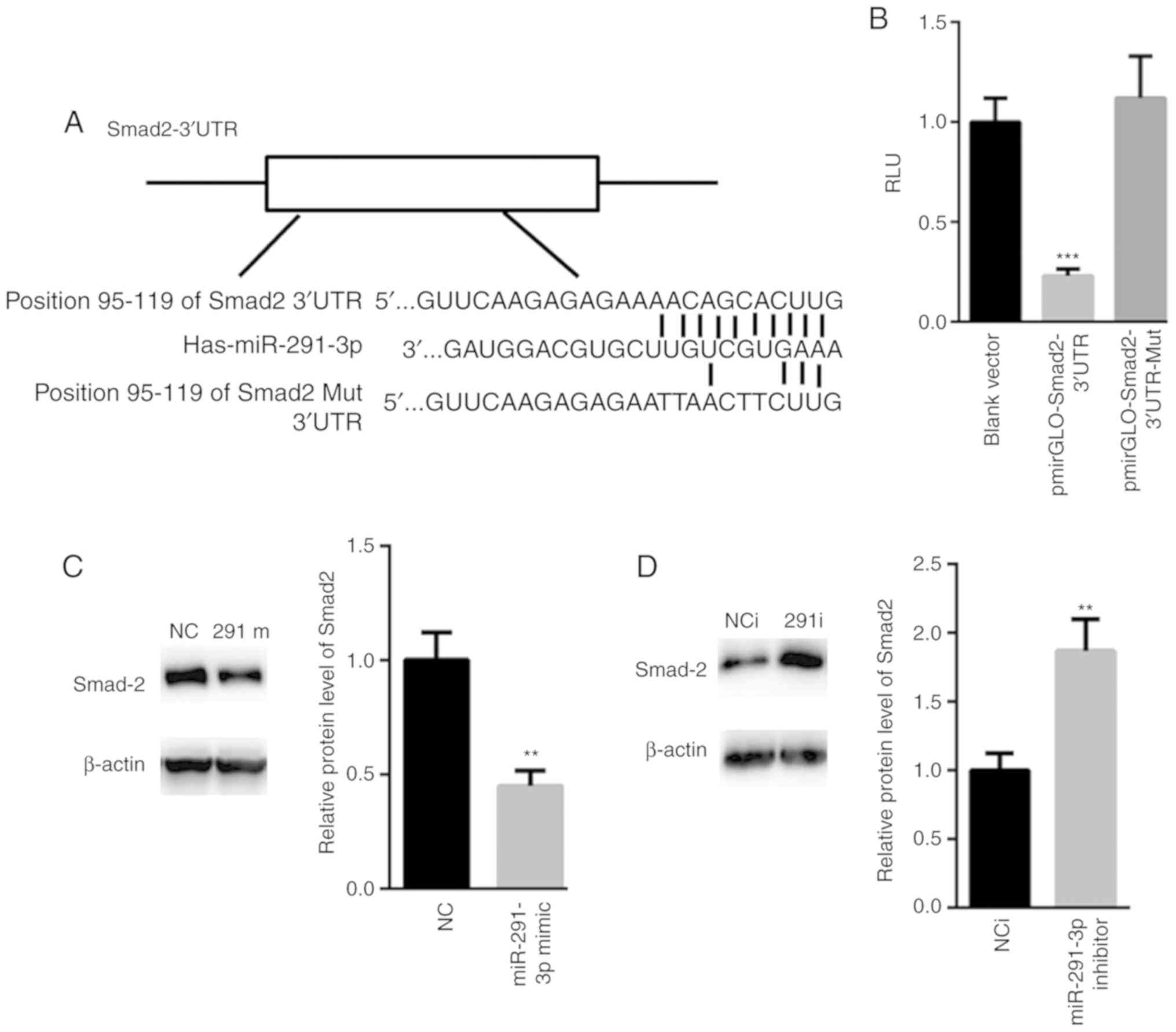
Smad2 is a target gene of
miR-291-3p
Based on these above observations, the possible
target genes of miR-291-3p were explored. TargetScan analysis
demonstrated a conserved binding site of miR-291-3p in the 3′UTR of
Smad2 (position 95–119 of Smad2 3′UTR; Fig. 3A). Dual luciferase reporter assay
showed that overexpression of miR-291-3p significantly suppressed
the relative luciferase activity of pmirGLO-Smad2-3′UTR, but not
the relative luciferase activity of pmirGLO-Smad2-3′UTR-Mut
(Fig. 3B). Western blot assay also
showed that overexpression of miR-291-3p suppressed the expression
of Smad2 (Fig. 3C), while its
inhibition increased the protein levels of Smad2 (Fig. 3D).
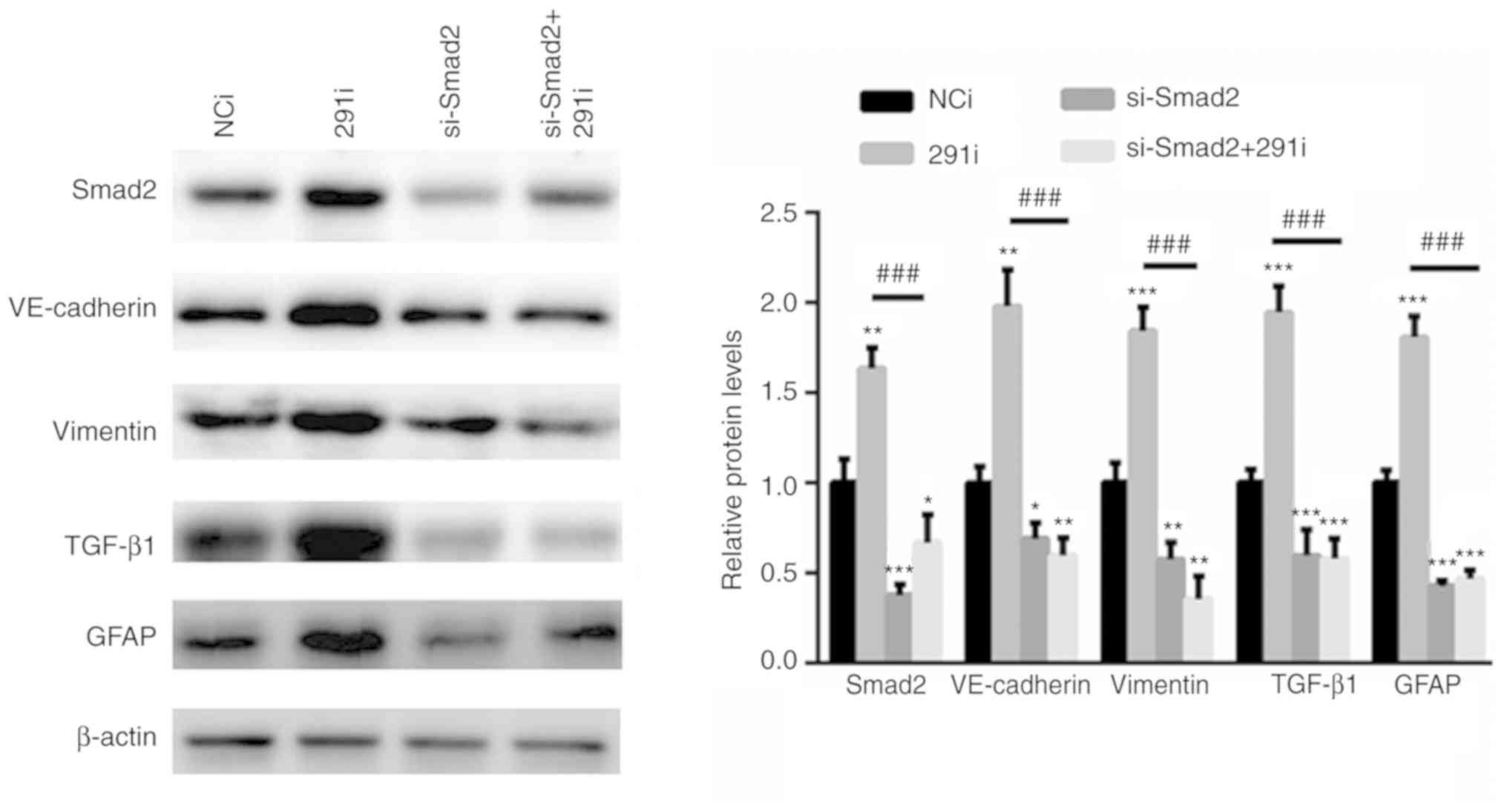
miR-291-3p suppresses TGF-β signaling
via Smad2
To further evaluate whether miR-291-3p suppressed
TGF-β signaling via Smad2, a specific siRNA targeting Smad2 was
selected. As shown in Fig. 4,
silencing of Smad2 significantly inhibited the activation of TGF-β
signaling. In comparison, inhibition of miR-291-3p increased the
activation of TGF-β signaling in Fig.
2D. However, knockdown of Smad2 together with miR-291-3p
inhibitor, significantly abolished miR-291-3p inhibition-induced
activation of TGF-β signaling (Fig.
4).
Discussion
Liver fibrosis is a major characteristic of most
chronic liver diseases (30,31). During progression, liver fibrosis can
develop into excessive scarring and organ failure, as demonstrated
in liver cirrhosis and primary liver cancer (32). It has been suggested that the fibrous
ECM is significantly accumulated and increased in the fibrotic
liver (33). Hepatic stellate cells
are the major contributors for ECM after liver injury. Once HSCs
are activated, they become proliferative resulting in enhanced ECM
production, thereby leading to liver fibrosis.
Although liver fibrosis can be reversible, how to
effectively prevent or reverse the process is still a large
challenge for clinicians (34).
Additionally, sound diagnostic biomarkers for liver fibrosis are
still rare. These limitations largely restrict the treatment
options (35). Currently, liver
biopsy is the gold standard to identify liver fibrosis (11). However, it is largely questioned due
to its painful and invasive characteristics (31). Therefore, early identification of
liver fibrosis is of great importance to develop effective
antifibrotic therapies.
Due to their stability and wide gene-regulating
capacity, miRNAs are attractive therapeutic targets (36,37). In
the progression of liver fibrosis, increasing evidence has shown
the abnormal expression of miRNAs, indicating the importance of
maintaining homeostasis via miRNAs (14,38). The
present study showed for the first time, to the best of our
knowledge, that miR-291-3p was decreased in the peripheral blood
and liver tissues of patients with HF. ROC analysis showed that
miR-291-3p could differentiate patients with HF from healthy
controls.
Based on the above findings, the study further
explored the possible association between miR-291-3p and liver
fibrosis. TGF-β1 is a major cytokine involved in liver fibrosis
(39). In the progression of liver
fibrosis, TGF-β1 could stimulate and activate HSCs to proliferate
and constantly secrete ECM via autocrine and paracrine mechanisms,
resulting in sustained fibrosis (40). The present study identified that
overexpression of miR-291-3p suppressed the activation of TGF-β1
signaling, while inhibition of miR-291-3p enhanced TGF-β1 signaling
activation. Furthermore, it indicated that Smad2 was a target gene
of miR-291-3p. More importantly, silencing of Smad2 could abolish
miR-291-3p inhibition-induced TGF-β1 signaling activation.
It is true that increased GFAP expression is
observed in liver fibrosis compared with normal control (28). However, it has also been stated that
high GFAP expression level is associated with low fibrosis
(28). These data suggested that
GFAP may be a more useful marker than alpha-smooth muscle actin
(α-SMA) for early activation of HSCs and represent an early
indicator of hepatic fibrogenesis (41). The present study did not examine
whether low miR-291 expression is associated with high fibrosis
since not enough samples were collected. The expression of GFAP in
cultured HSCs and the expression of miR-291-3p was not tested
according to the severity of HF was examined. Thus, further study
is necessary to explore whether miR-291-3p may represent as an
early or late indicator of hepatic fibrogenesis.
At present, the ‘gold standard’ for diagnosis of
liver fibrosis is still liver biopsy, but liver biopsy is an
invasive operation, which has some limitations, such as sampling
error, subjective deviation of different pathological readers
(42). Therefore, non-invasive
diagnosis of liver fibrosis has become a research topic in recent
years, among which aspartate aminotransferase (AST) to platelet
ratio index (APRI) and fibrosis-4 (FIB-4) are widely used (42). However, it is reported that the
accuracy of diagnosis of hepatic fibrosis by APRI and FIB-4 alone
is not more than 75% (43). The rate
of misdiagnosis and missed diagnosis is still high (43). However, the sensitivity and
specificity for peripheral blood miR-291-3p were 85.4 and 93.2%,
respectively. The samples in the current study were limited and
future work with large patient samples is required to validate the
present findings. Studies exploring the diagnostic value of
combination of APRI, FIB-4 and peripheral blood microRNA-291-3p for
liver fibrosis in patients with chronic hepatitis B, are needed in
order to improve the diagnostic accuracy of liver fibrosis in
patients with chronic hepatitis B.
In summary, reduced peripheral blood miR-291-3p may
serve a role in the progression of HF via targeting Smad2. However,
there are still some limitations in the current study. On the one
hand, the current study was limited due to the small sample size of
patient samples. On the other hand, whether miR-291-3p could be the
therapeutic target of HF deserves further study.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from The
First People's Hospital of Kunshan Affiliated with Jiangsu
University (KAJS-2017052′).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WY performed the experiments, analyzed the data and
wrote the mansucript. WZ, YZ, HN and LG performed RT-qPCR
experiments. MF designed the experiments, analyzed the data and
gave final approval for the publication of the final version of the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the First People's Hospital of Kunshan Affiliated with
Jiangsu University and was performed in compliance with the
Declaration of Helsinki. All patients have provided written
informed consent for this study.
Patient consent for publication
All patients provided written informed consent for
the publication of data in the current study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Xu B, Zhou NM, Cao WT and Li XJ:
Evaluation of elastography combined with serological indexes for
hepatic fibrosis in patients with chronic hepatitis B. World J
Gastroenterol. 24:4272–4280. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cargnoni A, Farigu S, Cotti Piccinelli E,
Bonassi Signoroni P, Romele P, Vanosi G, Toschi I, Cesari V, Barros
Sant'Anna L, Magatti M, et al: Effect of human amniotic epithelial
cells on pro-fibrogenic resident hepatic cells in a rat model of
liver fibrosis. J Cell Mol Med. 22:1202–1213. 2018.PubMed/NCBI
|
|
3
|
Chen J, Li X, Hu Y, Liu W, Zhou Q, Zhang
H, Mu Y and Liu P: Gypenosides ameliorate carbon
tetrachloride-induced liver fibrosis by inhibiting the
differentiation of hepatic progenitor cells into myofibroblasts. Am
J Chin Med. 45:1061–1074. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen J, Yu Y, Li S, Liu Y, Zhou S, Cao S,
Yin J and Li G: MicroRNA-30a ameliorates hepatic fibrosis by
inhibiting Beclin1-mediated autophagy. J Cell Mol Med.
21:3679–3692. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wells RG: The epithelial-to-mesenchymal
transition in liver fibrosis: Here today, gone tomorrow?
Hepatology. 51:737–740. 2010.PubMed/NCBI
|
|
6
|
Fabris L, Brivio S, Cadamuro M and
Strazzabosco M: Revisiting epithelial-to-mesenchymal transition in
liver fibrosis: Clues for a better understanding of the ‘reactive’
biliary epithelial phenotype. Stem Cells Int. 2016:29537272016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lim YS, Kim KA, Jung JO, Yoon JH, Suh KS,
Kim CY and Lee HS: Modulation of cytokeratin expression during in
vitro cultivation of human hepatic stellate cells: Evidence of
transdifferentiation from epithelial to mesenchymal phenotype.
Histochem Cell Biol. 118:127–136. 2002.PubMed/NCBI
|
|
8
|
Lim YS, Lee HC and Lee HS: Switch of
cadherin expression from E- to N-type during the activation of rat
hepatic stellate cells. Histochem Cell Biol. 127:149–160. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ding YF, Wu ZH, Wei YJ, Shu L and Peng YR:
Hepatic inflammation-fibrosis-cancer axis in the rat hepatocellular
carcinoma induced by diethylnitrosamine. J Cancer Res Clin Oncol.
143:821–834. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
El-Mezayen NS, El-Hadidy WF, El-Refaie WM,
Shalaby TI, Khattab MM and El-Khatib AS: Hepatic stellate
cell-targeted imatinib nanomedicine versus conventional imatinib: A
novel strategy with potent efficacy in experimental liver fibrosis.
J Control Release. 266:226–237. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shi J, Zhao J, Zhang X, Cheng Y, Hu J, Li
Y, Zhao X, Shang Q, Sun Y, Tu B, et al: Activated hepatic stellate
cells impair NK cell anti-fibrosis capacity through a
TGF-β-dependent emperipolesis in HBV cirrhotic patients. Sci Rep.
7:445442017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tang LY, Heller M, Meng Z, Yu LR, Tang Y,
Zhou M and Zhang YE: Transforming growth factor-β (TGF-β) directly
activates the JAK1-STAT3 axis to induce hepatic fibrosis in
coordination with the SMAD pathway. J Biol Chem. 292:4302–4312.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang JJ, Tao H, Liu LP, Hu W, Deng ZY and
Li J: miR-200a controls hepatic stellate cell activation and
fibrosis via SIRT1/Notch1 signal pathway. Inflamm Res. 66:341–352.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou G, Lin W, Fang P, Lin X, Zhuge L, Hu
Z and Jin L: MiR-10a improves hepatic fibrosis by regulating the
TGFβl/Smads signal transduction pathway. Exp Ther Med.
12:1719–1722. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang Y, Du J, Niu X, Fu N, Wang R, Zhang
Y, Zhao S, Sun D and Nan Y: MiR-130a-3p attenuates activation and
induces apoptosis of hepatic stellate cells in nonalcoholic
fibrosing steatohepatitis by directly targeting TGFBR1 and TGFBR2.
Cell Death Dis. 8:e27922017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu F, Chen B, Fan X, Li G, Dong P and
Zheng J: Epigenetically-regulated microRNA-9-5p suppresses the
activation of hepatic stellate cells via TGFBR1 and TGFBR2. Cell
Physiol Biochem. 43:2242–2252. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Long J, Menggen Q, Wuren Q, Shi Q and Pi
X: MiR-219-5p inhibits the growth and metastasis of malignant
melanoma by targeting BCL-2. Biomed Res Int. 2017:90325022017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Q, Zhu L, Jiang Y, Xu J, Wang F and
He Z: miR-219-5p suppresses the proliferation and invasion of
colorectal cancer cells by targeting calcyphosin. Oncol Lett.
13:1319–1324. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guo J, Li M, Meng X, Sui J, Dou L, Tang W,
Huang X, Man Y, Wang S and Li J: MiR-291b-3p induces apoptosis in
liver cell line NCTC1469 by reducing the level of RNA-binding
protein HuR. Cell Physiol Biochem. 33:810–822. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Meng X, Guo J, Fang W, Dou L, Li M, Huang
X, Zhou S, Man Y, Tang W, Yu L and Li J: Liver microRNA-291b-3p
promotes hepatic lipogenesis through negative regulation of
adenosine 5′-monophosphate (AMP)-activated protein kinase α1. J
Biol Chem. 291:10625–10634. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guo J, Dou L, Meng X, Chen Z, Yang W, Fang
W, Yang C, Huang X, Tang W, Yang J and Li J: Hepatic MiR-291b-3p
mediated glucose metabolism by directly targeting p65 to upregulate
PTEN expression. Sci Rep. 7:398992017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Weiskirchen R and Gressner AM: Isolation
and culture of hepatic stellate cells. Methods Mol Med. 117:99–113.
2005.PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Carotti S, Morini S, Corradini SG, Burza
MA, Molinaro A, Carpino G, Merli M, De Santis A, Muda AO, Rossi M,
et al: Glial fibrillary acidic protein as an early marker of
hepatic stellate cell activation in chronic and posttransplant
recurrent hepatitis C. Liver Transpl. 14:806–814. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bai X, Saab AS, Huang W, Hoberg IK,
Kirchhoff F and Scheller A: Genetic background affects human glial
fibrillary acidic protein promoter activity. PLoS One.
8:e668732013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Romão LF, Sousa Vde O, Neto VM and Gomes
FC: Glutamate activates GFAP gene promoter from cultured astrocytes
through TGF-beta1 pathways. J Neurochem. 106:746–756. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zheng JY, Sun J, Ji CM, Shen L, Chen ZJ,
Xie P, Sun YZ and Yu RT: Selective deletion of apolipoprotein E in
astrocytes ameliorates the spatial learning and memory deficits in
Alzheimer's disease (APP/PS1) mice by inhibiting TGF-β/Smad2/STAT3
signaling. Neurobiol Aging. 54:112–132. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hassan S, Syed S and Kehar SI: Glial
fibrillary acidic protein (GFAP) as a mesenchymal marker of early
hepatic stellate cells activation in liver fibrosis in chronic
hepatitis C infection. Pak J Med Sci. 30:1027–1032. 2014.PubMed/NCBI
|
|
29
|
Hu Z, Qin F, Gao S, Zhen Y, Huang D and
Dong L: Paeoniflorin exerts protective effect on radiation-induced
hepatic fibrosis in rats via TGF-β1/Smads signaling pathway. Am J
Transl Res. 10:1012–1021. 2018.PubMed/NCBI
|
|
30
|
Jiang Y, Wang C, Li YY, Wang XC, An JD,
Wang YJ and Wang XJ: Mistletoe alkaloid fractions alleviates carbon
tetrachloride-induced liver fibrosis through inhibition of hepatic
stellate cell activation via TGF-β/Smad interference. J
Ethnopharmacol 158 Pt A. 230–238. 2014. View Article : Google Scholar
|
|
31
|
Wang J, Chu ES, Chen HY, Man K, Go MY,
Huang XR, Lan HY, Sung JJ and Yu J: microRNA-29b prevents liver
fibrosis by attenuating hepatic stellate cell activation and
inducing apoptosis through targeting PI3K/AKT pathway. Oncotarget.
6:7325–7338. 2015.PubMed/NCBI
|
|
32
|
Lamb CL, Cholico GN, Pu X, Hagler GD,
Cornell KA and Mitchell KA: 2,3,7,8-Tetrachlorodibenzo-p-dioxin
(TCDD) increases necroinflammation and hepatic stellate cell
activation but does not exacerbate experimental liver fibrosis in
mice. Toxicol Appl Pharmacol. 311:42–51. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu YW, Chiu YT, Fu SL and Huang YT:
Osthole ameliorates hepatic fibrosis and inhibits hepatic stellate
cell activation. J Biomed Sci. 22:632015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nunez Lopez O, Bohanon FJ, Wang X, Ye N,
Corsello T, Rojas-Khalil Y, Chen H, Chen H, Zhou J and
Radhakrishnan RS: STAT3 inhibition suppresses hepatic stellate cell
fibrogenesis: HJC0123, a potential therapeutic agent for liver
fibrosis. RSC Adv. 6:100652–100663. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Perumal N, Perumal M, Halagowder D and
Sivasithamparam N: Morin attenuates diethylnitrosamine-induced rat
liver fibrosis and hepatic stellate cell activation by co-ordinated
regulation of Hippo/Yap and TGF-β1/Smad signaling. Biochimie.
140:10–19. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang Z, Zha Y, Hu W, Huang Z, Gao Z, Zang
Y, Chen J, Dong L and Zhang J: The autoregulatory feedback loop of
microRNA-21/programmed cell death protein 4/activation protein-1
(MiR-21/PDCD4/AP-1) as a driving force for hepatic fibrosis
development. J Biol Chem. 288:37082–37093. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhou L, Liu S, Han M, Ma Y, Feng S, Zhao
J, Lu H, Yuan X and Cheng J: miR-185 inhibits fibrogenic activation
of hepatic stellate cells and prevents liver fibrosis. Mol Ther
Nucleic Acids. 10:91–102. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Roderburg C, Luedde M, Vargas Cardenas D,
Vucur M, Mollnow T, Zimmermann HW, Koch A, Hellerbrand C,
Weiskirchen R, Frey N, et al: miR-133a mediates TGF-β-dependent
derepression of collagen synthesis in hepatic stellate cells during
liver fibrosis. J Hepatol. 58:736–742. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xiao YH, Liu DW and Li Q: Effects of drug
serum of anti-fibrosis I herbal compound on calcium in hepatic
stellate cell and its molecular mechanism. World J Gastroenterol.
11:1515–1520. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu LX, Zhang HY, Zhang QQ and Guo XH:
Effects of insulin-like growth factor binding protein-related
protein 1 in mice with hepatic fibrosis induced by thioacetamide.
Chin Med J (Engl). 123:2521–2526. 2010.PubMed/NCBI
|
|
41
|
Zakaria S, Youssef M, Moussa M, Akl M,
El-Ahwany E, El-Raziky M, Mostafa O, Helmy AH and El-Hindawi A:
Value of α-smooth muscle actin and glial fibrillary acidic protein
in predicting early hepatic fibrosis in chronic hepatitis C virus
infection. Arch Med Sci. 6:356–365. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Castera L: Invasive and non-invasive
methods for the assessment of fibrosis and disease progression in
chronic liver disease. Best Pract Res Clin Gastroenterol.
25:291–303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Crisan D, Radu C, Lupsor M, Sparchez Z,
Grigorescu MD and Grigorescu M: Two or more synchronous combination
of noninvasive tests to increase accuracy of liver fibrosis
assessement in chronic hepatitis C; results from a cohort of 446
patients. Hepat Mon. 12:177–184. 2012. View Article : Google Scholar : PubMed/NCBI
|