Introduction
Myocardial infarction (MI) has a high rate of
mortality and morbidity worldwide and due to the growth of an aging
population the healthcare burden of MI is increasing (1). MI is characterized by inflammation and
cardiomyocyte apoptosis, and can lead to left ventricular
enlargement, thoracalgia, breathing difficulty, asthenia, fatigue,
diaphoresis, cardiopalmus, heart failure and coma (2,3).
Coronary artery occlusion is one of the main causes of MI. With the
discovery of an increasing number of therapeutic approaches,
long-term survival after MI is increasing worldwide (4). Experimental and clinical studies have
shown that cardiomyocyte apoptosis following MI is caused by
hypoxic injury, ischemia-reperfusion injury and oxidative stress.
Mitigation of these processes is a feasible way to treat MI
(5).
Cardiomyocyte dysfunction is regarded as a leading
cause of cardiac abnormality and may be responsible for the high
mortality of patients with heart failure (6). Evidence has shown that myocardial
ischaemia-induced cardiomyocyte dysfunction may be a contributor to
cardiac cell death (7). MI is the
main cause of death in patients with cardiovascular disease
(8). Myocardial damage in hypoxia
leads to various chemical and physical changes, including scarring,
inflammation, necrosis, cell apoptosis and cardiac remodeling
(9). An increasing number of studies
have highlighted the significance of cardiomyocyte apoptosis and
inflammatory response in MI development, due to their role in
regulating heart and cardiac muscle function (9,10). A
previous study has shown that increased levels of inflammatory
mediators in cardiac dysfunction, especially increased levels of
tumor necrosis factor-α (TNF-α) in the locally infarcted
myocardium, directly lead to cardiomyocyte apoptosis and myocardial
dysfunction (11). Though signaling
pathways, including inflammatory and calcium signaling, and
reactive oxygen species (ROS) have been shown to play important
roles in hypoxia-induced cardiomyocyte apoptosis, the precise
molecular mechanisms underlying MI-induced cardiac damage remain
unclear (12).
Lidocaine, also known as N-diethylaminoacetyl-2,
6-dimethylaniline, is one of the most commonly used local
anesthetics (13) and an
anti-arrhythmic drug in a number of heart conditions (14,15).
Lidocaine has a variety of pharmacological effects, including
immunomodulation, anti-inflammatory, anti-oxidative effects and
anti-tumor effects (16–21). One study assessing lidocaine toxicity
showed that it can inhibit major cell signaling pathways, including
AKT and ERK (22). Studies have
indicated that lidocaine has a protective effect against
cardiomyocyte injury (23,24). Lidocaine-induced blockade of
voltage-gated sodium channels has been shown to rescue the function
of ischemic myocardium in vivo (23) and has been confirmed to protect mice
from myocardial damage due to ischemia-reperfusion injury (24). In particular, Okamoto et al
(25) previously reported that
hypoxia inducible factor 1α overexpression reduces
lidocaine-induced renal cell-derived RCC4 cell and neuronal SH-SY5Y
cell death. However, lidocaine has not been previously investigated
as a potential treatment for hypoxia-induced cardiomyocyte cell
death, and the protective mechanisms of lidocaine in this process
remain unclear. Based on existing research data (23–25), it
was hypothesized that lidocaine may have a protective effect
against hypoxia-induced apoptosis and cardiac damage through the
activation of the mitogen activated protein kinase (MAPK)
/ERK/NF-κB signaling pathway. The purpose of this study was to
explore this hypothesis and the underlying mechanisms of lidocaine
action to provide the basis for developing new treatments for
MI.
Materials and methods
Cell culture and hypoxia
treatment
The rat cardiac myoblast cell line H9c2 was obtained
from the American Type Culture Collection and they are used here as
model for cardiac myocytes. H9c2 cells were cultured in DMEM
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal
bovine serum (Invitrogen; Thermo Fisher Scientific, Inc.), 100 U/ml
penicillin and 100 mg/ml of streptomycin (Sigma-Aldrich; Merck
KGaA). Cells were cultured at 37°C in a humidified atmosphere of 5%
CO2. Culture medium was changed every 2 days.
To induce hypoxia, H9c2 cells
(5×104/well) at 80% confluence were placed in a hypoxic
chamber (Thermo Fisher Scientific, Inc.) containing 1%
O2, 5% CO2 and 94% N2 for 48 h.
Cells were cultured with a range of lidocaine (Sigma-Aldrich, Merck
KGaA; dissolved in sterile water) concentrations (0.5, 1, 5, 10 mM)
for 48 h from the onset of hypoxia. The concentrations of lidocaine
used were based on a previous study (26). Cells in the control group were
incubated at 37°C in a humidified atmosphere of 5%
CO2.
Cellular injury assessment
H9c2 cells were treated with lidocaine (0.5, 1, 5,
10 mM) for 48 h under hypoxic conditions before a rat cardiac
troponin (cTnI) ELISA kit (cat. no. CSB-E08594r; Cusabio Technology
LLC) and a rat creatine kinase-muscle/brain (CK-MB) ELISA kit (cat.
no. CSB-E14403r; Cusabio Technology LLC) were used to evaluate cTnI
and CK-MB release into the cell culture medium. Data were presented
as the fold change of the control group (6). During these experiments, the activity
of mitochondria in the different treatment groups was measured
using Mitochondrial Viability Stain (cat. no. ab129732; Abcam)
according to manufacturer's protocol (6).
Cell viability assay
Cell viability was determined using the MTT assay
(Beyotime Institute of Biotechnology). H9c2 cells were cultured in
96-well plates, and ~5,000 cells per well adhered to the culture
dish wall. After incubation of cells with 0.5, 1, 5 and 10 mM
lidocaine for 48 h under hypoxic conditions, MTT (5 mg/ml) was
added to each well. The cells were cultured for a further 4 h
before 100 µl of dimethyl sulfoxide (Sigma-Aldrich; Merck KGaA) was
added per well. Finally, absorbance was measured at 490 nm using an
automated micro-plate reader (BioTek Instruments, Inc.).
Quantitative analysis of
apoptosis
Cell apoptosis was quantified using the FITC-Annexin
V/PI detection kit (Beijing Biosea Biotechnology Co. Ltd.)
according to manufacturer's protocols. Briefly, cells were treated
with lidocaine (0.5, 1, 5, 10 mM) for 48 h under hypoxic
conditions. A total of 1×105 cells were collected from
each sample and resuspended in 200 µl of binding buffer containing
10 µl of FITC-Annexin V. The samples were then incubated at room
temperature for 30 min, and 300 µl of PBS and 5 µl of propidium
idodide (PI) were added. The samples were immediately analyzed
using a flow cytometer (Beckman Coulter, Inc.). FlowJo software
(version 7.2.4; FlowJo LLC) was used to analyze the data and
calculate levels of early and late stage apoptosis.
Western blot analysis
Total protein from H9c2 cells was isolated using
RIPA lysis buffer (Beyotime Institute of Biotechnology). The
protein concentration in whole cell extracts was quantified using a
BCATM Protein Assay kit (Pierce, Thermo Fisher Scientific Inc.).
Protein (0.1 mg) from each sample was separated by 12% sodium
dodecyl sulfate polyacrylamide gel electrophoresis and then
transferred onto a nitrocellulose membrane. The membrane was
blocked with 5% skim milk for 1 h at room temperature and then
probed with primary antibodies: Bcl-2 (cat no. ab196495; 1:1,000;
Abcam), Bax (cat no. 14796; 1:1,000; Cell Signaling Technology,
Inc.), caspase-3 (cat no. 14220; 1:1,000; Cell Signaling
Technology, Inc.), NF-κB p65 (cat no. 8242; 1:1,000; Cell Signaling
Technology, Inc.), phosphorylated (p)-p65 (cat no. 3033; 1:1,000;
Cell Signaling Technology, Inc.), ERK1/2 (cat no. 4695; 1:1,000;
Cell Signaling Technology, Inc.), p-ERK1/2 (cat no. 4376; 1:1,000;
Cell Signaling Technology, Inc.) and β-actin (cat no. 4970;
1:1,000; Cell Signaling Technology, Inc.), at 4°C overnight. The
membranes were then incubated for 1 h at room temperature with the
horseradish peroxidase-conjugated secondary antibody (cat no. 7074;
1:2,000; Cell Signaling Technology, Inc.). Positive bands from each
group of samples were visualized using enhanced chemiluminescent
reagent (ECL Advance Western Blotting Detection Kit; GE Healthcare
Life Sciences) (27). The intensity
of each band was quantified using Image Lab™ Software (version
5.2.1; Bio-Rad Laboratories Inc.).
Reverse transcription quantitative PCR
(RT-qPCR)
Total RNA was collected from cells using
TRIzol® reagent (Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. cDNA was generated
using a Transcriptor First Strand cDNA Synthesis kit (Roche
Diagnostics) according to the manufacturer's protocol. The
temperature protocol for the reverse transcription reaction
consisted of primer annealing at 25°C for 5 min, cDNA synthesis at
42°C for 60 min and termination at 80°C for 2 min. FastStart
Universal SYBR Green Master (Roche Diagnostics) was used to analyze
cDNA levels. The thermocycling conditions were conducted as
follows: Initial denaturation 95°C for 5 min, followed by 40 cycles
of denaturation at 95°C for 15 sec and annealing/elongation at 60°C
for 30 sec. The primer sequences used for qPCR were as follows:
GAPDH forward, 5′-CTTTGGTATCGTGGAAGGACTC-3′ and reverse,
5′-GTAGAGGCAGGGATGATGTTCT-3′; IL-1β forward,
5′-TGTGAAATGCCACCTTTTGA-3′ and reverse, 5′-TGAGTGATACTGCCTGCCTG-3′;
TNF-α forward, 5′-GAACTGGCAGAAGAGGCACT-3′ and reverse,
5′-GGTCTGGGCCATAGAACTGA-3′ and IL-6 forward,
5′-CCGGAGAGGAGACTTCACAG-3′ and reverse, 5′-CAGAATTGCCATTGCACA-3′.
The target gene expression level was normalized to GAPDH. The
2−ΔΔCq method (28) was
used to determine relative gene expression.
ELISA for inflammatory cytokines
Cells were treated with lidocaine (0.5, 1, 5, 10 mM)
for 48 h under hypoxic conditions and the supernatants collected by
centrifugation (500 × g; 5 min; 4°C) for the determination of
levels of the inflammatory cytokines TNF-α (Rat TNF-α ELISA kit;
cat. no. PT516), interleukin (IL)-6 (Rat IL-6 ELISA kit; cat. no.
PI328) and IL-1β (Rat IL-1β ELISA kit; cat. no. PI303). All kits
were obtained from Beyotime Institute of Biotechnology. ELISAs were
performed according to the manufacturer's protocols and repeated
three times.
Statistical analysis
The experimental results were expressed as the mean
± SD from three independent experiments. Experimental data were
analyzed using SPSS 16.0 software (SPSS, Inc.). Statistical
significance was evaluated by one-way analysis of variance followed
by Tukey's test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of lidocaine on hypoxia-induced
cell damage
Rat cardiac myoblast cell line H9c2 was used in this
study and they were used in the present study as a model for
cardiac myocytes. The release of two biomarkers of cardiomyoblast
injury, CK-MB and cTnI, was measured to assess the effect of
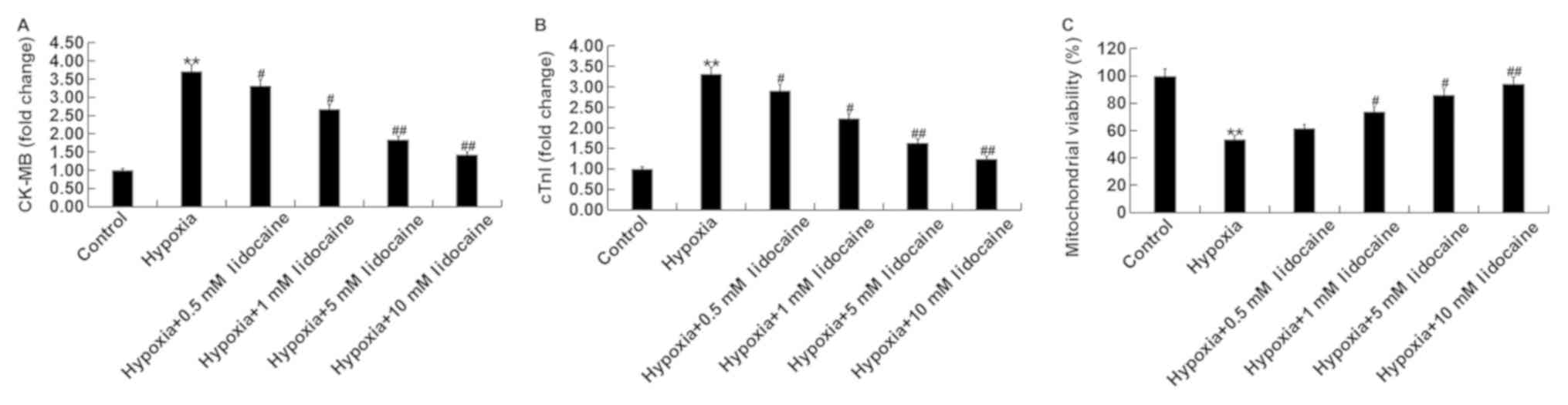
lidocaine on hypoxia-induced cell injury. Fig. 1A and B shows that the expression of
both biomarkers, CK-MB and cTnI, were significantly increased
relative to the control group due to hypoxia. Lidocaine
significantly reduced the hypoxia-induced expression levels of
CK-MB and cTnI in a dose-dependent manner. In addition, the results
showed that hypoxia treatment of H9c2 cells significantly reduced
mitochondrial viability, and lidocaine treatment significantly
increased mitochondrial viability under hypoxic conditions in a
dose-dependent manner relative to the control group (Fig. 1C).
Effect of lidocaine on cell viability
of hypoxic cardiomyoblasts
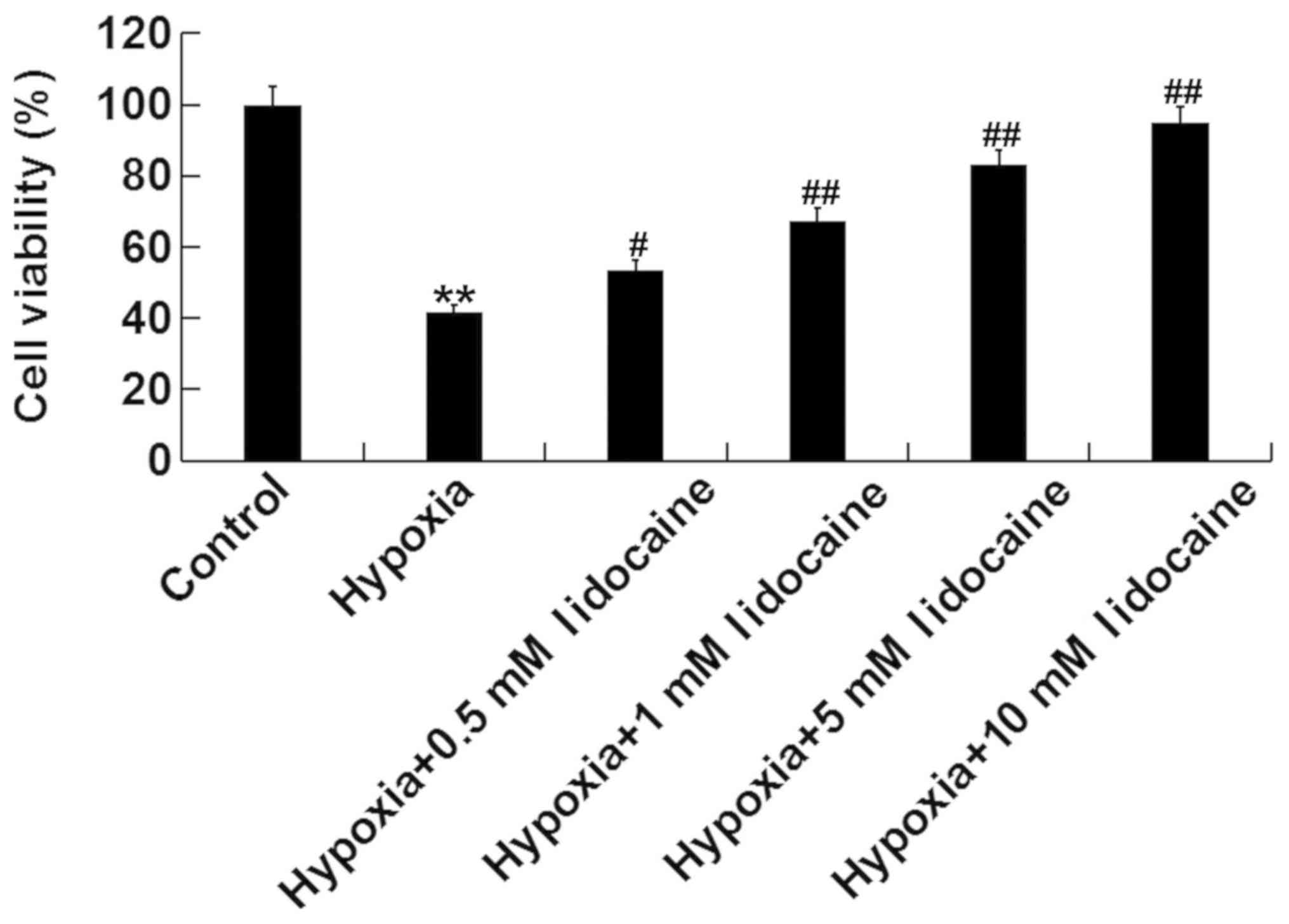
MTT assay was used to determine the effect of
lidocaine on H9c2 cell viability under hypoxia. As shown in
Fig. 2, H9c2 cell viability under
hypoxia was significantly reduced relative to the control group,
but lidocaine could significantly improve H9c2 cell viability under
hypoxia in a dose-dependent manner.
Effect of lidocaine on hypoxia-induced
apoptosis of cardiomyoblasts
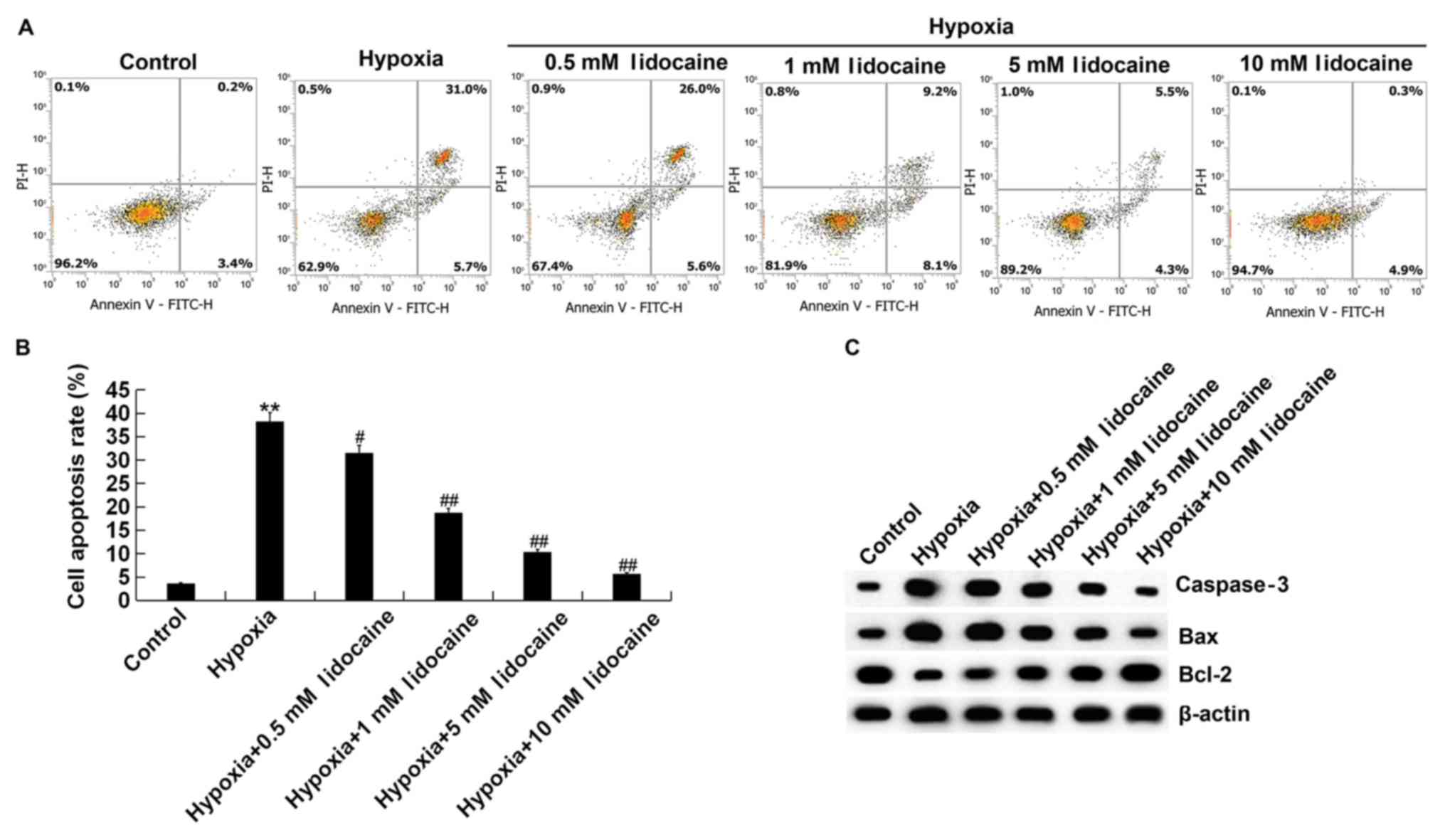
A FITC-Annexin V/PI detection kit was used to study
the effects of lidocaine on hypoxia-induced apoptosis. As shown in
Fig. 3A and B, compared with the
control group, hypoxia significantly induced H9c2 cell apoptosis,
and lidocaine significantly decreased hypoxia-induced H9c2 cell
apoptosis in a dose-dependent manner. To further determine the
protective effect of lidocaine against hypoxia-induced H9c2 cell
apoptosis, the expression levels of apoptosis-related proteins were
determined experimentally. Western blotting showed that
hypoxia-induction markedly increased Bax expression and decreased
Bcl-2 expression relative to the control group, but lidocaine
dose-dependently increased Bcl-2 and decreased Bax expression in
the hypoxic cells. Western blotting was also used to detect the
protein level of caspase-3 in H9c2 cells. Levels of caspase-3 were
markedly increased in hypoxic cells, but were reduced in these
cells after lidocaine treatment in a dose-dependent manner
(Fig. 3C). In summary, the results
showed that lidocaine inhibited hypoxia-induced cardiomyoblast
apoptosis, indicating that the drug had anti-apoptotic effects.
Lidocaine treatment inhibits the
hypoxia-induced inflammatory response in cardiomyoblasts
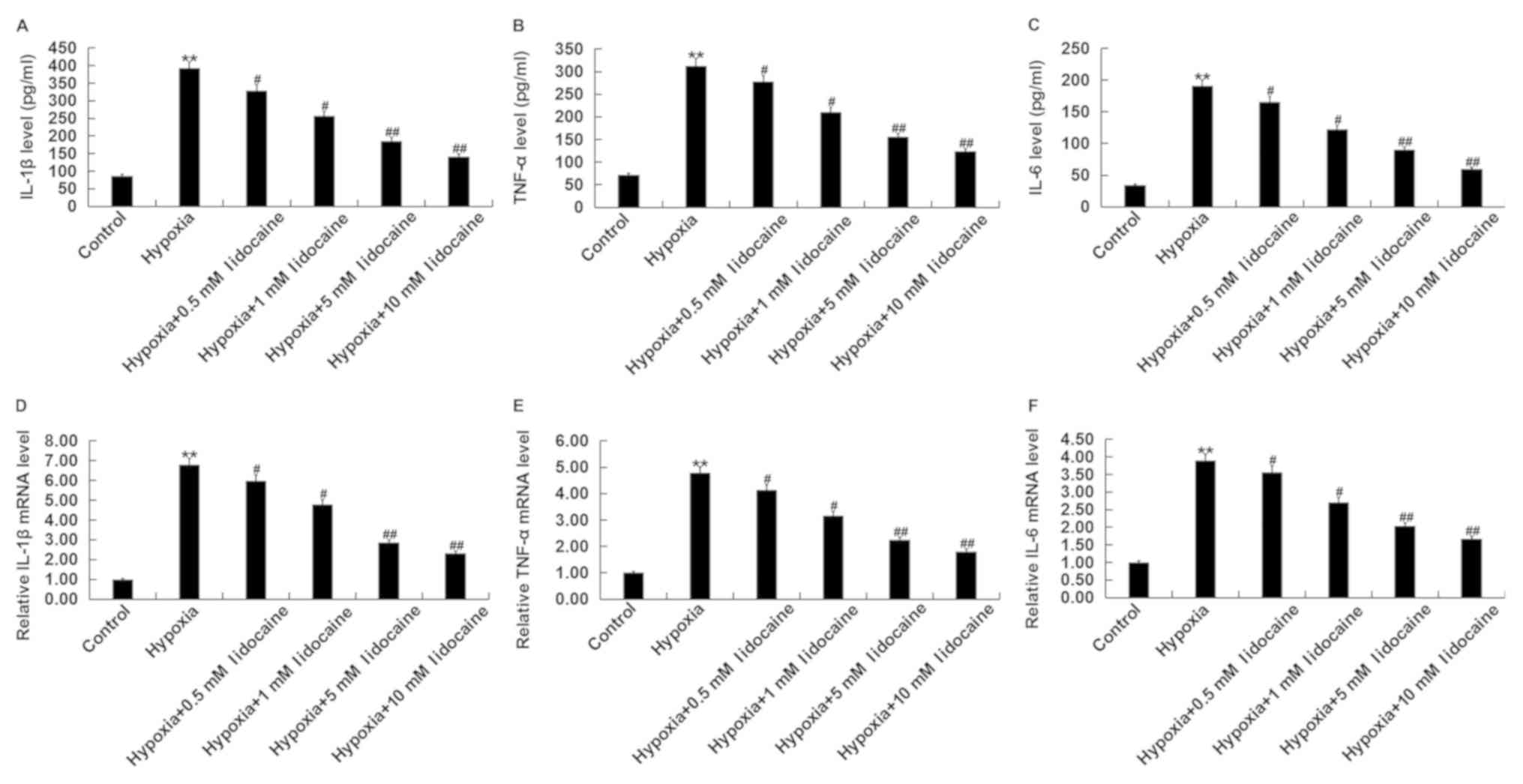
The therapeutic effect of lidocaine on hypoxic
cardiac myocytes was determined by measuring the levels of
inflammatory cytokines IL-1β, TNF-α and IL-6. The results of ELISA
assay showed that the release of inflammatory cytokines increased
significantly in H9c2 cells under hypoxic conditions when compared
with control. Lidocaine treatment significantly decreased the level
of inflammatory cytokines released by hypoxic H9c2 cells in a
dose-dependent manner (Fig. 4A-C).
Similar results were obtained from the RT-qPCR analysis of cytokine
expression at the mRNA level (Fig.
4D-F). Together, these findings indicated that treatment with
lidocaine inhibited the hypoxia-induced inflammatory response in
cardiomyoblasts.
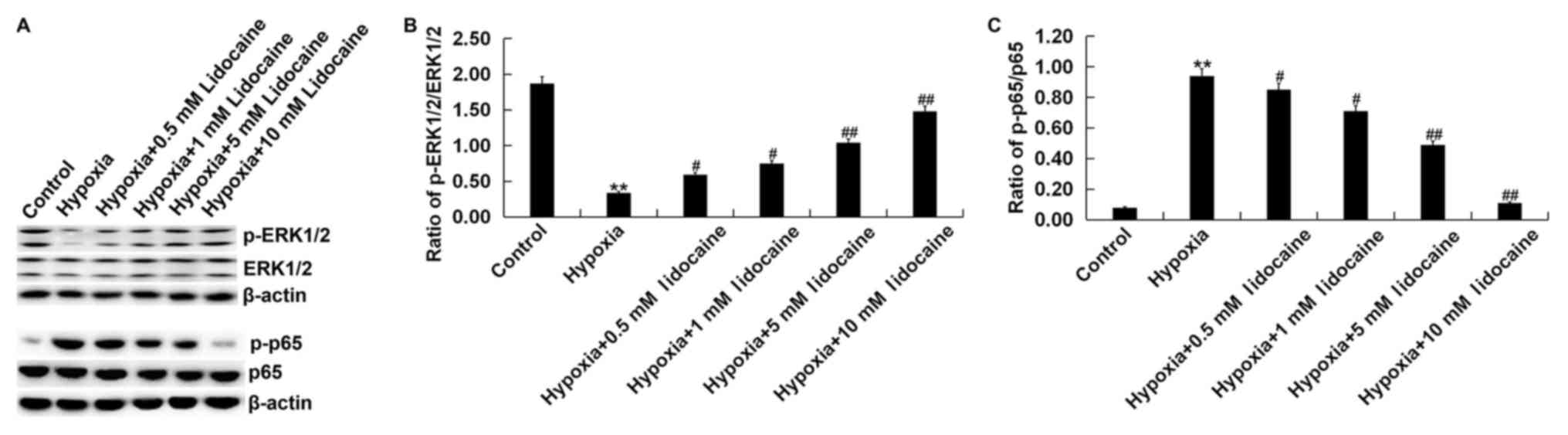
Effect of lidocaine on the
MAPK/ERK/NF-κB signaling pathway in hypoxic cardiomyoblasts
The MAPK/ERK/NF-κB signaling pathway was
investigated in order to demonstrate the potential protective
mechanism of lidocaine against hypoxia-induced cardiomyoblast
injury. Western blot analysis showed that hypoxia inhibited
MAPK/ERK/NF-κB signaling in H9c2 cells, as the cellular
p-ERK1/2/ERK1/2 ratio was downregulated, whilst the p-p65/p65 ratio
was upregulated compared with the control. Hypoxia-induced changes
in expression of these phosphorylated proteins were alleviated by
lidocaine treatment (Fig. 5).
Discussion
MI is pathologically defined as myocardial cell
death caused by prolonged ischemia (29). The two most prominent features that
can enhance each other during MI-induced cardiac injury are the
physiological defects of the ischemic tissue and the sustained
inflammatory response, which ultimately lead to heart failure. One
of the major complications of MI treatment is hypoxia-induced
cardiomyocyte death (30). Previous
studies have shown that protection of cardiomyocytes from
hypoxia-induced injury could lead to a potential treatment for MI
(31,32). The findings of the present study are
consistent with previous studies (31,32).
After 48 h of hypoxia H9c2 cell viability was significantly
impaired and cell apoptosis was induced, indicating that hypoxia
does indeed cause cell damage in vitro.
cTnI and CK-MB are two well-known bio-markers of
myocardial cell damage (6,33). The present study found that after
treatment with lidocaine, the levels of cTnI and CK-MB in
hypoxia-treated cells were significantly reduced when compared with
controls, indicating that treatment with lidocaine reduced
myocardial cell damage. In addition, it was found in the present
study that lidocaine promoted cell viability and inhibited
apoptosis in a dose-dependent manner to protect H9c2 cells from
damage.
Lidocaine is used in the treatment of acute
myocardial infarction and other heart diseases complicated by rapid
ventricular arrhythmia (34). To
evaluate the molecular mechanisms underlying the potential benefits
of lidocaine in MI, hypoxic H9c2 cells were used as a cellular
model of MI. The findings of the present study indicated that a
hypoxic environment significantly inhibited myocardial cell
viability and induced apoptosis, which were reversed by lidocaine
treatment. To further confirm the anti-apoptotic activity of
lidocaine on hypoxia induced H9c2 cells, proteins associated with
apoptosis, including Caspase 3, Bax and Bcl-2, were detected in the
present study. The Bcl-2 family consists of a group of proteins
which regulate apoptosis (35). In
particular, increases in the Bax/Bcl-2 expression ratio has been
reported to activate caspase-3 protease activity in cardiomyocytes,
leading to programmed cell death (36). Consistent with the findings of Zhang
et al (37) and Li et
al (38), the present study
demonstrated that hypoxia-induction markedly increased Bax and
Caspase-3 expression whilst decreasing Bcl-2 expression compared
with the control group. However, lidocaine dose-dependently
increased Bcl-2 and decreased Bax and Caspase-3 expression in
hypoxic H9c2 cells.
Accumulating evidence demonstrates that the
inflammatory response serves critical roles in MI pathogenesis
(5,39). Increased expression of a multiple
endogenous inflammatory cytokines can lead to myocardial
dysfunction. The findings of the present study were consistent with
a previous study (9), which
indicated that hypoxia promotes an inflammatory response in
cardiomyocytes due to increased secretion of the cytokines IL-6,
IL-1β and TNF-α. The results of the present study suggested that
lidocaine treatment might effectively reduce hypoxia-induced
inflammation.
Myocardial ischemia and hypoxia activate several
protein kinase pathways (40).
Increased activation of the p38-MAPK, ERK1/2 and JNK pathways
occurs during ischemia-reperfusion (41). Research has shown that during
myocardial damage, pro-inflammatory cytokine levels are elevated in
a manner that is dependent on NF-κB activation (42). It has been shown that, in treatment
of the ischemic myocardium, activation of the MAPK/ERK/NF-κB
pathway is essential in preventing cardiomyocyte death, which can
be achieved by ischemic post-conditioning or administration of
certain pharmacological agents (6,41,43). The
results of the present study suggested that lidocaine activated the
MAPK/ERK/NF-κB signaling pathway through a significant upregulation
in ERK1/2 phosphorylation and a downregulation of p-p65 levels, and
may protect cells against hypoxia-induced damage.
In summary, the present study highlighted the
protective effect of lidocaine against hypoxia-induced damage to
cardiomyoblasts and suggested a role for lidocaine treatment in MI.
However, this study was only preliminary and further in
vitro and in vivo study with a greater range of
lidocaine concentrations is necessary to confirm these results.
Acknowledgements
The authors would like to thank Dr Gao Bo, director
of the Department of Cardiology, Tianjin Hospital (Tianjin, China)
for the help and guidance he provided.
Funding
No funding was received.
Availability of data and materials
All datasets used and/or generated during the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
HBJ contributed to study design, data collection,
statistical analysis, data interpretation and manuscript
preparation. JY contributed to data collection and statistical
analysis.
Ethical approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Moran AE, Forouzanfar MH, Roth GA, Mensah
GA, Ezzati M, Flaxman A, Murray CJ and Naghavi M: The global burden
of ischemic heart disease in 1990 and 2010: The Global Burden of
disease 2010 study. Circulation. 129:1493–1501. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Boateng S and Sanborn T: Acute myocardial
infarction. Dis Mon. 59:83–96. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lu L, Liu M, Sun R, Zheng Y and Zhang P:
Myocardial Infarction: Symptoms and treatments. Cell Biochem
Biophys. 72:865–867. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Johansson S, Rosengren A, Young K and
Jennings E: Mortality and morbidity trends after the first year in
survivors of acute myocardial infarction: A systematic review. BMC
Cardiovasc Disord. 17:532017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen-Scarabelli C, Saravolatz L Jr, Murad
Y, Shieh WS, Qureshi W, Di Rezze J, Abrencillo R, Gardin T, Gidwani
UK, Saravolatz L, et al: A critical review of the use of carvedilol
in ischemic heart disease. Am J Cardiovasc Drugs. 12:391–401. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gong L, Chang H, Zhang J, Guo G, Shi J and
Xu H: Astragaloside IV protects rat cardiomyocytes from
hypoxia-induced injury by down-regulation of miR-23a and miR-92a.
Cell Physiol Biochem. 49:2240–2253. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yan L, Yang H, Li Y, Duan H, Wu J, Qian P,
Li B and Wang S: Regulator of calcineurin 1-1L protects
cardiomyocytes against hypoxia-induced apoptosis via mitophagy. J
Cardiovasc Pharmacol. 64:310–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Task Force on the management of ST-segment
elevation acute myocardial infarction of the European Society of
Cardiology (ESC), ; Steg PG, James SK, Atar D, Badano LP,
Blömstrom-Lundqvist C, Borger MA, Di Mario C, Dickstein K, Ducrocq
G, et al: ESC Guidelines for the management of acute myocardial
infarction in patients presenting with ST-segment elevation. Eur
Heart J. 33:2569–2619. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yuan M, Zhang L, You F, Zhou J, Ma Y, Yang
F and Tao L: MiR-145-5p regulates hypoxia-induced inflammatory
response and apoptosis in cardiomyocytes by targeting CD40. Mol
Cell Biochem. 431:123–131. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Marchant DJ, Boyd JH, Lin DC, Granville
DJ, Garmaroudi FS and McManus BM: Infammation in myocardial
diseases. Circ Res. 112:126–144. 2012. View Article : Google Scholar
|
|
11
|
Sun M, Dawood F, Wen WH, Chen M, Dixon I,
Kirshenbaum LA and Liu PP: Excessive tumor necrosis factor
activation after infarction contributes to susceptibility of
myocardial rupture and left ventricular dysfunction. Circulation.
110:3221–3228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tare M, Bensley JG, Moss TJ, Lingwood BE,
Kim MY, Barton SK, Kluckow M, Gill AW, De Matteo R, Harding R, et
al: Exposure to intrauterine inflammation leads to impaired
function and altered structure in the preterm heart of fetal sheep.
Clin Sci (Lond). 127:559–569. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mandel I, Podoxenov Y, Suhodolo I,
Podoxenov A, Svirko Y, Kamenschikov N, Mikheev S, Sementsov A,
Dzuman A and Maslov L: Hypoxic and hyperoxic preconditioning in
myocardial protection against ischemia-reperfusion injury:
Experimental study. J Cardiothor Vasc Anes. 30 (Suppl 1):S6–S7.
2016. View Article : Google Scholar
|
|
14
|
Carson IW, Lyons SM and Shanks RG:
Anti-arrhythmic drugs. Br J Anaest. 51:659–670. 1979. View Article : Google Scholar
|
|
15
|
Khan SU, Winnicka L, Saleem MA, Rahman H
and Rehman N: Amiodarone, lidocaine, magnesium or placebo in shock
refractory ventricular arrhythmia: A Bayesian network
meta-analysis. Heart Lung. 46:417–424. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Caracas HC, Maciel JV, Martins PM, de
Souza MM and Maia LC: The use of lidocaine as an anti-inflammatory
substance: A systematic review. J Dent. 37:93–97. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fukuda S, Nojima J, Motoki Y, Yamaguti K,
Nakatomi Y, Okawa N, Fujiwara K, Watanabe Y and Kuratsune H: A
potential biomarker for fatigue: Oxidative stress and
anti-oxidative activity. Biol Psych. 118:88–93. 2016. View Article : Google Scholar
|
|
18
|
Li H, Li C, Zhang H, Zhang L, Cheng R, Li
M, Guo Y, Zhang Z, Lu Z, Zhuang Y, et al: Effects of lidocaine on
regulatory Tcells in atopic dermatitis. J Allergy Clin Immun.
137:613–617.e5. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bundscherer AC, Malsy M, Bitzinger DI,
Wiese CH, Gruber MA and Graf BM: Effects of Lidocaine on HT-29 and
SW480 colon cancer cells in vitro. Anticancer Res. 37:1941–1945.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ye L, Zhang Y, Chen YJ and Liu Q:
Anti-tumor effects of lidocaine on human gastric cancer cells in
vitro. Bratisl Lek Listy. 120:212–217. 2019.PubMed/NCBI
|
|
21
|
Župčić M, Graf Župčić S, Duzel V, Šimurina
T, Šakić L, Fudurić J, Peršec J, Milošević M, Stanec Z, Korušić A
and Barišin S: A combination of levobupivacaine and lidocaine for
paravertebral block in breast cancer patients undergoing
quadrantectomy causes greater hemodynamic oscillations than
levobupivacaine alone. Croat Med J. 58:270–280. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maurice JM, Gan Y, Hibner M and Huang Y:
Bupivacaine causes cytotoxicity via extracellular signal-regulated
kinase (ERK) and Akt pathways in mouse myoblast C2C12 Cells. J
Minim Invas Gyn. 16 (Suppl):S11–S12. 2009. View Article : Google Scholar
|
|
23
|
Müller-Edenborn B, Kania G, Osto E, Jakob
P, Krasniqi N, Beck-Schimmer B, Blyszczuk P and Eriksson U:
Lidocaine enhances contractile function of ischemic myocardial
regions in mouse model of sustained myocardial ischemia. PLoS One.
11:e01546992016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kaczmarek DJ, Herzog C, Larmann J,
Gillmann HJ, Hildebrand R, Schmitz M, Westermann A, Harendza T,
Werdehausen R, Osthaus AW, et al: Lidocaine protects from
myocardial damage due to ischemia and reperfusion in mice by its
antiapoptotic effects. Anesthesiology. 110:1041–1049. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Okamoto A, Sumi C, Tanaka H, Kusunoki M,
Iwai T, Nishi K, Matsuo Y, Harada H, Takenaga K, Bono H and Hirota
K: HIF-1-mediated suppression of mitochondria electron transport
chain function confers resistance to lidocaine-induced cell death.
Sci Rep. 7:38162017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chang YC, Hsu YC, Liu CL, Huang SY, Hu MC
and Cheng SP: Local anesthetics induce apoptosis in human thyroid
cancer cells through the mitogen-activated protein kinase pathway.
PLoS One. 9:e895632014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fantetti KN, Gray EL, Ganesan P, Kulkarni
A and O'Donnell LA: Interferon gamma protects neonatal neural
stem/progenitor cells during measles virus infection of the brain.
J Neuroinflamm. 13:1072016. View Article : Google Scholar
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Choi CW, Hwang JH, Chang YS, Shin SM, Park
WS and Lee M: Effects of alpha-phenyl-N-tert-butyl nitrone (PBN)on
brain cell membrane function and energy metabolism during transient
global cerebral hypoxia-ischemia and reoxygenation-reperfusion in
newborn piglets. J Korean Med Sci. 19:413–418. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vorobieva VV and Shabanov PD: Vibration
model for hypoxic type of cell metabolism evaluated on rabbit
cardiomyocytes. Bull Exp Biol Med. 147:768–771. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Han F, Chen Q, Su J, Zheng A, Chen K, Sun
S, Wu H, Jiang L, Xu X, Yang M, et al: MicroRNA-124 regulates
cardiomyocyte apoptosis and myocardial infarction through targeting
Dhcr24. J Mol Cell Cardiol. 132:178–188. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang J, Qiu W, Ma J, Wang Y, Hu Z, Long
K, Wang X, Jin L, Tang Q, Tang G, et al: miR-27a-5p attenuates
hypoxia-induced rat cardiomyocyte injury by inhibiting Atg7. Int J
Mol Sci. 20(pii): E24182019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Deng F, Wang S, Zhang L, Xie X, Cai S, Li
H, Xie GL, Miao HL, Yang C, Liu X and Xia Z: Propofol through
upregulating caveolin-3 attenuates post-hypoxic mitochondrial
damage and cell death in H9C2 cardiomyocytes during hyperglycemia.
Cell Physiol Biochem. 44:279–292. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Martí-Carvajal AJ, Simancas-Racines D,
Anand V and Bangdiwala S: Prophylactic lidocaine for myocardial
infarction. Cochrane Database Syst Rev. CD0085532015.PubMed/NCBI
|
|
35
|
Daido S, Tamiya T, Ono Y, Terada K,
Mizumatsu S and Ohmoto T: Expression of Bcl-2, Bcl-x and Bax
proteins in astrocytomas in relation to patient survival. Biochem
Biophys Res Commun. 18:123–129. 2001.
|
|
36
|
Tamatani M, Ogawa S, Nuñez G and Tohyama
M: Growth factors prevent changes in Bcl-2 and Bax expression and
neuronal apoptosis induced by nitric oxide. Cell Death Differ.
5:911–919. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang J, Xia Y, Xu Z and Deng X: Propofol
suppressed Hypoxia/Reoxygenation-induced apoptosis in HBVSMC by
regulation of the expression of Bcl-2, Bax, Caspase3, Kir6.1, and
p-JNK. Oxid Med Cell Longev. 2016:15187382016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li H, Lv B, Kong L, Xia J, Zhu M, Hu L,
Zhen D, Wu Y, Jia X, Zhu S and Cui H: Nova1 mediates resistance of
rat pheochromocytoma cells to hypoxia-induced apoptosis via the
Bax/Bcl-2/caspase-3 pathway. Int J Mol Med. 40:1125–1133. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Frangogiannis NG: The inflammatory
response in myocardial injury, repair, and remodelling. Nat Rev
Cardiol. 11:255–265. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ferdinandy P, Schulz R and Baxter GF:
Interaction of cardiovascular risk factors with myocardial
ischemia/reperfusion injury, preconditioning, and postconditioning.
J Pharmacol Rev. 59:418–458. 2007. View Article : Google Scholar
|
|
41
|
Milano G, Morel S, Bonny C, Samaja M, von
Segesser LK, Nicod P and Vassalli G: A Peptide inhibitor of c-Jun
NH2-terminal kinase reduces myocardial ischemia-reperfusion injury
and infarct size in vivo. Am J Physiol Heart Circ Physiol.
292:H1828–H1835. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wei H, Li H, Wan SP, Zeng QT, Cheng LX,
Jiang LL and Peng YD: Cardioprotective effects of Malvidin against
isoproterenol-induced myocardial infarction in rats: A mechanistic
study. Med Sci Monit. 23:2007–2016. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen Y, Ba L, Huang W, Liu Y, Pan H,
Mingyao E, Shi P, Wang Y, Li S, Qi H, et al: Role of carvacrol in
cardioprotection against myocardial ischemia/reperfusion injury in
rats through activation of MAPK/ERK and Akt/eNOS signaling
pathways. Eur J Pharmacol. 796:90–100. 2017. View Article : Google Scholar : PubMed/NCBI
|