Introduction
Lung cancer is the leading cause of cancer-related
mortality in both men and women (1). Non-small cell lung cancer (NSCLC)
represents 80% of lung cancers, and most patients are diagnosed
with stage IIIB and IV disease. Five-year survival rates for these
patients remain below 10%. Current treatments, including
chemotherapy, radiotherapy and surgery, have provided only limited
improvement in the natural history of the disease (2). This dismal clinical and
epidemiological picture underscores the need for novel treatment
strategies to target this aggressive disease.
Over the last decade, the epidermal growth factor
receptor (EGFR) has emerged as one of the most important signaling
components involved in cell growth and survival. EGFR and its
family members, ErbB2, ErbB3 and ErbB4, are receptor tyrosine
kinases which send signals into the cell to regulate many critical
processes including development, tissue homeostasis, and
tumorigenesis (3). The binding of
a ligand (e.g. EGF) to the extracellular region of the EGFR induces
receptor dimerization and activation of the intracellular region.
The intracellular tyrosine kinase domain then phosphorylates
several tyrosine residues of the receptor and relays the signal to
downstream signaling pathways (4).
Extracellular signal-regulated kinase (ERK)1/2, which is one of the
three major groups of mitogen-activated protein kinases (MAPKs) in
mammals, is activated by the EGFR tyrosine kinase and plays an
essential role in cell proliferation. The phosphatidylinositol
3-kinase (PI3K)/Akt pathway which is activated by the EGFR has been
implicated in both cell proliferation and survival (5). Furthermore, recent large-scale
retrospective analyses have reported that EGFR is overexpressed in
62% of NSCLCs (6). EGFR is
implicated in the development and progression of the majority of
common human epithelial cancers; therefore, various agents have
been developed to block EGFR activation in cancer cells. Small
molecule kinase inhibitors targeting the ErbB family have been the
subject of intensive drug development and clinical trials in cancer
therapy (7). Gefitinib
(Iressa®) and erlotinib (Tarceva®) are
members of a class of quinazolium-derived agents that inhibit the
EGFR pathway by binding in a reversible fashion to the EGFR ATP
pocket domain (8). Both agents are
approved for treatment of patients with advanced NSCLC and have
provided hope for better survival. However, their efficacy is still
limited, predominantly due to drug resistance (9). Despite initial and sometimes dramatic
responses of specific NSCLC cases to EGFR TKIs, nearly all patients
develop resistance and relapse.
Recently, the T790M mutation (10,11)
and amplification of MET (12)
were identified as two main mechanism of acquired resistance.
Although together they account for approximately 50% of cases with
acquired resistance, the mechanism involved in the remaining 50% of
cases remains unknown (13).
The type I insulin-like growth factor receptor
(IGF-1R) signaling pathway is another important growth-regulatory
pathway that is prevalent in a variety of cancer types, including
NSCLC (14). IGF-1R is a
heterotetrameric receptor (two extracellular 125-kDa α chains and
two transmembrane 95-kDa β chains) that auto-phosphorylates after
ligand binding and activates several downstream signaling routes,
including the PI3K and MAPK pathways. Signaling through IGF-1R
stimulates proliferation, promotes angiogenesis and metastasis, and
inhibits apoptosis (15,16). Elevated IGF-1R expression and
activity have been associated with multiple aspects of cancer
progression including enhanced carcinogenesis, tumorigenesis,
metastasis, resistance to chemotherapeutics and other molecularly
targeted drugs and to transformation (17). Major signaling pathways activated
by the IGF-1R and EGFR include the PI3K and MAPK pathways. Because
these receptors appear to be so similar in their signaling
mechanisms, it raises the possibility that IGF-1R signaling may be
involved in tumor resistance to EGFR-TKIs. Investigations to date
have also demonstrated that persistent activation of PI3K/Akt
signaling is involved in acquired resistance to EGFR TKIs. In this
study, we evaluated the effects of a combined treatment of the
IGF-1R inhibitor AG1024 with gefitinib on the human non-small cell
lung cancer PC9/G cell line with acquired resistance to gefitinib,
and investigated the possible mechanisms through inhibition of
IGF-1R.
Materials and methods
Cell lines and culture
Human non-small lung cancer cell lines PC9 and PC9/G
were kindly provided by Dr Caicun Zhou, Department of Oncology,
Shanghai Pulmonary Disease Hospital affiliated with Tongji
University, Shanghai, China. Both were cultured in RPMI-1640 medium
(Gibco-BRL, Grand Island, NY, USA) containing 10% fetal bovine
serum (FBS), 1% penicillin -streptomycin (GIBCO, Invitrogen) at
37°C in a humidified atmosphere with 5% CO2 and 95% air.
Subcultures were produced by trypsinization and were reseeded for
experiments.
Cell proliferation assay and combination
index (CI)
The proliferative activity of PC9 and PC9/G cells
for the different treatments was assessed using the Cell Counting
Kit-8 (CCK-8) (Dojindo, Japan). Cells were plated in 96-well plates
at 2,000 cells/well in complete medium and cultured for 24 h. The
media were then replaced with RPMI-1640, 1% FBS with or without
inhibitors. Each condition in each experiment was studied in five
replicate wells. After incubation for 72 h, 10 μl of CCK-8
was added to each well, and the plates were further incubated for 4
h in an incubator. The absorbance at 450 nm was read
spectrophotometrically using a microplate reader. The combination
effect was evaluated by the CCK-8 assay. CI values <1, 1 and
>1 indicated synergism, additive effect and antagonism,
respectively.
Apoptosis assay
Cell apoptosis was determined by the Annexin V
fluorescein isothiocyanate (FITC) Apoptosis Kit I (BD Biosciences,
Franklin Lakes, NJ, USA) according to the manufacturer’s protocol.
Briefly, after treatment with different inhibitors for 24 h, cells
were harvested, washed with PBS and resuspended in binding buffer.
Cells were stained with Annexin V-FITC and 5 μl propidium
iodide (PI), and incubated for 15 min at room temperature in the
dark. Binding buffer 1X (400 μl) was added, and the cells
were analyzed by flow cytometry (Beckman Coulter).
Western blot analysis
After treatment with the different inhibitors for 24
h, cells were harvested and washed with ice-cold PBS, lysed in
lysis buffer at 0°C for 10 min, and centrifuged. Briefly, the
lysates containing 30 μg protein were electrophoresed on 10%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) polyacrylamide gels and transferred to PVDF membranes.
Antibodies were obtained from the following sources: EGFR
phospho-antibody (py1068) was purchased from Epitomics, Inc.
(Burlingame, CA, USA), EGF receptor rabbit antibody, Akt rabbit
antibody, phospho-Akt (Ser473) mouse mAb, p44/42 MAPK (Erk1/2)
rabbit antibody and P-p44/42MAPK (Thr202/Tyr204) rabbit antibody
were purchased from Cell Signaling Technology (Boston, MA, USA).
All target proteins were immunoblotted with appropriate primary and
horseradish peroxidase-conjugated secondary antibodies.
Immunoreactive bands were visualized by the Enhanced
Chemiluminescence system (CWBIO Biotechnology, China). One Step
Western Kit HRP (rabbit) and One Step Western Kit HRP (mouse) were
purchased from CWBIO Biotechnology. β-actin was used as an internal
control.
Statistical analysis
The experiments were repeated three times. Data are
expressed as means ± SD. For comparison of two groups, the
two-tailed, paired t-test was used. A value of P<0.05 was
considered statistically significant. Statistical tests were
performed using SAS 13.0 (SAS Institute, Cary, NC, USA).
Results
Acquired resistance of PC9/G cells to
gefitinib
The PC9 cells were induced to mutate by the mutagen
N-methyl-N’-nitro-N-nitroso-guanidine (MNNG) and then selected. The
resistant cell line to gefitinib, PC9/G, was obtained by limited
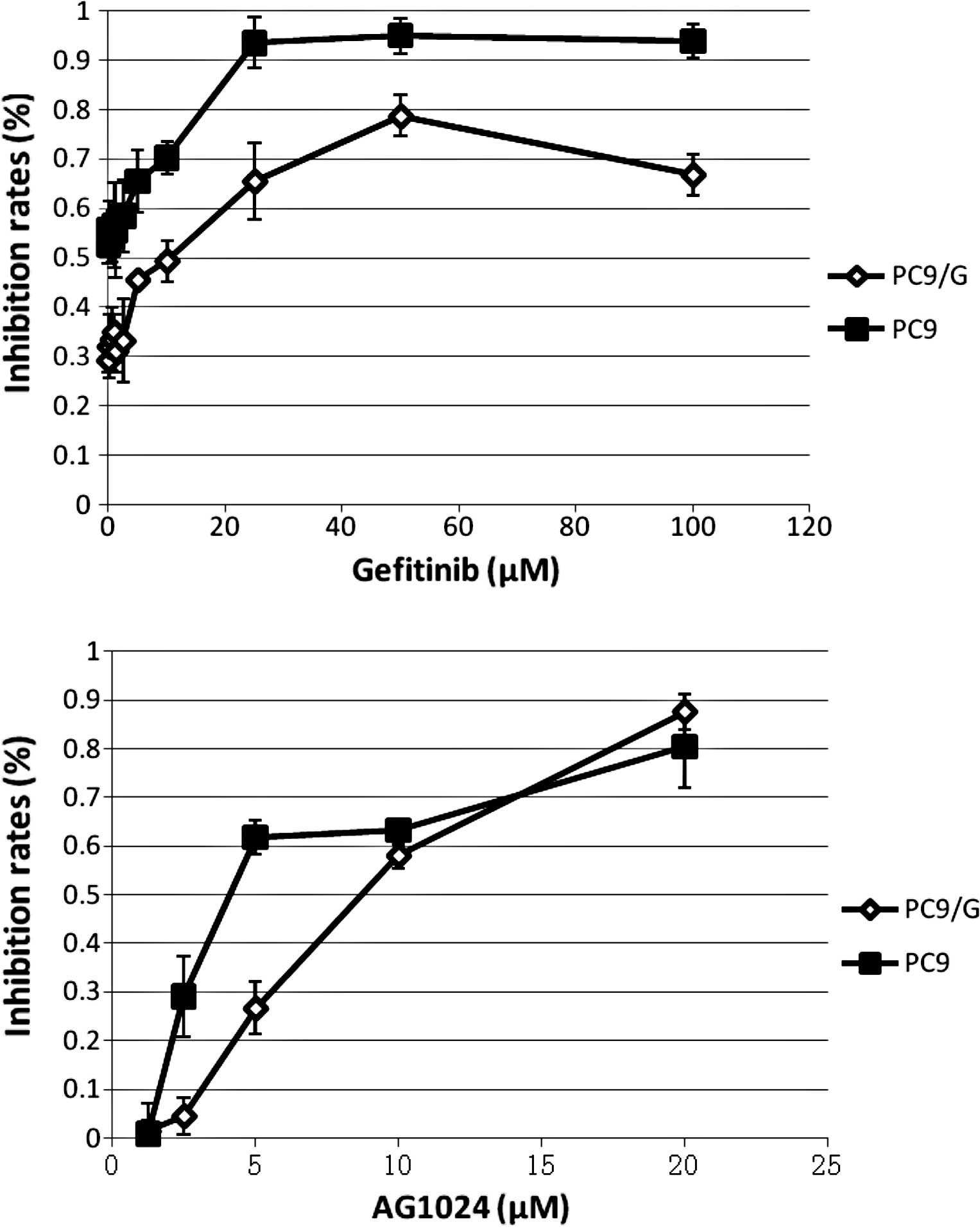
dilution. The sensitivity and IC50 for gefitinib and
AG1024 were determined by the CCK-8 assay in the PC9 and PC9/G
cells. Cells were treated with various concentrations of gefitinib
and AG1024 for 72 h. IC50 values for gefitinib in the
PC9 and PC9/G cells were 0.04±0.02 and 9.66±3.14 μmol/l,
respectively. PC9/G cells showed a more than 100-fold higher
IC50 for gefitinib than the parental cells. In addition,
IC50 values for AG1024 in the PC9 and PC9/G cells were
5.10±0.60 and 7.56±0.63 μmol/l, respectively, indicating
that the PC9/G cells were also more resistant to AG1024 than the
PC9 cells.
AG1024 enhanced the growth inhibitory
effects of gefitinib in the PC9/G cells
Both agents inhibited cell proliferation to
different degrees (Fig. 1). To
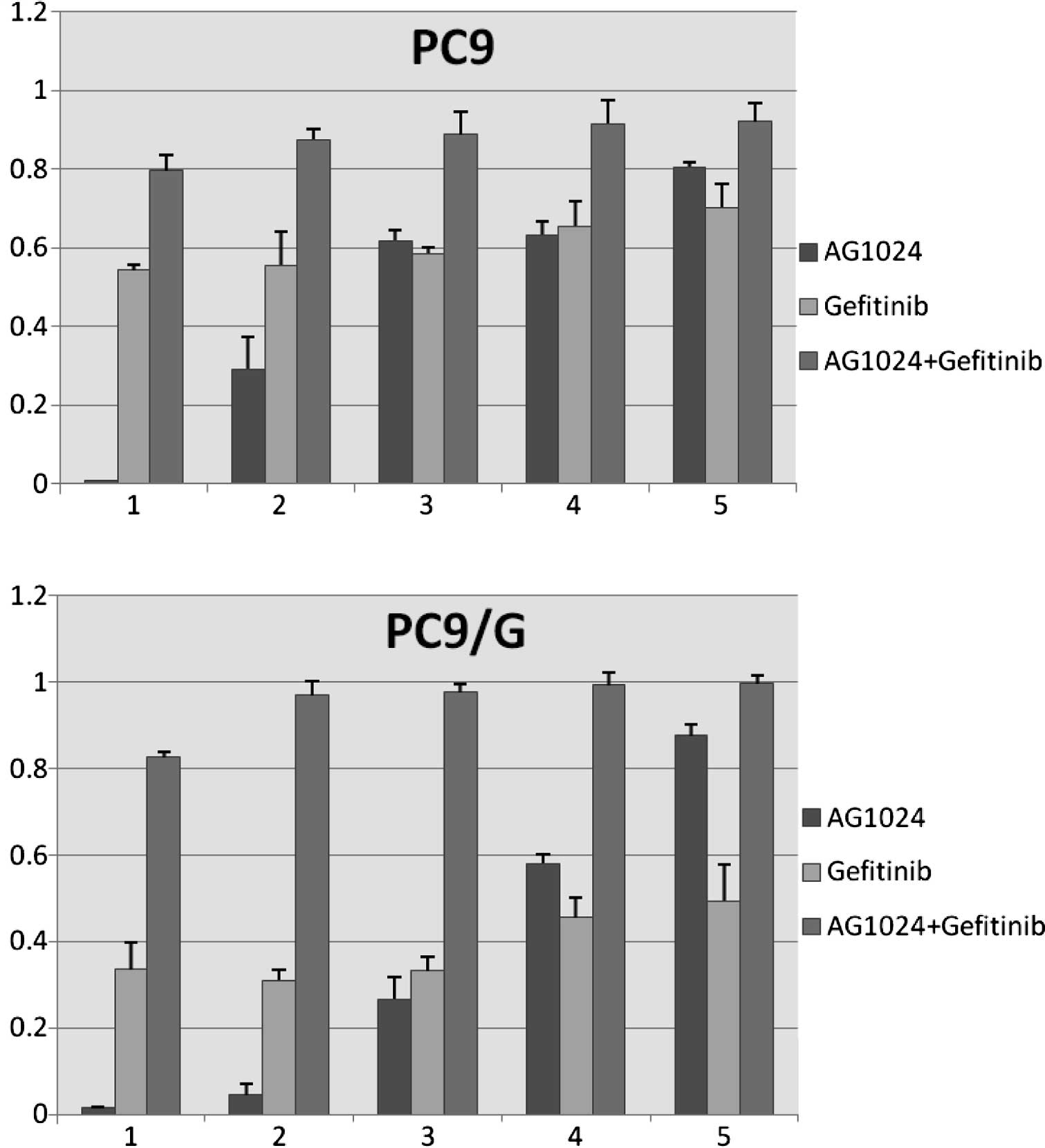
evaluate the cell growth inhibitory effect of the combination of
AG1024 and gefitinib, we treated both cells with gefitinib and
AG1024 for 72 h at a concentration ratio of 1:2. The combined
treatment revealed that this co-targeting approach achieved a
greater growth inhibition (Fig.
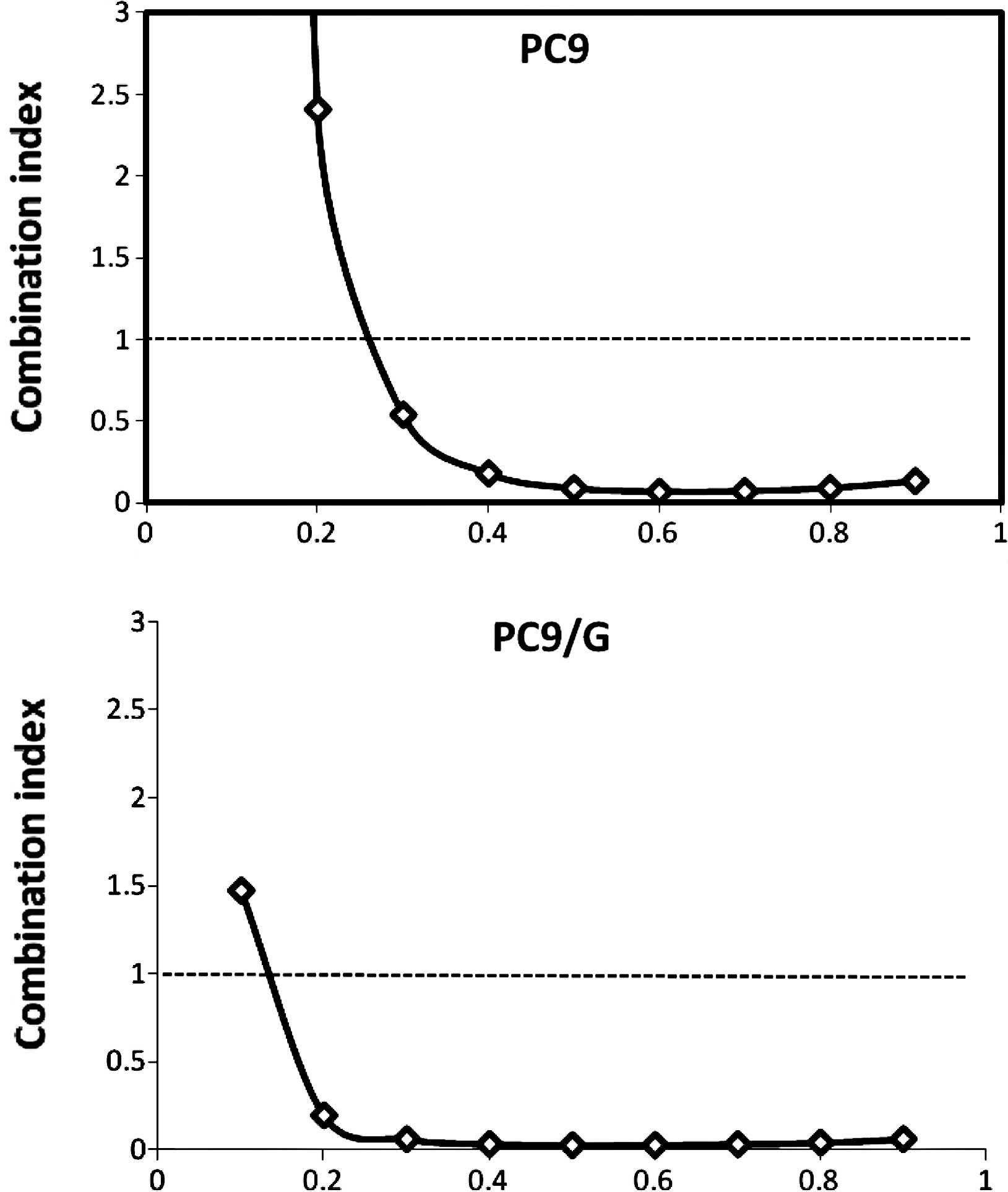
2). The combination index (CI) values were determined using the
Chou and Talalay method (18), a
well-established mathematical analysis to determine the
pharmacologic interaction between two drugs. We evaluated the
interactions between inhibitors as additive (CI=1), antagonistic
(CI>1) or synergistic (CI<1). CI values significantly <1
in PC9 (CI=0.092) and PC9/G (CI=0.022) cells at the 50% inhibition
level were observed, indicative of a synergistic effect. A similar
synergestic effect was observed over the entire range of tested
concentrations (Fig. 3).
Adding an anti-IGF-1R strategy to
gefitinib treatment increases the levels of apoptosis
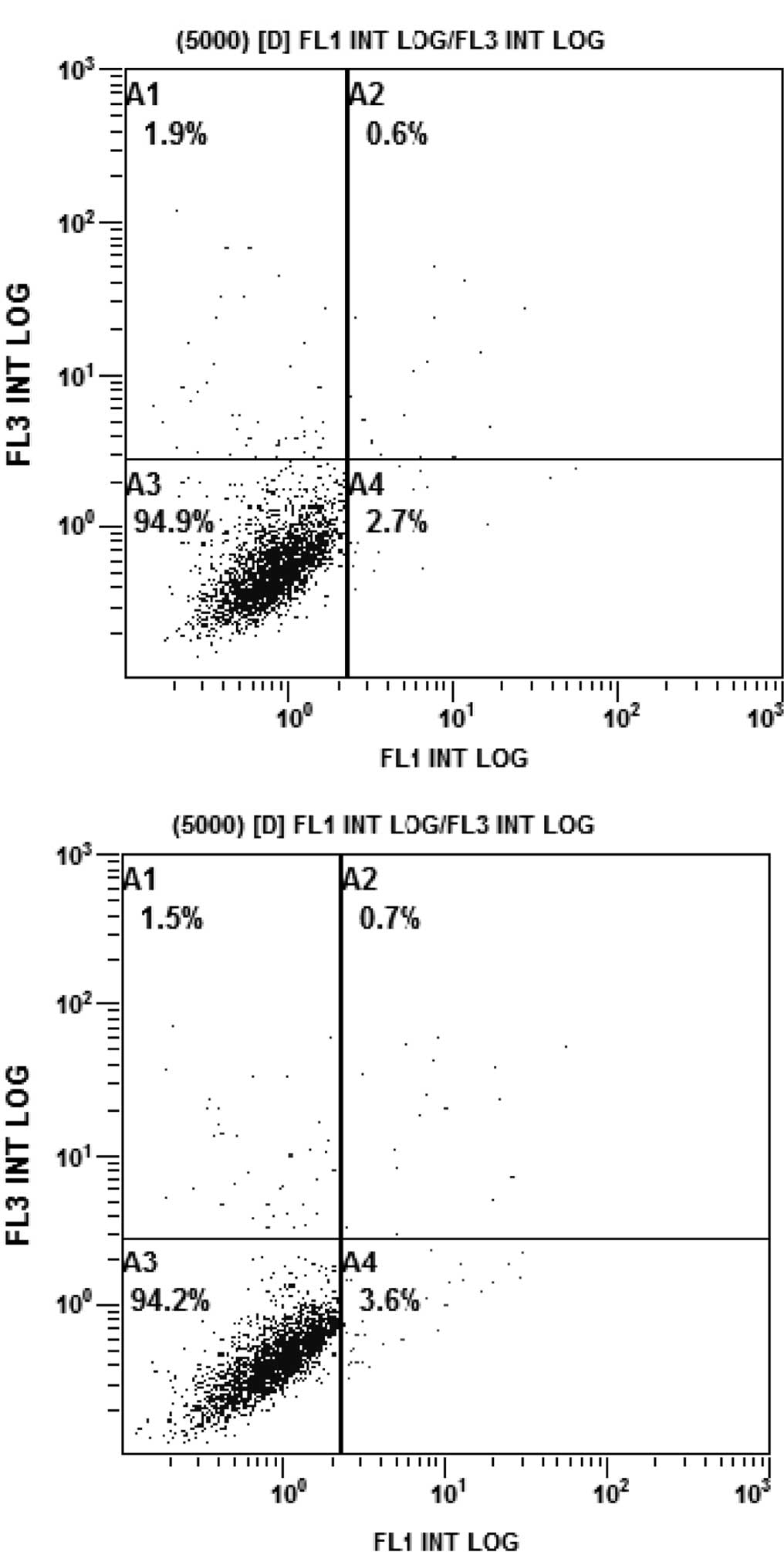
The rates of apoptosis of the PC9/G cells in the
different treatment groups were analyzed by flow cytometry. PC9/G
cells were treated with AG1024 (10 μmol/l) and gefitinib (5
μmol/l) for 24 h, alone or in combination. Consistent with
the results of the CCK-8 assay, the PC9/G cells displayed apoptotic
features after treatment with AG1024 and gefitinib alone. Addition
of AG1024 to gefitinib significantly increased the levels of
apoptosis in the PC9/G cells (Fig.
4).
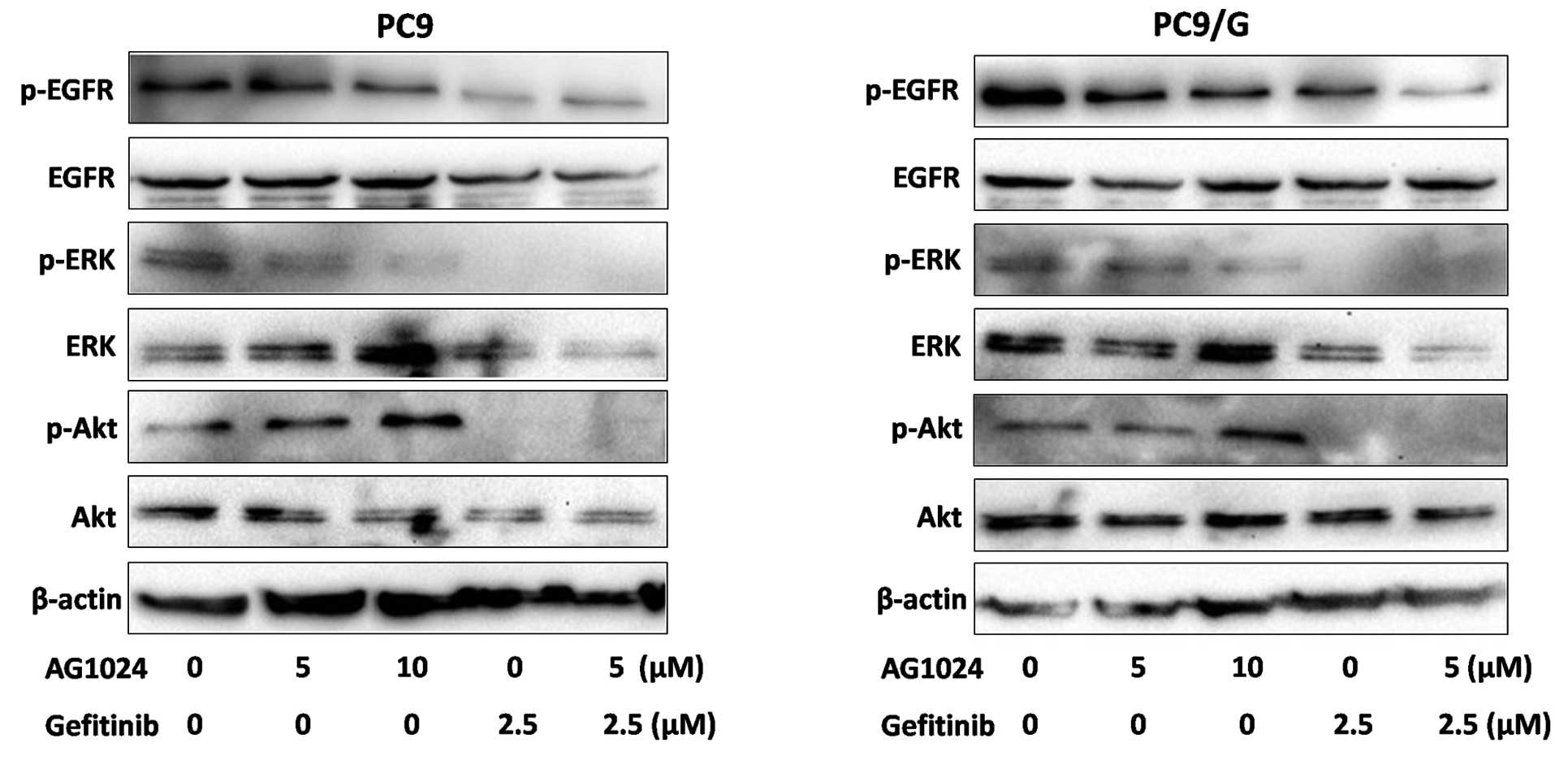
Blockade of IGF-1R in combination with
gefitinib inhibits the expression of p-Akt and p-ERK in PC9/G
cells
To examine the effect of AG1024 and gefitinib on the
EGFR downsteam signaling pathway, we measured the level of
phosphorylation and expression of signaling molecules by Western
blotting (Fig. 5). After
treatments for 24 h, phosphorylation levels of EGFR were decreased
by gefitinib in the parental cells, while only moderately affected
in the resistant cells. Both agents reduced the expression of
p-ERK, but AG1024 was unable to reduce the expression of p-Akt.
When treated with AG1024 and gefitinib in combination, the
phosphorylation of Akt and ERK was completely abolished.
Discussion
Despite remarkable advances in oncology medicine,
the prognosis of lung cancer patients has not greatly improved over
the past few decades. One of the most significant developments in
cancer research in recent years has been the clinical validation of
molecularly targeted drugs that inhibit the action of pathogenic
tyrosine kinases. Treatment of appropriately selected patients with
these drugs can alter the natural history of their disease and
improve survival (7). Beneficial
responsiveness to these EGFR tyrosine kinase inhibitors in patients
with NSCLC was found to be closely associated with EGFR mutations
such as del746–750 and L858R in the kinase domain (19,20).
In the present study, we established gefitinib-resistant NSCLC
cells from the PC9 cell line, which harbors the delE746-A750
mutation in EGFR exon 19 and is highly sensitive to gefitinib, by
inducing mutation with the mutagen MNNG. The gefitinib-resistant
cells were subsequently selected for the experiments. A subclone of
the gefitinib-resistant cell line was obtained by limited dilution,
and its sensitivity for gefitinib was determined by the CCK-8
assay. We found that PC9/G cells showed more than a 100-fold higher
IC50 for gefitinib than the parental cells. A previous
study showed that the gene expression profile was also
significantly different between PC9/G cells and the parental cells,
as determined by a DNA microarray (21). Gene function annotation and
grouping showed that fatty acid metabolism and oxidative
phosphorylation-related genes were down-regulated, while glycolysis
genes were up-regulated in this cell line. Moreover, the
insulin-like receptor and positive regulative factors for the NF-κB
cascade were up-regulated. These result suggest that the resistance
to gefitinib may be due to activation of an alternative signaling
pathway and the downstream molecules of the signaling pathway.
The IGF-1R and its associated signaling system has
provoked considerable interest over recent years as a novel
therapeutic target in cancer (22). Two classes of IGF-1R inhibitors are
currently under clinical development for cancer therapy: mAbs and
small molecule tyrosine kinase inhibitors, such as AG1024. In our
study, gefitinib and AG1024 used as single agents showed
antiproliferative activity in PC9 and PC9/G cells, and their
combination resulted in a synergistic enhancement of growth
inhibition. The mechanism of this action is associated with
reversible G1 arrest induced by gefitinib (23). The antiproliferative effect was
mainly found to be cytostatic, but high doses of the drug are
needed to induce apoptosis in normal mammary epithelial cells and
primary cultures of mammary carcinoma cells (24). Inhibiting the IGF-1R pathway by the
addition of AG1024 improves the induction of apoptosis in PC9/G
cells at a level higher than that upon treatment with gefitinib
alone.
In addition, Western blot analysis showed that both
gefitinib and AG1024 affect phosphorylation levels of ERK to
different degrees, but that AG1024 is unable to reduce
phosphorylation levels of Akt. Combination treatment induces a
further reduction in the activation of the Akt and ERK signaling
pathways downstream of EGFR. Notably, PC9 cells expressed high
levels of phosphorylated and total EGFR, which were inhibited by
gefitinib. In contrast, the phosphorylation levels of EGFR were
slightly affected by the treatment in PC9/G cells. Therefore, we
suggest that the maintenance of this survival pathway may be
related to acquired resistance to gefitinib. IGF-1R signaling can
activate downstream signaling pathways such as ERK/MAPK and
PI3K/Akt, which are also modulated by EGFR (25). A previous study has suggested that
crosstalk and interaction between the IGF-1R and EGFR is important,
and clear evidence exists for the involvement of constitutive
activation of the PI3K/Akt signaling pathway in lung carcinogenesis
and resisitance to tyrosine kinase inhibitors (26). For example, EGFR-amplified cells
with loss of PTEN exhibit resistance to EGFR inhibitors, even
though inhibiting EGFR led to down-regulation of ERK signaling
(27).
Some studies, for example in NSCLC and breast
cancer, have suggested that IGF-1R expression is associated with
improved survival, suggesting that the relationship of the IGF-1R
system to outcome is more complex than initially thought (28). The mechanism of the IGF-1R pathway
involvement in cellular resistance to gefitinib remains unclear.
Further development of rational clinical strategies require greater
clarification of the key signaling factors which are potential
targets for cancer therapies (29). The data presented here support
further research into NSCLC therapeutic strategies combining
gefitinib with anti-IGF-1R agents.
In conclusion, the present study suggests that the
IGF-1R pathway contributes to the acquired resistance of gefitinib
in NSCLC. Combination therapy with AG1024 and gefitinib markedly
inhibited the growth of PC9/G cells and showed a synergistic effect
in inducing apoptosis. Combined blockade of EGFR and IGF-1R
signaling may be considered as a new effective therapeutic approach
for NSCLC patients to overcome gefitinib resistance.
Abbreviations:
|
NSCLC,
|
non-small cell lung cancer;
|
|
IGF-1R,
|
insulin-like growth factor-1
receptor;
|
|
EGFR,
|
epidermal growth factor receptor;
|
|
ERK,
|
extracellular signal-regulated
kinase;
|
|
PI3K,
|
phosphatidylinositol 3-kinase
|
References
|
1
|
Jemal A, Murray T, Samuels A, Ghafoor A,
Ward E and Thun MJ: Cancer statistics, 2003. CA Cancer J Clin.
53:5–26. 2003. View Article : Google Scholar
|
|
2
|
Carney DN: Lung cancer - time to move on
from chemotherapy. N Engl J Med. 346:126–128. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bose R and Zhang X: The ErbB kinase
domain: structural perspectives into kinase activation and
inhibition. Exp Cell Res. 315:649–658. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Citri A and Yarden Y: EGF-ERBB signalling:
towards the systems level. Nat Rev Mol Cell Biol. 7:505–516. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Takeuchi K and Ito F: EGF receptor in
relation to tumor development: molecular basis of responsiveness of
cancer cells to EGFR-targeting tyrosine kinase inhibitors. FEBS J.
277:316–326. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hirsch FR, Varella-Garcia M, Bunn PA Jr,
et al: Epidermal growth factor receptor in non-small-cell lung
carcinomas: correlation between gene copy number and protein
expression and impact on prognosis. J Clin Oncol. 21:3798–3807.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baselga J: Targeting tyrosine kinases in
cancer: the second wave. Science. 312:1175–1178. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wakeling AE, Guy SP, Woodburn JR, et al:
ZD1839 (Iressa): an orally active inhibitor of epidermal growth
factor signaling with potential for cancer therapy. Cancer Res.
62:5749–5754. 2002.PubMed/NCBI
|
|
9
|
Van der Veeken J, Oliveira S, Schiffelers
RM, Storm G, van Bergen En Henegouwen PM and Roovers RC: Crosstalk
between epidermal growth factor receptor- and insulin-like growth
factor-1 receptor signaling: implications for cancer therapy. Curr
Cancer Drug Targets. 9:748–760. 2009.PubMed/NCBI
|
|
10
|
Pao W, Miller V, Zakowski M, et al: EGF
receptor gene mutations are common in lung cancers from ‘never
smokers’ and are associated with sensitivity of tumors to gefitinib
and erlotinib. Proc Natl Acad Sci USA. 101:13306–13311. 2004.
|
|
11
|
Kobayashi S, Boggon TJ, Dayaram T, et al:
EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung cancer
by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yao Z, Fenoglio S, Gao DC, et al: TGF-beta
IL-6 axis mediates selective and adaptive mechanisms of resistance
to molecular targeted therapy in lung cancer. Proc Natl Acad Sci
USA. 107:15535–15540. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sachdev D and Yee D: The IGF system and
breast cancer. Endocr Relat Cancer. 8:197–209. 2001. View Article : Google Scholar
|
|
15
|
Camirand A, Zakikhani M, Young F and
Pollak M: Inhibition of insulin-like growth factor-1 receptor
signaling enhances growth-inhibitory and proapoptotic effects of
gefitinib (Iressa) in human breast cancer cells. Breast Cancer Res.
7:R570–R579. 2005. View
Article : Google Scholar
|
|
16
|
Pollak MN, Schernhammer ES and Hankinson
SE: Insulin-like growth factors and neoplasia. Nat Rev Cancer.
4:505–518. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Knowlden JM, Jones HE, Barrow D, Gee JM,
Nicholson RI and Hutcheson IR: Insulin receptor substrate-1
involvement in epidermal growth factor receptor and insulin-like
growth factor receptor signalling: implication for gefitinib
(‘Iressa’) response and resistance. Breast Cancer Res Treat.
111:79–91. 2008.PubMed/NCBI
|
|
18
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: the combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lynch TJ, Bell DW, Sordella R, et al:
Activating mutations in the epidermal growth factor receptor
underlying responsiveness of non-small-cell lung cancer to
gefitinib. N Engl J Med. 350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Paez JG, Jänne PA, Lee JC, et al: EGFR
mutations in lung cancer: correlation with clinical response to
gefitinib therapy. Science. 304:1497–1500. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Su B, Su CX, Zhang HP, Dun QF, Zhao YM and
Zhou CC: Selection and establishment of gefitinib-resistant PC9
cell line and its gene expression profile. Tumor. 28:552–557.
2008.
|
|
22
|
Hewish M, Chau I and Cunningham D:
Insulin-like growth factor 1 receptor targeted therapeutics: novel
compounds and novel treatment strategies for cancer medicine.
Recent Pat Anticancer Drug Discov. 4:54–72. 2009. View Article : Google Scholar
|
|
23
|
Busse D, Doughty RS, Ramsey TT, et al:
Reversible G(1) arrest induced by inhibition of the epidermal
growth factor receptor tyrosine kinase requires up-regulation of
p27(KIP1) independent of MAPK activity. J Biol Chem. 275:6987–6995.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ciardiello F, Caputo R, Bianco R, et al:
Antitumor effect and potentiation of cytotoxic drug activity in
human cancer cells by ZD-1839 (Iressa), an epidermal growth factor
receptor-selective tyrosine kinase inhibitor. Clin Cancer Res.
6:2053–2063. 2000.
|
|
25
|
Jorissen RN, Walker F, Pouliot N, Garrett
TP, Ward CW and Burgess AW: Epidermal growth factor receptor:
mechanisms of activation and signalling. Exp Cell Res. 284:31–53.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Balsara BR, Pei J, Mitsuuchi Y, et al:
Frequent activation of AKT in non-small cell lung carcinomas and
preneoplastic bronchial lesions. Carcinogenesis. 25:2053–2059.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bianco R, Shin I, Ritter CA, et al: Loss
of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells
counteracts the antitumor action of EGFR tyrosine kinase
inhibitors. Oncogene. 22:2812–2822. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cappuzzo F, Toschi L, Tallini G, et al:
Insulin-like growth factor receptor 1 (IGFR-1) is significantly
associated with longer survival in non-small-cell lung cancer
patients treated with gefitinib. Ann Oncol. 17:1120–1127. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dobashi Y, Koyama S, Kanai Y and Tetsuka
K: Kinase-driven pathways of EGFR in lung carcinomas: perspectives
on targeting therapy. Front Biosci. 16:1714–1732. 2011. View Article : Google Scholar : PubMed/NCBI
|