Contents
Introduction
The p53 signaling pathway is activated by
various cellular stresses
p53-dependent cancer therapy via the
restoration of p53 function
p53-independent cancer therapy
Conclusion
Introduction
HNSCC is the sixth most common type of cancer
worldwide. More than 49,000 new cases of HNSCC cancer are predicted
to have occurred in the US in 2010 (1). The disease is multifactorial in its
pathogenesis and is associated with smoking, alcohol and infection
with the human papillomavirus (HPV) (2). However, the abrogation of p53
function is one of the most common molecular alterations in human
cancer cells, including HNSCC (3–5). The
prognosis for patients with tumors bearing p53 mutations is
often worse than for those with tumors lacking a wild-type
p53 (wtp53) gene (6). For predictive assays, which can be
used to evaluate prognosis in cancer therapy, the genetic status of
the p53 gene is one of the most critical candidates among
various cancer-related genes (7).
In addition, the spectrum of p53 deletions or mutations
observed among tumor cells suggests that the mutations vary in
their prognostic power. Disruptive p53 mutations in tumor
DNA are reported to be associated with reduced survival following
surgical treatment of HNSCC (2).
It has been previously reported that the radio-,
heat- and chemo-sensitivities of HNSCC cells are
p53-dependent, and are closely correlated with induction of
apoptosis in vitro (8–10)
and in vivo (11–13). Consequently, the restoration of
wtp53 function and p53-independent therapy have been
developed as therapeutic strategies to target tumors with abrogated
wtp53 functions.
In this review, cancer therapies aimed at targeting
signaling pathways controlled by p53 are described. These include
p53-gene therapy, chemical chaperones, p53 C-terminal
peptides and small molecules that can target p53. In addition,
therapeutic strategies independent of p53 status in cancer
cells are discussed. These include high-linear energy transfer
(LET) heavy-ion radiation, and enhancement of cancer therapies with
other strategies, including an RNA-silencing therapy targeted at
DNA repair pathways, and a molecular-targeting therapy for the
survival pathway Akt-mTOR.
The p53 signaling pathway is activated by
various cellular stresses
The p53 protein was identified in simian virus 40
(SV40) transformed cells where it is associated with the large T
antigen (14), and was initially
considered to be an oncogene. Subsequently, the p53 gene was
revealed to be mutated in various human tumors (15), while its protein product was
reported to act as a tumor suppressor (16). p53-deleted and
p53-mutated cells make up approximately 50% of the cells in
advanced cancers (17).
p53 is normally in ‘standby’ mode. The p53 protein
is a powerful transcription factor and plays a pivotal role in the
pathway controlling apoptosis, cell growth and cell proliferation
in response to cellular stresses. These include genotoxic and
non-genotoxic stresses, such as DNA damage, hypoxia, oncogene
overexpression and viral infection (18–20).
The p21/WAF1 (wtp53 activated fragment 1) gene
product, a p53 target gene, inactivates the proliferating cell
nuclear antigen (PCNA), which can regulate DNA replication
(21), and induce a
p53-dependent G1 arrest through the inhibition of cyclin/CDK
activity (22,23). During cell cycle arrest,
p53-regulated pathways, including those involving growth
arrest and DNA damage inducible 45 (Gadd45) and the p53
ribonucleotide reductase small subunit 2 (p53R2), are
significant in the repair of damaged DNA (24,25).
In the absence of competent repair activity, DNA damage induces
apoptosis through interactions with other genes in
p53-regulated pathways, including the Bcl-2-associated X
protein (Bax) (26),
p53-upregulated modulator of apoptosis (PUMA) (27), Fas/APO-1 (28) and p53-activated gene 608 (PAG608)
(29). By contrast, the
p53-regulated MDM2 (murine double minute 2) (30) functions to produce negative
feedback, which regulates p53 activity (26).
In the presence of cellular stresses, p53 is
subjected to a complex and diverse array of covalent
post-translational modifications. These include phosphorylation
(31), acetylation (32), poly(ADP-ribosyl)ation (33), ubiquitylation and sumoylation
(34,35). In response to cellular stress,
Ser15/Ser20 in p53 are phosphorylated and MDM2 is separated from
the phosphorylated p53, leading to the stabilization and activation
of p53 (36–38). Therefore, p53 can bind to the
promoter of the p21 or p53R2 genes associated with
DNA repair, and induce their expression. However, if there are
numerous DNA lesions or too much cellular damage, G1 arrest and DNA
repair will not be successful. In this situation, p53 can be
phosphorylated at Ser46, and bind to the promoter of the
p53-regulated apoptosis-inducing protein 1 (p53AIP1) gene,
leading to apoptosis (39,40). Moreover, PUMA is reported to be
required not only for p53-dependent apoptosis induced by DNA
damage, hypoxia and oncogenes, but also for apoptosis induced by
p53-independent stimuli, including serum withdrawal,
glucocorticoids, kinase inhibitors and phorbol esters (27). By contrast, p53 molecules are
inactivated and degraded by activated MDM2 molecules, which are
phosphorylated at multiple sites by other protein kinases. In
addition, p53 is reported to bind to other proteins, including heat
shock proteins (HSPs), functioning as stress proteins (41,42),
and Bcl-X (43). Consequently,
these modifications of p53 molecules can regulate or affect the
fate of cells following exposure to stresses, including cancer
therapies.
p53-dependent cancer therapy via the
restoration of p53 function
Recently, Poeta et al reported an association
between a p53 mutation in a patient with HNSCC and survival
following surgical treatment (2).
The results demonstrated that p53-deleted and
p53-mutated HNSCC patients were significantly associated
with short survival periods. These data indicate that p53
mutations could be a useful evaluation or stratification factor in
prospective clinical trials. However, in the study, chemotherapy
was administered only as an adjuvant measure in combination with
postoperative radiation therapy, or prior to study entry in a few
cases. There are no data on tumor response to chemotherapy. It
would be clinically useful to determine whether p53
mutations are associated with a response to treatments that attack
p53-specific pathways. A study described that sensitization to
radiation, heat and chemical therapies was observed in cells
containing wtp53, but not in cells containing mutated
p53 (mp53) in vitro and in vivo
(8–10). Furthermore, in attempts to treat
cancer using more than one treatment modality, a synergistic
depression of tumor growth was found only in tumors containing
wtp53 (44). These findings
suggest that hyperthermic enhancement of tumor growth inhibition
with irradiation may result in p53-dependent apoptosis via
caspase-3 activation in HNSCC cells. Therefore, an analysis of
p53-gene status in cancer cells could be considered as a
useful predictive assay for estimating the possible effectiveness
of combined therapies involving radiation, heat and anti-cancer
agents. Thus, it is very reasonable to enhance p53-dependent
apoptosis pathways through the restoration of p53 function even for
mp53 HNSCC cells as a more effective therapeutic strategy. A
number of approaches have been employed to achieve this outcome
(illustrated in Fig. 1).
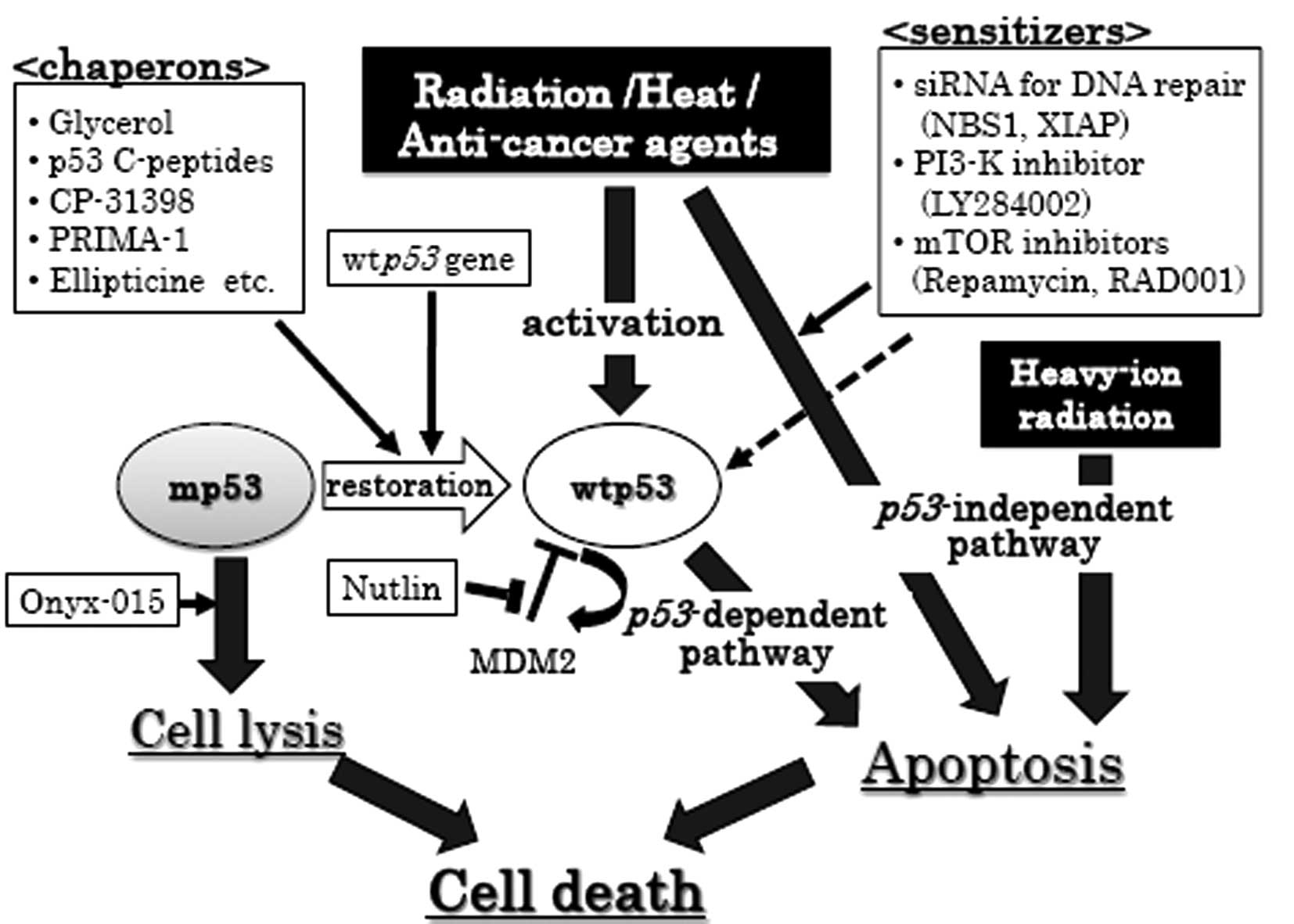 | Figure 1.p53-dependent and -independent
therapeutic strategies for cancer cells. Circles, p53 status
of cancer cells; black squares, cancer therapeutic tool; white
squares, enhancer for cancer therapeutic tool; thin arrows,
enhancement; dashed arrows, partial enhancement; thick arrows,
therapeutic pathway. MDM2, murine double minute 2; XIAP,
X-chromosome-linked inhibitor of apoptosis protein; wtp53,
wildtype p53; mp53, mutant p53; siRNA, small interference RNA. |
A p53 gene therapy-based approach
As previously mentioned, the activation of
endogenous wtp53 by radiation and/or chemotherapy in
wtp53 cancer cells leads to p53-mediated apoptosis.
In recent years, the introduction of exogenous wtp53 into
cancer cells, either by gene delivery or by direct protein
delivery, has been explored. Although preliminary studies in cell
cultures and in animal models have indicated the effectiveness and
the low toxicity of these approaches (45–47),
their efficacy in clinical trials is currently controversial.
Clinical studies in lung, bladder, ovarian and breast cancer
revealed the absence of additional beneficial effects compared to
conventional treatments (48). On
the other hand, encouraging results were reported for phase I and
II clinical trials on 135 patients with advanced HNSCC. In this
study, patients were treated with a combination of recombinant
adenovirus-p53 (Gendicine) administration and radiotherapy.
The results demonstrated that 64% of the patients achieved complete
regression, and 32% achieved partial regression. No serious side
effects were observed (49).
Although such results are encouraging, further improvements in
methods are required to accomplish the safe and effective delivery
of wtp53 in vivo (50).
Onyx-015, an adenovirus based
therapy
In the absence of wtp53 activity in cancer cells,
the generation of a mutated viral vector for tumor cell lysis
(Onyx-015) was exploited. McCormick et al from Onyx
Pharmaceuticals hypothesized that an adenovirus with the E1B
region deleted could only replicate and generate cellular lysis in
cells lacking functional p53, due to the putative requirement for
p53 inactivation for adenoviral replication. Accordingly, the
Onyx-015 reagent, a p53-targeting oncolytic mutant adenovirus, has
been developed for clinical application (51). However, evaluation of numerous
clinical trials performed thus far have indicated that the
administration of Onyx-015 as a single agent produces only a
marginal benefit, whereas its administration in combination with
conventional therapy is more effective (52).
Chaperones for the restoration of the p53
molecule
Glycerol
Another approach in preclinical development involves
restoring tumor-suppressing function to mp53. Studies have
demonstrated that glycerol, as a chemical chaperone, can restore
normal p53 function in mp53 HNSCC cells (53). Glycerol has been previously
reported to act as a chemical chaperone due to its ability to
refold proteins and restore normal activity; this type of activity
was able to alter or restore a functional protein conformation from
conformation forms found in a human disease state (54,55).
Consistent with this observation, glycerol is capable of restoring
p53-dependent radiosensitivity in mp53-HNSCC cells
via Bax-mediated induction of apoptosis (53,56,57).
Glycerol can also restore heat-induced p53-dependent
apoptosis in A-172/mp53 cells (58) and CDDP-induced tumor growth
inhibition in 8305c (59) and
SAS/mp53 cells (13), by
binding to p53 consensus sequences (p53CON) located upstream of
p53-regulated genes. These results suggest that glycerol
could be effective in causing conformational changes that restore
wtp53 functioning to mp53, leading to enhanced results with radio-,
hyperthermic- and/or chemo-therapies through the induction of
apoptosis via a restored wtp53 function.
p53 C-terminal peptides. One of the
significant features of p53 tumor-suppressor activity is its
ability to bind to p53CON; the majority of mutations in p53
are localized in the binding domain of p53CON (60,61).
This sequence-specific DNA-binding ability of p53 is allosterically
regulated by its C-terminal domain (62,63),
and can be activated in vitro by C-terminal truncation or
anti-p53 monoclonal antibody PAb421 binding, which recognizes a
C-terminal epitope (64). In
addition, small peptides corresponding to the C-terminal residues
369–383 of p53 are capable of activating latent p53, and permit
specific DNA-binding in vitro (65). Thus, the sequence-specific DNA
binding activity of mp53 proteins could be rescued by PAb421
(62,66,67).
The inhibition of cell proliferation (68) and the induction of apoptosis
(69) were effectively induced in
mp53 cancer cells transfected with C-terminal peptides following
X-ray-irradiation or heat-treatment (70,71).
CP-31398 and other small
molecules
In other studies evaluating binding to p53CON
sequences, CP-31398 was identified as a small molecule with the
ability to restore the wild-type conformation to mp53 protein by
stabilizing the active conformation of the DNA binding domain
(72,73). Further studies have confirmed that
CP-31398 treatment causes: i) stabilization of wtp53 levels; ii)
apoptosis-related changes; and iii) induction of p53 target genes.
Moreover, CP-31398 was demonstrated to increase the levels of wtp53
protein by inhibiting the MDM2-mediated ubiquitylation and
degradation of p53 (73). The
observation that CP-31398 stabilizes wtp53 suggests that CP-31398
interacts with newly synthesized p53 molecules in vivo and
changes its folding behavior (74). Subsequently, PRIMA-1 (75) and ellipticine (76) were also found to be capable of
inducing mp53-dependent cell death. On the other hand,
Nutlin was developed to rescue wtp53 from degradation
mediated by MDM2 (77). More
recently, p53 family members could be activated, were
capable of serving as substitutes for p53 in tumor cells, and were
able to induce cell death. These observations may provide a novel
tool for the correction of mp53 conformation and loss of function,
and may be applicable to p53-targeted cancer therapy.
p53-independent cancer therapy
High-LET heavy-ion radiation
High-LET charged particle radiation has several
potential advantages over X-rays, including an excellent dose
distribution, a higher relative biological effectiveness (RBE), a
reduction in the oxygen enhancement ratio, less variation in cell
cycle-related radiosensitivity, and the existence of less efficient
repair mechanisms for cellular radiation injury (78–80).
As a result, high-LET charged particle radiation could have serious
lethal effects, even on radioresistant tumors (81). Heavy ion beams can also allow a
high radiation dose to be delivered to a tumor with minimal
irradiation of the surrounding normal tissues. High-LET radiation
also induces apoptosis effectively regardless of the genetic status
of the p53 gene in cancer cells (82,83).
Heavy ion radiotherapies consequently appear to be attractive for
use in numerous types of human cancer. The lack of a
p53-regulated pathway is a common feature in a large number
of tumors, suggesting that it is a significant factor in the
pathogenesis of human cancers. As previously reported, cellular
sensitivity to radiation and/or heat depends on the p53-gene
status in HNSCC cells (9,18) and other cells (82,84).
Therefore, the aim of a number of studies has been to induce
apoptosis by reinstating normal functioning of the mp53 gene
(58). However, it has not been
practical in the clinic to monitor the status of the p53
gene or other useful genetic markers. Thus, attention has been
given to therapies using high-LET radiation, which have highly
lethal effects on radioresistant tumors (81), and which can induce apoptosis
effectively regardless of the p53-gene status (82,83).
It has been suggested that high-LET radiation delivered to HNSCC
cells may enhance apoptosis through the activation of caspase-3
through caspase-9, even in the presence of mp53 (85).
RNA-silencing therapy targeting DNA
repair pathway
RNA interference has become a valuable tool for the
selective suppression of the expression of a target gene. The mRNAs
produced by a targeted gene are cleaved by an RNA-induced silencing
complex, which includes small interference RNAs (siRNAs) and a
nuclease. Cell cycle signaling or DNA repair proteins, including
ATM, ATR and DNA-PKcs, have become the targets of interest in
investigations involving the siRNA-mediated enhancement of
radiation sensitivity (86,87).
Studies have demonstrated the potential use of siRNA as a novel
radiation sensitizer for improving the effectiveness of radiation
therapy in cancer.
The NBS1 protein is essential for the initial
processing of the DNA double-strand break via the homologous
recombination (HR) repair pathway (88,89).
NBS1 forms a complex with MRE11 and RAD50 in the nucleus. This
complex binds to ATM-phosphorylated γH2AX and is recruited to the
area surrounding damaged DNA ends (90). MRE11/RAD50/NBS1 complexes can be
visualized in the form of foci in an irradiated area of the nucleus
(91). Studies indicate that NBS1
appears to regulate radiation sensitivity in cells through its role
in the HR repair system. In addition, it has recently been
demonstrated that the siRNA-mediated reduction of NBS1 appears to
lead to an increase in radiation-induced mutagenesis in human cells
(92).
Ionizing radiation induces a signaling pathway which
activates the transcription factor NF-κB. NF-κB then regulates the
transcriptional activation of a number of genes involved in cell
proliferation, angiogenesis, metastasis and the suppression of
apoptosis (93). Therefore, the
radiation-induced activation of NF-κB could promote oncogenesis and
resistance to cancer therapy (94,95).
Moreover, it is possible that the NBS1 protein may play a role in
the NF-κB pathway, which is activated by radiation; NBS1-deficient
cells exhibit a defective activation of NF-κB following exposure to
radiation (96). Thus, inhibition
of NBS1 could result in depression of NF-κB activation and in the
transcriptional activation of NF-κB-regulated genes. One of the
proteins regulated by NF-κB, X-chromosome-linked inhibitor of
apoptosis protein (XIAP), plays a pivotal role in cancer
progression, and is a strong candidate among cancer therapy targets
(97).
Sensitization to radiation results from
NBS1-siRNA mediated suppression of DNA repair functions and
X-ray-induced cell survival signaling pathways, which operate
through NF-κB and XIAP (98). NBS1
is also involved in the heat-induced cellular responses to DNA
damage, and it has been suggested that NBS1-siRNA is a
potential candidate for a sensitizer for heat treatments, which
could be effective regardless of cellular p53-gene status
(99,100). Moreover, siRNA that can target
XIAP can lead to an effective enhancement of X-ray-induced
apoptosis in human cancer cells with mp53 (98,101). The results described in these
studies suggest that siRNA designed to target DNA repair functions
could lead to novel methods that could increase radiation and/or
heat sensitivity, even in human mp53-bearing cancer
cells.
Molecular-targeting therapy for
Akt-mTOR pathway
The PI3K/Akt pathway is a major cell survival
pathway and plays a critical role in oncogenesis and tumor cell
growth (102). Recent studies
have reported that Akt activation contributes to resistance to
radiation, chemotherapy and tyrosine kinase inhibitors by promoting
survival signals, which protect cancer cells from undergoing
apoptosis (103,104). The inhibition of PI3K/Akt through
pharmacological or genetic means induces anti-proliferative effects
in HNSCC cells in vitro and in vivo (105,106). Akt is activated by heat and
radiation through a phosphatidylinositol-3-kinase (PI3K)-mediated
phosphorylation pathway (107).
Radio-sensitization induced by LY294002, a specific inhibitor of
PI3K, has been reported in in vitro (108) and in vivo experiments
(109). LY294002 inhibits
anti-apoptosis signaling through the induction of Hsp27 and Hsp72,
and cell survival signaling through Akt and survivin. LY294002
appears to be a noteworthy candidate as a p53-independent
heat sensitizer in hyperthermic cancer therapy (110).
The mammalian target of rapamycin, mTOR, is a
289-kDa serine-threonine kinase, which acts as a downstream
effector for Akt (111). It
regulates key processes, including cell growth and proliferation,
cell cycle progression and protein translation through two distinct
pathways; one involving the ribosomal p70S6 kinase (p70S6K), and
one involving the eukaryotic translation initiation factor 4E
(eIF4E) binding proteins (4E-BPs) (112). Akt activation is closely
associated with the upregulation of mTOR activity. It has been
suggested that dysregulation of mTOR contributes to cancer
progression (111), and
therefore, mTOR may be a potential therapeutic target which could
inhibit or block the PI3K/Akt pathway. Inhibitors of mTOR are
currently under development; rapamycin and its derivatives CCI-770,
AP23573 and RAD001. The anti-proliferative effects of mTOR
inhibitors have been observed in various tumor cells in
vitro and in vivo (113–115). These mTOR inhibitors are
generally regarded as cytostatic agents, since they induce G1 cell
cycle arrest, but not apoptosis (113). However, recent studies have
demonstrated that mTOR inhibitors can enhance the cytotoxic effects
of chemotherapeutic agents and radiation in a number of human
cancers (116–118). Furthermore, a study has
demonstrated that rapamycin in combination with radiation was able
to augment the cytostatic effects of radiation regardless of
cellular p53-gene status in lung cancer and HNSCC cells,
suggesting that inhibition of the mTOR signaling may be a promising
strategy for radio-sensitization regardless of p53-gene
status, with respect to cell lethality and cell growth depression
(119).
Conclusion
Over the past few decades, despite the introduction
of new multimodal therapies, there has been a failure to achieve a
high efficacy in tumor therapy. This may be caused by a primary or
acquired resistance to the DNA damaging agents used in chemotherapy
and radiotherapy, and remains a formidable and poorly understood
problem. This review discussed the effect of p53-targeting
cancer therapies on p53 signaling pathways, including
p53-gene therapy, chemical chaperones, p53 C-terminal
peptides, p53-targeting chemicals and inhibitors targeting several
signaling pathways, in an effort to induce cell death in cancer
cells. High-LET radiation can induce apoptosis effectively
regardless of the genetic status of the p53-gene in cancer
cells. Identification of the p53 status in the target cells
is imperative, and the knowledge of additional oncogenic events
contributing to specific types of cancer could significantly aid in
the selection of appropriate therapeutic protocols. The restoration
of p53 functioning could be helpful when pathways upstream of p53
expression are defective, but not if defects are downstream of p53
signaling. The re-expression and re-activation of p53 in human
cancer cells could increase tumor susceptibility to radiation or
chemotherapy, enhance the efficacy of standard therapeutic
protocols, and lead to individually designed therapies (52). Further investigations in this area
will hopefully lead to more effective cancer treatments in the near
future.
Acknowledgements
This work was supported in part by
Grants-in-Aid for Scientific Research from the Ministry of
Education, Culture, Sports, Science and Technology of Japan.
References
|
1.
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300
|
|
2.
|
Poeta ML, Manola J, Goldwasser MA, et al:
TP53 mutations and survival in squamous-cell carcinoma of the head
and neck. N Engl J Med. 357:2552–2561. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Guimaraes DP and Hainaut P: TP53: a key
gene in human cancer. Biochimie. 84:83–93. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Gasco M and Crook T: The p53 network in
head and neck cancer. Oral Oncol. 39:222–231. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Lowe SW: Cancer therapy and p53. Curr Opin
Oncol. 7:547–553. 1995. View Article : Google Scholar
|
|
7.
|
Velculescu VE and El-Deiry WS: Biological
and clinical importance of the p53 tumor suppressor gene. Clin
Chem. 42:858–868. 1996.PubMed/NCBI
|
|
8.
|
Ota I, Ohnishi K, Takahashi A, et al:
Transfection with mutant p53 gene inhibits heat-induced apoptosis
in a head and neck cell line of human squamous cell carcinoma. Int
J Radiat Oncol Biol Phys. 47:495–501. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Takahashi A: Different inducibility of
radiation- or heat-induced p53-dependent apoptosis after acute or
chronic irradiation in human cultured squamous cell carcinoma
cells. Int J Radiat Biol. 77:215–224. 2001. View Article : Google Scholar
|
|
10.
|
Ohnishi K, Ota I, Takahashi A, Yane K,
Matsumoto H and Ohnishi T: Transfection of mutant p53 gene
depresses X-ray- or CDDP-induced apoptosis in a human squamous cell
carcinoma of the head and neck. Apoptosis. 7:367–372. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Asakawa I, Yoshimura H, Takahashi A, et
al: Radiation-induced growth inhibition in transplanted human
tongue carcinomas with different p53 gene status. Anticancer Res.
22:2037–2043. 2002.PubMed/NCBI
|
|
12.
|
Tamamoto T, Yoshimura H, Takahashi A, et
al: Heat-induced growth inhibition and apoptosis in transplanted
human head and neck squamous cell carcinomas with different status
of p53. Int J Hyperthermia. 19:590–597. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Yuki K, Takahashi A, Ota I, et al:
Sensitization by glycerol for CDDP-therapy against human cultured
cancer cells and tumors bearing mutated p53 gene. Apoptosis.
9:853–859. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Lane DP and Crawford LV: T antigen is
bound to a host protein in SV40-transformed cells. Nature.
278:261–263. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Nigro JM, Baker SJ, Preisinger AC, et al:
Mutations in the p53 gene occur in diverse human tumour types.
Nature. 342:705–708. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Finlay CA, Hinds PW and Levine AJ: The p53
proto-oncogene can act as a suppressor of transformation. Cell.
57:1083–1093. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Hollstein M, Sidransky D, Vogelstein B and
Harris CC: p53 mutations in human cancers. Science. 253:49–53.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Efeyan A and Serrano M: p53: guardian of
the genome and policeman of the oncogenes. Cell Cycle. 6:1006–1010.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Slee EA, O'Connor DJ and Lu X: To die or
not to die: how does p53 decide? Oncogene. 23:2809–2818. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Vousden KH and Lu X: Live or let die: the
cell's response to p53. Nat Rev Cancer. 2:594–604. 2002.
|
|
21.
|
Bambara RA and Jessee CB: Properties of
DNA polymerases delta and epsilon, and their roles in eukaryotic
DNA replication. Biochim Biophys Acta. 1088:11–24. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
El-Deiry WS, Tokino T, Velculescu VE, et
al: WAF1, a potential mediator of p53 tumor suppression. Cell.
75:817–825. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Deng C, Zhang P, Harper JW, Elledge SJ and
Leder P: Mice lacking p21CIP1/WAF1 undergo normal development, but
are defective in G1 checkpoint control. Cell. 82:675–684. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Kastan MB, Zhan Q, el-Deiry WS, et al: A
mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is
defective in ataxia-telangiectasia. Cell. 71:587–597. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Smith ML, Kontny HU, Zhan Q, Sreenath A,
O'Connor PM and Fornace AJ Jr: Antisense GADD45 expression results
in decreased DNA repair and sensitizes cells to u.v.-irradiation or
cisplatin. Oncogene. 13:2255–2263. 1996.PubMed/NCBI
|
|
26.
|
Miyashita T and Reed JC: Tumor suppressor
p53 is a direct transcriptional activator of the human bax gene.
Cell. 80:293–299. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Yu J and Zhang L: No PUMA, no death:
implications for p53-dependent apoptosis. Cancer Cell. 4:248–249.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Owen-Schaub LB, Zhang W, Cusack JC, et al:
Wild-type human p53 and a temperature-sensitive mutant induce
Fas/APO-1 expression. Mol Cell Biol. 15:3032–3040. 1995.PubMed/NCBI
|
|
29.
|
Israeli D, Tessler E, Haupt Y, et al: A
novel p53-inducible gene, PAG608, encodes a nuclear zinc finger
protein whose overexpression promotes apoptosis. EMBO J.
16:4384–4392. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Haupt S, Louria-Hayon I and Haupt Y: P53
licensed to kill? Operating the assassin. J Cell Biochem. 88:76–82.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Barak Y and Oren M: Enhanced binding of a
95 kDa protein to p53 in cells undergoing p53-mediated growth
arrest. EMBO J. 11:2115–2121. 1992.PubMed/NCBI
|
|
32.
|
Brooks CL and Gu W: Ubiquitination,
phosphorylation and acetylation: the molecular basis for p53
regulation. Curr Opin Cell Biol. 15:164–171. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Valenzuela MT, Guerrero R, Nunez MI, et
al: PARP-1 modifies the effectiveness of p53-mediated DNA damage
response. Oncogene. 21:1108–1116. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Schmidt D and Muller S: Members of the
PIAS family act as SUMO ligases for c-Jun and p53 and repress p53
activity. Proc Natl Acad Sci USA. 99:2872–2877. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Melchior F and Hengst L: SUMO-1 and p53.
Cell Cycle. 1:245–249. 2002. View Article : Google Scholar
|
|
36.
|
Siliciano JD, Canman CE, Taya Y, Sakaguchi
K, Appella E and Kastan MB: DNA damage induces phosphorylation of
the amino terminus of p53. Genes Dev. 11:3471–3481. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Shieh SY, Ahn J, Tamai K, Taya Y and
Prives C: The human homologs of checkpoint kinases Chk1 and Cds1
(Chk2) phosphorylate p53 at multiple DNA damage-inducible sites.
Genes Dev. 14:289–300. 2000.PubMed/NCBI
|
|
38.
|
Urban G, Golden T, Aragon IV, et al:
Identification of a functional link for the p53 tumor suppressor
protein in dexamethasone-induced growth suppression. J Biol Chem.
278:9747–9753. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Oda K, Arakawa H, Tanaka T, et al:
p53AIP1, a potential mediator of p53-dependent apoptosis, and its
regulation by Ser-46-phosphorylated p53. Cell. 102:849–862. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Saito S, Goodarzi AA, Higashimoto Y, et
al: ATM mediates phosphorylation at multiple p53 sites, including
Ser(46), in response to ionizing radiation. J Biol Chem.
277:12491–12494. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Ohnishi T, Matsumoto H, Takahashi A,
Shimura M and Majima HJ: Accumulation of mutant p53 and hsp72 by
heat treatment, and their association in a human glioblastoma cell
line. Int J Hyperthermia. 11:663–671. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Matsumoto H, Wang X and Ohnishi T: Binding
between wild-type p53 and hsp72 accumulated after UV and gamma-ray
irradiation. Cancer Lett. 92:127–133. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Petros AM, Gunasekera A, Xu N, Olejniczak
ET and Fesik SW: Defining the p53 DNA-binding
domain/Bcl-x(L)-binding interface using NMR. FEBS Lett.
559:171–174. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Takahashi A, Ota I, Tamamoto T, et al:
p53-dependent hyper-thermic enhancement of tumour growth inhibition
by X-ray or carbon-ion beam irradiation. Int J Hyperthermia.
19:145–153. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Fujiwara T, Cai DW, Georges RN,
Mukhopadhyay T, Grimm EA and Roth JA: Therapeutic effect of a
retroviral wild-type p53 expression vector in an orthotopic lung
cancer model. J Natl Cancer Inst. 86:1458–1462. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Scardigli R, Bossi G, Blandino G,
Crescenzi M, Soddu S and Sacchi A: Expression of exogenous wt-p53
does not affect normal hematopoiesis: implications for bone marrow
purging. Gene Ther. 4:1371–1378. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Bossi G, Mazzaro G, Porrello A, Crescenzi
M, Soddu S and Sacchi A: Wild-type p53 gene transfer is not
detrimental to normal cells in vivo: implications for tumor gene
therapy. Oncogene. 23:418–425. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Vecil GG and Lang FF: Clinical trials of
adenoviruses in brain tumors: a review of Ad-p53 and oncolytic
adenoviruses. J Neurooncol. 65:237–246. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Pearson S, Jia H and Kandachi K: China
approves first gene therapy. Nat Biotechnol. 22:3–4. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Bossi G, Lapi E, Strano S, Rinaldo C,
Blandino G and Sacchi A: Mutant p53 gain of function: reduction of
tumor malignancy of human cancer cell lines through abrogation of
mutant p53 expression. Oncogene. 25:304–309. 2006.PubMed/NCBI
|
|
51.
|
Bischoff JR, Kirn DH, Williams A, et al:
An adenovirus mutant that replicates selectively in p53-deficient
human tumor cells. Science. 274:373–376. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Bossi G and Sacchi A: Restoration of
wild-type p53 function in human cancer: relevance for tumor
therapy. Head Neck. 29:272–284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Ohnishi K, Ota I, Takahashi A and Ohnishi
T: Glycerol restores p53-dependent radiosensitivity of human head
and neck cancer cells bearing mutant p53. Br J Cancer.
83:1735–1739. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Welch WJ and Brown CR: Influence of
molecular and chemical chaperones on protein folding. Cell Stress
Chaperones. 1:109–115. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Thomas PJ, Qu BH and Pedersen PL:
Defective protein folding as a basis of human disease. Trends
Biochem Sci. 20:456–459. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
56.
|
Ohnishi K, Ota I, Yane K, et al: Glycerol
as a chemical chaperone enhances radiation-induced apoptosis in
anaplastic thyroid carcinoma cells. Mol Cancer. 1:42002. View Article : Google Scholar
|
|
57.
|
Imai Y, Ohnishi K, Yasumoto J, et al:
Glycerol enhances radio-sensitivity in a human oral squamous cell
carcinoma cell line (Ca9-22) bearing a mutant p53 gene via
Bax-mediated induction of apoptosis. Oral Oncol. 41:631–636. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Ohnishi T, Ohnishi K and Takahashi A:
Glycerol restores heat-induced p53-dependent apoptosis of human
glioblastoma cells bearing mutant p53. BMC Biotechnol. 2:62002.
View Article : Google Scholar : PubMed/NCBI
|
|
59.
|
Yuki K, Takahashi A, Ota I, et al:
Glycerol enhances CDDP-induced growth inhibition of thyroid
anaplastic carcinoma tumor carrying mutated p53 gene. Oncol Rep.
11:821–824. 2004.PubMed/NCBI
|
|
60.
|
Bargonetti J, Friedman PN, Kern SE,
Vogelstein B and Prives C: Wild-type but not mutant p53
immunopurified proteins bind to sequences adjacent to the SV40
origin of replication. Cell. 65:1083–1091. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
61.
|
Cho Y, Gorina S, Jeffrey PD and Pavletich
NP: Crystal structure of a p53 tumor suppressor-DNA complex:
understanding tumorigenic mutations. Science. 265:346–355. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
62.
|
Halazonetis TD and Kandil AN:
Conformational shifts propagate from the oligomerization domain of
p53 to its tetrameric DNA binding domain and restore DNA binding to
select p53 mutants. EMBO J. 12:5057–5064. 1993.PubMed/NCBI
|
|
63.
|
Hupp TR, Meek DW, Midgley CA and Lane DP:
Regulation of the specific DNA binding function of p53. Cell.
71:875–886. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
64.
|
Hupp TR and Lane DP: Allosteric activation
of latent p53 tetramers. Curr Biol. 4:865–875. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
65.
|
Hupp TR, Sparks A and Lane DP: Small
peptides activate the latent sequence-specific DNA binding function
of p53. Cell. 83:237–245. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
66.
|
Hupp TR, Meek DW, Midgley CA and Lane DP:
Activation of the cryptic DNA binding function of mutant forms of
p53. Nucleic Acids Res. 21:3167–3174. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
67.
|
Abarzua P, LoSardo JE, Gubler ML, et al:
Restoration of the transcription activation function to mutant p53
in human cancer cells. Oncogene. 13:2477–2482. 1996.PubMed/NCBI
|
|
68.
|
Selivanova G, Iotsova V, Okan I, et al:
Restoration of the growth suppression function of mutant p53 by a
synthetic peptide derived from the p53 C-terminal domain. Nat Med.
3:632–638. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
69.
|
Kim AL, Raffo AJ, Brandt-Rauf PW, et al:
Conformational and molecular basis for induction of apoptosis by a
p53 C-terminal peptide in human cancer cells. J Biol Chem.
274:34924–34931. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
70.
|
Ohnishi K, Inaba H, Yasumoto J, Yuki K,
Takahashi A and Ohnishi T: C-terminal peptides of p53 molecules
enhance radiation-induced apoptosis in human mutant p53 cancer
cells. Apoptosis. 9:591–597. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
71.
|
Selivanova G, Ryabchenko L, Jansson E,
Iotsova V and Wiman KG: Reactivation of mutant p53 through
interaction of a C-terminal peptide with the core domain. Mol Cell
Biol. 19:3395–3402. 1999.PubMed/NCBI
|
|
72.
|
Foster BA, Coffey HA, Morin MJ and
Rastinejad F: Pharmacological rescue of mutant p53 conformation and
function. Science. 286:2507–2510. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
73.
|
Takimoto R, Wang W, Dicker DT, Rastinejad
F, Lyssikatos J and el-Deiry WS: The mutant p53-conformation
modifying drug, CP-31398, can induce apoptosis of human cancer
cells and can stabilize wild-type p53 protein. Cancer Biol Ther.
1:47–55. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
74.
|
Rippin TM, Bykov VJ, Freund SM, Selivanova
G, Wiman KG and Fersht AR: Characterization of the p53-rescue drug
CP-31398 in vitro and in living cells. Oncogene. 21:2119–2129.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
75.
|
Bykov VJ, Issaeva N, Shilov A, et al:
Restoration of the tumor suppressor function to mutant p53 by a
low-molecular-weight compound. Nat Med. 8:282–288. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
76.
|
Peng Y, Li C, Chen L, Sebti S and Chen J:
Rescue of mutant p53 transcription function by ellipticine.
Oncogene. 22:4478–4487. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
77.
|
Vassilev LT, Vu BT, Graves B, et al: In
vivo activation of the p53 pathway by small-molecule antagonists of
MDM2. Science. 303:844–848. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
78.
|
Blakely EA and Kronenberg A: Heavy-ion
radiobiology: new approaches to delineate mechanisms underlying
enhanced biological effectiveness. Radiat Res. 150:S126–S145. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
79.
|
Guida P, Vazquez ME and Otto S: Cytotoxic
effects of low- and high-LET radiation on human neuronal progenitor
cells: induction of apoptosis and TP53 gene expression. Radiat Res.
164:545–551. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
80.
|
Demizu Y, Kagawa K, Ejima Y, et al: Cell
biological basis for combination radiotherapy using heavy-ion beams
and high-energy X-rays. Radiother Oncol. 71:207–211. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
81.
|
Debus J, Jackel O, Kraft G and
Wannenmacher M: Is there a role for heavy ion beam therapy? Recent
Results Cancer Res. 150:170–182. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
82.
|
Takahashi A, Matsumoto H, Yuki K, et al:
High-LET radiation enhanced apoptosis but not necrosis regardless
of p53 status. Int J Radiat Oncol Biol Phys. 60:591–597. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
83.
|
Takahashi A, Matsumoto H, Furusawa Y,
Ohnishi K, Ishioka N and Ohnishi T: Apoptosis induced by high-LET
radiations is not affected by cellular p53 gene status. Int J
Radiat Biol. 81:581–586. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
84.
|
Takahashi A, Ohnishi K, Wang X, et al: The
dependence of p53 on the radiation enhancement of thermosensitivity
at different LET. Int J Radiat Oncol Biol Phys. 47:489–494. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
85.
|
Yamakawa N, Takahashi A, Mori E, et al:
High LET radiation enhances apoptosis in mutated p53 cancer cells
through caspase-9 activation. Cancer Sci. 99:1455–1460. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
86.
|
Peng Y, Zhang Q, Nagasawa H, Okayasu R,
Liber HL and Bedford JS: Silencing expression of the catalytic
subunit of DNA-dependent protein kinase by small interfering RNA
sensitizes human cells for radiation-induced chromosome damage,
cell killing, and mutation. Cancer Res. 62:6400–6404. 2002.
|
|
87.
|
Collis SJ, Swartz MJ, Nelson WG and
DeWeese TL: Enhanced radiation and chemotherapy-mediated cell
killing of human cancer cells by small inhibitory RNA silencing of
DNA repair factors. Cancer Res. 63:1550–1554. 2003.PubMed/NCBI
|
|
88.
|
Tauchi H, Kobayashi J, Morishima K, et al:
Nbs1 is essential for DNA repair by homologous recombination in
higher vertebrate cells. Nature. 420:93–98. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
89.
|
Sakamoto S, Iijima K, Mochizuki D, et al:
Homologous recombination repair is regulated by domains at the N-
and C-terminus of NBS1 and is dissociated with ATM functions.
Oncogene. 26:6002–6009. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
90.
|
Tauchi H, Matsuura S, Kobayashi J,
Sakamoto S and Komatsu K: Nijmegen breakage syndrome gene, NBS1,
and molecular links to factors for genome stability. Oncogene.
21:8967–8980. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
91.
|
Nelms BE, Maser RS, MacKay JF, Lagally MG
and Petrini JH: In situ visualization of DNA double-strand break
repair in human fibroblasts. Science. 280:590–592. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
92.
|
Zhang Y, Lim CU, Williams ES, et al: NBS1
knockdown by small interfering RNA increases ionizing radiation
mutagenesis and telomere association in human cells. Cancer Res.
65:5544–5553. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
93.
|
Lee SJ, Dimtchev A, Lavin MF, Dritschilo A
and Jung M: A novel ionizing radiation-induced signaling pathway
that activates the transcription factor NF-kappaB. Oncogene.
17:1821–1826. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
94.
|
Orlowski RZ and Baldwin AS Jr: NF-kappaB
as a therapeutic target in cancer. Trends Mol Med. 8:385–389. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
95.
|
Yamagishi N, Miyakoshi J and Takebe H:
Enhanced radiosensitivity by inhibition of nuclear factor kappa B
activation in human malignant glioma cells. Int J Radiat Biol.
72:157–162. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
96.
|
Habraken Y, Jolois O and Piette J:
Differential involvement of the hMRE11/hRAD50/NBS1 complex, BRCA1
and MLH1 in NF-kappaB activation by camptothecin and X-ray.
Oncogene. 22:6090–6099. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
97.
|
LaCasse EC, Baird S, Korneluk RG and
MacKenzie AE: The inhibitors of apoptosis (IAPs) and their emerging
role in cancer. Oncogene. 17:3247–3259. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
98.
|
Ohnishi K, Scuric Z, Schiestl RH, Okamoto
N, Takahashi A and Ohnishi T: siRNA targeting NBS1 or XIAP
increases radiation sensitivity of human cancer cells independent
of TP53 status. Radiat Res. 166:454–462. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
99.
|
Ohnishi K, Scuric Z, Yau D, et al:
Heat-induced phosphorylation of NBS1 in human skin fibroblast
cells. J Cell Biochem. 99:1642–1650. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
100.
|
Okamoto N, Takahashi A, Ota I, et al:
siRNA targeted for NBS1 enhances heat sensitivity in human
anaplastic thyroid carcinoma cells. Int J Hyperthermia.
27:297–304
|
|
101.
|
Ohnishi K, Nagata Y, Takahashi A,
Taniguchi S and Ohnishi T: Effective enhancement of X-ray-induced
apoptosis in human cancer cells with mutated p53 by siRNA targeting
XIAP. Oncol Rep. 20:57–61. 2008.PubMed/NCBI
|
|
102.
|
Nicholson KM and Anderson NG: The protein
kinase B/Akt signalling pathway in human malignancy. Cell Signal.
14:381–395. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
103.
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
104.
|
Bjornsti MA and Houghton PJ: The TOR
pathway: a target for cancer therapy. Nat Rev Cancer. 4:335–348.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
105.
|
Wang F, Arun P, Friedman J, Chen Z and Van
Waes C: Current and potential inflammation targeted therapies in
head and neck cancer. Curr Opin Pharmacol. 9:389–395. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
106.
|
Raimondi AR, Molinolo A and Gutkind JS:
Rapamycin prevents early onset of tumorigenesis in an oral-specific
K-ras and p53 two-hit carcinogenesis model. Cancer Res.
69:4159–4166. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
107.
|
Shaw M, Cohen P and Alessi DR: The
activation of protein kinase B by H2O2 or
heat shock is mediated by phosphoinositide 3-kinase and not by
mitogen-activated protein kinase-activated protein kinase-2.
Biochem J. 336:241–246. 1998.
|
|
108.
|
Rosenzweig KE, Youmell MB, Palayoor ST and
Price BD: Radiosensitization of human tumor cells by the
phosphatidylinositol3-kinase inhibitors wortmannin and LY294002
correlates with inhibition of DNA-dependent protein kinase and
prolonged G2-M delay. Clin Cancer Res. 3:1149–1156. 1997.
|
|
109.
|
Gupta AK, Cerniglia GJ, Mick R, et al:
Radiation sensitization of human cancer cells in vivo by inhibiting
the activity of PI3K using LY294002. Int J Radiat Oncol Biol Phys.
56:846–853. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
110.
|
Ohnishi K, Yasumoto J, Takahashi A and
Ohnishi T: LY294002, an inhibitor of PI-3K, enhances heat
sensitivity independently of p53 status in human lung cancer cells.
Int J Oncol. 29:249–253. 2006.PubMed/NCBI
|
|
111.
|
Guertin DA and Sabatini DM: Defining the
role of mTOR in cancer. Cancer Cell. 12:9–22. 2007. View Article : Google Scholar
|
|
112.
|
Hay N and Sonenberg N: Upstream and
downstream of mTOR. Genes Dev. 18:1926–1945. 2004. View Article : Google Scholar
|
|
113.
|
Beuvink I, Boulay A, Fumagalli S, et al:
The mTOR inhibitor RAD001 sensitizes tumor cells to DNA-damaged
induced apoptosis through inhibition of p21 translation. Cell.
120:747–759. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
114.
|
Majumder PK, Febbo PG, Bikoff R, et al:
mTOR inhibition reverses Akt-dependent prostate intraepithelial
neoplasia through regulation of apoptotic and HIF-1-dependent
pathways. Nat Med. 10:594–601. 2004. View
Article : Google Scholar
|
|
115.
|
Boulay A, Zumstein-Mecker S, Stephan C, et
al: Antitumor efficacy of intermittent treatment schedules with the
rapamycin derivative RAD001 correlates with prolonged inactivation
of ribosomal protein S6 kinase 1 in peripheral blood mononuclear
cells. Cancer Res. 64:252–261. 2004. View Article : Google Scholar
|
|
116.
|
Mabuchi S, Altomare DA, Cheung M, et al:
RAD001 inhibits human ovarian cancer cell proliferation, enhances
cisplatin-induced apoptosis, and prolongs survival in an ovarian
cancer model. Clin Cancer Res. 13:4261–4270. 2007. View Article : Google Scholar
|
|
117.
|
Cao C, Subhawong T, Albert JM, et al:
Inhibition of mammalian target of rapamycin or apoptotic pathway
induces autophagy and radiosensitizes PTEN null prostate cancer
cells. Cancer Res. 66:10040–10047. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
118.
|
Albert JM, Kim KW, Cao C and Lu B:
Targeting the Akt/mammalian target of rapamycin pathway for
radiosensitization of breast cancer. Mol Cancer Ther. 5:1183–1189.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
119.
|
Nagata Y, Takahashi A, Ohnishi K, et al:
Effect of rapamycin, an mTOR inhibitor, on radiation sensitivity of
lung cancer cells having different p53 gene status. Int J Oncol.
37:1001–1010
|














