Introduction
Genetic and epigenetic alterations underlie the
pathogenesis of cancer. The disruption of epigenetic mechanisms
leads to abnormal development and is involved in malignant
transformation (1). Epigenetic
changes include DNA methylation and histone modifications, which
may affect gene expression by changing chromatin structure or
modifying the DNA without altering the native nucleotide sequence.
Since epigenetic changes are potentially reversible, they make
attractive targets for therapeutic intervention (2).
Cytosine methylation usually only occurs in the CG
dinucleotides which are unevenly distrubuted in the genome. The
vast majority are located in the repetitive elements and
heterochromatin. CpG islands in the promoter have a frequency of
approximately five times greater than the rest of the genome
(3). The CpG islands of
transcriptionally active genes in normal cells are unmethylated.
Methylation of this region leads to the modification of chromatin
and transcriptional silencing of the gene.
Chromatin structure plays an integral role in the
control of gene expression. The basic repeating unit of chromatin
is the nucleosome which functions as a DNA packaging unit and
transcripitional regulator (4).
The amino terminal tails of the histones protrude out of the
nucleosome and are subject to a variety of chemical modifications,
including phosphorylation, acetylation, ubiquitination and
methylation. These modifications affect the access of regulatory
factors and complexes to chromatin and influence gene expression.
Various sites of lysine methylation on the histones play essential
roles in regulating chromatin structure and gene transcription.
Methylation at lysine on histone H3 has recently been shown to be a
marker of heterochromatin in all organisms (5). The methylation of H3K4, H3R17 and
H4R3 has also been associated with the activation of transcription.
By contrast, methylation of H3K9 has been correlated with gene
silencing (6–9). H3K9 methylation is recognized by
heterochromatin-associated proteins and is required to maintain the
heterochromatic state. Degrees of methylation are differentially
regulated and exert various functional outcomes. Studies have shown
that modifications of histone H3 also contribute to gene silencing
by switching between acetylation and methylation of the lysine 9
residue (10).
Epigenetic mechanisms play an important role in
colon cancer and aberrant methylation of the p16INK4A
gene is commonly observed (11,12).
The p16INK4A gene [also known as cyclin-dependent kinase
inhibitor 2A (CDKN2A)] is a member of the INK4A/ARF family
of suppressors of cyclin-dependent kinases (CDKs) which plays an
important role in the G1-S transition by binding to CDK4 and CDK6
and inhibiting the progression of the cell cycle (13,14).
Inactivation of the p16INK4A gene is considered to be
the second most common defect in human cancer (15,16).
The p16INK4A gene is inactivated by homozygous
deletions, point mutations and preferentially by methylation of the
gene promoter (17,18). p16INK4A promoter
methylation and subsequent gene silencing have been reported in
various cancer types (19–23). The differences between the
methylation states of the histone H3 lysine 9 have been associated
with repression (24) and
activation (25) of important
genes. In this study we aimed to investigate the expression of the
p16INK4A gene and the methylation status of the promoter
region and the histone H3 lysine 9 residue. To our knowledge this
is the first study in the literature to analyze these parameters in
matched tumor samples.
Materials and methods
In this study tumor specimens from 71 patients with
colorectal cancer (mean age, 61.04±14.8) and matched normal tissue
samples were analyzed. The characteristics of the patients are
summarized in Table I. The study
was approved by the Istanbul Faculty of Medicine Ethics Committee
(2006-504).
 | Table IClinical characteristics of the
patients. |
Table I
Clinical characteristics of the
patients.
|
Characteristics | No. of
patients | % |
|---|
| Stage (n=46) | | |
| I | 3 | 6.5 |
| II | 5 | 10.8 |
| III | 16 | 34.7 |
| IV | 19 | 41.3 |
| Tumor location
(n=71) | | |
| Colon | 31 | 43.6 |
| Rectum | 30 | 42.2 |
| Rectosigmoid | 10 | 14 |
| Lymph node status
(n=46) | | |
| Negative | 11 | 23.9 |
| Positive | 35 | 76.08 |
| Distant metastasis
(n=46) | | |
| Negative | 27 | 58.6 |
| Positive | 19 | 41.3 |
| Differentiation
(n=36) | | |
| High | 5 | 13.8 |
| Middle | 28 | 77.7 |
| Low | 3 | 8.3 |
Sodium bisulfite modification of DNA
Genomic DNA was extracted from the tissue samples.
The quality of DNA was evaluated spectrophotometrically at 260/280
nm. The bisulfite conversion was performed according to the
manufacturer’s instructions using the Methylamp™ DNA Modification
kit (Epigentek, Brooklyn, NY, USA). During this process all
unmethylated cytosines are deaminated, sulfonated and converted to
uracil, while 5-methylcytosines remain unaltered. The
bisulfite-modified DNA was stored at −20°C until use. DNA from
placenta treated with SssI methyltransferase (New England
Biolabs, Ipswich, MA, USA) was used as a standard for the
methylated reaction.
Methylation-specific PCR
To increase the sensitivity of methylation
detection, we used a 2-step methylation-specific PCR approach, in
which the outside primer pairs selectively amplify
bisufite-modified DNA, irrespective of the methylation status. A
second reaction then uses specially designed nested primer pairs to
discriminate between the methylated and unmethylated alleles. The
sequences of the outside primers were: forward;
5′-GGAGGAAGAAAGAGGAGGGGT-3 and reverse; 5′-CTACCTAATTCCAATTCCCCT-3′
(Integrated DNA Technologies, Coralville, IA, USA). Amplification
was performed on a thermal cycler (Techne, Inc., Burlington, NJ,
USA) as follows: denaturation for 5 min at 94°C and 35 cycles of
denaturation at 94°C for 30 sec, annealing at 64°C for 30 sec and
extension at 72°C for 30 sec. PCR reactions were carried out in a
total volume of 25 μl containing 1X reaction buffer, 2 mM
MgCl2, 200 μM deoxynucleotide triphosphate, 400 pmol of
each primer, 1.5 unit Taq polymerase (Fermentas, Vilnius,
Lithuania) and 3 μl modified DNA.
The second round PCR reaction (quantitative step)
was performed using real-time PCR (LightCycler®; Roche,
Mannheim, Germany). The PCR mix contained 1 μl of PCR product from
the first step, 1X hybridization probes, 2 mM MgCl2, 0.5
μM of each primer and 0.2 μM of each probe in a total volume of 15
μl. Following denaturation at 95°C for 10 min, amplification was
performed by 45 cycles of denaturation at 94°C for 10 sec,
annealing at 50°C for 10 sec and extension at 72°C for 10 sec.
Standard curves were constructed using serial dilutions of
methylated placental DNA. The sequences of the primers were:
forward; 5′-TAGAGGGTGGGGCGGATCGCG-3′ and reverse;
5′-CCAAAATCGCCCGCCATCCC-3′. The fragment of the p16INK4A
gene amplified by the second primer pair covers 12 CpG
dinucleotides. The sequences of probes were
5′-CCGCCGCCCGCTACCTACTCT-FL, 5′-LC640-CCCTCTCCGCAACCGCCGAAC-PH. The
two probes were designed to hybridize to the same strand between
two unlabeled primers. Fluorescence monitoring of amplification is
based on the concept that a signal is generated when fluorescence
resonance energy transfer occurs between two adjacent
fluorescently-labeled sequence-specific hybridization probes. The
emitted fluorescence is measured and is proportional to the
quantity of specific target sequences in the reaction mixture. The
methylation levels were calculated by comparing the data from the
samples with the standards using the LightCycler Relative
Quantification Software (Roche).
Chromatin immunoprecipitation (ChIP)
For the ChIP assay the method available on the UC
Davis Genome Center web site (26)
was used with certain modifications. Briefly, tissue samples were
treated with 1% formaldehyde for 15 min to cross-link histones to
DNA. After washing, the pellets were resuspended in the lysis
buffer [5 mM PIPES (pH 8.0), 85 mM KCl, 0.5% NP-40, protease
inhibitors (aprotinin, leupeptin, PMSF)] and nuclei lysis buffer
[50 mM Tris-Cl (pH 8.1), 10 mM EDTA, 1% SDS, protease inhibitors],
respectively. Each sample was sonicated 7 times for 8 sec each. The
lysate was then divided into 3 fractions; which were incubated
overnight at 4°C with 2 μg of the appropriate antibodies for H3K9
mono-, di- and trimethylation (Millipore, Billerica, MA, USA),
respectively. The pellets were washed with dialysis buffer [2 mM
EDTA, 50 mM Tris-Cl (pH 8.0)] and IP wash buffer [100 mM Tris-Cl
(pH 9.0), 500 mM LiCl, 1% NP-40, 1% deoxycholic acid] and were
resuspended in the elution buffer (50 mM NaHCO3, 1%
SDS). DNA was extracted using the phenol-chloroform method, ethanol
precipitated and resuspended in 20 μl of water. The PCR
amplification of DNA was performed using the LightCycler 1.2
(Roche) and the double-stranded DNA binding dye SYBR-Green I as the
fluorescent molecule. The PCR mix contained 1.2 μl of SYBR Green
mix, 2 mM MgCl2, 0.5 μM each primer and 4 μl DNA in a
total volume of 12 μl. Amplification was performed by denaturation
at 95°C for 10 min followed by 45 cycles of denaturation at 94°C
for 10 sec, annealing at 60°C for 10 sec and extension at 72°C for
10 sec. The sequences of the primers were: forward;
5′-AGACAGCCGTTTTACACGCAG-3′ and reverse;
5′-CACCGAGAAATCGAAATCACC-3′. Known concentrations of serial diluted
DNA were used for the standard curve. To increase the specificity
of SYBR Green I detection, we performed a melting curve analysis of
the amplification reaction. The area below each of the peaks
representing the relative amount of nucleic acids was calculated
using the LightCycler Data Analysis Software (Roche).
Analysis of p16INK4A gene
expression
RNA samples were available from tumors of 41
patients. p16INK4A mRNA levels were measured by
real-time quantitative RT-PCR using the LightCycler 480 instrument
(Roche Applied Science, Indianapolis, IN, USA) and UPL probes.
Total RNA was extracted from the tumor and normal
tissue samples using the SV Total RNA Isolation system (Promega
Corporation, Madison, WI, USA) as per the manufacturer’s
instructions. RNA from each sample was used to generate cDNA using
the RevertAid First Strand cDNA Synthesis kit (Fermentas). Briefly,
1 μg RNA and 1 μl of oligo(dT) were used as starting materials,
heated and kept at 70°C for 5 min. The samples were then chilled on
ice, the other components (5X reaction buffer, ribonuclease
inhibitor and dNTP mix) were added and the samples were incubated
at 37°C for 5 min. Then 1 μl reverse transcriptase (200 U/μl) was
added and the samples were incubated at 42°C for 60 min. The
reaction was inactivated at 70°C for 10 min.
The probes were designed using the Lightcycler Probe
Design Software 2.0 (Roche). The HPRT gene was used as the internal
control. The PCR mix of 20 μl was prepared by adding to 5 μl cDNA
template, 2.5 μM of each of the forward and reverse primers (final
concentration) and 10 μl of the probe master mix. The sequences of
the forward and reverse primers were: 5′-GTGGACCTGGCTGAGGAG-3′ and
5′-CTTTCAATCGGGGATGTCTG-3′. The PCR conditions were: one cycle at
95°C for 10 min, 45 cycles of denaturation at 95°C for 10 sec,
annealing at 60°C for 30 sec and elongation at 72°C for 1 sec with
subsequent cooling at 40°C for 30 sec. The results were analyzed by
basic relative quantification.
The tumor and normal samples were compared using the
Mann-Whitney U test; associations with clinical parameters were
evaluated using the Kruskal-Wallis test and Spearman’s rank
correlation.
Results
Tissue samples were collected from 71 patients with
colorectal cancer who underwent surgery for tumor resection.
Methylation of the p16INK4A gene promoter was analyzed
by quantitative real-time PCR in the tumors and matched normal
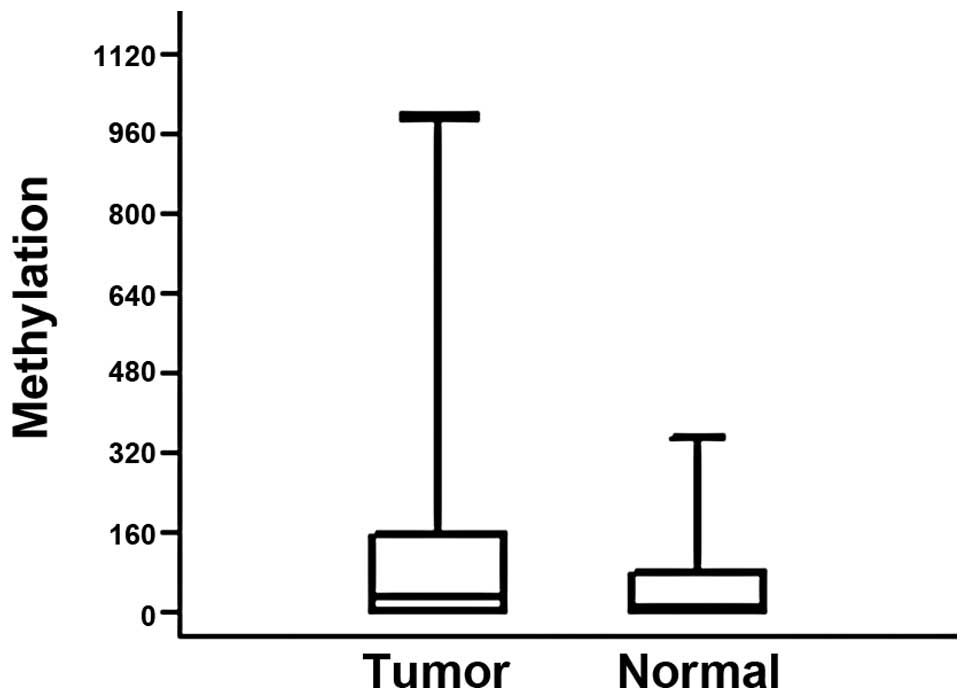
tissue samples from the patients. Methylation at the
p16INK4A promoter was higher in the tumors from 38
patients (53.5%) when compared with the normal tissue of the same
individuals (Fig. 1). The median
methylation levels were 24 for tumor tissue and 3.5 for normal
tissue. However, due to the wide range of the values the difference
was not significant (P=0.06). We also did not identify any
significant correlation between the methylation levels and clinical
parameters, including lymph node status, metastasis, stage and
tumor location.
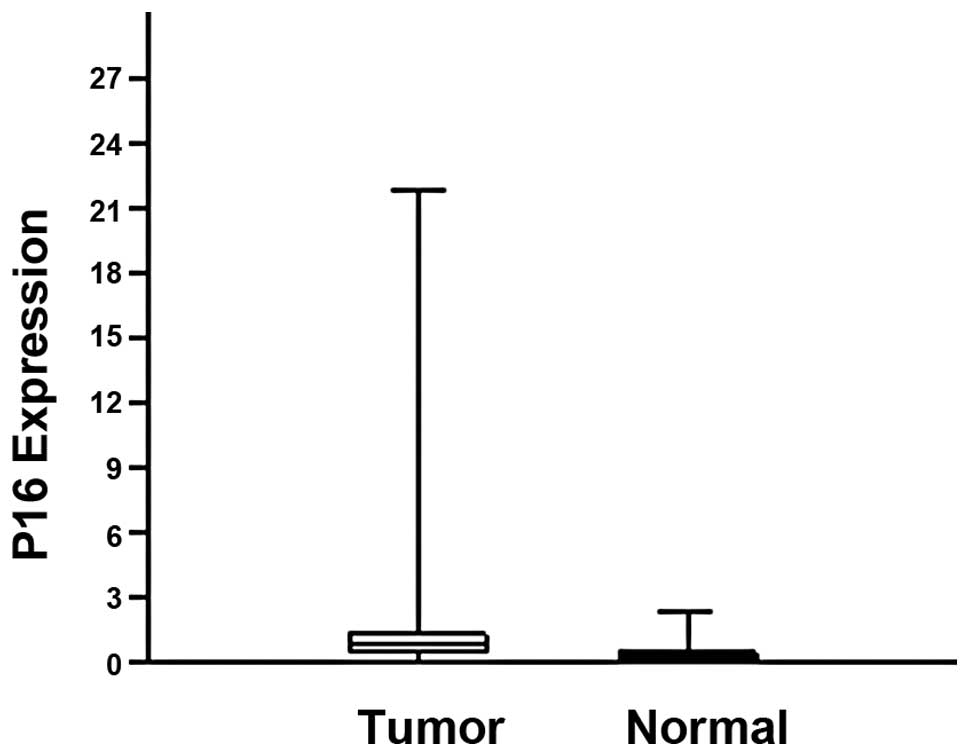
The expression of the p16INK4A gene was
investigated in 41 samples by real-time PCR. p16INK4A
expression was higher in the tumors than normal tissue in 31
patients (75.6%). The median expression levels were 0.869 and 0.312
for the tumor and normal tissue samples, respectively. The
difference was significant (P=0.0005; Fig. 2). No association was observed
between the methylation status and expression of the
p16INK4A gene both for the tumor (P=0.315) and normal
tissue (P=0.209) samples.
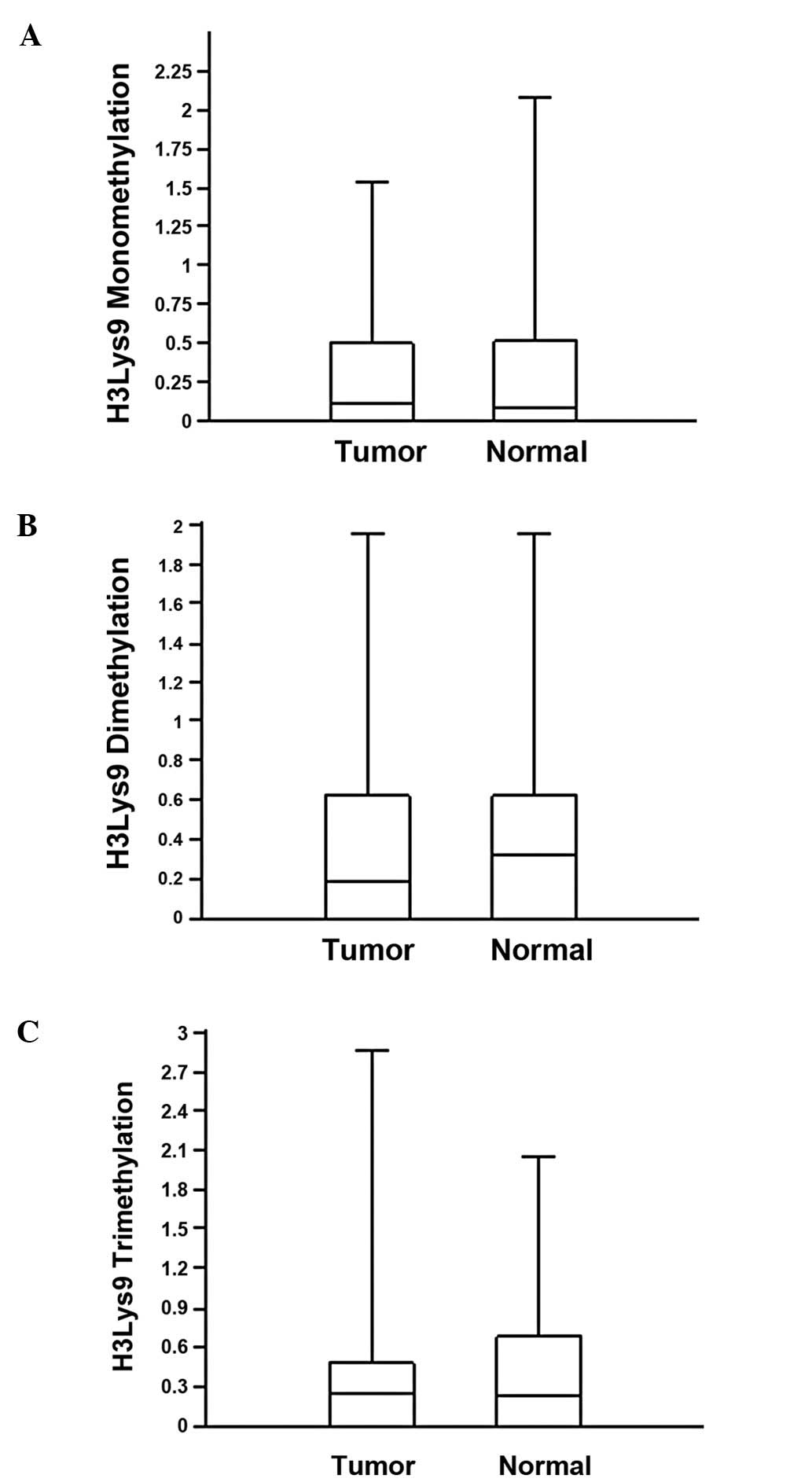
To determine the correlation between
p16INK4A methylation and chromatin remodelling, mono-,
di- and trimethylation of the H3K9 residue was analyzed by
chromatin immunoprecipitation and real-time PCR. The values of H3K9
mono-, di- and trimethylation for the normal and tumor tissues were
similar (Table II). The median
values of monomethylation for the tumor and normal tissue samples
were 0.112 and 0.08, respectively (Fig. 3A). Median values for di- and
trimethylation in the tumors and normal tissue were 0.187 vs. 0.325
and 0.257 vs. 0.234, respectively (Fig. 3B and C). The difference between the
methylation levels of H3K9 in the normal and tumor tissues was not
significant (Mann-Whitney U test, P>0.05). The expression level
of the p16INK4A gene was not associated with mono-, di-
or trimethylation for the tumor (P=0.404, P=0.457 and P=0.465,
respectively) or normal (P=0.288, P=0.62 and P=0.667, respectively)
samples.
| Table IIH3K9 methylation frequencies and
median values in the tumors and normal samples. |
Table II
H3K9 methylation frequencies and
median values in the tumors and normal samples.
| Monomethylation
| Dimethylation
| Trimethylation
|
|---|
| Sample | % | Median | % | Median | % | Median |
|---|
| Tumor | 56.3 | 0.112 | 63.3 | 0.187 | 61.9 | 0.257 |
| Normal | 53.5 | 0.08 | 61.9 | 0.325 | 57.7 | 0.234 |
Discussion
Epigenetic mechanisms involving DNA methylation and
histone modifications are closely associated but the critical
initiating events in silencing remain undefined. Malignant cells
are characterized by a localized increase in methylation in CpG
island-associated promoters (27,28).
Several lines of evidence indicate that alterations in histone
modifications are crucial in cancer development and progression
(29). In certain organisms all
DNA methylation is dependent on H3K9 methylation (30). However, it remains unclear whether
DNA methylation actually directs H3K9 methylation or whether it
acts independently to silence gene expression in the absence of
H3K9 methylation.
Methylation of the p16INK4A gene as an
epigenetic event has been reported in several tumor types (19–23,31).
In this study methylation levels of the p16INK4A gene
promoter and of the histone H3K9 were analyzed in colorectal tumors
and normal colon samples. To date, this is the first study to
investigate DNA and histone methylation in a series of matched
human tumors and normal tissue samples.
Expression of the p16INK4A gene was
significantly higher in tumor samples. This finding is consistent
with the observation that promoter methylation levels were not
different between the tumor and normal samples since methylation
may also be detected in normal colon cells (32). The frequency of the hypermethylated
tumors was 53% in this study. This value is in agreement with
recent reports (33–36) but there are also studies reporting
lower values in the literature (32,37,38).
These differences may be explained by the high sensitivity of the
methods used in our study. The use of primers and probes designed
for the methylated sequences and application of nested PCR
increases the specificity of the method markedly and makes it
possible to detect low levels of methylated sequences (39). We also observed methylation in
45.3% of the normal tissue samples. A number of recent reports have
confirmed that methylation is also observed in normal tissue
(32,40,41).
It has been suggested that methylation in normal tissue may be
associated with increasing age (40,42).
Suppression of p16INK4A expression by methylation may be
much lower or even absent in aging colon tissue (43) since increased p16INK4A
expression which accompanies promoter methylation has been reported
in aging cells (44,45). This phenomenon is thought to play a
role in the aging process by promoting senescence and the decline
in gene function (46).
The epigenetic reprogramming of cancer cells occurs
early in oncogenesis and is difficult to study in clinical samples.
The correlation between DNA methylation and histone lysine
methylation during the development of colon cancer remains unclear.
There are numerous studies reporting findings in various fields.
One of the first studies suggested a direct association between DNA
methylation and histone lysine methylation in a cell line (47). In plants DNA methylation is
sufficent for gene silencing (48). In prostate cancer cells, the effect
of H3K27 trimethylation on gene silencing is independent of DNA
methylation (49). It has also
been demonstrated that silencing of p16INK4A and H3K9
methylation precede DNA methylation (50). Histone H3K9 trimethylation is
essential for the cells to survive radiation damage. Impairment of
lysine 9 methylation may increase cancer risk by altering the
efficiency of DNA repair (51).
When we compared H3K9 methylation levels in the tumor and normal
tissue samples no significant difference was observed. Analysis of
H3K9 methylation indicated that various levels of methylation exist
within the tissue. The rate of monomethylation in the tissue
samples was lowest while the di- and trimethylation levels were
similar. Conflicting results in the literature show that epigenetic
mechanisms may also differ from organism to organism or from gene
to gene. In the present study we could not detect any correlation
between DNA methylation and H3K9 methylation. We also did not
observe any correlation between the p16INK4A promoter or
H3K9 methylation and the clinical parameters. The lack of
association with clinical characteristics is in accordance with
recent reports (16,32).
In our study expression of the p16INK4A
gene was higher in the tumor tissue than in normal tissue in 75% of
the patients. These data are in agreement with studies showing that
p16INK4A expression in colon carcinoma is frequently
observed (31,52). It has even been reported that the
majority of colorectal tumors express the p16INK4A
protein (53). Coexistence of
p16INK4A methylation and expression has also been
demonstrated (54,55). Similar findings have been confirmed
by various studies in which p16INK4A expression in
excess of 80% of the colon tumors has been observed (16,56,57).
To the best of our knowledge, this is the first study comparing
methylation and histone modifications of the p16INK4A
gene in colon tumors and corresponding normal tissue. Our results
suggest that although epigenetic alterations of the
p16INK4A gene are frequent in colon tumors they appear
to accompany other genetic alterations which play a causative role
in colon carcinogenesis.
Acknowledgements
This study was supported by the
Istanbul University Research Fund, project no. T-839/02062006.
References
|
1
|
Tsai HC and Baylin SB: Cancer epigenetics:
linking basic biology to clinical medicine. Cell Res. 21:502–517.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baylin SB and Jones PA: A decade of
exploring the cancer epigenome: biological and translational
implications. Nat Rev Cancer. 11:726–734. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yao JY, Zhang L, Zhang X, He ZY, Ma Y, Hui
LJ, Wang X and Hu Y: H3K27 trimethylation is an early epigenetic
event of p16INK4A silencing for regaining tumorigenesis
in fusion reprogrammed hepatoma cells. Biol Chem. 285:18828–18837.
2010.PubMed/NCBI
|
|
4
|
Shones DE, Cui K, Cuddapah S, Roh TY,
Barski A, Wang Z, Wei G and Zhao K: Dynamic regulation of
nucleosome positioning in the human genome. Cell. 132:887–898.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rea S, Eisenhaber F, O’Carroll D, Strahl
BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD
and Jenuwein T: Regulation of chromatin structure by site-specific
histone H3 methyltransferases. Nature. 406:593–599. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Noma K, Allis CD and Grewal SI:
Transitions in distinct histone H3 methylation patterns at the
heterochromatin domain boundaries. Science. 293:1150–1155. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Peters AH, O’Carroll D, Scherthan H,
Mechtler K, Sauer S, Schöfer C, Weipoltshammer K, Pagani M, Lachner
M, Kohlmaier A, et al: Loss of the Suv39h histone
methyltransferases impairs mammalian heterochromatin and genome
stability. Cell. 107:323–337. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stallcup MR: Role of protein methylation
in chromatin remodeling and transcriptional regulation. Oncogene.
20:3014–3020. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang Y and Reinberg D: Transcription
regulation by histone methylation: interplay between different
covalent modifications of the core histone tails. Genes Dev.
15:2343–2360. 2001. View Article : Google Scholar
|
|
10
|
Nielsen SJ, Schneider R, Bauer UM,
Bannister AJ, Morrison A, O’Carroll D, Firestein R, Cleary M,
Jenuwein T, Herrera RE and Kouzarides T: Rb targets histone H3
methylation and HP1 to promoters. Nature. 412:561–565. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Herman JG, Merlo A, Mao L, Lapidus RG,
Issa JP, Davidson NE, Sidransky D and Baylin SB: Inactivation of
the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA
methylation in all common human cancers. Cancer Res. 55:4525–4530.
1995.PubMed/NCBI
|
|
12
|
Ahuja N, Mohan AL, Li Q, Stolker JM,
Herman JG, Hamilton SR, Baylin SB and Issa JP: Association between
CpG island methylation and microsatellite instability in colorectal
cancer. Cancer Res. 57:3370–3374. 1997.PubMed/NCBI
|
|
13
|
Collado M, Blasco M and Serrano M:
Cellular senescence in cancer and ageing. Cell. 130:223–233. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Clark SJ and Melki J: DNA methylation and
gene silencing in cancer: which is the guilty party? Oncogene.
21:5380–5387. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gonzalez-Zulueta M, Bender CM, Yang AS,
Nguyen T, Beart RW, Van Tornout JM and Jones PA: Methylation of the
5′ CpG island of the p16/CDKN2 tumor suppressor gene in normal and
transformed human tissues correlates with gene silencing. Cancer
Res. 4531–4535. 1995.
|
|
16
|
Malhotra P, Kochhar R, Vaiphei K, Wig JD
and Mahmood S: Aberrant promoter methylation of p16 in colorectal
adenocarcinoma in North Indian patients. World J Gastrointest
Oncol. 7:295–303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liggett WH Jr and Sidransky D: Role of the
p16 tumor suppressor gene in cancer. J Clin Oncol. 16:1197–1206.
1998.PubMed/NCBI
|
|
18
|
Matsuda Y: Molecular mechanism underlying
the functional loss of cyclin dependent kinase inhibitors p16 and
p27 in hepatocellular carcinoma. World J Gastroenterol.
14:1734–1740. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Esteller M: The necessity of a human
epigenome project. Carcinogenesis. 27:1121–1125. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou J, Cao J, Lu Z, Liu H and Deng DA:
115-bp MethyLight assay for detection of p16 (CDKN2A) methylation
as a diagnostic biomarker in human tissues. BMC Med Genet.
12:672011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang LW, Pan HS, Lin YH, Seow KM, Chen HJ
and Hwang JL: P16 methylation is an early event in cervical
carcinogenesis. Int J Gynecol Cancer. 21:452–456. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang Y, Wang R, Song H, Huang G, Yi J,
Zheng Y, Wang J and Chen L: Methylation of multiple genes as a
candidate biomarker in non-small cell lung cancer. Cancer Lett.
303:21–28. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zainuddin N, Kanduri M, Berglund M,
Lindell M, Amini RM, Roos G, Sundström C, Enblad G and Rosenquist
R: Quantitative evaluation of p16(INK4a) promoter methylation using
pyrosequencing in de novo diffuse large B-cell lymphoma. Leuk Res.
35:438–443. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rosenfeld JA, Wang Z, Shones DE, Zhao K,
DeSalle R and Zhang MQ: Determination of enriched histone
modifications in non-genic portions of the human genome. BMC
Genomics. 10:1432009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Barski A, Cuddapah S, Cui K, Roh TY,
Schones DE, Wang Z, Wei G, Chepelev I and Zhao K: High-resolution
profiling of histone methylations in the human genome. Cell.
129:823–837. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Farnham Lab Chromatin Immunoprecipitation
Protocol for Tissues: UC Davis Genome Center. 2006, http://farnham.genomecenter.ucdavis.edu/protocols/tissues.htmluri.
Accessed April 19, 2012.
|
|
27
|
Baylin SB: DNA methylation and gene
silencing in cancer. Nat Clin Prac Oncol. 2(Suppl 1): S4–S11. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gal-Yam EN, Saito Y, Egger G and Jones PA:
Cancer epigenetics: modifications, screening, and therapy. Annu Rev
Med. 59:267–280. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Enroth S, Rada-Iglesias A, Andersson R,
Wallerman O, Wanders A, Påhlman L, Komorowski J and Wadelius C:
Cancer associated epigenetic transitions identified by genome-wide
histone methylation binding profiles in human colorectal cancer
samples and paired normal mucosa. BMC Cancer. 11:4502011.
View Article : Google Scholar
|
|
30
|
Bird A: DNA methylation patterns and
epigenetic memory. Genes Dev. 16:6–21. 2002. View Article : Google Scholar
|
|
31
|
Esteller M, Corn PG, Baylin SB and Herman
JG: A gene hypermethylation profile of human cancer. Cancer Res.
61:3225–3229. 2001.PubMed/NCBI
|
|
32
|
Shima K, Nosho K, Baba Y, Cantor M,
Meyerhardt JA, Giovannucci EL, Fuchs CS and Ogino S: Prognostic
significance of CDKN2A (p16) promoter methylation and loss of
expression in 902 colorectal cancers: Cohort study and literature
review. Int J Cancer. 128:1080–1094. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Krtolica K, Krajnovic M, Usaj-Knezevic S,
Babic D, Jovanovic D and Dimitrijevic B: Comethylation of p16 and
MGMT genes in colorectal carcinoma: correlation with
clinicopathological features and prognostic value. World J
Gastroenterol. 13:1187–1194. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sakamoto J, Fujiya M, Okamoto K, Nata T,
Inaba Y, Moriichi K, Tanabe H, Mizukami Y, Watari J, Ashida T and
Kohgo Y: Immunoprecipitation of nucleosomal DNA is a novel
procedure to improve the sensitivity of serum screening for the p16
hypermethylation associated with colon cancer. Cancer Epidemiol.
34:194–199. 2010. View Article : Google Scholar
|
|
35
|
Guan RJ, Fu Y, Holt PR and Pardee AB:
Association of K-ras mutations with p16 methylation in human colon
cancer. Gastroenterology. 116:1063–1071. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Krakowczyk L, Strzelczyk JK, Adamek B,
Zalewska-Ziob M, Arendt J, Półtorak S, Maciejewski B and Wiczkowski
A: Methylation of the MGMT and p16 genes in sporadic colorectal
carcinoma and corresponding normal colonic mucosa. Med Sci Monit.
14:BR219–BR225. 2008.PubMed/NCBI
|
|
37
|
Psofaki V, Kalogera C, Tzambouras N,
Stephanou D, Tsianos E, Seferiadis K and Kolios G: Promoter
methylation status of hMLH1, MGMT, and CDKN2A/p16 in colorectal
adenomas. World J Gastroenterol. 16:3553–3560. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Barault L, Charon-Barra C, Jooste V, de la
Vega MF, Martin L, Roignot P, Rat P, Bouvier AM, Laurent-Puig P,
Faivre J, et al: Hypermethylator phenotype in sporadic colon
cancer: study on a population-based series of 582 cases. Cancer
Res. 68:8541–8546. 2008. View Article : Google Scholar
|
|
39
|
Deligezer U, Esin Akisik EE and Dalay N: A
novel application of melting curves: utility of peak area
calculation for relative methylation quantification. Clin Chem Lab
Med. 45:867–873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yoshino M, Suzuki M, Tian L, et al:
Promoter hypermethylation of the p16 and Wif-1 genes as an
independent prognostic marker in stage 1A non-small cell lung
cancers. Int J Oncol. 35:1201–1209. 2009.PubMed/NCBI
|
|
41
|
Steinmann K, Sandner A, Schagdarsurengin U
and Dammann RH: Frequent promoter hypermethylation of tumor-related
genes in head and neck squamous cell carcinoma. Oncol Rep.
22:1519–1526. 2009.PubMed/NCBI
|
|
42
|
Santini V, Kantarjian HM and Issa JP:
Changes in DNA methylation in neoplasia: pathophysiology and
therapeutic implications. Ann Intern Med. 134:573–586. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sauer J, Jang H, Zimmerly EM, Kim KC, Liu
Z, Chanson A, Smith DE, Mason JB, Friso S and Choi SW: Ageing,
alcohol consumption and folate are determinants of genomic DNA
methylation, p16 promoter methylation and the expression of p16 in
the mouse colon. Br J Nutr. 104:24–30. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Keyes MK, Jang H, Mason JB, Liu Z, Crott
JW, Smith DE, Friso S and Choi SW: Older age and dietary folate are
determinants of genomic and p16-specific DNA methylation in mouse
colon. J Nutr. 137:1713–1717. 2007.PubMed/NCBI
|
|
45
|
Collins CJ and Sedivy JM: Involvement of
the INK4a/Arf gene locus in senescence. Aging Cell. 2:145–150.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Krishnamurthy J, Ramsey MR, Ligon KL,
Torrice C, Koh A, Bonner-Weir S and Sharpless NE:
p16INK4a induces an age-dependent decline in islet
regenerative potential. Nature. 443:453–457. 2006.
|
|
47
|
Kondo Y, Shen L and Issa JP: Critical role
of histone methylation in tumor suppressor gene silencing in
colorectal cancer. Mol Cell Biol. 23:206–215. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xiao W, Custard KD, Brown RC, Lemmon BE,
Harada JJ, Goldberg RB and Fischer RL: DNA methylation is critical
for Arabidopsis embryogenesis and seed viability. Plant Cell.
18:805–814. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kondo Y, Shen L, Cheng AS, Ahmed S,
Boumber Y, Charo C, Yamochi T, Urano T, Furukawa K, Kwabi-Addo B,
et al: Gene silencing in cancer by histone H3 lysine 27
trimethylation independent of promoter DNA methylation. Nat Genet.
40:741–750. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bachman KE, Park BH, Rhee I, Rajagopalan
H, Herman JG, Baylin SB, Kinzler KW and Vogelstein B: Histone
modifications and silencing prior to DNA methylation of a tumor
suppressor gene. Cancer Cell. 3:89–95. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau
LA, Whetstine JR and Price BD: Histone H3 methylation links DNA
damage detection to activation of the Tip60 tumor suppressor. Nat
Cell Biol. 11:1376–1382. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
King-Yin Lam A, Ong K and Ho YH:
Colorectal mucinous adenocaecinoma: the clinicopathologic features
and significance of p16 and p53 expression. Dis Colon Rectum.
49:1275–1283. 2006.PubMed/NCBI
|
|
53
|
Kim BN, Yamamoto H, Ikeda K, Damdinsuren
B, Sugita Y, Ngan CY, Fujie Y, Ogawa M, Hata T, Ikeda M, et al:
Methylation and expression of p16INK4 tumor suppressor
gene in primary colorectal cancer tissues. Int J Oncol.
26:1217–1226. 2005.PubMed/NCBI
|
|
54
|
Tada T, Watanabe T, Kazama S, Kanazawa T,
Hata K, Komuro Y and Nagawa H: Reduced p16 expression correlates
with lymphatic invasion in colorectal cancers.
Hepatogastroenterology. 50:1756–1760. 2003.PubMed/NCBI
|
|
55
|
Shim YH, Kang GH and Ro JY: Correlation of
p16 hypermethylation with p16 protein loss in sporadic gastric
carcinomas. Lab Invest. 80:689–695. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lam AK, Ong K, Giv MJ and Ho YH: p16
expression in colorectal adenocarcinoma: marker of aggressiveness
and morphological types. Pathology. 40:580–585. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Palmqvist R, Rutegârd JN, Bozoky B,
Landberg G and Stenling R: Human colorectal cancers with an intact
p16/cyclin D1/pRb pathway have up-regulated p16 expression and
decreased proliferation in small invasive tumor clusters. Am J
Pathol. 157:1947–1953. 2000. View Article : Google Scholar
|