Introduction
Spinal muscular atrophy (SMA) is an autosomal
recessive disease that is a result of the degeneration and
progressive death of α motor neurons in the anterior horns of the
spinal cord and brainstem nuclei (1). SMA is characterized by progressive
muscle weakness, and is the most common genetic cause of infant
mortality (1). SMA presents with an
extremely variable phenotype. The clinical spectrum of SMA is
continuous; however, variations in the age of onset and symptoms
have led to a classification system of the clinical variants of SMA
that is based on maximum patient motor skill achievement. This
classification system has resulted in an improved guideline design
for the clinical management and follow-up of SMA, which is based on
the patient's maximum motor skills (2,3). Sixty
percent of SMA cases are type I, which is characterized by proximal
muscle weakness, affecting the legs most severely, areflexia, and
abdominal breathing that produces a bell-shaped chest deformity
with breathing complications. SMA type I is also known as
Werdnig-Hoffman disease (OMIM, #253,300) and has a prevalence of
4.1/100,000 live births, with patient mortality typically occurring
at ~2 years of age (4). SMA type II,
the intermediate form (OMIM, #253,550) of the disease, has an onset
age of 6–18 months. Patients with SMA type II may be able to sit,
but develop hypotonia, hyporeflexia and lingual fasciculation, and
98% of affected individuals survive to 5 years of age, although
overall lifespan is reduced (2,3). SMA
type III (Kugelberg-Welander type; OMIM, #253,400) has an onset age
of 18 months, and patients affected with this SMA type are able to
sit and walk (5). SMA type IV
presents in adults >21 years; these patients exhibit a normal
acquisition of all fine motor skills prior to the onset of
neuromuscular symptoms and usually have a normal lifespan (6).
The overall prevalence of SMA is 1/6,000–1/10,000,
with a carrier frequency range of 1/40–1/60 (7,8).
Homozygous loss of the survival of motor neuron 1 (SMN1)
gene accounts for 94–95% of all SMA cases. The remaining 5% of
cases consist of compound heterozygotes with the SMN1
deletion in one allele and an alternative mutation in the remaining
allele. This mutational event occurs in <1/4,000 affected
individuals and follows the Hardy-Weinberg equilibrium (9–11). The
chromosomal region 5q11.2–13.3 is susceptible to non-allelic
homologous chromosomal rearrangements. The two variants of the
SMN gene, a telomeric (SMN1 or SMNT) (OMIM,
#600,354) and a centromeric gene (SMN2 or SMNc)
(OMIM, #601,627), differ in a single nucleotide (840C>T), which
results in the alternative splicing of exon 7 in the SMN2
gene in 75–90% of the transcripts produced (2,12–16). The
severity of SMA is primarily determined by the copy number of the
SMN2 gene. The SMN protein forms a complex of molecules that
regulates motoneuron survival by maintaining normal axonal
transport and the growth, maturation and formation of axons and
neurites (17–19).
SMA is diagnosed using various molecular procedures;
the majority are polymerase chain reaction (PCR)-based methods and
rely on an analysis of the gene dosage. In the present study, the
frequency of SMN1 gene deletion was determined in healthy
carriers and non-carriers from northeastern and central Mexican
Mestizo populations. The results may aid in establishing a basis
for implementing carrier screening programs across the country
following further validation.
Materials and methods
This pilot study (no. GN08–005) was conducted at the
Department of Genetics, School of Medicine, Autonomous University
of Nuevo León (Monterrey, Mexico). The study was approved by the
committee for ethics, research and biosecurity of the Autonomous
University of Nuevo León.
A total of 420 individuals aged >18 years, with
parents and four grandparents of Mexican origin, were recruited
from northeastern (n=287) and central Mexico (n=133). Informed
consent was obtained from all participants prior to peripheral
blood collection by venipuncture in accordance with the ethical
guidelines established by the Declaration of Helsinki of the World
Medical Association in 1964 and modified in 1989. Genomic DNA was
obtained from peripheral blood cells using a QIAamp® Blood Mini kit
employing the automated system QIAcube (Qiagen GmbH, Hilden,
Germany).
Molecular detection of the SMN1
deletion
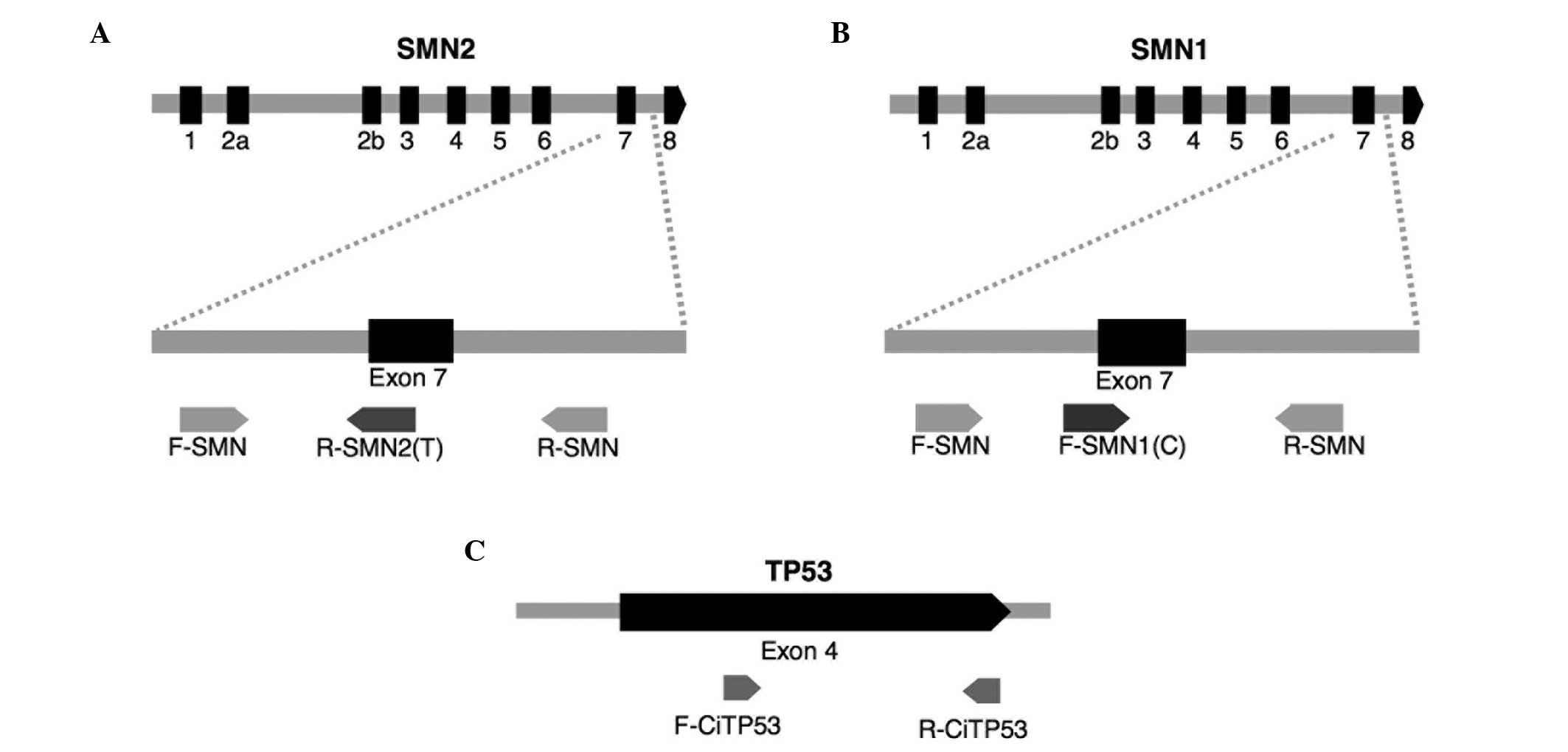
A molecular assay was designed to simultaneously
determine the relative gene dosage of the SMN1 deletion and
SMN2 in relation to the TP53 gene, which was used as
an internal control. This method was based on amplification
refractory mutation system PCR (ARMS-PCR) (20) using six primers and capillary
electrophoresis (Fig. 1). The four
primers (Applied Biosystems Life Technologies, Foster City, CA,
USA) used to amplify the SMN1 and SMN2 genes were as
follows: SMN1, forward 5′-TGT GAA ACA AAA TGC TTT TTA ACA
TCC-3′ and reverse 5′-AAA ACA TTT GTT TTC CAA AAC CAT AAA-3′;
SMN2, forward 5′-FAM-TTC CTT TAT TTT CCT TACAGGG TGT C-3′
and reverse 5′-FAM-CAC CTT CCT TCT TTT TGA TTT TGT ATA-3′. The
primers used to amplify the TP53 gene were as follows:
Forward 5′-GGT CCA GAT GAA GCT CCC AGA AT-3′ and reverse 5′-FAM-TCA
CAG ACT TGG CTG TCC CAG AAT-3′. Primers labeled 5′-FAM were 5′
fluorescently labeled with the fluorophore 6-FAM.
The method was validated by assaying triplicate DNA
samples from six individuals with SMA type I that were homozygous
for the deletion of the SMN1 gene, based on a
PCR-restriction fragment length polymorphism analysis. In addition,
samples from healthy carriers of the deletion were used in the
validation assay.
PCR procedure
PCR reactions were conducted in a reaction system
containing 20 ng genomic DNA, 0.031 µM TP53 primers, 0.038
µM forward and 0.42 µM reverse external primers for
SMN1/SMN2, 0.022 µM primer specific to the ‘C’
nucleotide (c.840C of SMN1), 0.030 µM primer specific to the
‘T’ nucleotide (c.840T of SMN2), 0.75 U GoTaq DNA polymerase
(Promega Corporation, Madison, WI, USA), 1.5 mM MgCl2
and 0.3 mM dNTPs (Promega Corporation), with a final volume of 15
µl. The cycling procedure included one cycle of initial
denaturation at 95°C for 1 min, 29 cycles of denaturation at 95°C
for 40 sec, annealing at 53°C for 40 sec and extension at 72°C for
1 min; and a final 72°C extension step for 20 min. Immediately, 1
µl of the amplicon was mixed with 0.3 µl GeneScan™-500 LIZ® Size
Standard (Applied Biosystems Life Technologies) and 8.7 µl
formamide. This mixture was placed in a Hybaid PCR Express HBPX110
thermocycler (Thermo Fisher Scientific, Renfrew, UK) for 5 min at
95°C, then at −20°C for 5 min. Following denaturation, the samples
were analyzed using a 3100 Avant Genetic Analyzer (Applied
Biosystems Life Technologies).
Estimation of gene copy number
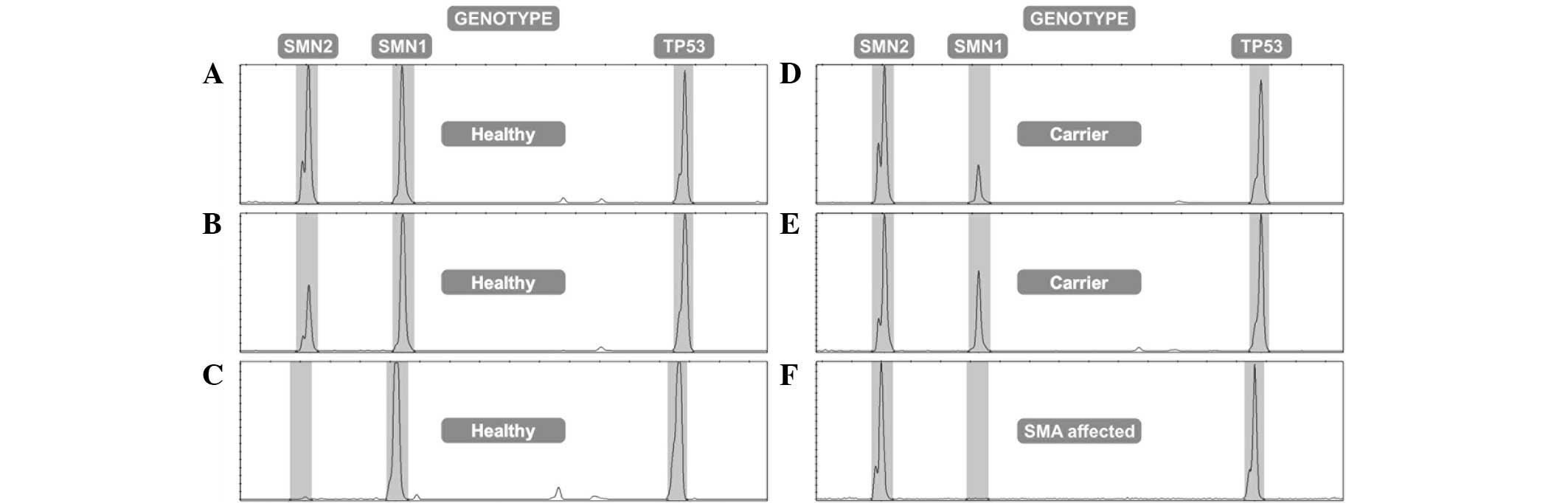
Three peaks corresponding to the SMN2 (135
bp), SMN1 (149 bp) and TP53 (194 bp) genes were
obtained using electropherograms generated by ABI Prism 3100 AVANT
genetic analyzer (Applied Biosystems Life Technologies) (Fig. 2). The area of each peak was
determined based on the GeneMapper software, version 3.1 with a
peak detection algorithm (Applied Biosystems Life Technologies) and
following a previously described protocol (21) with modifications (allele size
detection adjusted for SMN1, SMN2 and TP53
amplicons). The ratio of the peak area of the SMN1 gene to
the TP53 gene represented the relative copy number of the
target gene SMN1. For this analysis, a TP53 gene
dosage of 100% (two gene copies of TP53) was assumed to be
present in all subjects, as none presented with Li-Fraumeni
syndrome (22). Based on this
analysis, subjects with at least two copies of SMN1 were
considered to be healthy non-carriers, those with one copy of
SMN1 were considered true carriers, and those with no copies
of the SMN1 gene were considered to be affected with SMA. A
limitation of this detection method is that it did not distinguish
whether the two copies of the SMN1 gene were in cis or
trans.
Statistical analysis
Fisher's exact test was applied to compare the
northeastern and central Mexican Mestizo populations. The
χ2 test was applied to compare the carrier frequency
among the populations in the present study, worldwide populations
and the Hispanic population.
Data were analyzed using SPSS statistical software,
version 22.0 (IBM SPSS, Armonk, NY, USA). P<0.05 was considered
to indicate a statistically significant difference.
Results
A total of 420 DNA samples were obtained from male
and female participants aged 18–71 years, with four grandparents of
Mexican descent. Among this population, 68.33% (n=287) were from
northeastern and 31.67% (n=133) from central Mexico. The
SMN1 copy number was standardized using DNA from affected
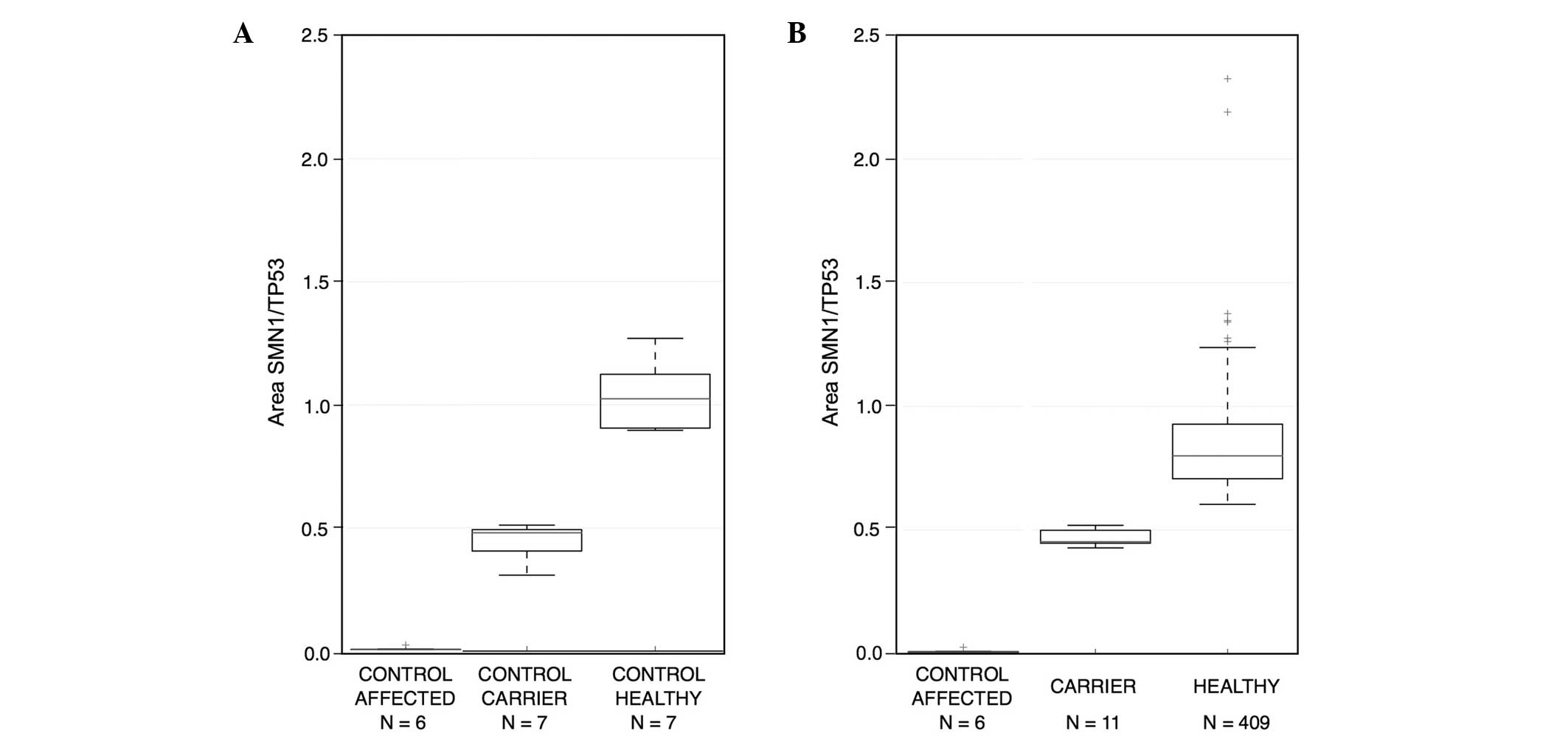
and healthy carrier individuals. The peak electropherogram area of
the SMN1 gene in the affected patients was 0.00–0.05,
indicating a mean loss of the SMN1 gene among homozygous
carriers. In heterozygous carriers, the range of the peak area was
0.3–0.52, representing half of the subjects. The range was >0.6
for healthy individuals, which indicated complete gene dosage
(Fig. 3). If it was not possible to
determine whether a sample corresponded to a carrier or a
non-carrier (SMN1/TP53 ratio between 0.52–0.6), the
sample was reanalyzed in triplicate, and the mean of the three
measurements was used.
Among the study population, 11 subjects (2.62%)
carried one copy of SMN1 and 409 subjects (97.38%) carried
two copies (Table I). Nine of the
single-copy carriers (3.14%) were from northeastern and two (1.50%)
were from central Mexico. No significant difference was observed
between these groups using two-tailed Fisher's exact test
(P=0.5143; Table I). The overall
frequency of carriers among the two Mexican population groups was
11/420 (or 1/38). Thus, the carrier frequency detected in the
present pilot study was 2.62%.
 | Table I.Frequencies of SMN1 gene
deletion carriers. |
Table I.
Frequencies of SMN1 gene
deletion carriers.
|
| Northeastern | Central | Total |
|---|
|
|
|
|
|
|---|
| Carrier status | n | % | n | % | n | % |
|---|
| Carriers | 9 | 3.14 | 2 | 1.50 | 11 | 2.62 |
| Non-carriers | 278 | 96.86 | 131 | 98.50 | 409 | 97.38 |
| Total | 287 |
| 133 |
| 420 |
|
Discussion
The results of the present pilot study indicate that
the carrier frequency of SMN1 in northeastern and central
Mexico is 1/38, which is comparable to the reported frequencies in
other ethnic populations, including African-American, Asian,
Korean, Jewish and Israeli populations (Table II). However, the SMA carrier
frequency in this Mexican population is significantly higher
compared with that in a Hispanic population from the USA previously
examined by Hendrickson et al (4). In the northeastern Mexican population,
the frequency detected was 1/32 (3.14%).
| Table II.Distribution of carrier frequencies
with an SMN1 deletion among nine populations worldwide and
in the present study. |
Table II.
Distribution of carrier frequencies
with an SMN1 deletion among nine populations worldwide and
in the present study.
|
| Carriers |
|
|
|
|---|
|
|
|
|
|
|
|---|
| Population | Yes (%) | No (%) | Total | Method | Reference |
|---|
| Mexican | 11 (2.62) | 409 (97.38) | 420 | PCR-ARMS | Present study |
| Hispanic | 8 (0.8) | 1,022 (99.2) | 1,030 | RT-qPCR | Hendrickson et
al, 2009 (4) |
|
African-American | 18 (1.8) | 997 (98.2) | 1,015 | RT-qPCR | Hendrickson et
al, 2009 (4) |
| Asian | 18 (1.8) | 1,009 (98.2) | 1,027 | RT-qPCR | Hendrickson et
al, 2009 (4) |
| Korean | 2 (2.0) | 98 (98.0) | 100 | MLPA | Yoon et al,
2010 (19) |
| Jewish | 22 (2.2) | 980 (97.8) | 1,002 | RT-qPCR | Hendrickson et
al, 2009 (4) |
| Israeli | 159 (2.5) | 6,235 (97.5) | 6,394 | MLPA | Ben-Shachar et
al, 2011 (23) |
| Caucasian | 28 (2.7) | 1,000 (97.3) | 1,028 | RT-qPCR | Hendrickson et
al, 2009 (4) |
| Arabian | 2 (4.0) | 48 (96.0) | 50 | Multiplex-PCR | Majumdar et
al, 2005 (8) |
| Iranian | 10 (5.0) | 190 (95.0) | 200 | qPCR | Hasanzad et
al, 2009 (9) |
| Total | 278 | 12,080 | 12,358 |
|
|
In Hispanic populations in the USA, the SMA carrier
frequency is comparatively low, at 1/125 (24,25).
However, the term Hispanic is used as a general term to refer to
all persons of Hispanic descent and encompasses a diverse group of
individuals (26). A previous study
of 7,655 Hispanic individuals in the USA, in which family history
and ethnicity was considered (27),
observed that SMN1 one-copy frequency did not differ
significantly from that observed in an existing study of 1,030
individuals (P=0.1869) (4). A recent
study demonstrated that the Mexican population is genetically
diverse. Therefore, detailed studies of population structure,
including geographical data, may be required in order to assess the
frequency and prevalence of genetic diseases in native and Mestizo
Mexican populations (28).
The universal implementation of expanded newborn
screenings for metabolic inherited diseases has been tailored to
specific populations and countries. If the carrier status of an
individual for a genetic disease is known, several options are
available: i) Choice of a partner who is a non-carrier; ii)
pre-implantation diagnosis; iii) prenatal diagnosis; or iv)
acceptance of the 25% probability of a child being affected. In
cases of infants born affected by the disease, early recognition
allows for more effective treatment, which justifies the use of SMA
as a target disease for population screening (6,29,30). The
identification of SMA carriers may facilitate a reduction in the
prevalence of SMA.
To the best of our knowledge, the frequency of SMA
carriers in the Mexican populations investigated in the present
study has not been reported previously. A carrier frequency of 1/38
(2.62%) was determined in the present study. No significant
differences in carrier frequency were identified between
populations from the northeastern and central regions of Mexico.
Therefore, the frequency of the deletion of the SMN1 gene
remains homogeneous across the Mexican populations examined.
Further studies with larger sample sizes may provide an improved
understanding of whether this frequency distribution is
representative of the entire Mexican population; however, this
pilot study provides an initial approximation of the carrier
frequency for Mexican individuals. Furthermore, these results may
be used to improve evaluation procedures, and thus justify future
studies on a larger scale to validate neonatal screening in the
Mexican population. It is hypothesized that carrier testing may be
a useful technique to prevent SMA, which frequently results in
mortality and is currently untreatable. Similar studies have been
conducted in other populations, including Israeli, Caucasian,
Ashkenazi Jewish, Asian and African-American populations (Table II).
The present study is relevant to the study of
carrier and patient frequencies in mixed populations of complex
ethnic components. The Mexican population is a convenient model for
genetic studies due to its notable ethnic diversity in native and
Mestizo populations. A previous study reported significantly
different ancestry between individuals from separate geographic
regions in Mexico (28). In order to
avoid errors such as stratification, false negatives and
irreproducibility, the results of the present study may be used as
a reference in genetic studies of gene frequency, prevalence and
association in characterized populations. In the present study of
SMA carrier frequency in the northeastern and central Mexico
populations, no statistically significant differences were
observed; however, further studies are required to evaluate the
remaining regions of Mexico.
The technique applied in the present study is an
alternative to existing molecular tests to determine the copy
number of the SMN1 gene in order to detect SMA carriers and
affected individuals.
Acknowledgements
The authors acknowledge the medical students Alan
Ureña, Dolores Álvarez and Fabiola Yazmín Agüero Zapata for their
collaborative contribution in recovering patient samples, Mauricio
de la Rosa Garza for technical support, and Celia Nohemi Sanchez
Dominguez for critically reviewing the manuscript. The authors
thank Dr Guillermo Barrera Jr for database management. The present
study was partially funded by a grant from CONACYT/2008/SNI and by
the Research Department of the Autonomous University of Nuevo León.
American Journal Experts reviewed the grammar of the
manuscript.
References
|
1
|
Markowitz JA, Singh P and Darras BT:
Spinal muscular atrophy: A clinical and research update. Pediatr
Neurol. 46:1–12. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Scheffer H, Cobben JM, Matthijs G and
Wirth B: Best practice guidelines for molecular analysis in spinal
muscular atrophy. Eur J Hum Genet. 9:484–491. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Prior TW: Perspectives and diagnostic
considerations in spinal muscular atrophy. Genet Med. 12:145–152.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hendrickson BC, Donohoe C, Akmaev VR, et
al: Differences in SMN1 allele frequencies among ethnic groups
within North America. J Med Genet. 46:641–644. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jedrzejowska M, Milewski M, Zimowski J, et
al: Phenotype modifiers of spinal muscular atrophy: The number of
SMN2 gene copies, deletion in the NAIP gene and probably gender
influence the course of the disease. Acta Biochim Pol. 56:103–108.
2009.PubMed/NCBI
|
|
6
|
Monani UR and De Vivo DC:
Neurodegeneration in spinal muscular atrophy: From disease
phenotype and animal models to therapeutic strategies and beyond.
Future Neurol. 9:49–65. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ogino S, Leonard DG, Rennert H, Ewens WJ
and Wilson RB: Genetic risk assessment in carrier testing for
spinal muscular atrophy. Am J Med Genet. 110:301–307. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Majumdar R, Rehana Z, Al Jumah M and
Fetaini N: Spinal muscular atrophy carrier screening by multiplex
polymerase chain reaction using dried blood spot on filter paper.
Ann Hum Genet. 69:216–221. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hasanzad M, Golkar Z, Kariminejad R, et
al: Deletions in the survival motor neuron gene in Iranian patients
with spinal muscular atrophy. Ann Acad Med Singapore. 38:139–141.
2009.PubMed/NCBI
|
|
10
|
Hasanzad M, Azad M, Kahrizi K, et al:
Carrier frequency of SMA by quantitative analysis of the SMN1
deletion in the Iranian population. Eur J Neurol. 17:160–162. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dobrowolski SF, Pham HT, Downes FP, Prior
TW, Naylor EW and Swoboda KJ: Newborn screening for spinal muscular
atrophy by calibrated short-amplicon melt profiling. Clin Chem.
58:1033–1039. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wirth B, Herz M, Wetter A, et al:
Quantitative analysis of survival motor neuron copies:
Identification of subtle SMN1 mutations in patients with spinal
muscular atrophy, genotype-phenotype correlation and implications
for genetic counseling. Am J Hum Genet. 64:1340–1356. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tizzano E and Baiget M: Molecular basis of
spinal muscular atrophy: The SMN gene. Neurologia. 15:393–400.
2000.(In Spanish). PubMed/NCBI
|
|
14
|
Sumner CJ: Therapeutics development for
spinal muscular atrophy. NeuroRx. 3:235–245. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Singh NN and Singh RN: Alternative
splicing in spinal muscular atrophy underscores the role of an
intron definition model. RNA Biol. 8:600–606. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu-Jin Q, Juan D, Er-zhen L, et al: Subtle
mutations in the SMN1 gene in Chinese patients with SMA:
p.Arg288Met mutation causing SMN1 transcript exclusion of exon7.
BMC Med Genet. 13:862012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Monani UR, McPherson JD and Burghes AH:
Promoter analysis of the human centromeric and telomeric survival
motor neuron genes (SMNC and SMNT). Biochim Biophys Acta.
1445:330–336. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Anhuf D, Eggermann T, Rudnik-Schöneborn S
and Zerres K: Determination of SMN1 and SMN2 copy number using
TaqMan technology. Hum Mutat. 22:74–78. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yoon S, Lee CH and Lee KA: Determination
of SMN1 and SMN2 copy numbers in a Korean population using
multiplex ligation-dependent probe amplification. Korean J Lab Med.
30:93–96. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ye S, Dhillon S, Ke X, Collins AR and Day
IN: An efficient procedure for genotyping single nucleotide
polymorphisms. Nucleic Acids Res. 29:E88. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cheng CY, Kao WH, Patterson N, et al:
Admixture mapping of 15,280 African Americans identifies obesity
susceptibility loci on chromosomes 5 and X. PLoS Genet.
5:e10004902009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guthrie PA, Gaunt TR, Abdollahi MR, et al:
Amplification ratio control system for copy number variation
genotyping. Nucleic Acids Res. 39:e542011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ben-Shachar S, Orr-Urtreger A, Bardugo E,
Shomrat R and Yaron Y: Large-scale population screening for spinal
muscular atrophy: Clinical implications. Genet Med. 13:110–114.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wirth B: An update of the mutation
spectrum of the survival motor neuron gene (SMN1) in autosomal
recessive spinal muscular atrophy (SMA). Hum Mutat. 15:228–237.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Muralidharan K, Wilson RB, Ogino S, Nagan
N, Curtis C and Schrijver I: Population carrier screening for
spinal muscular atrophy a position statement of the association for
molecular pathology. J Mol Diagn. 13:3–6. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics for Hispanics/Latinos, 2012. CA: Cancer J Clin.
62:283–298. 2012.PubMed/NCBI
|
|
27
|
Sugarman EA, Nagan N, Zhu H, et al:
Pan-ethnic carrier screening and prenatal diagnosis for spinal
muscular atrophy: Clinical laboratory analysis of >72,400
specimens. Eur J Hum Genet. 20:27–32. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Moreno-Estrada A, Gignoux CR,
Fernández-López JC, et al: Human genetics. The genetics of Mexico
recapitulates Native American substructure and affects biomedical
traits. Science. 344:1280–1285. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Basel-Vanagaite L, Taub E, Drasinover V,
et al: Genetic carrier screening for spinal muscular atrophy and
spinal muscular atrophy with respiratory distress 1 in an isolated
population in Israel. Genet Test. 12:53–56. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Arkblad E, Tulinius M, Kroksmark AK,
Henricsson M and Darin N: A population-based study of genotypic and
phenotypic variability in children with spinal muscular atrophy.
Acta Paediatr. 98:865–872. 2009. View Article : Google Scholar : PubMed/NCBI
|