Introduction
Advanced glycation end products (AGEs) result from
the Maillard reaction, which is a non-enzymatic, irreversible
process (1). Some studies suggest
that AGEs accelerate atherosclerosis in type-2 diabetic patients
with coronary heart disease (2,3).
In diabetic patients, previous studies have shown that
proliferation of vascular smooth muscle cells (VSMCs), a key factor
in the development of atherosclerotic lesions, are significantly
stimulated by the accumulation of AGEs and their interaction with
the receptor for advanced glycation end products (RAGE) (4,5).
Activation of RAGE not only accelerates early lesion formation but
sustains lesion progression in the diabetic apoE-null mouse model
(6,7).
Autophagy is an evolutionarily conserved process
involving degradation of long-lived proteins. It is important for
balancing sources of energy at critical times (8,9).
There are many genes involved in the process of autophagy. Among
them, the microtubule-associated protein 1 light chain 3 (LC3) is
critical to autophagosome formation. When autophagy is induced, the
cytoplasmic form of LC3 (LC3-I) becomes membrane-associated
(LC3-II). LC3-II has been used extensively as a marker protein for
autophagy (10). In advanced
atherosclerotic plaques, autophagy is notably activated by several
pathological conditions, such as oxidized lipids, inflammation,
oxidative stress and metabolic stress conditions (11–13). However, the relationship between
AGEs and autophagy in atherosclerotic plaques is rarely reported.
Therefore, we hypothesized that autophagy is a pathological
mechanism involved in AGE-accelerated atherosclerosis, especially
in AGE-mediated proliferation of VSMCs.
Autophagy involves both the Akt and mitogen
activated protein kinase (MAPK) pathways. The ERK pathway, as one
of MAPK family members, phosphorylates the Gα-interacting protein
to accelerate the rate of GTP hydrolysis to induce autophagy
(14). In response to starvation,
inflammation and oxidative stress, the PI3K/Akt/mTOR pathway
negatively regulates autophagy in VSMCs (15).
The relationship between AGEs and autophagy has not
been fully elucidated. To study the underlying mechanisms of
AGE-induced autophagy, we examined the activation and the function
of autophagy in rat A7R5 VSMCs treated with AGEs.
Materials and methods
Materials
Monoclonal rabbit anti-Beclin-1 antibody was
obtained from Epitomics (CA, USA). Polyclonal rabbit anti-LC3B
antibody was purchased from Novus (CO, USA). Monoclonal rabbit
antibodies including anti-Erk, anti-phospho-Erk, anti-JNK,
anti-phospho-JNK, anti-p38 and anti-phospho-p38, anti-AKT,
anti-phospho-AKT, anti-mTOR and anti-phospho-mTOR were obtained
from Cell Signaling Technology (MA, USA). Polyclonal rabbit
anti-RAGE antibody was purchased from Millipore (Boston, MA, USA).
The MTT, BSA, 3-MA, MAPK inhibitors including PD98059, SP600125 and
SB203580, were obtained from Sigma (St. Louis, MO, USA). HRP-marked
anti-GAPDH antibody was purchased from Kangchen (Shanghai, China).
Rat IGF-1 was obtained from R&D (MN, USA). RAGE RNAi was
designed and purchased from Shanghai GenePharma (Shanghai,
China).
Cell culture
Rat A7R5 vascular smooth muscle cells (BioHermes,
China) were cultured in low-glucose Dulbecco's modified Eagle's
medium (Gibco, USA) supplemented with 10% fetal bovine serum
(Sijiqing, China) in a humidified 5% CO2/95% air
atmosphere at 37°C.
Preparation of AGEs
AGEs were prepared as previously reported (16). Briefly, BSA was incubated with 0.5
M glucose in phosphate-buffered saline (PBS) in the dark for 16
weeks at 37°C. The unincorporated sugars were removed by dialyzing
against PBS (pH 7.4). Control nonglycated BSA was incubated in the
absence of glucose under the same conditions. Endotoxin levels were
checked using an endotoxin testing kit (Chromogenic TAL Endpoint
Assay kit, China). The AGE-BSA solutions were confirmed to be
endotoxin free (<2.5 U/ml of endotoxin).
Western blot analysis
Cells were solubilized in a lysis buffer containing
50 mM Tris (pH 7.4), 150 mM NaCl, 1% NP-40, 5% deoxycholic acid,
0.1% SDS, 1 mM EDTA, 10 mM NaF, 1 mM Na3VO4,
1 mM dithiothreitol, 1 mM PMSF, 2 μg/ml leupeptin for 30 min. Total
protein concentrations were measured by a BCA Protein Assay kit
(Applygen Technologies Inc., China). After the samples were
heat-denatured, they were analyzed on a 10 or 15% trisglycine
gradient gel, transferred to PVDF membranes and blocked with 5%
nonfat milk in Tris-buffered solution (TBS) for 1 h at room
temperature. The membranes were incubated with primary antibody
overnight at 4°C. After being washed three times, membranes were
incubated with secondary antibodies for 1.5 h at room temperature.
Antibodies were detected by enhanced chemiluminescence (ECL)
reagents and imaged using an Image Quant LAS-4000 (Fujifilm, Tokyo,
Japan). The band densities were determined using Multi-Gauge
Software (Fujifilm).
Immunofluorescence
Cells were fixed with 4% paraformaldehyde at room
temperature for 15 min. After washing with PBS three times, cells
were permeabilized with 0.25% Triton X-100 in PBS for 5 min, and
then incubated in a blocking buffer containing 10% goat serum,
followed by incubation with anti-LC3 antibody (1:200) in PBS
containing 10% goat serum overnight at 4°C. After incubating with
0.1% DAPI for 5 min at room temperature and another washing step
with PBS, secondary Rhodanmine Red-X labelled antibody (1:100) was
applied for 60 min. After washing with PBS, coverslips were
transferred onto glass slides. Images were captured on a wide-field
fluorescent microscopy (Zeiss).
Electron microscopy
Ultrastructural analysis was performed to examine
autophagy. Rat A7R5 VSMCs were grown in 6-well plates, treated with
100 μg/ml AGEs for 6 h, fixed with a solution containing 3%
glutaraldehyde and then sent to Zhejiang University for electron
microscopic analysis.
RNA interference
For function-blocking experiments, we used small
interfering RNA molecules (siRNA) targeted to RAGE mRNA. RAGE siRNA
was purchased from Shanghai GenePharma. The siRNA was designed
against RAGE (sense, 5′-GCCGGAAAUUGUGAAUCCUTT-3′; antisense,
5′-AGGA UUCACAAUUUCCGGCTT-3′). The negative control siRNA was
non-targeting (sense, 5′-UUCUCCGAACGUGUCACG UTT-3′; antisense,
5′-ACGUGACACGUUCGGAGAATT-3′). Cells were transfected with si-RAGE
using Lipofectamine 2000 (Invitrogen, USA) for 48 h according to
the manufacturer's instructions. The final concentration of siRNA
was 100 pM. The efficacy of RNA interference was determined by
Western blotting.
Measurement of proliferation of
VSMCs
To assess cell proliferation, rat A7R5 VSMCs were
plated in a 96-well plate. After 24 h, the medium was changed, and
the cells were incubated with fresh medium containing AGEs (100
μg/ml) or 3-MA (2 mM) for another 48 h. Then, 20 μl of MTT solution
(final concentration, 5 mg/ml was added to each well for 4 h at
37°C. The medium was then discarded and 100 μl of DMSO was added to
each well. The absorbance was measured at 490 nm.
Statistical analysis
All data were obtained from at least 3 individual
experiments. Values are expressed as the mean ± SEM. Statistical
analysis between groups was performed by one-way ANOVA. The
statistical significance was set at p<0.05.
Results
AGE-induced autophagy in rat A7R5
VSMCs
To determine whether AGEs can affect the level of
autophagy in VSMCs, cells were treated with AGEs or BSA (100 μg/ml)
for various times (0, 0.5, 1, 2, 6, 12 and 24 h). The expression of
LC3-II and the ratio of LC3-II to LC3-I were significantly
increased after treatment with AGEs, peaking at 6 h. In contrast,
treatment with BSA did not change the expression of LC3-II or the
LC3-II to LC3-I ratio (Fig. 1A).
Cells were also treated with AGEs or BSA at various concentrations
(0, 1, 10 and 100 μg/ml) for 6 h. The expression of LC3-II and the
LC3-II to LC3-I ratio were notably increased in a dose-dependent
manner in AGE-treated cells (Fig.
1B).
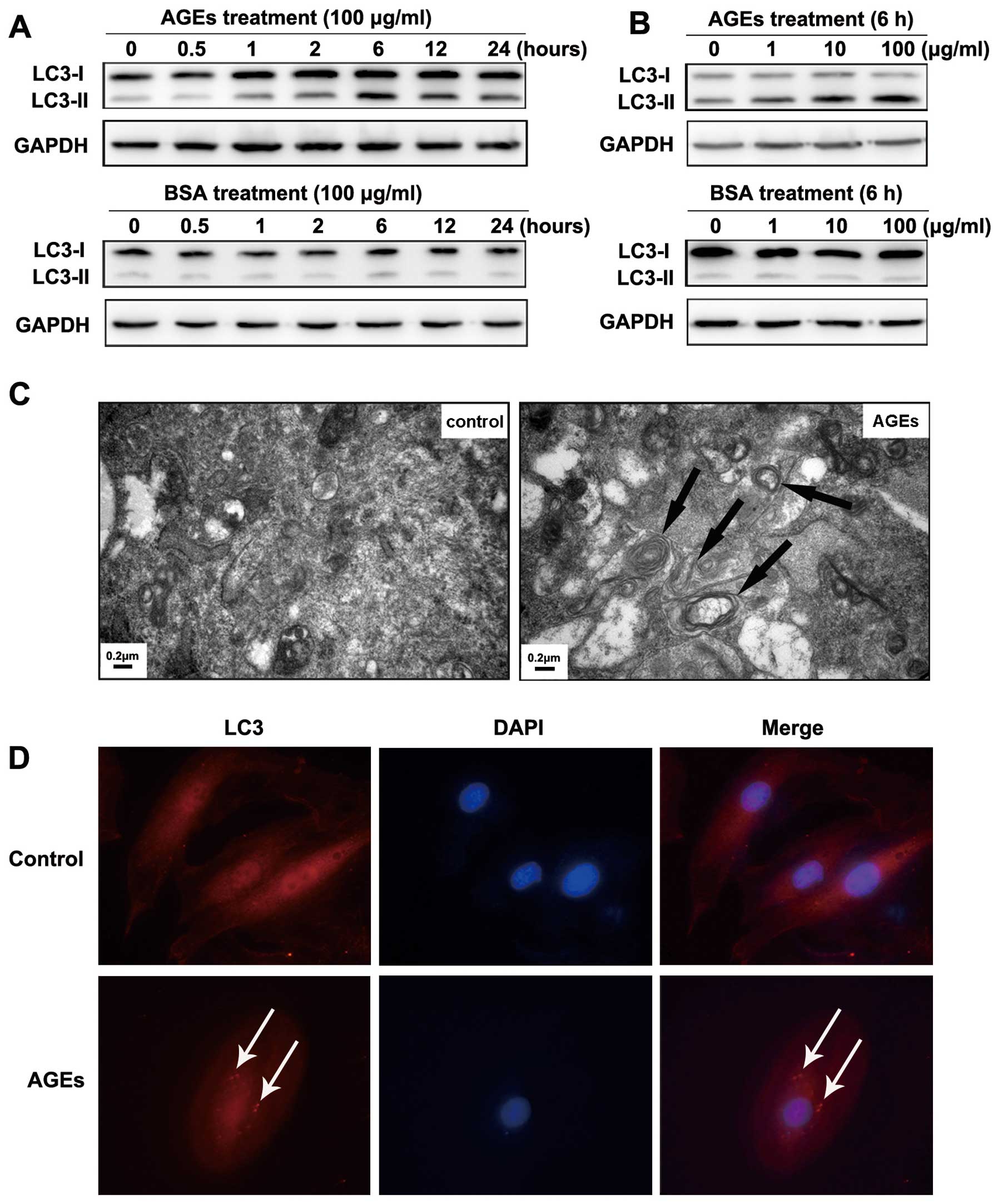 | Figure 1AGE-induced autophagy in A7R5 VSMCs
and autophagy is involved in AGE-induced proliferation of VSMCs.
(A) Western blot analysis of LC3-I and LC3-II protein levels
treated with BSA or AGEs. Cells were treated with 100 μg/ml BSA or
AGEs for 0, 0.5, 1, 2, 6, 12 and 24 h. (B) Cells were treated for 6
h with 0, 1, 10, 100 μg/ml BSA or AGEs. The results show a time-
and dose-dependent effect of AGEs treatment on the expression of
LC3-II and the LC3-II to LC3-I ratio. (C) Representative electron
micrographs of VSMCs treated with 100 μg/ml BSA or AGEs for 6 h.
Typical autophagic vacuoles containing cellular material or
membranous structures (bold arrows) were frequently found in cells
treated with AGEs but not BSA. (D) The localization of LC3 as
detected by immunofluorescence. In BSA-treated cells, LC3 was
distributed homogeneously in the cytoplasm, whereas the cells
treated with AGEs showed LC3 dots (red fluorescence) around the
nucleus (blue fluorescence). |
To directly visualize autophagy, we used
transmission electron microscopy to examine autophagic vacuoles
(autophagosomes). We treated cells with 100 μg/ml BSA or AGEs for 6
h. In BSA-treated cells, autophagic vacuoles were rarely detected.
However, we found that autophagic vacuoles containing cellular
material or membranous structures were increased in AGE-treated
cells (bold arrows in Fig.
1C).
To examine the localization of autophagosomes in rat
A7R5 VSMCs, we detected the autophagosome-specific protein LC3 (red
fluorescence) by immunofluorescence imaging. Cells were treated
with 100 μg/ml BSA or AGEs for 6 h. In BSA-treated cells, LC3 was
distributed homogeneously in the cytoplasm. In contrast, LC3 was
found in dots around the nucleus in cells treated with AGEs (blue
fluorescence) (indicated by white arrows in Fig. 1D).
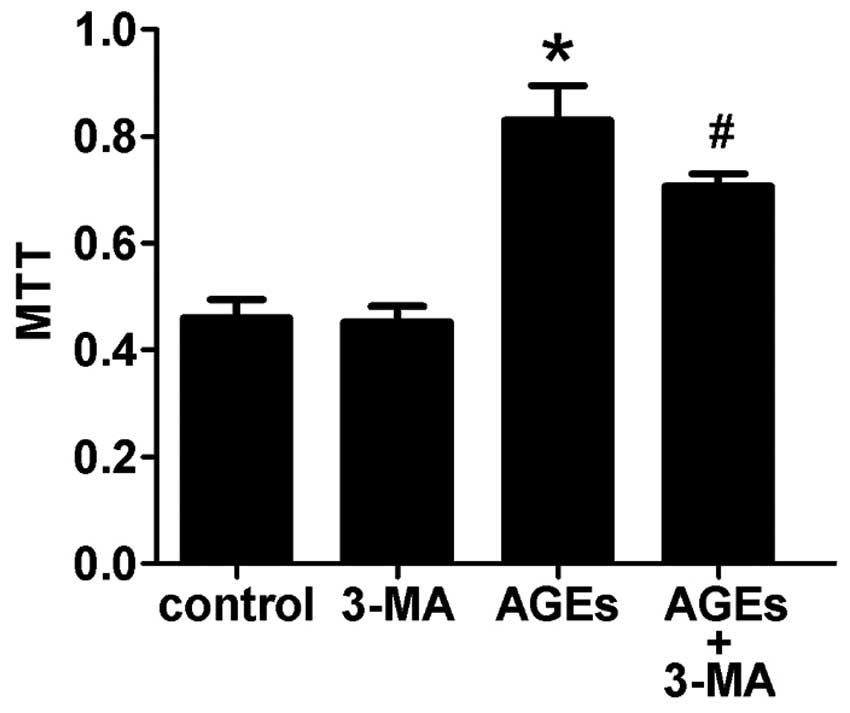
Autophagy is involved in AGE-induced
proliferation
Compared with the control group, cells treated with
100 μg/ml AGEs for 48 h showed increased proliferation of VSMCs.
Furthermore, pretreating cells with 3-MA, an autophagy inhibitor,
for 30 min could attenuate this effect (Fig. 2). This indicates that autophagy is
involved in AGE-induced proliferation of VSMCs.
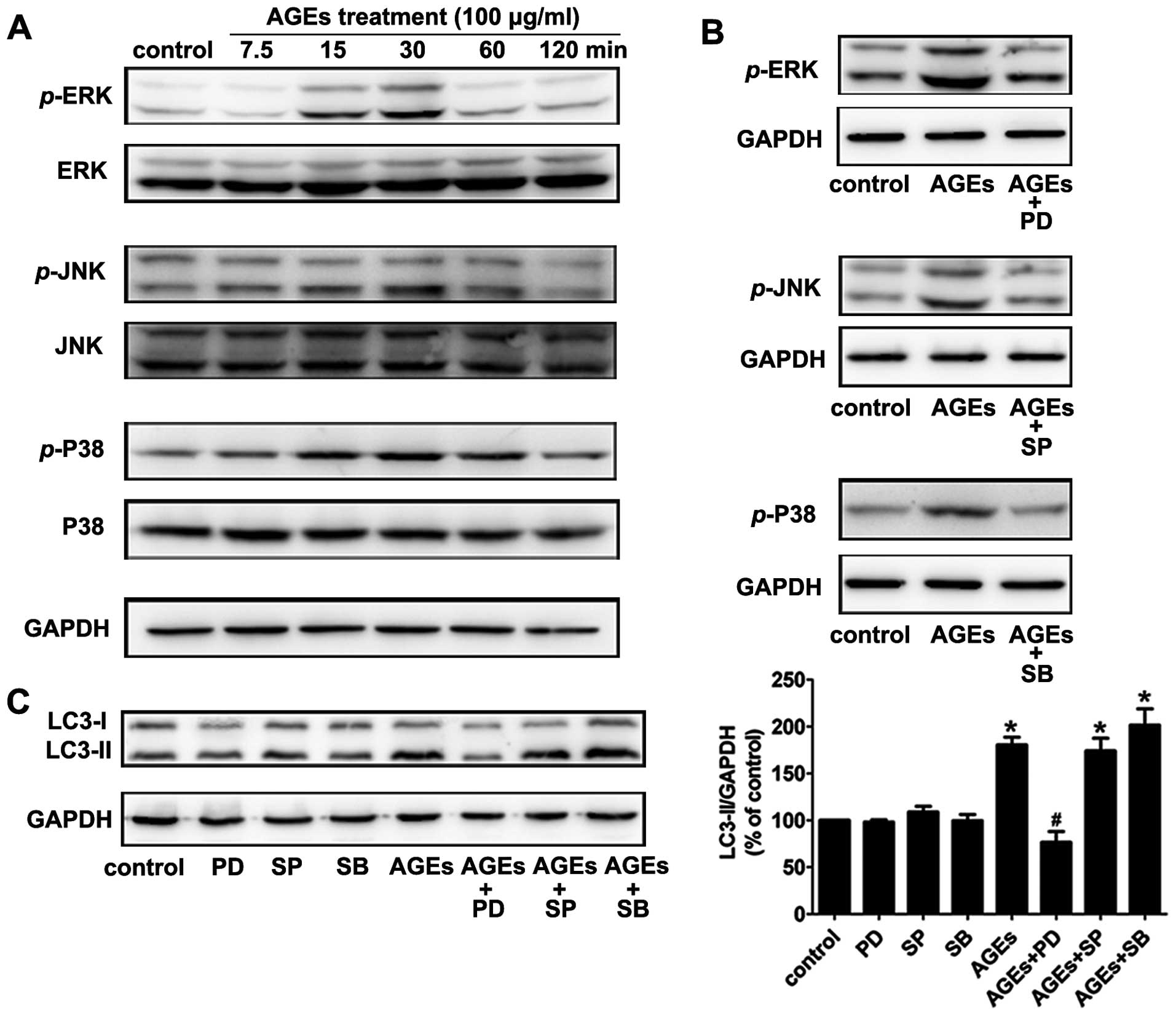
The ERK pathway is involved in
AGE-induced autophagy
MAPK pathways are important for cells to respond to
numerous extracellular signals. To investigate the mechanisms
involved in AGE-induced autophagy, we examined the phosphorylation
level of various MAPK family proteins (ERK, p38, JNK) in VSMCs by
western blotting. Cells were treated with 100 μg/ml AGEs for 0,
7.5, 15, 30, 60 and 120 min. As is shown in Fig. 2, AGEs stimulated phosphorylation
of MAPKs (ERK, p38, JNK) in rat A7R5 VSMCs in a time-dependent
manner. Phosphorylation peaked at 15–30 min and then declined
(Fig. 3A). Pretreating cells with
the ERK inhibitor PD98059 (20 μM) for 30 min blocked 90% of ERK
activation. The p38 MAPK inhibitor SB203580 (10 μM) and JNK MAPK
inhibitor SP600125 (20 μM) also blocked the activation of the
corresponding MAPKs (Fig.
3B).
In our experiments, the ERK inhibitor PD98059, but
not the p38 inhibitor SB203580 or JNK inhibitor SP600125,
suppressed the AGE-induced expression of LC3-II (Fig. 3C). The results indicate that the
ERK signal transduction pathway is involved in AGE-induced
autophagy.
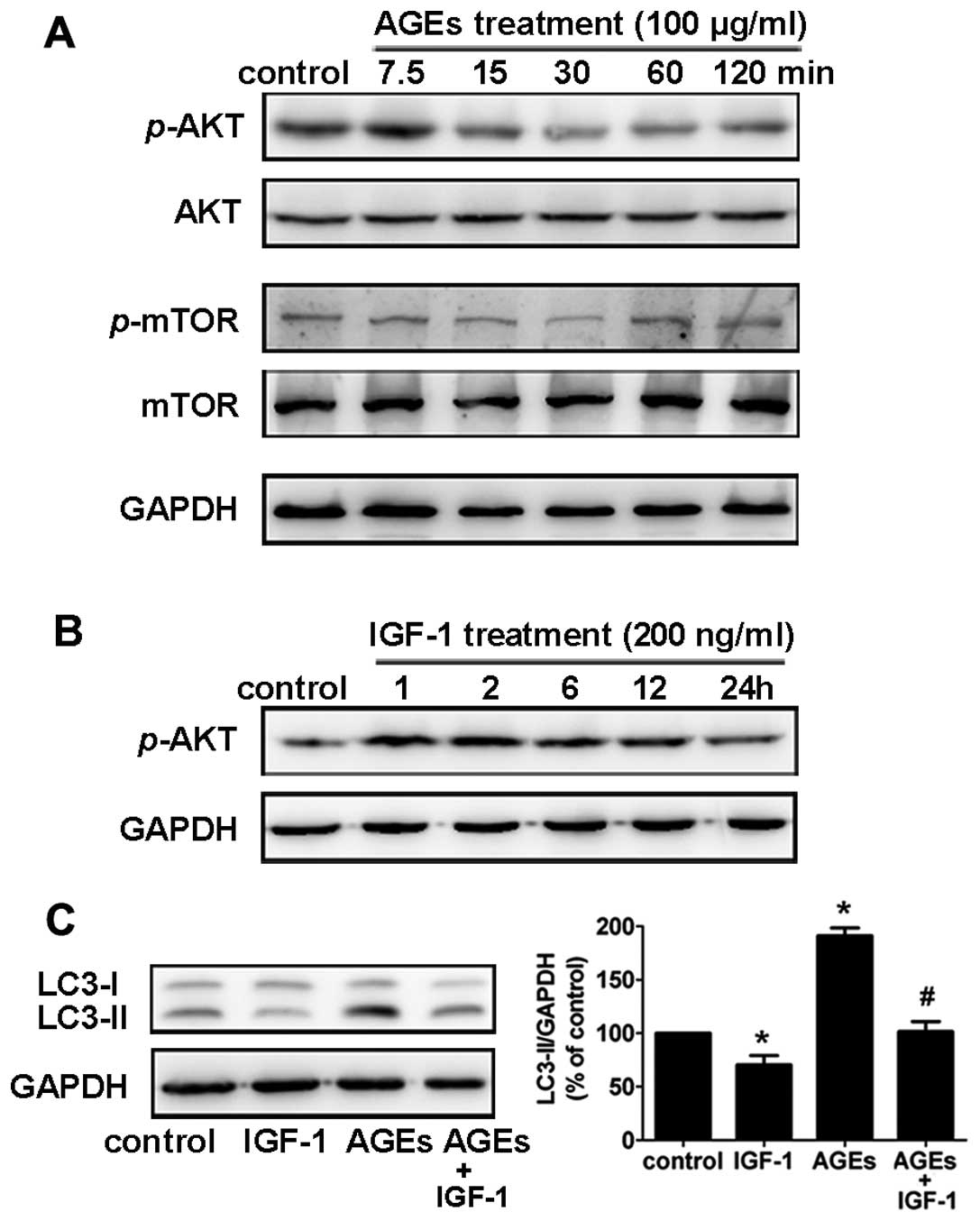
The Akt/mTOR signaling pathway is
involved in AGE-induced autophagy
The Akt/mTOR signaling pathway, which promotes cell
growth and survival in response to mitogenic signals, is a major
pathway that negatively regulates autophagy. In rat A7R5 VSMCs,
phosphorylation of Akt and mTOR decreased 30 min to 2 h after
treatment with 100 μg/ml AGEs (Fig.
4A). To examine the role of the Akt/mTOR pathway in AGE-induced
autophagy, we used insulin-like growth factor 1 (IGF-1) to activate
the Akt pathway (17). We found
that rat A7R5 VSMCs pretreated with IGF-1 (200 ng/ml) for 1 h
showed a notable increase in Akt phosphorylation (Fig. 4B).
In addition, pretreatment with IGF-1 (200 ng/ml)
suppressed the AGE-induced LC3-II expression in Rat A7R5 VSMCs
(Fig. 4C). This result suggests
that the Akt/mTOR signaling pathway is involved in AGE-induced
autophagy in rat A7R5 VSMCs.
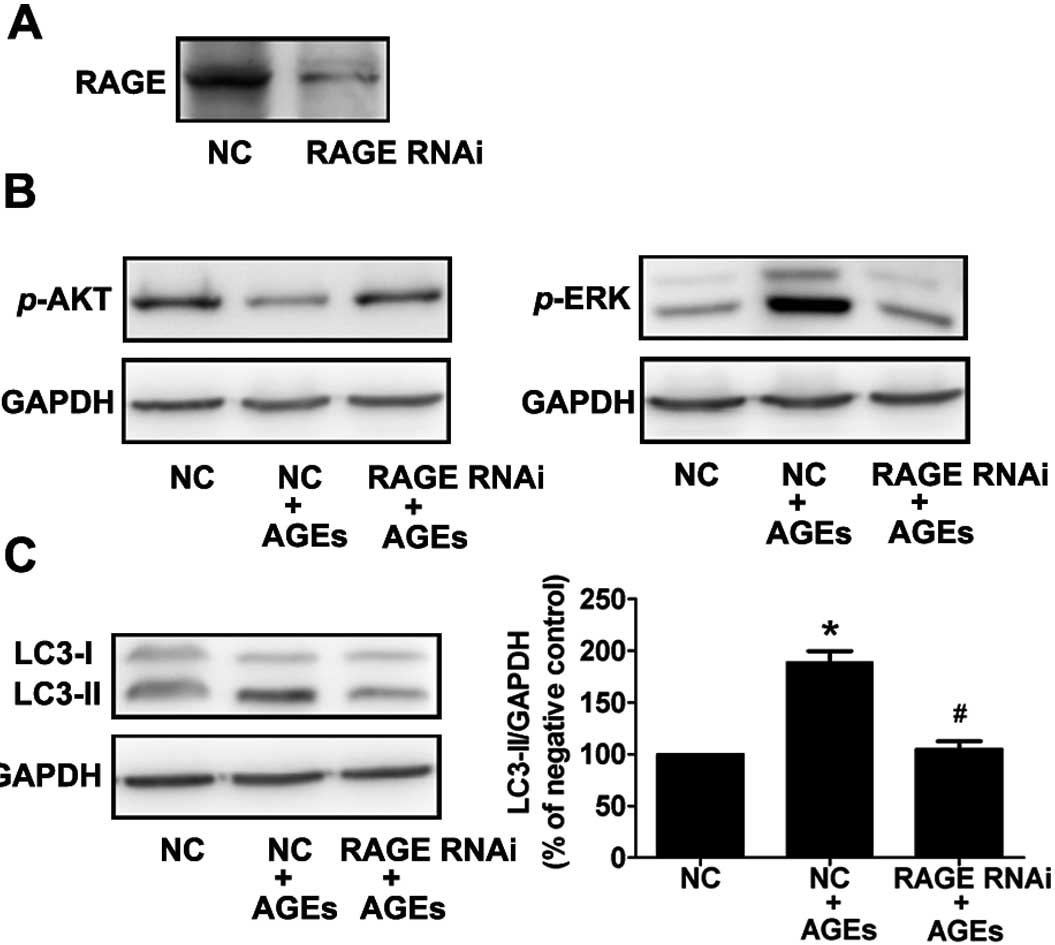
RAGE plays an essential role in
AGE-induced autophagy
To further demonstrate the importance of RAGE in
AGE-induced autophagy, cells were treated with siRNA to RAGE, and
the levels of activated ERK, Akt, and LC3-II were detected by
western blot analysis using phosphospecific antibodies. Compared to
the control (scrambled) siRNA-transfected cells, cells transfected
with RAGE siRNA exhibited a 90% reduction in RAGE protein
expression (Fig. 5A). In
AGE-treated cells, activation of ERK and inhibition of AKT was
reversed by RNA interference of RAGE (Fig. 5B). This result indicates that AGEs
activate ERKs and suppress Akt via RAGE.
Furthermore, we found that the AGE-induced
expression of LC3-II was significantly reduced in cells transfected
with RAGE siRNA compared with control transfected cells (Fig. 5C). These data illustrate that RAGE
plays an essential role in AGE-induced autophagy in A7R5 VSMCs.
Discussion
Previous studies have demonstrated that AGE-induced
proliferation of VSMCs is a key factor in the pathogenesis of
acceleration of atherosclerosis in diabetic patients (4,18).
Yoon et al reported previously that AGEs increased
proliferation of VSMCs via ERK and p38 dependent pathways (19). Several recent experimental studies
suggest the proliferation of VSMCs in diabetic models is related to
several cytokines and growth factors, including platelet derived
growth factor (PDGF) (20) and
basic fibroblast growth factor (bFGF) (21). In this study, we found that
autophagy is also involved in AGE-induced proliferation of VSMCs.
The interaction between AGEs and RAGE significantly increased
autophagy in VSMCs via the ERK and Akt pathways. This may
contribute to proliferation of VSMCs in the pathophysiological
process of atherosclerosis in diabetic patients.
Several recent studies have attempted to elucidate
the pathological mechanisms underlying disorders that involve AGEs.
AGEs have been shown to induce proliferation and migration of
VSMCs, increase generation of reactive oxygen species (22), decrease nitric oxide
bioavailability (23) and
up-regulate the production of various cytokines or growth factors
(24,25), such as TNF-α, PDGF and VCAM-1.
However, there is no direct evidence for a relationship between
AGEs and autophagy. In our study, we found the expression of the
autophagosome specific isoform LC3-II protein was enhanced in a
time- and dose-dependent manner in cells treated with AGEs. In
addition, we observed autophagic vacuoles in AGE-treated cells by
transmission electron microscopy. Our results demonstrate that AGEs
could induce autophagy, suggesting a novel, pathobiological
function for AGEs in diabetes.
Autophagy has been recognized as a cellular defense
mechanism that is used to remove protein aggregates and damaged
organelles (26). The autophagic
vacuole, or autophagosome, contains portions of the cytoplasm and
organelles and is surrounded by multiple membrane layers. In the
fibrous caps of atherosclerotic plaques, autophagic vacuoles have
been detected by electron microscopy in VSMCs (27). However, it is still not clear
whether autophagy is beneficial or detrimental in atherosclerotic
plaques. In this study, the autophagy inhibitor, 3-MA, could
attenuate the effects of AGE-induced proliferation of VSMCs. This
result indicates that AGE-induced autophagy is involved in
AGE-stimulated proliferation of VSMCs. This may provide a new
therapeutic strategy for preventing atherosclerosis in diabetic
patients.
RAGE is a major receptor for AGEs. The binding of
AGEs to RAGE leads to activation of cell signaling pathways, such
as the MAPK, p21ras, NF-κB, and JAK/Stat pathways
(28,29). We found that AGEs induced
phosphorylation of ERK, JNK, and p38 and inhibited phosphorylation
of Akt. Furthermore, autophagy in A7R5 VSMCs treated with AGEs was
reduced when cells were pretreated with the ERK inhibitor PD98059
but not by SB203580 or SP600125. Activation of the Akt pathway
using IGF-1 inhibited AGE-induced autophagy. These results imply
that the ERK and Akt pathways have opposing functions in
AGE-induced autophagy: the ERK pathway positively regulates
autophagy whereas the Akt pathway negatively regulates autophagy.
However, since we could not exclude the possibility that other
signaling pathways participate in autophagy, further investigation
is needed. We also found that RNAi of RAGE in VSMCs inhibited
AGE-induced ERK and LC3-II activity and recovered Akt activity.
These findings further underscore the importance of the interaction
between AGEs and RAGE in AGE-induced autophagy.
In summary, our studies demonstrate that AGEs could
induce proliferation of VSMCs through mechanisms involving
regulation of autophagy through the ERK and Akt signaling pathways.
This suggests that regulating the AGEs-RAGE-autophagy pathway can
attenuate proliferation of VSMCs and therefore may reduce the
development of atherosclerosis in diabetic patients. Further
studies are needed to dissect the relationship between AGEs and
autophagy in animal models and to explore possible drug-targeting
methods to regulate this pathway.
Acknowledgements
This work was supported by grants from the
Administration of Traditional Chinese Medicine of Zhejiang Province
(2008CA062, P.L. Lu) and the National Natural Science Foundation of
China (81100159, D.W. Lai). The research was performed in the
Biomedical Research Center, Sir Run Run Shaw Hospital, Zhejiang
University school of Medicine, Hangzhou.
References
|
1
|
S YamagishiM TakeuchiY InagakiK NakamuraT
ImaizumiRole of advanced glycation end products (AGEs) and their
receptor (RAGE) in the pathogenesis of diabetic microangiopathyInt
J Clin Pharmacol Res23129134200315224502
|
|
2
|
BK KilhovdTJ BergKI BirkelandP ThorsbyKF
HanssenSerum levels of advanced glycation end products are
increased in patients with type 2 diabetes and coronary heart
diseaseDiabetes
Care2215431548199910.2337/diacare.22.9.154310480523
|
|
3
|
DP BarlovicA Soro-PaavonenKA
Jandeleit-DahmRAGE biology, atherosclerosis and diabetesClin Sci
(Lond)1214355201110.1042/CS2010050121457145
|
|
4
|
VJ DzauRC Braun-DullaeusDG SeddingVascular
proliferation and atherosclerosis: New perspectives and therapeutic
strategiesNat Med812491256200210.1038/nm1102-124912411952
|
|
5
|
N SakataJ MengS TakebayashiEffects of
advanced glycation end products on the proliferation and
fibronectin production of smooth muscle cellsJ Atheroscler
Thromb7169176200010.5551/jat1994.7.16911480459
|
|
6
|
LG BucciarelliT WendtW QuRAGE blockade
stabilizes established atherosclerosis in diabetic apolipoprotein
E-null
miceCirculation10628272835200210.1161/01.CIR.0000039325.03698.3612451010
|
|
7
|
DX BuV RaiX ShenActivation of the ROCK1
branch of the transforming growth factor-beta pathway contributes
to RAGE-dependent acceleration of atherosclerosis in diabetic
ApoE-null miceCirc
Res10610401051201010.1161/CIRCRESAHA.109.20110320133903
|
|
8
|
DJ KlionskyAutophagy: from phenomenology
to molecular understanding in less than a decadeNat Rev Mol Cell
Biol8931937200710.1038/nrm224517712358
|
|
9
|
T YorimitsuDJ KlionskyAutophagy: molecular
machinery for self-eatingCell Death Differ12Suppl
215421552200510.1038/sj.cdd.4401765
|
|
10
|
N MizushimaT YoshimoriHow to interpret LC3
immunoblottingAutophagy3542545200710.4161/auto.460017611390
|
|
11
|
W MartinetDM SchrijversJP TimmermansH
BultInteractions between cell death induced by statins and
7-ketocholesterol in rabbit aorta smooth muscle cellsBr J
Pharmacol15412361246200810.1038/bjp.2008.18118469840
|
|
12
|
R KiffinU BandyopadhyayAM CuervoOxidative
stress and autophagyAntioxid Redox
Signal8152162200610.1089/ars.2006.8.152
|
|
13
|
D HeymannAutophagy: A protective mechanism
in response to stress and inflammationCurr Opin Investig
Drugs7443450200616729721
|
|
14
|
P CodognoS PattingreC BauvyAmino acids
interfere with the ERK1/2-dependent control of macroautophagy by
controlling the activation of Raf-1 in human colon cancer HT-29
cellsJ Biol Chem2781666716674200310.1074/jbc.M21099820012609989
|
|
15
|
HW ChiuSY HoHR GuoYJ WangCombination
treatment with arsenic trioxide and irradiation enhances autophagic
effects in U118-MG cells through increased mitotic arrest and
regulation of PI3K/Akt and ERK1/2 signaling
pathwaysAutophagy5472483200910.4161/auto.5.4.7759
|
|
16
|
FF HouGM ChertowJ KayInteraction between
beta 2-microglobulin and advanced glycation end products in the
development of dialysis related-amyloidosisKidney
Int5115141519199710.1038/ki.1997.2089150467
|
|
17
|
CS MitsiadesN MitsiadesV PoulakiActivation
of NF-kappaB and upregulation of intracellular anti-apoptotic
proteins via the IGF-1/Akt signaling in human multiple myeloma
cells: therapeutic
implicationsOncogene2156735683200210.1038/sj.onc.1205664
|
|
18
|
MA CreagerA GoldinJA BeckmanAM
SchmidtAdvanced glycation end products - Sparking the development
of diabetic vascular
injuryCirculation114597605200610.1161/CIRCULATIONAHA.106.62185416894049
|
|
19
|
YW YoonTS KangBK LeePathobiological role
of advanced glycation endproducts via mitogen-activated protein
kinase dependent pathway in the diabetic vasculopathyExp Mol
Med40398406200810.3858/emm.2008.40.4.39818779652
|
|
20
|
M KawanoT KoshikawaT KanzakiN MorisakiY
SaitoS YoshidaDiabetes mellitus induces accelerated growth of
aortic smooth muscle cells: association with overexpression of PDGF
beta-receptorsEur J Clin
Invest238490199310.1111/j.1365-2362.1993.tb00745.x8462625
|
|
21
|
N TaniguchiH KanetoM AsahiInvolvement of
glycation and oxidative stress in diabetic
macroangiopathyDiabetes45Suppl
38183199610.2337/diab.45.3.S818674900
|
|
22
|
G BastaG LazzeriniS Del TurcoGM RattoAM
SchmidtR De CaterinaAt least 2 distinct pathways generating
reactive oxygen species mediate vascular cell adhesion molecule-1
induction by advanced glycation end productsArterioscler Thromb
Vasc Biol2514011407200510.1161/01.ATV.0000167522.48370.5e
|
|
23
|
B XuR ChibberD RuggieroE KohnerJ RitterA
FerroImpairment of vascular endothelial nitric oxide synthase
activity by advanced glycation end productsFASEB
J1712891291200312738813
|
|
24
|
T MiyataO HoriJ ZhangThe receptor for
advanced glycation end products (RAGE) is a central mediator of the
interaction of AGE-beta2microglobulin with human mononuclear
phagocytes via an oxidant-sensitive pathway. Implications for the
pathogenesis of dialysis-related amyloidosisJ Clin
Invest9810881094199610.1172/JCI118889
|
|
25
|
Y YamamotoS YamagishiCC HsuH
YamamotoAdvanced glycation endproducts-receptor interactions
stimulate the growth of human pancreatic cancer cells through the
induction of platelet-derived growth factor-BBiochem Biophys Res
Commun222700705199610.1006/bbrc.1996.0807
|
|
26
|
T ShintaniDJ KlionskyAutophagy in health
and disease: a double-edged
swordScience306990995200410.1126/science.109999315528435
|
|
27
|
MM KockxGR De MeyerN BuyssensMW KnaapenH
BultAG HermanCell composition, replication, and apoptosis in
atherosclerotic plaques after 6 months of cholesterol
withdrawalCirc Res83378387199810.1161/01.RES.83.4.3789721694
|
|
28
|
HM LanderJM TaurasJS OgisteO HoriRA MossAM
SchmidtActivation of the receptor for advanced glycation end
products triggers a p21(ras)-dependent mitogen-activated protein
kinase pathway regulated by oxidant stressJ Biol
Chem2721781017814199710.1074/jbc.272.28.178109211935
|
|
29
|
HJ HuttunenC FagesH RauvalaReceptor for
advanced glycation end products (RAGE)-mediated neurite outgrowth
and activation of NF-kappa B require the cytoplasmic domain of the
receptor but different downstream signaling pathwaysJ Biol
Chem2741991919924199910.1074/jbc.274.28.1991910391939
|