Introduction
Prostate cancer (PC) is the most frequently
diagnosed cancer and the second-leading cause of cancer-related
deaths in men worldwide (1,2).
The general prognosis for diagnosed PC remains inadequate and
advanced PC is difficult to treat successfully (3,4).
Therefore, a better understanding of the nature and underlying
causes of PC will lead to improved treatment.
c-Jun-NH2-terminal kinases (JNKs), is
believed to participate in most aspects of cellular function,
including replication, growth, metabolism, differentiation, and
apoptosis (5–7). JNKs, belonging to the group of
stress-activated protein kinases (SAPKs), are encoded by three
genes, namely JNK-1 and JNK-2 that are ubiquitously expressed, and
JNK-3, whose expression is restricted to the brain, heart and
testis (7–9). The role of JNKs in apoptosis is
unclear because these proteins have been assigned both pro- and
anti-apoptotic properties. JNK becomes activated through
phosphorylation and some tumour cell lines contain constitutively
active JNK (10–12). Anti-sense JNK oligonucleotides
have been reported to inhibit the growth of such tumor cells and
induce apoptosis (13,14). In contrast, activated JNK is
required for UV-induced apoptosis and is suggested to be essential
for cytochrome c release from mitochondria in mouse fibroblasts
(15). The role of JNKs in
several cancers has been extensively examined in many studies over
the past decade (16). Reducing
JNK levels by genetic means in established breast cancer cell lines
will lead to a better understanding of its role in maintaining the
malignant phenotype (12). PC
progression is accelerated by reducing the level of apoptosis in
cancerous cells, suggesting that defects in apoptosis may
participate in prostate carcinogenesis. JNK-1 is a member of a
highly conserved mitogen-activated protein kinase superfamily (MAP
kinases) which also includes p38, and ERK. JNK phosphorylates and
augments the transactivating activity of many transcription
factors, which control the expression of various genes. Previous
studies in our laboratory and in others laboratories have shown
that JNK-1 is involved in and required for apoptosis signaling
induced by various agents (7,17).
We have shown that JNK activity is required for breast cells to
grow and surve (12). However,
the molecular mechanism by which JNK is involved in apoptosis in PC
and the mechanism by which JNK is regulated during apoptosis have
not been fully characterized.
To the best of our knowledge, programmed cell death,
or apoptosis, is a way for the organism to tightly control the cell
number and tissue size, and to prevent cancer from developing.
Apoptosis, is a regulated, complex process through which normal
cells in the body die after a specified life span. Cells that are
defective, damaged or redundant are eliminated. Multicellular
organisms use apoptosis, to eliminate abnormal or unwanted cells
(18,19). All cancer cells, as a result of
accumulated mutations fail to execute apoptosis, allowing them to
live indefinitely and grow uncontrollably. Breakdown of the
cellular apoptosis regulatory machinery is a hallmark of cancer.
Although the normal apoptotic pathways are defective, many cancer
cells are resistant or develop resistance to these agents. Several
families of proteins that can block apoptosis in normal cells have
been studied in cancer cells (20). These include the Bcl-2 family of
proteins, p53 tumor suppressor protein and the inhibitors of
apoptosis proteins (IAPs) (20,21). IAP are a family of functionally-
and structurally-related proteins, which serve as endogenous
inhibitors of programmed cell death (20,21). The human IAP family consists of 8
members, and IAP homologs have been identified in numerous
organisms. The first members of the IAPs identified were IAPs,
Cp-IAP and Op-IAP, which bind to and inhibit caspases as a
mechanism that contributes to its efficient infection and
replication cycle in the host. Later, 5 more human IAPs were
discovered which included XIAP, c-IAP1, c-IAP2, NAIP and survivin
(22,23). XIAP, which binds caspases-9, -3
and -7, inhibiting their activation and prevents apoptosis
(21). Also c-IAP1 and c-IAP2
have been shown to bind caspases, although how the IAPs inhibit
apoptosis mechanistically at the molecular level is not completely
understood. In the present study, we used RNA interference to
eliminate JNK-1 expression in PC-3 cells and studied which
apoptotic proteins are involved or affected after knockdown of
JNK-1. We showed by western blotting, Annexin V, and cell viability
that the siRNA complementary to JNK-1 mRNA was able to inhibit the
expression of their respective target, promoting apoptosis and cell
death. In addition, protein expression of Bcl-2, XIAP and survivin
were affected after blocking of JNK-1 protein expression by
siRNA-JNK-1 treatment. Our results suggest that siRNA against JNK-1
induced-cell death occurs via the activation of caspases.
Interference RNA-specific depletion of JNK-1 in PC-3 cells greatly
reduced JNK-1 activity induced by TPA and completely blocked both
cell growth and cell survival, suggesting that inhibition of JNK-1
represents a necessary and early step for the induction of
apoptosis in the prostate carcinoma cell line PC-3.
Materials and methods
Cell lines and culture
The human prostate carcinoma cell line PC-3 was a
gift from Dr Dan Mercola (The Sidney Kimmel Cancer Center, La
Jolla, CA, USA). The cells were cultured in RPMI-1640 medium
supplemented with 100 ml/l fetal bovine serum (FBS),
8x105 U/l penicillin and 0.1 g/l streptomycin in a
humidified incubator containing 50 ml/l CO2 at 37°C
(14,15). The antibodies used for western
blotting included those against protein kinase JNK-1, survivin,
XIAP, Bcl-2, p21 and VEGF (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA), against caspases-3, -8, -9, cytochrome c, β-actin
(Sigma, St. Louis, MO, USA), and cytochrome oxidase subunit IV (Cox
IV; Invitrogen, Carlsbad, CA, USA). Western blotting was performed
as previously described (23–26).
siRNA preparation and transfection of
small interfering RNA
Human PC-3 cells were seeded one day prior to
transfection at 30x103 cells/cm2. The cells
were transfected with 1 μg (12-well plate) of JNK-1, sense
(5′-AAGCCCAGTAATATAGTA GTA-3′), antisense
(5′-ACGTGACACGTTCGGAGAATT-3′) together with 6 μl RNAiFect
Transfection Reagent according to the manufacturer’s instructions
(Qiagen, Germantown, MD, USA). Optimal transfection conditions were
determined by transfection with Alexa Fluor 555 labeled
non-silencing siRNA, consisting of a scrambled sequence with no
homology to mammalian genes and (sense, 5′-AATTCTCCGAACGT
GTCACGT-3′ and antisense, 5′-ACGTGACACGTTCGGAG AATT-3′). The cells
were incubated at standard culture conditions (37°C, 5%
CO2) and after 8 h, the siRNA transfection medium was
replaced with a fresh medium. A significant decrease in protein
level was observed 48 h following siRNA addition. Silencing of JNK
by siRNA was confirmed by western blot analysis. All siRNA and
reagent used were obtained from Qiagen.
Western blot analysis
PC-3 carcinoma cells (5x105) were seeded
onto 6-well plates. Forty-eight hours after transfection, cells
were collected and washed twice by cold PBS, and each well was
treated with 50 ml lysis buffer (2 mmol/l Tris-HCl, pH 7.4, 50
mmol/l NaCl, 25 mmol/l EDTA, 50 mmol/l NaF, 1.5 mmol/l
Na3VO4, 1% Triton X-100, 0.1% SDS,
supplemented with protease inhibitors 1 mmol/l
phenylmethylsulfonylfluoride, 10 mg/l pepstatin, 10 mg/l aprotinin
and 5 mg/l leupeptin) (Sigma). Protein concentrations were
determined using the Bradford protein assay. Equal amounts of
protein (50 μg) were separated on a 15% SDS polyacrylamide
gel and transferred to a nitrocellulose membrane (Hybond C;
Amersham Pharmacia Biotech, Freiburg, Germany). Membranes were
blocked in 5% non-fat dry milk in TBS for 1 h at room temperature
and probed with rabbit anti-JNK-1 (sc-1648) antibody dilution,
1:500 (Santa Cruz Biotechnology, Inc.) overnight at 4°C. After
washing 3 times with TBS containing 0.1% Tween-20, membranes were
incubated with anti-rabbit IgG-horseradish-peroxidase (1:5,000;
Santa Cruz Biotechnology, Inc.), and developed by luminal mediated
chemiluminescence (Appylgen Technologies, Inc., China). To confirm
equal protein loading, membranes were re-proped with a 1:1,000
dilution of an anti-actin antibody (Santa Cruz Biotechnology,
Inc.). Densitometric analyses were performed using the Scion Image
software (12).
Measurement of caspase activity
Cells were washed with ice-cold PBS and lysed with
100 μl of lysis buffer [50 mM Tris-HCl (pH 7.5), 150 mM
NaCl, 1 mM EGTA, 1 mM NaF, 1% Nonidet P-40, 1 mM PMSF, protease
inhibitor cocktail] for 30 min at 4°C. Protein extracts were
collected after centrifugation at 14,000 rpm for 10 min. Protein
concentration was determined using the Bio-Rad protein assay kit
(Bio-Rad, USA). Equal amounts (50 μg) of protein extracts
were mixed with an assay buffer [20 mM HEPES (pH 7.4), 100 mM NaCl,
0.1% CHAPS, 10 mM DTT, 1 mM EDTA, 10% sucrose], added to 96-well
microtiter plates, and incubated with 100 μM of fluorogenic
substrates, specific for caspase-3 (Ac-DEVDAMC), Ac-IETD-AMC,
(caspase-8) and Ac-LEHD-AMC (caspase-9) for 1 h at 37°C. Free
aminomethylcoumarin (AMC) resulting from cleavage of the
aspartate-AMC bond by caspase was measured at an excitation
wavelength of 380 nm and an emission wavelength of 460 nm using a
spectrofluorometer (Geminin E; Molecular Devices, USA) (27).
Measurement of DNA fragmentation
The degree of DNA fragmentation was determined using
a Cell Death ELISA kit (Roche Diagnostics, Indianapolis, IN, USA).
PC-3 cells (50,000) were treated with apoptotic compounds for the
indicated periods of time. Cells were harvested by centrifugation
and lysed in 0.5 ml of the lysis buffer provided with the kit. Two
microliters of the extract was used for the ELISA, which was
performed as instructed by the manufacturer. The OD at 405 nm was
measured and the fold induction of apoptosis was calculated using
untreated cells as a control.
Reverse transcription polymerase chain
reaction
PC-3 cells (5x105) were seeded onto
6-well plates and treated with scrambled siRNA or siRNA-JNK-1.
Total-RNA was extracted 48 h after transfection using TRIzol
reagent. Reverse transcription was performed using a one step
RT-PCR kit. All assays were performed in triplicate and results
were plotted as average ± SD. The primers were: JNK-1, forward,
CGTCTGGTGGAAGGA GAGAG and reverse, TAATAACGGGGGTGGAGGAT; survivin,
forward, GCCATGAATTCATGGGTGCCCCGACG TGACGTTGC and reverse,
AGCTCTCTAGAGAGGCCTC AATCCATGGCA; p21/WAF-1, forward,
ATGAAATTCACCC CCTTTCC and reverse, CCCTAGGCTGTGCTCACTTC; XIAP,
forward, GCACGAGCAGGGTTTCTTTATACTGGTG and reverse,
CTTCTTCACAATACATGGCAGGGTTCCTC; Bcl-2, forward,
ACTTGTGGCCCAGATAGGCACCCAG and reverse, CGACTTCGCCGAGATGTCCAGCCAG;
and VEGF, forward, GGCAGAAGGAGGAGGGACAGAATC and reverse,
CATTTACACGTCTGCGGATCTTGT. The primers of human β-actin were
forward, 5′-TCACCAACTGGGACGACAT-3′ and reverse primer,
5′-GAAGTCCAGGGCGACGTAG-3′. Thermal cycle conditions were as
follows: 42°C for 30 min, 94°C for 2 min, followed by 28 cycles of
94°C for 15 sec, 55°C for 30 sec, 72°C for 1 min, with a final
extension at 72°C for 10 min. RT-PCR products were visualized by
ethidium bromide-stained agarose gels and the images were scanned
using UV light.
Cell viability was determined using the
3-(4–5 dimethylthiozol-2-yl) 2–5 diphenyl-tetrazolium bromide (MTT)
assay
PC-3 cells (2x104 cells/well) were
incubated in a 96-well plate containing 200 μl of medium.
The cells were divided into three groups: i) untransfected group;
ii) scrambled siRNA group; and iii) siRNA-JNK-1 group. Transfection
of siRNAs was conducted the following day as described previously.
The rate of cellular proliferation was measured for 24, 48 and 96
h. At the end of each time point, 20 μl of 5 mg/ml MTT
(Sigma) was added to each well. Four hours later, 200 μl of
DMSO was added to the MTT-treated wells and the absorption at 492
nm was determined with a spectrometer. Each experimental condition
was carried out in triplicate (28).
Flow cytometry for cell apoptosis
detection
PC-3 cells were plated in 6-well plates and were
divided into three groups: i) the scrambled siRNA group; ii) the
siRNA-JNK-1 group; and iii) the pCMV-JNK-1 group. Transfection of
siRNAs was performed the following day as described previously. The
siRNA transfection treatment remained the same. After 24 and 96 h,
cells were collected and washed twice with pre-chilled PBS. The
cell concentration was adjusted to
5x105–5x106 cells/well with 100 μl of
pre-chilled binding buffer. Five microliters of Annexin V-FITC and
5 μl of propidium iodide (PI) were added to each sample and
incubated for 10 min in the dark on ice. Pre-chilled binding buffer
(400 μl) was added at the end. The cell apoptosis was
determined by flow cytometry. Apoptosis of each group was assayed
three times (29).
Results
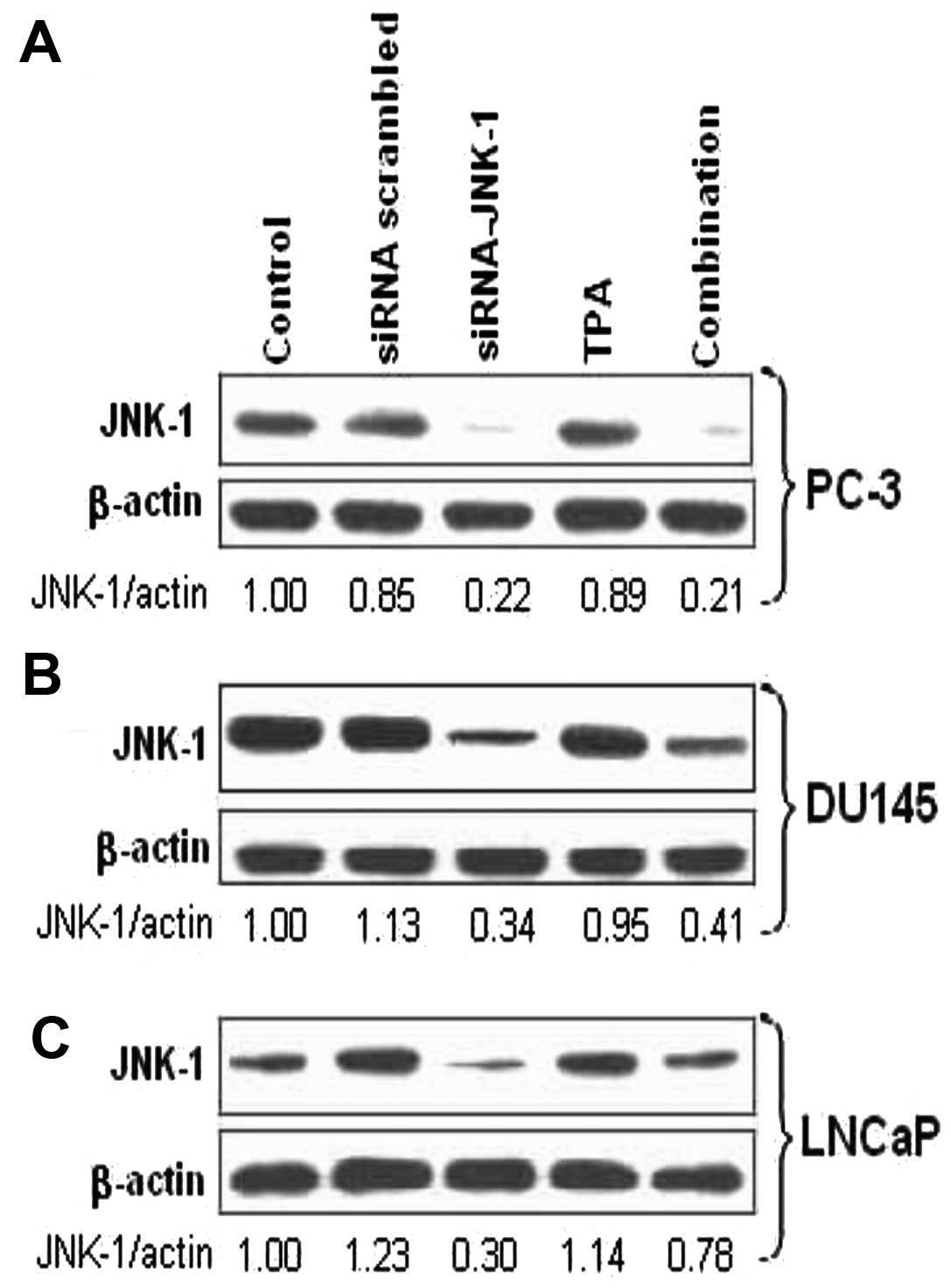
Endogenous JNK-1 expression in various
prostate cancer cells transfected with siRNA-JNK-1 or a scrambled
siRNA, or the combination of siRNA-JNK-1 and TPA
Treatment of prostate carcinoma cells with
siRNA-against JNK-1 but not with a scrambled siRNA, led to markedly
decreased JNK-1 protein level in PC-3, DU145 and LNCaP cells
(Fig. 1). Treatment of cells with
TPA further increased the expression of JNK-1 which was decreased
when cultured in combination with siRNA-JNK-1. The endogenous JNK-1
protein expression was detectable by western blot analysis in all
the prostate carcinoma cell lines treated (Fig. 1).
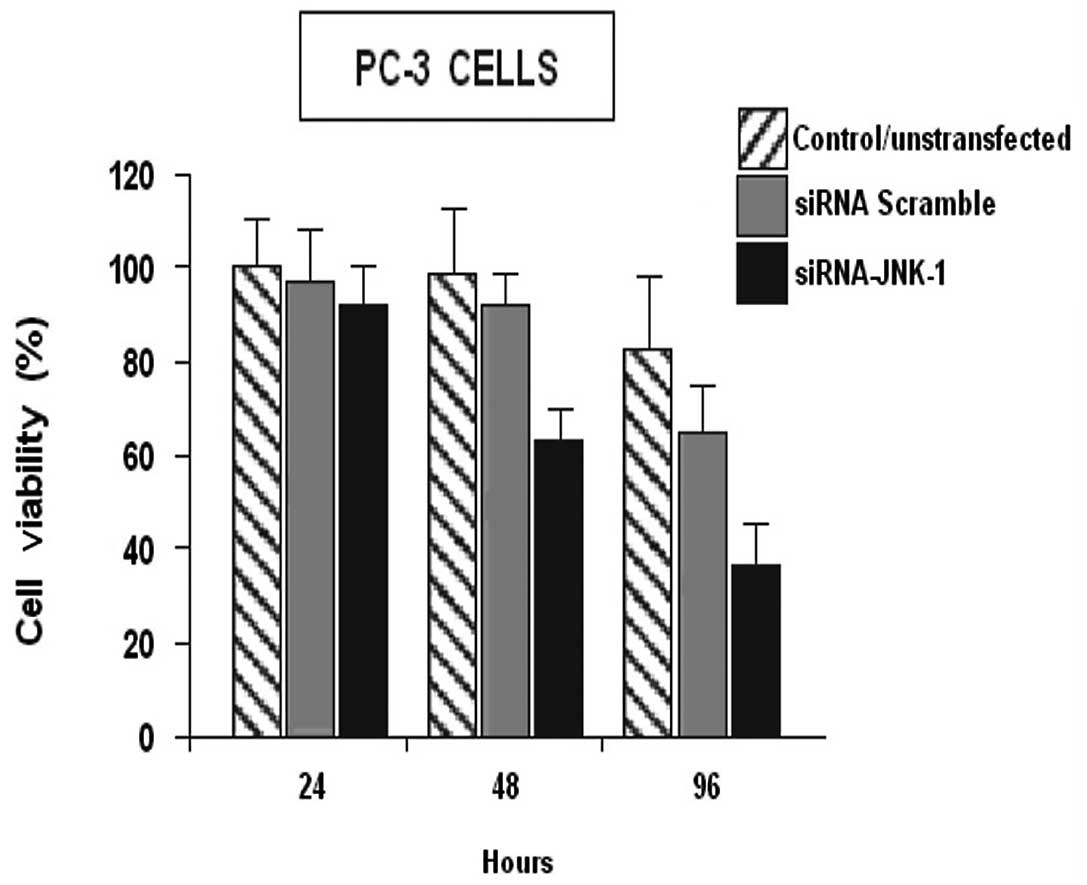
siRNA-JNK-1 inhibited cell viability in
the prostate PC-3 cell line
Decreased JNK-1 activity is associated with arrested
cell growth. Therefore, we examined whether the siRNA-JNK-1, would
affect cell viability in the PC-3 cell lines. PC-3 cells were
transfected with scrambled siRNA or with siRNA-JNK-1 as previously,
and the amount of viable cells was determined by the MTT assay for
24, 48 and 96 h (Fig. 2).
The ability of surviving cells to mitotically expand
into colonies forming a viable cell mass may be assessed by the
ability of the cell mass to reduce tetrazolium dyes. Viability was
assessed for 24, 48 and 96 h respectively, following siRNA-JNK-1
treatment (Fig. 2). At 24 h after
treatment, the survival of PC-3 cells was 91%. At 48 h after
treatment the survival of PC-3 cells was 66.3%. At 96 h of
treatment the survival of PC-3 cells was 40.7%. siRNA-JNK-1
significantly decreased the percentage of viable PC-3 cells,
compared to the control PC-3 cells or with the cell overexpressing
JNK-1 (pCMV-JNK-1). These observations suggest, that the continuous
expression of JNK-1 is necessary for normal proliferation of PC-3
cells.
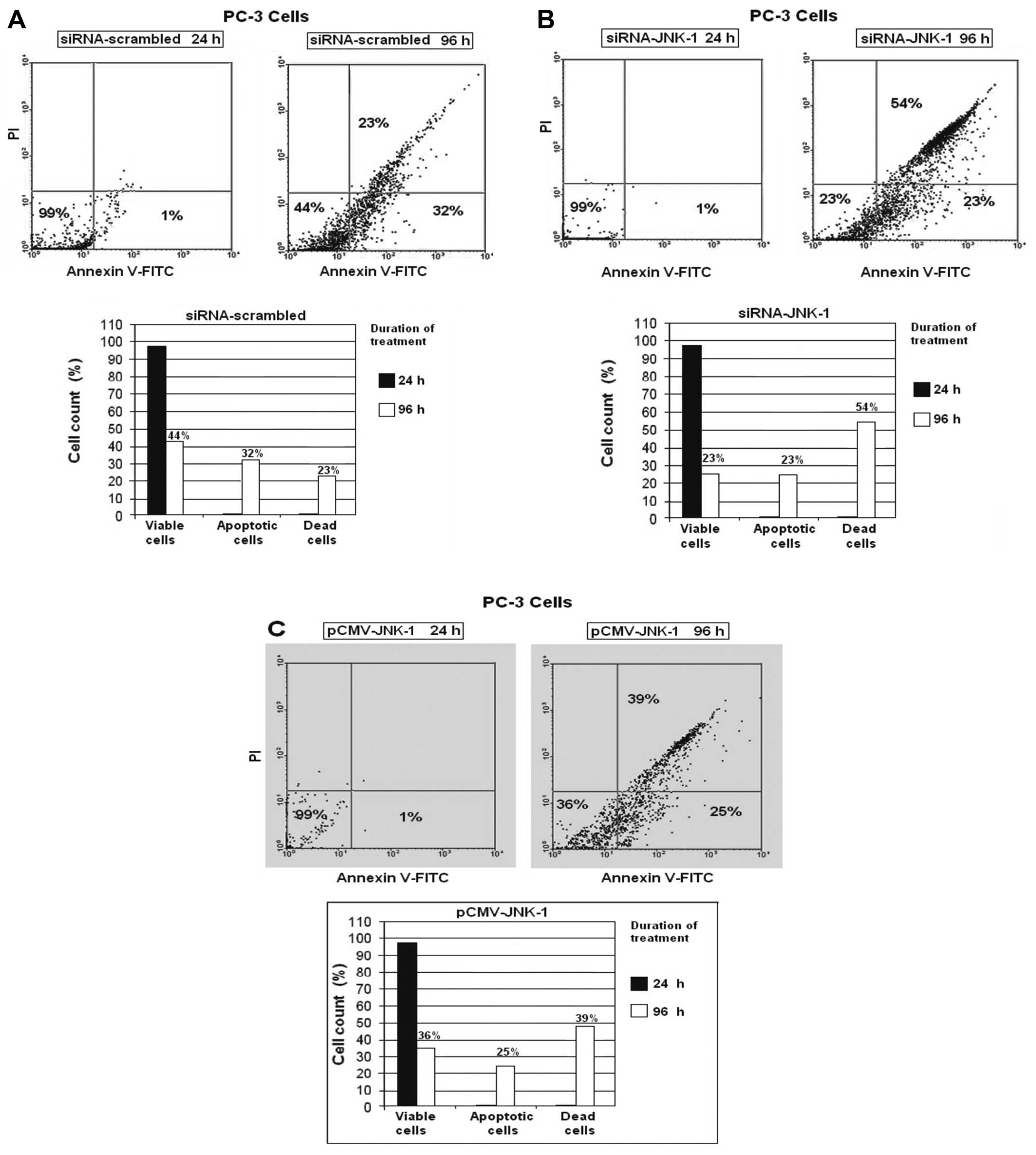
Transfection with siRNA against JNK-1,
strongly induces apoptosis in PC-3 cells
Detection of apoptosis of PC-3 cells was conducted
using flow cytometric analysis. To determine whether the decrease
in cell viability caused by the siRNAJNK-1 was due to an increase
in apoptosis or a decrease in cell proliferation, we determined the
amount of apoptotic cells from the scrambled transfected, control
siRNA-transfected and pCMV-JNK-1 transfected cells by Annexin
V-FITC and propidium iodide (PI) labeling followed by
fluorescence-activated cell sorting. In the flow cytometric
analysis, a double labeling technique, using Annexin V-FITC and PI,
was utilized. The lower left (LL) quadrant (Annexin
V−/PI−) is regarded as the population of live
cells; the lower right quadrant (LR) (Annexin
V+/PI−) is considered the cell population at
early apoptotic stage; the upper right (UR) quadrant (Annexin
V+/PI+) represents the cell population at the
late apoptotic stage and the upper left (UL) quadrant (Annexin
V−/PI+) is considered the necrotic cell
population. Flow cytometric data analysis revealed that after 96 h
of treatment of PC-3 cells with scrambled siRNA, 44% of cells were
in the LL quadrant; 32% of cells were in LR quadrant (early
apoptotic stage) and 23% of cells were in UR quadrant (late
apoptotic stage/dead cells) (Fig.
3A).
We observed that after 96 h of treatment of PC-3
cells with siRNA-against JNK-1 (siRNA-JNK-1) 23% of PC-3 cells were
in the LL quadrant; 23% of cells were in the LR quadrant (early
apoptotic stage) and 54% of cells were in the UR quadrant (late
apoptotic stage/dead cells) (Fig.
3B). After 96 h of treatment of PC-3 cells with a vector
expressing the JNK-1 protein (pCMV-JNK-1), 36% of PC-3 cells were
in the LL quadrant; 25% of cells were in the LR quadrant (early
apoptotic stage) and 39% of cells were in the UR quadrant (late
apoptotic stage/dead cells) (Fig.
3C). No significant percentage of cells was in the UL
quadrant.
Inhibition of JNK-1 expression decreases
the expression of some key cell cycle regulatory genes
To identify the mechanisms through which JNK-1
depletion may cause cell cycle arrest, apoptosis and cell death, we
focused on the function of JNK-1 as a transcription-regulatory
protein. We examined whether JNK-1 regulated the expression of key
genes coding for proteins involved in cell cycle control at the
G0/G1 phase. PC-3 cells were transfected with a pCMV-JNK-1 or
scrambled siRNA (control) or siRNA-JNK-1. Relative changes in mRNA
levels of a panel of candidate genes including JNK-1, survivin,
p21WAF1, XIAP, Bcl-2 and VEGF, were assayed by
quantitative real time RT-PCR (Fig.
4A). The mRNA levels of these genes were significantly
decreased after siRNA-JNK-1 treatment in assays performed on cDNA
samples from 3 independent transfections. Western blot analysis
showed that JNK-1, survivin, p21WAF1, XIAP and Bcl-2,
protein levels were decreased upon JNK-1 depletion (Fig. 4B).
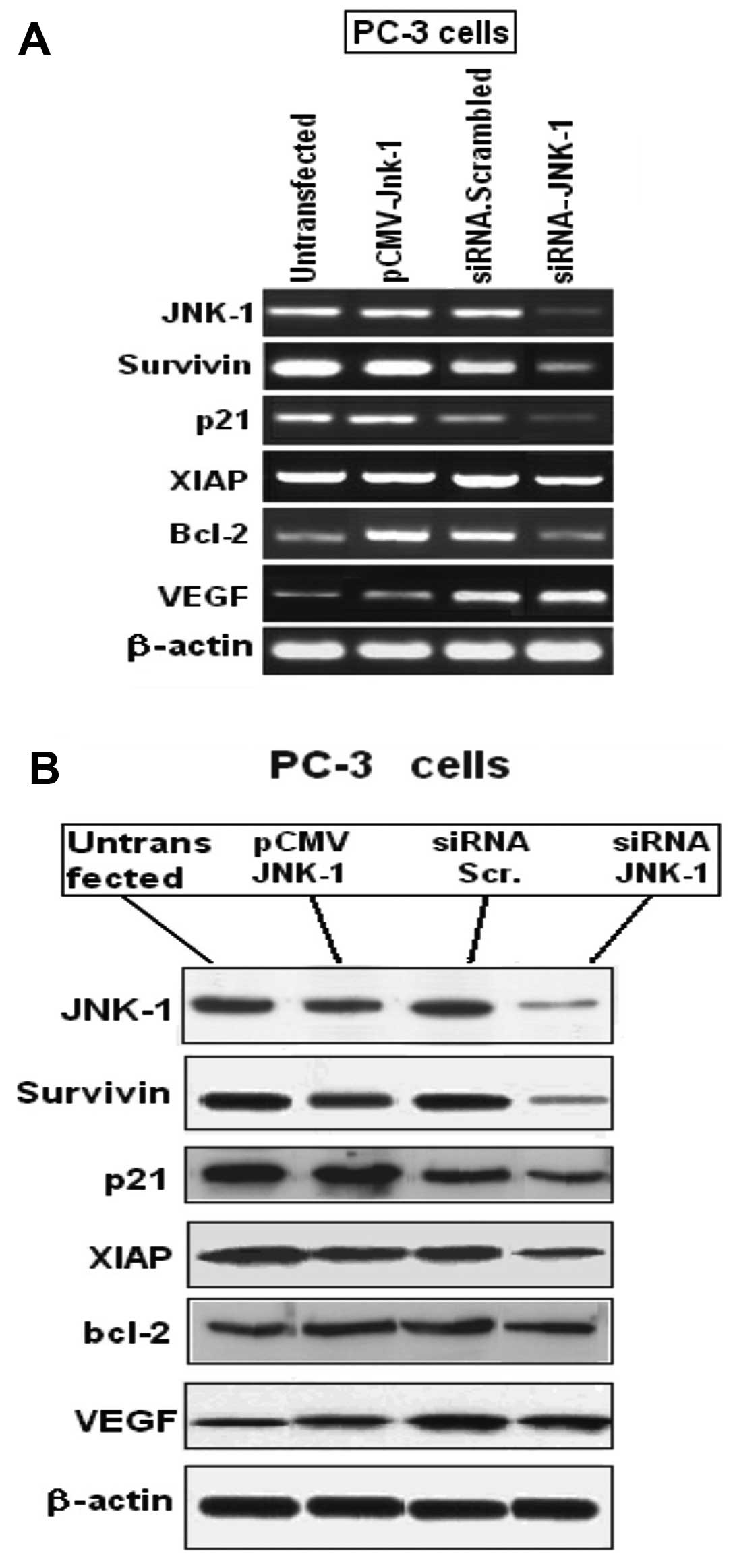 | Figure 4(A) JNK-1 depletion in PC-3 cells is
associated with decreased expression of JNK-1, survivin,
p21WAF1, XIAP and Bcl-2. A strong decrease in JNK-1 mRNA
expression shows the efficiency of siRNA-JNK-1 transfections. Using
β-actin as control, decrease in the mRNA levels of JNK-1, survivin,
p21WAF1, XIAP and Bcl-2 was seen. All changes in
expression are significant between control siRNA and siRNA-JNK-1
treatments (P<0.05). Values for control siRNA in each case is
normalized to 1 and not depicted. (B) Western blot analyses showed
that JNK-1, survivin, p21WAF1, XIAP and Bcl-2 expression
were decreased at the protein level. However, the expression of
VEGF was increased. One of three similar experiments is shown. |
Of all the JNK-1 target genes analyzed, survivin,
p21WAF1, and XIAP had the strongest decrease in
expression (Fig. 4). Since the
decreased expression of survivin, p21WAF1 and XIAP could
be associated with cell cycle arrest in G0/G1, we sought to
understand the mechanisms through which JNK-1 may regulate
expression of survivin, p21WAF1 and XIAP. Survivin and
XIAP belong to a family of proteins that suppress apoptosis by
inhibition of caspases in some cancer. Both confer resistance to
apoptosis induction by chemotherapeutic agents.
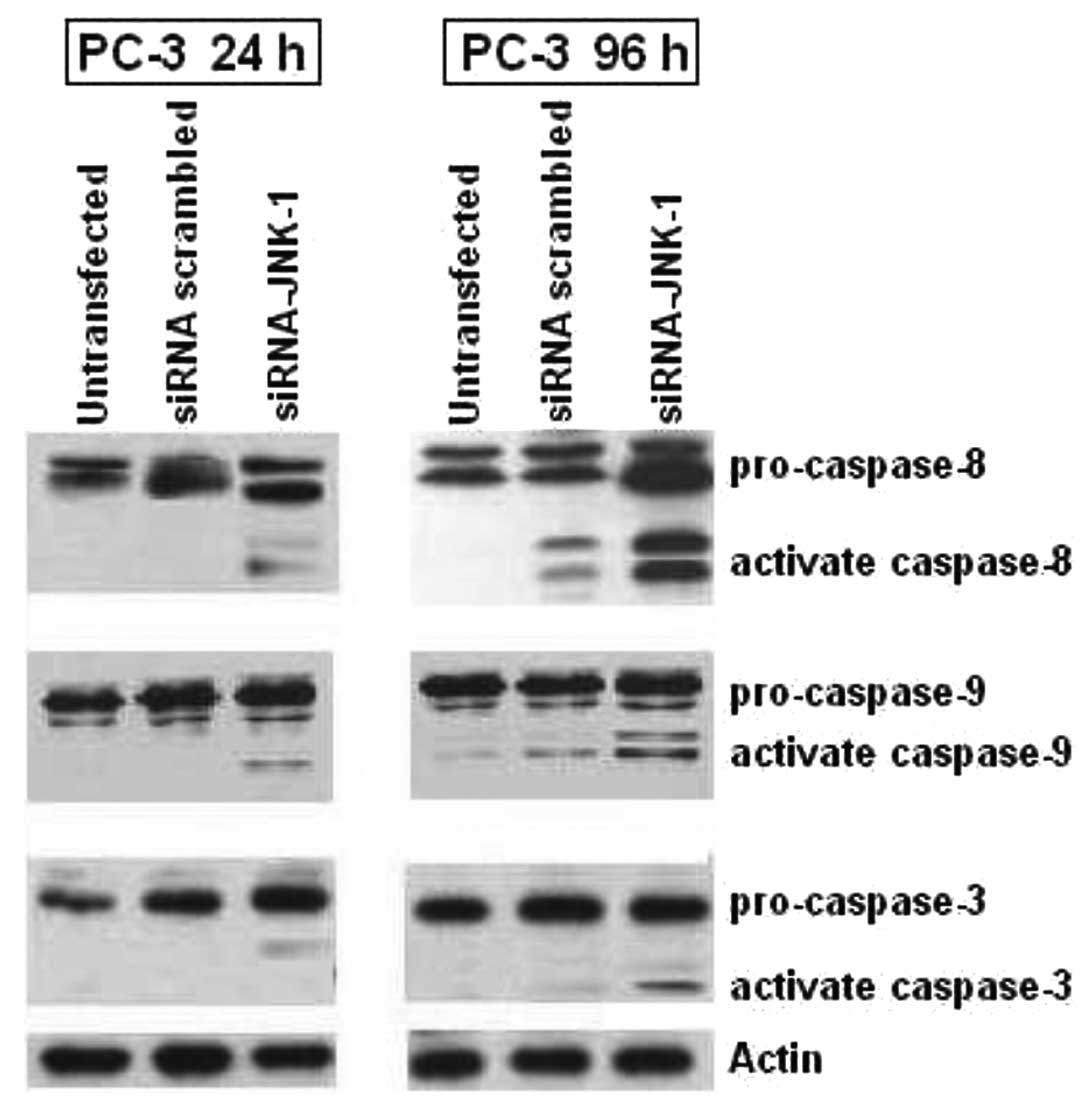
Activation of pro-caspases is accelerated
after inhibition of JNK-1 expression by siRNA-JNK-1 in PC-3
Caspases are important mediators of apoptosis.
Activation of caspases-8, -9 and -3 by enzymatic cleavages is a
hallmark event in caspase-dependent apoptosis. We investigate
caspases-8, -9 and -3 activation by western blotting Fig. 5. We determined if the apoptosis
induced by decreased activity of JNK-1 by siRNAJNK-1 in PC-3
prostate carcinoma cell line was accompanied by increased
activation of caspases (Fig. 5).
Consistent with the response to apoptosis and cell death observed
in cells treated with siRNA-JNK-1 (Fig. 3B), we observed that blocking of
JNK-1 expression induced cleavage of pro-caspase-8, which is an
initiator caspase linked to the receptor-mediated apoptotic
pathway. This effect was increased in PC-3 cells after 96 h of
treatment as compared to the PC-3 transfected with a scrambled
siRNA or with untransfected cells, respectively. The proteolytic
processing of caspase-9, was significantly induced in PC-3 cells
compared to cells treated with scrambled siRNA or untransfected
cells. In addition, the cleavage of procaspase-3 was weakly
increased by blocking JNK-1 expression in PC-3 cells compared with
untransfected or transfected cells. The data suggested involvement
of both caspases-9 and -8 pathways in siRNA-JNK-1-induced cell
death and that the apoptosis was at least in part due to the
decreased expression of JNK-1.
Discussion
It is well known that JNK is activated and
contributes to the induction of apoptosis with a variety of
extracellular stresses (30), but
its impact on prostate cancer cell survival remains controversial
(31,32). We have previously shown that
phosphatase PP4, acts in synergy with signals induced by cisplatin
and EGF, triggering an overexpression of JNK-1. Eventually this
would lead to a greater anti-apoptotic response by keeping low the
activity of one specific JNK-1 phosphatase and by inducing
activation of genes involved in the DNA damage response pathway
(23). In this study, we have
demonstrated that in PC-3 cells, siRNA-JNK-1 exerted multiple
effects on the viability and cell growth and evoked apoptosis via a
pathway dependent on caspases. Apoptosis is a carefully regulated
process of cell death that occurs as a normal part of development.
Inappropriately regulated apoptosis is implicated in disease
states, such as Alzheimer’s disease and cancer (33,34). In this regard, it has been shown
that the diverse JNK-mediated responses appear to be cell
type-specific (35). JNK-1
activity was shown to be critical for apoptosis in several cell
types including PC12 cells (15)
and U937 cells (36).
Accumulating evidence suggests that the MAP kinase family of
proteins are critical for the induction of cell death or survival
(15,37,38). The kinase JNK-1 is highly
expressed in most human cancers, including breast, prostate,
gastrointestinal cancer, lymphoma, melanoma, and myeloid leukemia
(37–39). Altered expression of JNK-1 or
JNK-2 seems to define a common event associated with the
pathogenesis of several human cancer types (40,41).
Our results strongly suggest that the siRNAs used
were specific for JNK-1 and validates the use of this approach to
alter expression of the endogenous protein. Indeed, expression of
human JNK-1 in siRNA knockdown PC-3 cells promotes apoptosis, which
further suggests the phenotypic changes in these structure is
specifically due to the loss of endogenous JNK-1. Previous studies
regarding prostate cancer demonstrated that the continued presence
of JNK-1/2 was required for cancer development in combination with
cisplatin (42,43) and that inactivation of JNK-1/2
resulted in the sustained regression of tumors (43,44). Although some studies previously
revealed that the effects of inactivation of JNK-1/2 in some cell
lines were modest, other groups using different approaches to
reduce the protein level of JNK-1/2 found that a decrease in
JNK-1/2 expression could inhibit the growth of several tumor cells,
including breast tumor cells (45,46). Our results are consistent with a
causal role of JNK-1 in transformation of PC-3 prostate carcinoma.
We demonstrated that siRNA against JNK-1 can effectively
downregulate JNK-1 overexpression with great specificity. We showed
that the siRNA-JNK-1 could successfully induce apoptosis and cell
death up to 54% as demonstrated by the Annexin V/PI assay in PC-3
cells treated with siRNA-JNK-1 96 h after transfection (Fig. 3). In addition, blocking of JNK-1
expression causes a decrease in the expression of survivin, p21,
XIAP and Bcl-2, all of which are anti-apoptotic proteins, promoting
the process of apoptosis (Fig.
4). Several reports have shown that JNK phosphorylates the
anti-apoptotic protein Bcl-2 and Bcl-x, although the consequences
of this phosphorylation are unclear. It is possible that
phosphorylation of these proteins impairs their anti-apoptotic
function, contributing to the induction of apoptosis (47). A cell viability assay, showed a
strong decrease in cell proliferation after siRNA-JNK-1 treatment.
These observations suggest, that the continuous expression of JNK-1
is necessary for normal proliferation of PC-3 cells. Therefore, we
determined whether the increased susceptibility of prostate cancer
PC-3 cells overexpressing endogenous JNK-1 to siRNA-JNK-1 induced
apoptosis was accompanied by the increased activation of caspases
(Fig. 5). Blocking of JNK-1
expression by siRNA induced a cleavage of pro-caspase-8, which is
an initiator caspase linked to the receptor-mediated apoptotic
pathway. The proteolytic processing of caspase-9, which has been
linked to the mitochondrial death pathway, was significantly
induced in PC-3 cells following treatment with siRNA-JNK-1 compared
to control cells. In addition, the cleavage of pro-caspase-3 was
very weak in two similar experiments. In fact, caspase-3 activity
is thought to be essential for DNA fragmentation. These results
indicate that the mitochondrial instability led to apoptosis
through the cytochrome c-mediated activation of caspases-9 and -8.
These data are consistent with the suggestion that the siRNA
against JNK-1 or JNK-2 is capable of inducing DNA fragmentation and
apoptosis in PC-3 cells.
In summary, we have shown that siRNA against JNK-1
promoted apoptosis in the human prostate carcinoma PC-3 cell line,
and this effect is partly due to the downregulation of Bcl-2,
survivin, p21WAF1, and XIAP proteins. In contrast,
treatment of PC-3 cells with scrambled siRNA did not have any
effect in the activity of JNK-1. Based on our present study, we
propose that blocking of JNK-1 expression selectively induces
apoptosis in PC-3 cells expressing endogenous-activated JNK-1
protein. Interference with the JNK-1 pathway attenuated the growth
and survival of PC-3 cells through the induction of selective
apoptosis. These results suggest that modulation of JNK-1 signaling
molecules by siRNA plays a critical role in JNK-1-induced apoptosis
in JNK-1-activated cells.
Acknowledgements
We would like to Dan Mercola (The
Sidney Kimmel Cancer Center, San Diego, CA, USA) for providing us
the vector for expressing JNK-1. This study was supported by the
Chilean National Science Foundation FONDECYT regular grant no.
1060774.
References
|
1.
|
M VermaP PatelM VermaBiomarkers in
prostate cancer
epidemiologyCancers337733798201110.3390/cancers304377324213111
|
|
2.
|
A JemalR SiegelJ XuE WardCancer
statistics, 2010Cancer J Clin60277300201010.3322/caac.20073
|
|
3.
|
A VickersC SavageH LiljaFinasteride to
prevent prostate cancer: should all men or only a high-risk
subgroup be treated?J Clin
Oncol2811121116201010.1200/JCO.2009.23.557220124185
|
|
4.
|
MS LuciaAK DarkePJ GoodmanFG La RosaHL
ParnesLG FordCA Coltman JrIM ThompsonPathologic characteristics of
cancers detected in The Prostate Cancer Prevention Trial:
implications for prostate cancer detection and
chemopreventionCancer Prev Res
(Phila)3167173200810.1158/1940-6207.CAPR-08-007819138952
|
|
5.
|
K ShimadaM NakamuraE IshidaM KishiN
KonishiRoles of p38- and c-jun NH2-terminal kinase-mediated
pathways in 2-methoxyestradiol-induced p53 induction and
apoptosisCarcinogenesis2410671075200310.1093/carcin/bgg05812807754
|
|
6.
|
Y FanH ChenB QiaoL LuoH MaOpposing effects
of ERK and p38 MAP kinases on HeLa cell apoptosis induced by
dipyrithioneMol Cells233038200717464209
|
|
7.
|
RJ DavisSignal transduction by the JNK
group of MAP
kinasesCell103239252200010.1016/S0092-8674(00)00116-111057897
|
|
8.
|
K SabapathyK HochedlingerSY NamA BauerM
KarinE WagnerDistinct roles for JNK1 and JNK2 in regulating JNK
activity and c-Jun-dependent cell proliferationMol
Cell15713725200410.1016/j.molcel.2004.08.02815350216
|
|
9.
|
CY KuanDD YangDR SamantaJ RogerRJ DavisP
RakicRA FlavellThe Jnk1 and Jnk2 protein kinases are required for
regional specific apoptosis during early brain
developmentNeuron22667676199910.1016/S0896-6273(00)80727-810230788
|
|
10.
|
H TsuikiM TnaniI OkamotoLC
KenyonConstitutively active forms of c-Jun NH2-terminal
kinase are expressed in primary glial tumorsCancer
Res63250255200312517805
|
|
11.
|
G LiaoQ TaoM KofronJS ChenA SchloemerRJ
DavisJC HsiehC WylieJ HeasmanCY KuanJun NH2-terminal
kinase (JNK) prevents nuclear β-catenin accumulation and regulates
axis formation in Xenopus embryosProc Natl Acad Sci
USA10316313163182006
|
|
12.
|
E ParraJ FerreiraKnockdown of the
c-Jun-N-terminal kinase expression by siRNA inhibits MCF-7 breast
carcinoma cell lines growthOncol
Rep2413391345201010.3892/or_0000099120878129
|
|
13.
|
F BostR McKayM BostO PotapovaNM DeanD
MercolaThe Jun kinase 2 isoform is preferentially required for
epidermal growth factor-induced transformation of human A549 lung
carcinoma cellsMol Cell Biol19193819491999
|
|
14.
|
YM YangF BostW CharbonoN DeanR McKayJS
RhimC DepatieD MercolaC-Jun NH2-terminal kinase mediates
proliferation and tumor growth of human prostate carcinomaClin
Cancer Res93913982003
|
|
15.
|
J LiuA LinRole of JNK activation in
apoptosis: a double-edged swordCell
Res153642200510.1038/sj.cr.729026215686625
|
|
16.
|
NJ KennedyRJ DavisRole of JNK in tumor
developmentCell Cycle2199201200312734425
|
|
17.
|
G PearsonF RobinsonTB GibsonB XuM
KarandikarK BermanMH CobbMitogen-activated protein (MAP) kinase
pathways: regulation and physiological functionsEndocr
Rev22153183200111294822
|
|
18.
|
S ElmoreApoptosis: a review of programmed
cell deathToxicol
Pathol35495516200710.1080/0192623070132033717562483
|
|
19.
|
A AshkenaziVM DixitDeath receptors:
signaling and
modulationScience28113051308199810.1126/science.281.5381.13059721089
|
|
20.
|
FM FosterTW OwensJ Tanianis-HughesRB
ClarkeK BrennanNJ BundredCH StreuliTargeting inhibitor of apoptosis
proteins in combination with ErbB antagonists in breast
cancerBreast Cancer Res11R41200910.1186/bcr232819563669
|
|
21.
|
Y WeiT FanM YuInhibitor of apoptosis
proteins and apoptosisActa Biochim Biophys Sin
(Shanghai)40278288200810.1111/j.1745-7270.2008.00407.x18401525
|
|
22.
|
NK SahZ KhanGJ KhanPS BisenStructural,
functional and therapeutic biology of survivinCancer
Lett244164171200610.1016/j.canlet.2006.03.00716621243
|
|
23.
|
J InostrozaL SaenzG CalafG CabelloE
ParraRole of the phosphatase PP4 in the activation of JNK-1 in
prostate carcinoma cell lines PC-3 and LNCaP resulting in increased
AP-1 and EGR-1 activityBiol
Res38163178200510.4067/S0716-9760200500020000616238095
|
|
24.
|
E ParraA OrtegaL SaenzDownregulation of
Egr-1 by siRNA inhibits growth of human prostate carcinoma cell
line PC-3Oncol Rep2215131518200919885607
|
|
25.
|
E ParraJ FerreiraA OrtegaOverexpression of
EGR-1 modulates the activity of NF-κB and AP-1 in prostate
carcinoma PC-3 and LNCaP cell linesInt J
Oncol39345352201121617851
|
|
26.
|
E ParraF FerreiraL SaenzDiverse effects of
siRNA-EGR-1 on NF-κB and AP-1 transcription factors in prostate
carcinoma cell lines PC-3 and LNCaPInt J Mol Med288478532011
|
|
27.
|
OA MareninovaKF SungP HongA LugeaSJ
PandolI GukovskyAS GukovskayaCell death in pancreatitis: caspases
protect from necrotizing pancreatitisJ Biol
Chem28133703381200610.1074/jbc.M51127620016339139
|
|
28.
|
DB LongleyTR WilsonM McEwanWL AllenU
McDermottL GalliganPG Johnstonc-FLIP inhibits chemotherapy-induced
colorectal cancer cell
deathOncogene25838848200610.1038/sj.onc.120912216247474
|
|
29.
|
M KingM Radicchi-MastroianniEffects of
caspase inhibition on camptothecin-induced apoptosis of HL-60
cellsCytometry492835200210.1002/cyto.1014112210608
|
|
30.
|
YT IpRJ DavisSignal transduction by the
c-Jun N-terminal kinase (JNK) from inflamation to developmentCurr
Opin Cell Biol10205219199810.1016/S0955-0674(98)80143-99561845
|
|
31.
|
PV MejiaJM BenitoA FernándezHD HanL
MangalaC Rodríguezc-Jun-NH2-kinase-1 inhibition leads to
antitumor activity in ovarian cancerClin Cancer Res161841922010
|
|
32.
|
J LiuY MinemotoA Linc-Jun N-terminal
protein kinase 1 (JNK1), but not JNK2, is essential for tumor
necrosis factor alpha-induced c-Jun kinase activation and
apoptosisMol Cell
Biol241084410856200410.1128/MCB.24.24.10844-10856.200415572687
|
|
33.
|
LF LinczDeciphering the apoptotic pathway:
all roads lead to deathImmunol Cell
Biol761199810.1046/j.1440-1711.1998.00712.x9553771
|
|
34.
|
JM BrownLD AttardiThe role of apoptosis in
cancer development and treatment responseNat Rev
Cancer5231237200515738985
|
|
35.
|
A WicovskyN MillerN DaryabR MarienfeldC
KneitzS KavuriM LeverkusB BaumannH WajantSustained JNK activation
in response to tumor necrosis factor is mediated by caspases in a
cell type-specific mannerJ Biol
Chem28221742183200710.1074/jbc.M60616720017121845
|
|
36.
|
A MeriinVL GabaiJ YaglomJY VictorI
ShifriniMY ShermanProteasome inhibitors activate stress kinases and
induce Hsp72. Diverse effects on apoptosisJ Biol
Chem27363736379199810.1074/jbc.273.11.63739497367
|
|
37.
|
E ParraActivation of MAP kinase family
members triggered by TPA or ionomycin occurs via the protein
phosphatase 4 pathway in Jurkat leukemia T cellsMol Med
Rep5773778201222200849
|
|
38.
|
P CodognoAJ MeijerAutophagy and signaling:
their role in cell survival and cell deathCell Death
Differ1215091518200510.1038/sj.cdd.440175116247498
|
|
39.
|
M ZhaoM YangL YangY YuM XieS ZhuR KangD
TangZ JiangW YuanHMGB1 regulates autophagy through increasing
transcriptional activities of JNK and ERK in human myeloid leukemia
cellsBMB Rep44601606201110.5483/BMBRep.2011.44.9.60121944254
|
|
40.
|
BK SabapathyT KallunkiJP DavidI GraefM
KarinEF Wagnerc-Jun NH2-terminal kinase (JNK)1 and JNK2 have
similar and stage-dependent roles in regulating T cell apoptosis
and proliferationJ Exp
Med193317328200110.1084/jem.193.3.31711157052
|
|
41.
|
C DongDD YangM WyskAJ WhitmarshRJ DavisRA
FlavellDefective T cell differentiation in the absence of
JNK1Science28220922095199810.1126/science.282.5396.20929851932
|
|
42.
|
R GjersetA HaghighiS LebedevaD MercolaGene
therapy approaches to sensitization of human prostate carcinoma to
cisplatin by adenoviral expression of p53 and by antisense jun
kinase oligonucleotide methodsMethods Mol
Biol175495520200111462854
|
|
43.
|
O PotapovaA HaghighiF BostC LiuMJ BirrerR
GjersetD MercolaThe Jun kinase/stress-activated protein kinase
pathway functions to regulate DNA repair and inhibition of the
pathway sensitizes tumor cells to cisplatinJ Biol
Chem2721404114044199710.1074/jbc.272.22.140419162025
|
|
44.
|
DL PersonsEM YazlovitskayaW CulJC
PellingCisplatin-induced activation of mitogen-activated protein
kinases in ovarian carcinoma cells: inhibition of extracellular
signal-regulated kinase activity increases sensitivity to
cisplatinClin Cancer Res5100710141999
|
|
45.
|
J HayakawaM OhmichiH KurachH IkegamiA
KimuraT MatsuokaH JikiharaD MercolaY MurataInhibition of
extracellular signal-regulated protein kinase or c-Jun N-terminal
protein kinase cascade, differentially activated by cisplatin,
sensitizes human ovarian cancer cell-lineJ Biol
Chem2743164831654199910.1074/jbc.274.44.31648
|
|
46.
|
YM YangF BostW CharbonoN DeanR McKayJS
RhimC DepatieD MercolaC-Jun NH2-terminal kinase mediates
proliferation and tumor growth in human prostrate carcinomaClin
Cancer Res93914012003
|
|
47.
|
S KornblauM WombleYH QiuCE JacksonW ChenM
KonoplevaEH EsteyM AndreeffSynergistic induction of apoptosis by
simultaneous disruption of the Bcl-2 and MEK/MAPK pathways in acute
myelogenous
leukemiaBlood9934613464200210.1182/blood.V99.9.346111964319
|