Introduction
Osteoarthritis (OA), the most common joint disorder,
is characterized by progressive degenerative structural changes in
articular cartilage and excessive production of several
inflammatory mediators (1). Among
these mediators, the pro-inflammatory cytokine interleukin (IL)-1β
plays a central role in inducing cartilage damage in arthritis
(2,3). IL-1β enhances the degradation of the
extracellular matrix (ECM), including proteoglycan, through the
activation of matrix metalloproteinases (MMPs). Degradation of the
ECM in articular cartilage is a central event leading to joint
destruction in several arthritic conditions, including OA (4). The pro-inflammatory effects may be
mediated through cyclooxygenase (COX)-2 induction, which produces
prostaglandin E2 (PGE2) that is responsible
for the pain and swelling of inflamed joints by enhancing MMP
expression and activity (5,6).
The expression and biological activity of the
pro-inflammatory factors mentioned are regulated by the
transcription factor nuclear factor κ-light chain-enhancer of
activated B cells (NF-κB). NF-κB regulates the expression of a
number of genes involved in immune responses and inflammation
(7). In unstimulated cells, NF-κB
is discovered in the cytoplasm (8). When these cells are stimulated by
cytokines such as IL-1β, which play important roles in the
initiation and development of OA, the NF-κB protein p65/p50 enters
into the nucleus where it regulates the expression of a number of
genes involved in inflammatory responses such as COX-2 (5,9).
At present, with the exception of anti-inflammatory
corticosteroids and non-steroidal anti-inflammatory drugs (NSAIDs)
which inhibit COX-2, a specific therapy based on fundamental
intracellular pathways of chondrocytes does not exist for the
medical management of OA (10);
therefore, safe and efficacious drugs are needed to treat this
debilitating disease.
Sphingosine-1-phosphate (S1P) is a member of an
important group of signaling sphingolipids recognized to play a
role in a diverse array of cellular processes, including apoptosis,
survival, motility, calcium signaling and differentiation in a
variety of cell types (11). S1P
is generated by the phosphorylation of sphingosine kinases 1
(Sphk-1) and 2 (Sphk-2). S1P exerts most of its activity as a
ligand of G-protein-coupled receptors (GPCRs) (12,13). At present, 5 members of the S1P
receptor family have been identified in mammals, S1P1–5,
possessing distinct expression profiles and affinities toward S1P
(13). Stradner et al
(14) demonstrated that the
expression of S1P1, S1P2 and S1P3
was observed in human articular chondrocytes.
Of the various cellular physiological actions of
S1P, we focused on its ability to markedly influence inflammation.
The S1P pathway has recently been associated with a variety of
inflammatory-based diseases, and several studies have reported on
the anti-inflammatory effects of S1P. Ogawa et al (15) discovered that the novel
S1P1 receptor agonist KRP-203 reduced experimental
autoimmune myocarditis in rats. Hughes et al (16) demonstrated that S1P significantly
reduced pro-inflammatory cytokine secretion such as ILs in
macrophages. S1P is known to have potent anti-inflammatory
actions.
In this study, we investigated the effects of S1P on
human arthritis, as well as cellular responses using an in
vitro model. Furthermore, the potential of S1P1 to
reduce the inflammation of human chondrocytes induced by IL-1β was
evaluated using a specific agonist and antagonist.
Materials and methods
Reagents
S1P was purchased from Cayman Chemical (Ann Arbor,
MI, USA) and prepared as a 2 mM solution in 0.3 M NaOH and was
further diluted in cell culture medium. SEW2871, an S1P receptor 1
(S1P1) agonist and W146, a S1P1 antagonist,
were purchased from Cayman Chemical. Recombinant human IL-1β was
purchased from Santa Cruz Biotechnology, Inc., (Santa Cruz, CA,
USA).
Human chondrocyte isolation and monolayer
cultures
Cartilage tissue samples were obtained from the
femoral condyle and tibial plateau of the knee from 6 OA patients
at the time of joint replacement surgery. Cartilage samples were
derived from human patients following full informed consent and
local ethics committee approval. Full-thickness cartilage slices
were obtained from above the subchondral bone from a relatively
lesion-free area. Human OA chondrocytes were harvested from the
discarded knee tissue as previously described (17). Briefly, the cartilage surfaces
were first rinsed with sterile NaCl/Pi. The cartilage
slices were chopped and incubated with 0.25% trypsin for 30 min,
followed by 0.1% collagenase (Sigma-Aldrich, St. Louis, MO, USA)
treatment for 6 h in Dulbecco’s modified Eagle’s medium (DMEM;
Invitrogen-Gibco-BRL, Grand Island, NY, USA) supplemented with 10%
(v/v) fetal bovine serum (FBS) (Invitrogen-Gibco-BRL) and
antibiotics. Cells were filtered through a 70-μm cell
strainer (Falcon, Franklin Lakes, NJ, USA), washed twice with
NaCl/Pi and then seeded into tissue culture flasks in
DMEM supplemented with 10% FBS and antibiotics. After ∼7 days,
confluent chondrocytes were split once and seeded at high density
and these first passage chondrocytes were used within 2 days in
subsequent experiments. Chondrocytes were incubated with DMEM
containing 1% FBS prior to treatment with S1P, SEW2871, W146 and
pro-inflammatory cytokines.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was extracted from human chondrocytes
using the Easy-spin™ total RNA extraction kit (Intron
Biotechnology, Seoul, Korea). cDNA synthesis was performed
following the instructions of the Takara PrimeScript™ first-strand
cDNA synthesis kit (Takara Bio, Inc., Tokyo, Japan). mRNA
expression of COX-2 and MMPs was analyzed by RT-PCR using the
specific primer sets. The primer sequences used for the RT-PCR were
as follows:
COX-2: forward, 5′AGTCCCTGAGCATCTACGGTTTG3′ and
reverse, 5′ CATCGCATACTCTGTTGTGTTCCC3′; MMP-1: forward,
5′TCCACTGCTGCTGCTGCTG3′ and reverse,
5′TTTCAACTTGCCTCCCATCATTCTTC3′; MMP-3: forward,
5′TGAACAATGGACAAAGGATACAACAGG3′ and reverse,
5′ATCATCTTGAGACAGGCGGAACC3′; MMP-14: forward,
5′GCTGGTTCTGGCGGGTGAG3′ and reverse, 5′TCTCGTAGGCAGTGTTGATGGAC3′;
β-actin (as an internal control): forward, 5′GCAAGCAGGAGTATGACGAG3′
and reverse, 5′CAAATAAAGCCATGCCAATC3′. The PCR conditions were as
follows: initial denaturation at 94°C for 2 min; 35 cycles of
denaturation at 94°C for 30 sec; amplification at 60°C for 30 sec;
extension at 72°C for 1 min. The size of the amplified products was
examined by agarose gel electrophoresis. Images were captured using
the Fusion FX7 acquisition system (Vilber Lourmat, Eberhardzell,
Germany). For semi-quantitive analyses, relative band intensities
against the internal control were calculated using Bio-1D (Vilber
Lourmat, Marne La Vallee, France).
Quantitative (q)RT-PCR
Total RNA was extracted from human chondrocytes
using the Easy-spin™ total RNA extraction kit. cDNA synthesis was
performed following the instructions of the Takara PrimeScript™
first-strand cDNA synthesis kit (Takara Bio, Inc.). For qRT-PCR, 1
μl of gene primers with SYBR-Green in 20 μl of
reaction volume was applied. The primer sequences used for the
real-time PCR were as follows: COX-2: forward,
5′AGTCCCTGAGCATCTACGGTTTG3′ and reverse, 5′
CATCGCATACTCTGTTGTGTTCCC3′; MMP-1: forward, 5′TCCACTGCTGCTGCTGCTG3′
and reverse, 5′TTTCAACTTGCCTCCCATCATTCTTC3′; β-actin (as an
internal control): forward, 5′GCAAGCAGGAGTATGACGAG3′ and reverse,
5′CAAATAAAGCCATGCCAATC3′. All reactions with iTaq SYBR-Green
Supermix were performed on the CFX96 real-time PCR detection system
(all were from Bio-Rad, Hercules, CA, USA).
Western blotting
Human chondrocytes were lysed in a lysis buffer.
Equal amounts of lysate protein were electrophoretically resolved
on a 10–15% SDS-PAGE, and the resolved proteins were transferred.
Immunoreactivity was detected through sequential incubation with
horseradish peroxidase-conjugated secondary antibodies and enhanced
chemiluminescence reagents. Images were captured using the Fusion
FX7 acquisition system. Densitometry of the signal bands was
analyzed using Bio-1D. The antibodies used for immunoblotting were
COX-2, p65 (Santa Cruz Biotechnology, Inc.) and β-actin
(Sigma-Aldrich).
Immunofluorescence staining
Human chondrocytes, cultured on glass slides, were
fixed with cold acetone and blocked by 5% FBS in TBST and incubated
with mouse NF-κB (active p65 subunit) antibody (Millipore) and goat
COX-2 antibody (Santa Cruz Biotechnology, Inc.) overnight at 4°C.
After washing with TBST, the cells were incubated with anti-mouse
IgG conjugated with Alexa Fluor® 488 (green) and
anti-goat IgG conjugated with Alexa Fluor® 350 (blue).
The cells were washed with TBST, mounted with fluorescence mounting
medium (Dako) and observed under a fluorescence microscope (Nikon
ECLIPSE 80i; Nikon Corporation).
Gelatin zymography
Gelatin zymography was performed for the detection
of MMP protein secretion and activation in conditioned medium.
Conditioned media were collected and centrifuged to remove cellular
debris, and the supernatant was collected and stored at −20°C. Each
sample suspension was mixed with SDS sample buffer without reducing
agent, followed by gelatin zymography. The sample was resolved by
SDS-PAGE gels containing 10% acrylamide and 1 mg/ml gelatin. The
gels were then washed twice in 2.5% (w/v) Triton X-100 in distilled
water for 30 min at room temperature and were incubated overnight
at 37°C in developing buffer containing 50 mM Tris-HCl (pH 7.5),
0.2 M NaCl and 5 mM CaCl2. The following morning, gels
were stained with 0.2% Coomassie Blue R-250 in 50% ethanol and 10%
acetic acid for 1 h at room temperature and destained with a buffer
consisting of 20% methanol, 10% acetic acid and 70% distilled water
for 30 min at room temperature to visualize these zones of
digestion as light areas against the darkly stained protein
background. The zymography gels were analyzed using the Fusion FX7
acquisition system.
Quantification of glycosaminoglycan (GAG)
release
Cartilage explants were treated with IL-1β with or
without S1P, SEW2871 and W146 for 72 h. GAG levels in the culture
medium were determined by reaction with 1,9-dimethylmethylene blue.
Twenty microliters of samples or chondroitin sulfate were mixed
with 180 μl DMB reagents (48 mg/ml DMB, 40 mM glycine, 40 mM
NaCl, 10 mM HCl and pH 3.0) for 10 min at room temperature.
Absorbance at 525 nm was measured with a spectrophotometer
(SpectraMax; Molecular Devices, Sunnyvale, CA, USA). All
measurements were performed in quadruplicate. Quantification was
performed using a standard curve of chondroitin sulfate in the
range of 0–5 μg/ml. Results were normalized to the protein
concentration for GAG release. Culture supernatant was also
measured for protein concentration by BCA reagent (Pierce
Biotechnology, Inc., Rockford, IL, USA).
Analysis of PGE2 levels:
enzyme-linked immunosorbent assay (ELISA)
Human chondrocytes were incubated with DMEM
containing 1% FBS prior to treatment with or without IL-1β (10
ng/ml), S1P, SEW2871 and W146 for 24 h. Culture supernatants were
collected and stored at −20°C. PGE2 levels in the
culture medium were quantified using an immunoenzymatic method
(PGE2 EIA kits; Cayman Chemical) according to the
manufacturer’s instructions.
Statistical evaluation
All data are expressed as the means ± SEM, and the
data were compared using the ANOVA and Duncan’s test using the SAS
statistical package.
Results
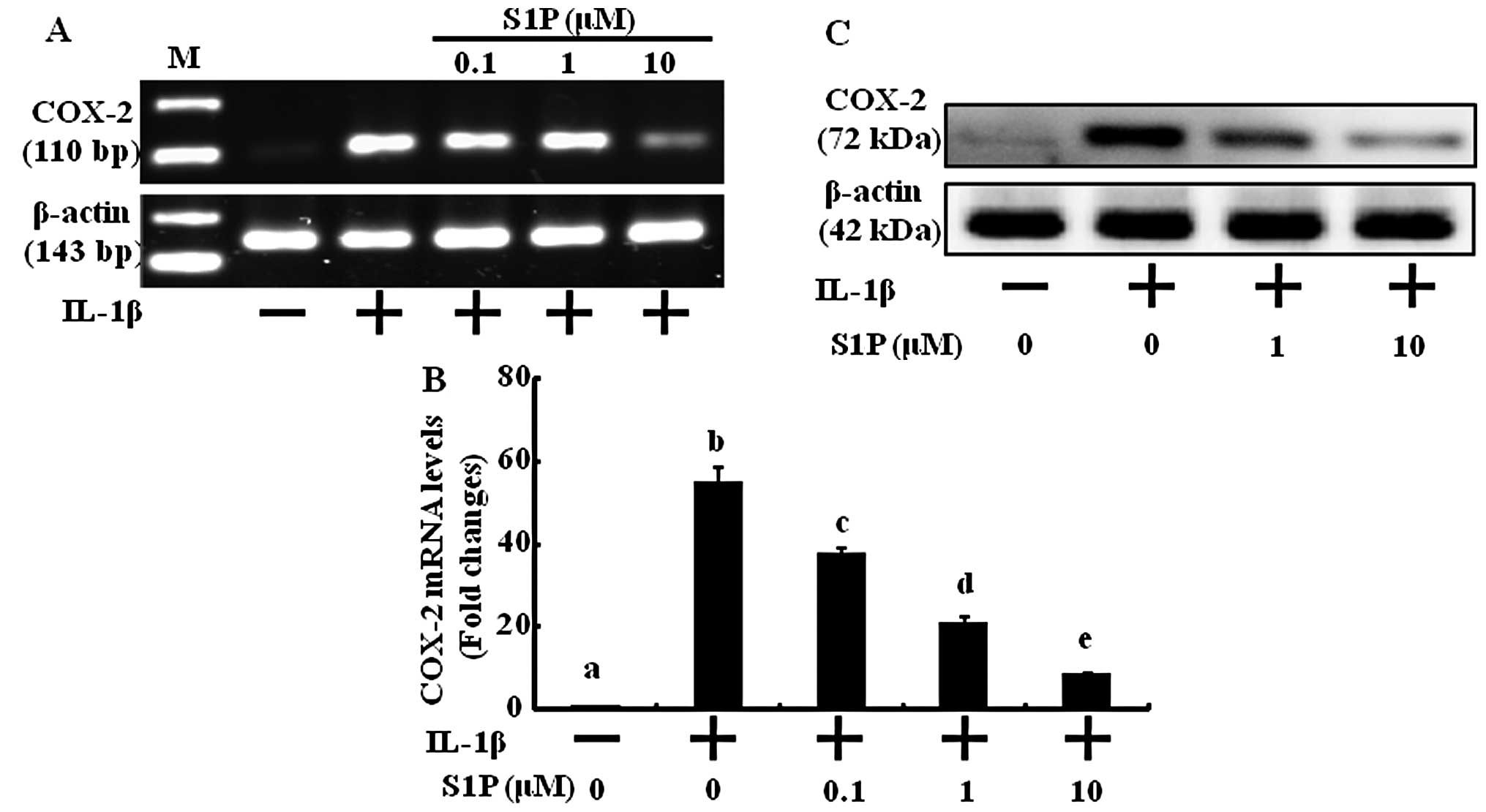
S1P inhibits IL-1β-induced COX-2
expression in human chondrocytes
The pro-inf lammatory cytokine IL-1β contributes to
the pathogenesis of OA and is a potent inducer of COX-2 expression
which plays a pivotal role in the pathogenesis of cartilage
inflammation. We treated human chondrocytes with various
concentrations of S1P to define its anti-inflammatory effects via
COX-2 expression inhibition. Under basal conditions, expression of
COX-2 mRNA and protein was undetectable. Treatment with IL-1β
resulted in significant increases in mRNA (Fig. 1A and B) and protein expression
(Fig. 1C). S1P prevented the
induction of COX-2 mRNA as well as protein expression by IL-1β in a
concentration-dependent manner (Fig.
1). The effect of S1P was significantly detected at 0.1
μM and was maximal at 10 μM. These results indicate
that S1P has anti-inflammatory effects by decreasing the expression
of COX-2 at the protein level as well as at the transcriptional
level.
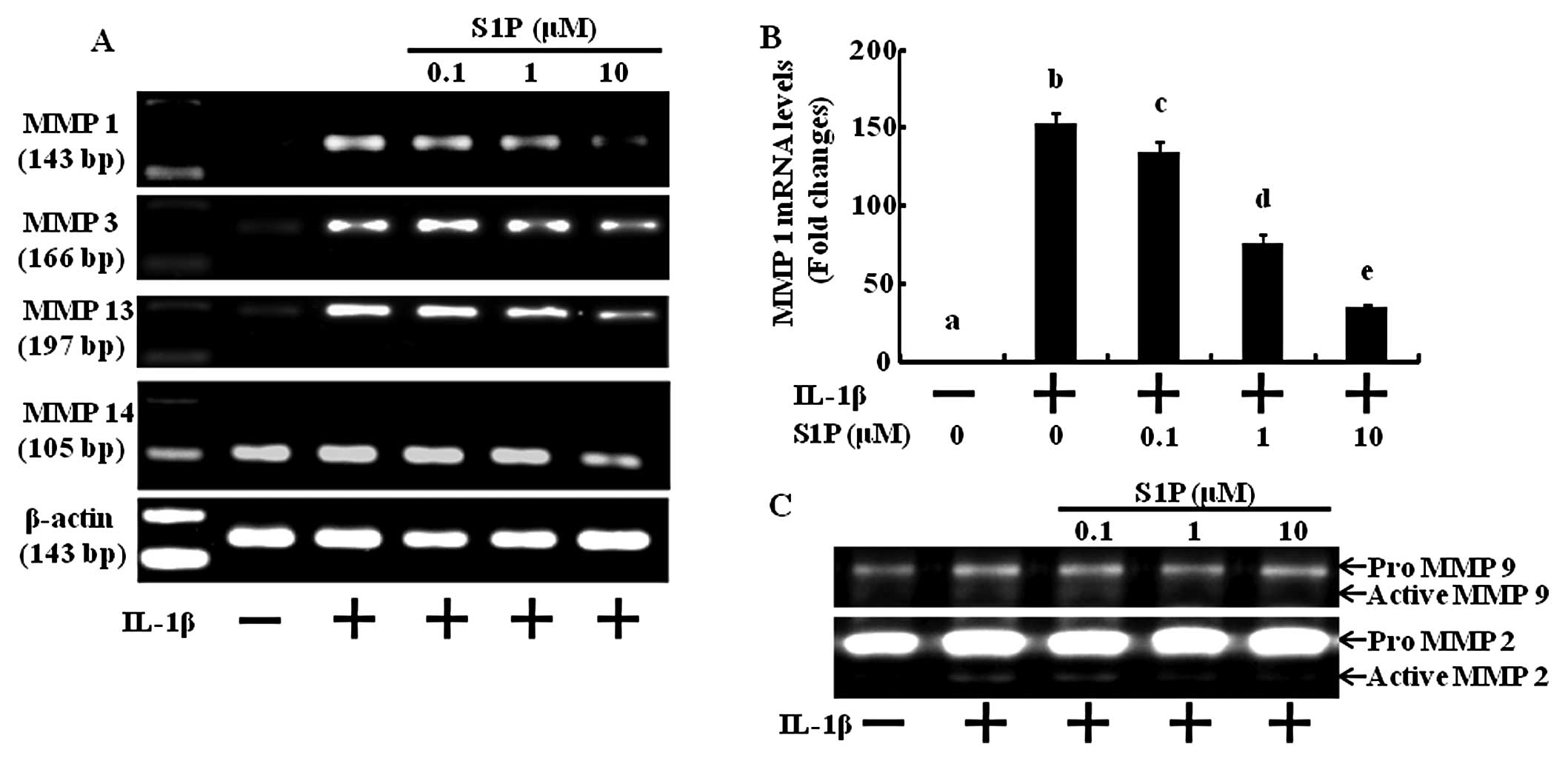
S1P decreases matrix degradation gene
products as well as MMP activity induced by IL-1β in human
chondrocytes
Since the loss of cartilage matrix is due to the
upregulation of MMP expression and activation (18), we investigated whether S1P
inhibits MMP gene expression and activation induced by
pro-inflammatory cytokines in human articular chondrocytes. In the
control situation, MMP mRNA was undetectable, but treatment with
IL-1β resulted in a substantial increase in mRNA expression
(Fig. 2A and B). Dose-dependent
treatment with S1P led to significant inhibition of all 4 MMP gene
products measured (MMP-1, -3, -13 and -14) (Fig. 2A and B). Next, we examined the
inhibitory effect of S1P on the MMP activation induced by IL-1β
using gelatin zymography (Fig.
2C). Under basal conditions, MMP-2 and -9 were not activated,
but the addition of IL-1β activated these MMPs (Fig. 2C). Increasing concentrations of
S1P significantly decreased the pro-inflammatory cytokine-induced
activities of MMP-2 and -9 (Fig.
2C). These results indicate that S1P inhibits MMP expression
and activation and may potentially block cartilage degradation.
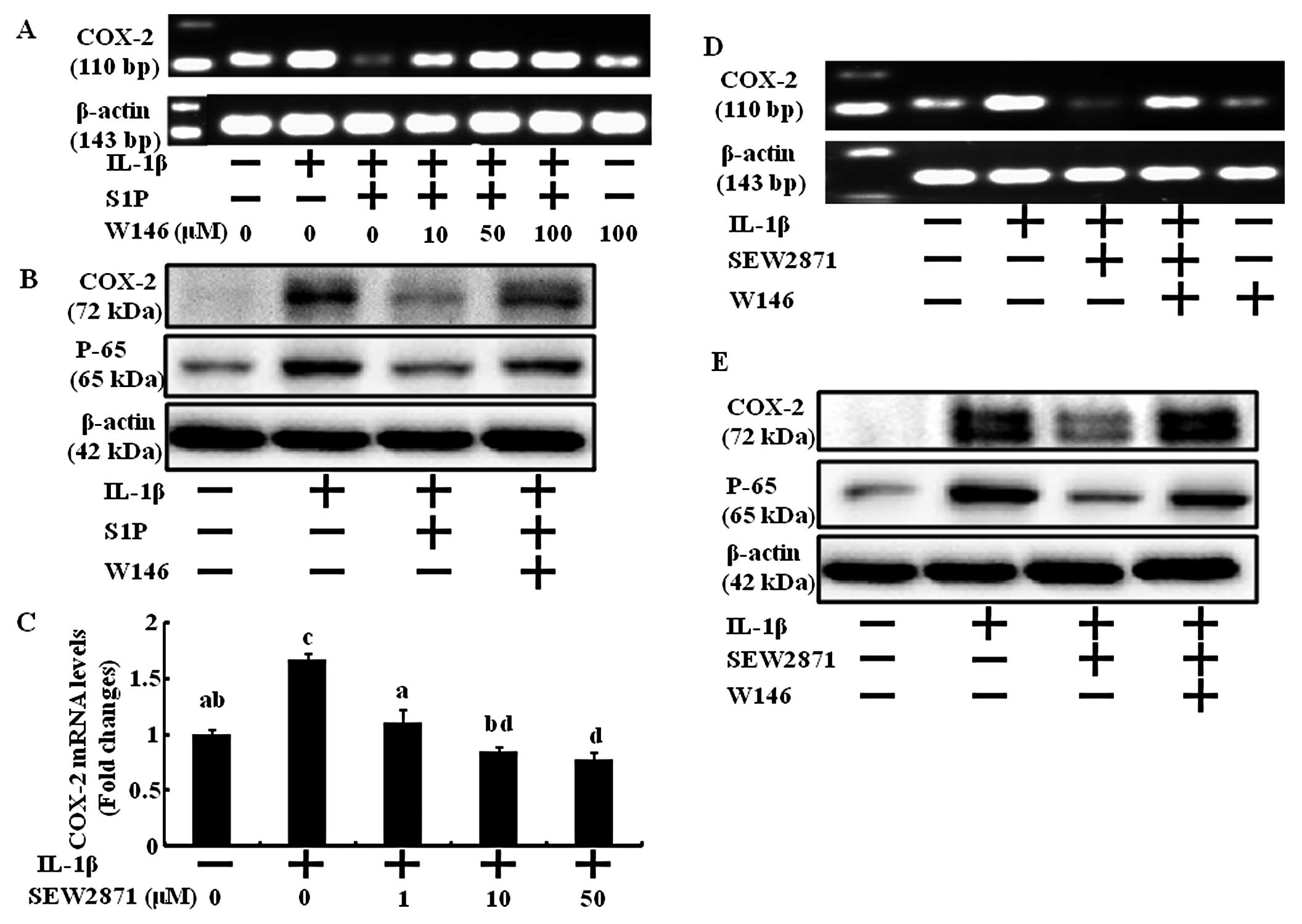
S1P1 receptor is involved in
the chondrocyte anti-inflammatory effect of S1P
The biological action of S1P is largely ascribed to
ligation to specific S1PRs that may evoke distinct biological
responses. According to Ogawa et al, the novel
S1P1 receptor agonist, KRP-203, reduces experimental
autoimmune myocarditis in rats (15). Therefore, among S1P-specific
receptors, we first investigated the involvement of S1P1
in anti-inflammation. Human chondrocytes, stimulated by IL-1β, were
treated with SEW2871, a selective S1P1 agonist, to
activate S1P1. With increasing concentrations of
SEW2871, COX-2 mRNA expression was significantly diminished
(Fig. 3C). In addition, the
anti-inflammatory action of S1P was prevented by blocking the
S1P1 receptor using W146, a selective S1P1
antagonist (Fig. 3A, B and F).
The inhibition of COX-2 mRNA and protein upregulation by IL-1β was
blocked by the addition of 10 μM of W146. Also, the
anti-inflammatory action of S1P1 receptor-specific
activation using SEW2871 was disturbed by blocking the
S1P1 receptor using W146 (Fig. 3D, E and G).
NF-κB is a transcription factor that regulates the
expression of several genes involved in immune responses and
inflammation including COX-2 and MMPs (7). S1P and SEW2871 both decreased the
IL-1-induced active form of NF-κB p65, but W146 reversed the
anti-inflammatory actions of S1P and SEW2871 (Fig. 3B and E–G).
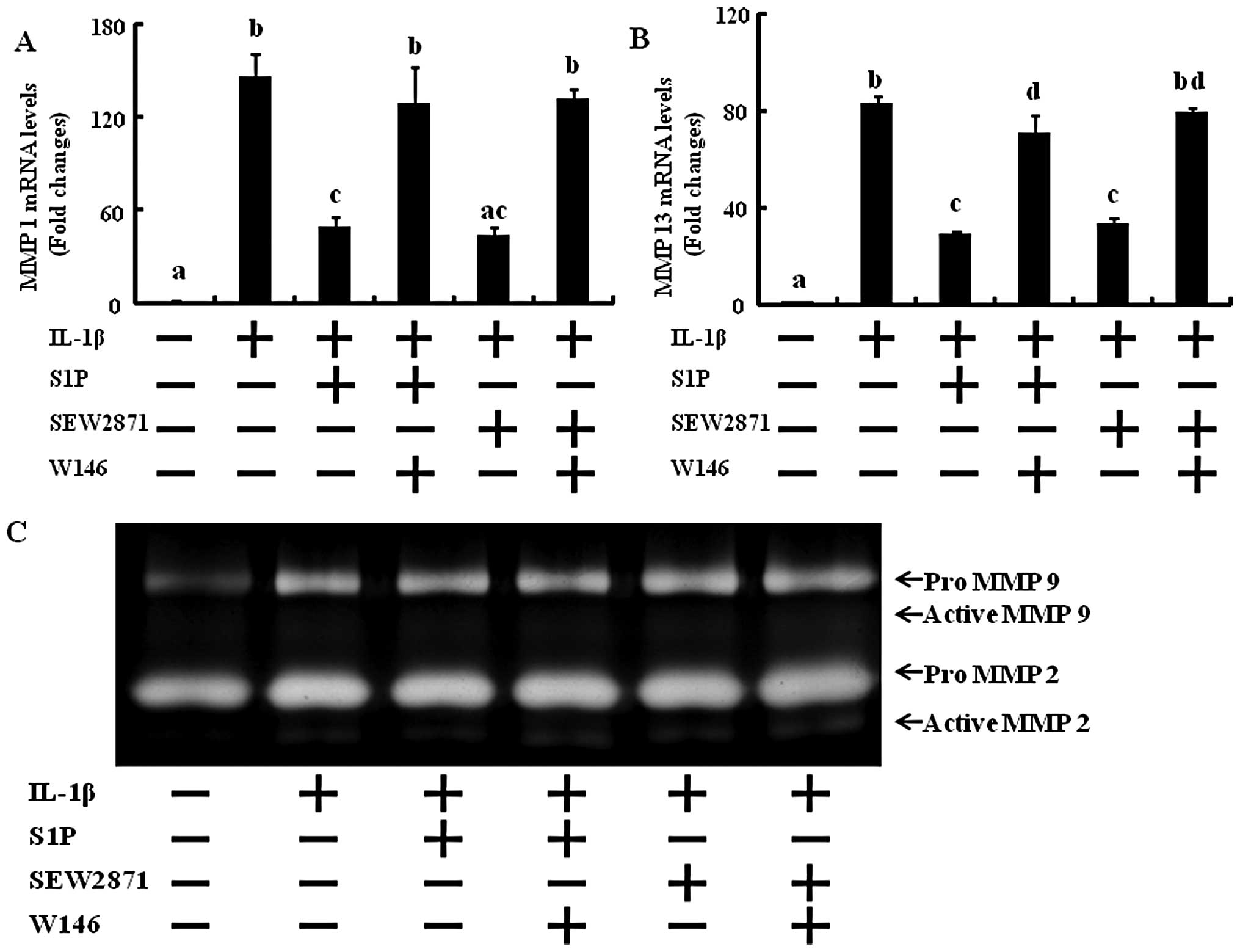
In order to determine whether only S1P1
selective activation affects MMP expression and activation and if
blocking the S1P1 receptor abolishes the effect of S1P
and SEW2871 on MMP expression and activation, RT-PCR and gelatin
zymography were performed. In human chondrocytes exposed to 10
ng/ml IL-1β and treated with SEW2871, the levels of MMP-1 and -13
were significantly decreased back to the levels of the control
(Fig. 4). W146 abolished the
action of S1P and SEW2871 that had decreased MMP-1 and -13 mRNA
levels (Fig. 4). These results
suggest that the regulation of NF-κB and MMP activation induced by
S1P is mediated through the S1P1 receptor.
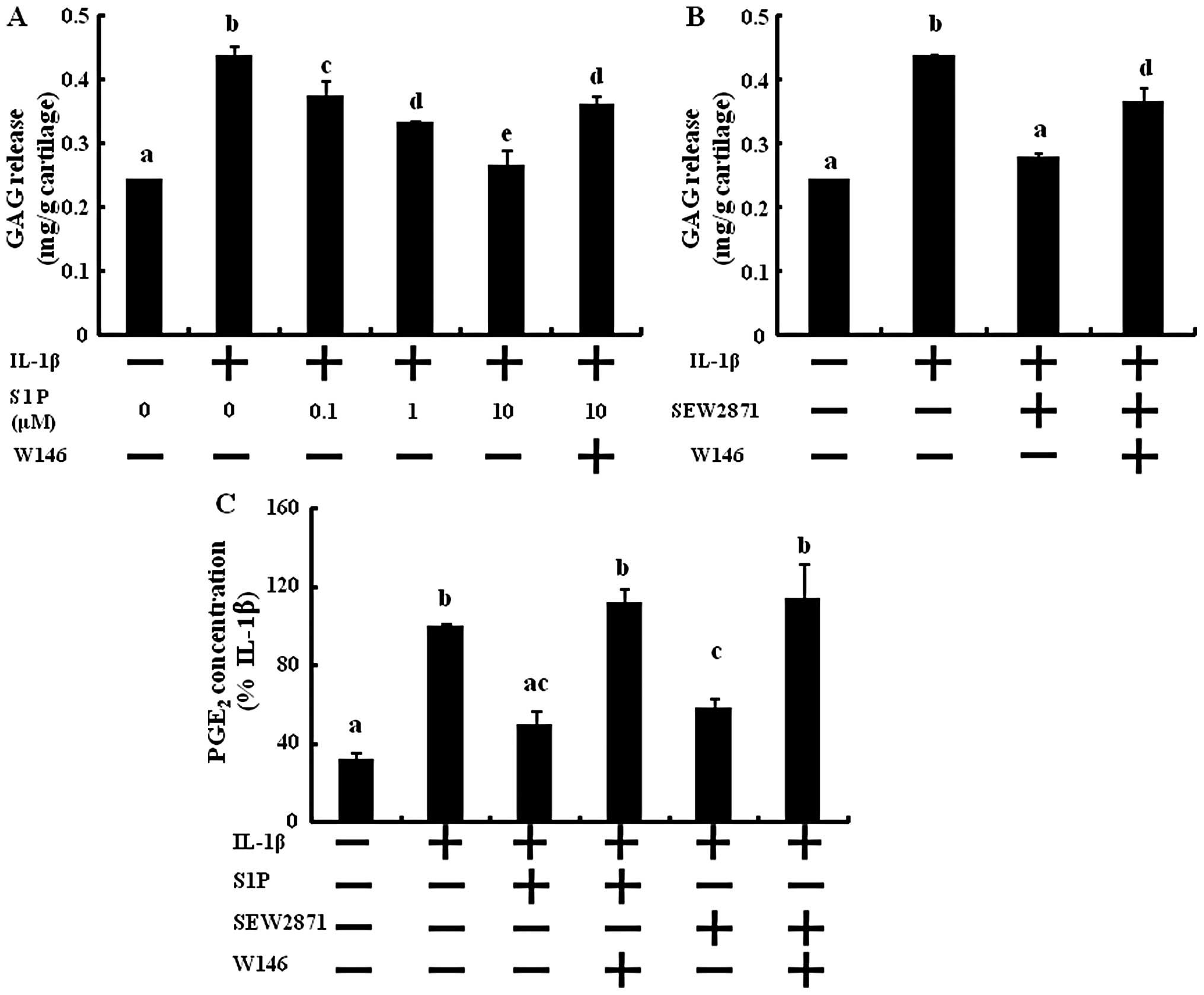
S1P protects cartilage explants from
IL-1-induced GAG loss and PGE2 production
Cartilage explants were treated with S1P in
combination with IL-1β. GAG release to the media was significantly
and dose-dependently inhibited by S1P (Fig. 5A). At 10 μM of S1P, GAG
release was reduced compared to the control. GAG release induced by
IL-1β was reduced by adding 50 μM of SEW2871 (Fig. 5B). However, W146 abolished the
inhibitory effect of the GAG degradation action of S1P and SEW2871
and the contents of GAG release were similar to that with the IL-1β
treatment. These results revealed that S1P protects cartilage
degradation from inflammatory cytokine-induced GAG degradation via
S1P1 receptor activation.
PGE2, which originates from the
activation of the COX pathways, is produced by OA chondrocytes and
may promote matrix degeneration (19,20). In ex vivo cultures of
cartilage explants, the production of PGE2 was increased
by the stimulation of IL-1β in the supernatants of the conditioned
media (Fig. 5C). After treatment
with S1P, PGE2 synthesis was significantly reduced to
the level of the control (Fig.
5C). Next, we investigated the action of S1P through the
S1P1 receptor. Only S1P1 selective activation
decreased PGE2 production, and blocking the
S1P1 receptor abolished the effect of S1P and SEW2871 on
PGE2 synthesis (Fig.
5C). These results suggest that S1P prevents inflammatory
cytokine-induced PGE2 synthesis through S1P1
receptor activation.
Discussion
The main goal of this study was to determine the
anti-inflammatory effects of S1P in human chondrocyte-induced
inflammation. The data presented demonstrated that S1P acts as an
anti-inflammatory regulator of chondrocytes and that the action of
S1P is mediated by the S1P1 receptor. Treatment with
exogenous S1P effectively inhibited COX-2 expression by reducing
the activation of NF-κB p65, which is considered one of the major
regulators of OA pathogenesis. Also, S1P significantly decreased
MMP-1, -3, -13 and -14 expression. Moreover, S1P protects cartilage
explants from IL-1β-induced PGE2 synthesis and GAG
degradation. S1P effectively prevented cartilage explants from
PGE2 synthesis and GAG release caused by IL-1β. These
S1P effects were abolished by the inhibition of the S1P1
receptor using W146. The S1P1 selective agonist,
SEW2871, had anti-inflammatory effects similar to S1P. These
anti-inflammatory effects of S1P via the S1P1 receptor
occurred through the regulation of PGE2 production
mediated by COX-2 expression and NF-κB and MMP activation.
OA is a painful and disabling disease that affects
millions of people. Arthritic joints display an altered metabolism
and an imbalance between anabolic growth factors and
pro-inflammatory cytokines, TNF-α and IL-1β produced by
inflammatory cells, synovial fibroblasts and chondrocytes in
affected joints (21). In fact,
IL-1β, a well-recognized pro-inflammatory cytokine, is increased
locally during the OA process (22). IL-1β induces a large cascade of
events that leads to cartilage damage, such as the synthesis of
MMPs and ECM proteins that are absent in normal cartilage and the
release of other inflammatory mediators including COX-2 (23). Several of these effects are
mediated by eicosanoids, which are products of arachidonic acid
metabolism. PGE2 is the predominant eicosanoid
synthesized by OA cartilage and mediates several IL-1β-induced
effects (24). The goal of
pharmacological treatment is usually to control symptoms of the
disease, pain and limitation of function, which is traditionally
accomplished by the use of analgesic agents and NSAIDs (25). However, while providing relief
from pain, none of these agents inhibit cartilage breakdown or
disease development; and they also have varying degrees of GI
toxicity (26). Therefore, new
and safe therapeutics which inhibit disease progression are
required.
The beneficial effects of S1P on inflammation in
different tissues and cells have been demonstrated (15,16). Therefore, S1P was used as an
anti-inflammatory agent for IL-1β-induced OA in a human chondrocyte
in vitro model. The results of this study demonstrated that
inflammation of human chondrocytes by IL-1β-induced expression of
COX-2 was effectively inhibited by S1P (Fig. 1). In contrast to our results,
Masuko et al demonstrated that S1P increased COX-2
expression and PGE2 production in chondrocytes. They
treated chondrocytes with S1P and the increase in COX-2 was 40
pg/ml (32). Similarly, we
observed that only S1P treatment increased the COX-2 expression and
PGE2 production slightly (not significant). However,
when the chondrocytes were pretreated with S1P prior to IL-1β
treatment, S1P significantly decreased IL-1β-induced COX-2
expression and PGE2 production (Figs. 1 and 5). Although S1P only slightly increased
COX-2 expression and PGE2 production under a normal
condition, S1P effectively functions as a COX-2 inhibitor in
arthritic joint tissue induced by IL-1β.
An immediate cause of the destruction of joint
tissue in OA is the augmentation of MMP family enzymes (27). In normal tissue, these enzymes are
expressed at low levels to maintain cartilage homeostasis, but in
pathological states such as OA, they are expressed at abnormally
high levels (28). S1P
significantly reduced MMP expression and activation (Fig. 2). MMPs are subdivided into several
subtypes. Among them, MMP-1 and -13 preferentially degrade native
type II collagen and are synthesized in increased amounts by OA
chondrocytes; thus, they are postulated to have an important role
in the destruction of cartilage (4). S1P was found to dose-dependently
decrease MMP-1 and -13 gene products, whereas proteoglycan loss
involves MMP-3 and -14 expression (4). Treatment with exogenous S1P reduced
MMP-3 and -14 gene products (Fig.
2). These results are consistent with the findings that high
levels of GAG released from cartilage explants caused by IL-1β were
diminished by S1P treatment (Fig.
5A), suggesting that S1P downregulates MMP-3 and -14 expression
at the transcriptional level, leading to suppression of GAG
degradation by pro-inflammatory cytokines. Together these data
suggest that S1P may prevent cartilage destruction in arthritis by
suppressing COX-2 and MMPs.
Several of the biological effects of IL-1β on
chondrocytes (i.e. upregulation of MMPs and COX-2) are also
mediated by NF-κB (29,30). In the present study, we
demonstrated that IL-1β enhanced the activation of NF-κB p65.
Interestingly, treatment with S1P downregulated the expression of
the active form of NF-κB p65 (Fig.
3B) suggesting that the anti-inflammatory action of S1P (i.e.
downregulation of MMPs and COX-2) is due to the inhibition of NF-κB
activation. These results are in accordance with reports revealing
that S1P suppresses NF-κB in germ cells (31).
Several signaling pathways that are activated in
response to the stimulation of cells by S1P are initiated by
activation of S1P-specific receptors. The results of this study
demonstrated that the anti-inflammatory action of S1P was
associated with the S1P1 receptor (Figs. 3–5). Treatment with S1P1
selective agonist SEW2871 inhibited the active form of NF-κB p65
(Fig. 3E), PGE2
production (Fig. 5C), COX-2
expression (Fig. 3A–C),
expression and activation of MMPs (Fig. 4) and GAG degradation (Fig. 5B) similar to S1P treatment
(Fig. 3C–E). In addition, the
S1P1 antagonist W146 significantly reversed the actions of S1P and
SEW2871 (Figs. 3–5). Collectively the evidence in the
current study indicates that the selective activation of S1P and
S1P1 may potentially improve the therapeutic effect in
arthritis.
In conclusion, we identified the anti-inflammatory
action of S1P via the S1P1 receptor by inhibiting NF-κB
p65 activation, COX-2 expression, MMP activation, PGE2
production and GAG degradation in articular chondrocytes. These
results suggest that S1P1 activation may be a
therapeutic target for OA. Therefore, S1P and the development of
S1P1 receptor subtype-specific ligands may result in a
promising new class of drugs for the treatment of OA.
Acknowledgements
This study was supported by the
National Research Foundation of the Korea Grant funded by the
Korean Government (2010-E00019).
References
|
1.
|
N ChabaneN ZayedH AfifHistone deacetylase
inhibitors suppress interleukin-1beta-induced nitric oxide and
prostaglandin E2 production in human chondrocytesOsteoarthritis
Cartilage1612671274200810.1016/j.joca.2008.03.00918417374
|
|
2.
|
MB GoldringKB MarcuCartilage homeostasis
in health and rheumatic diseasesArthritis Res
Ther11224200910.1186/ar259219519926
|
|
3.
|
MB GoldringF BerenbaumHuman chondrocyte
culture models for studying cyclooxygenase expression and
prostaglandin regulation of collagen gene expressionOsteoarthritis
Cartilage7386388199910.1053/joca.1998.0219
|
|
4.
|
HA KimY YeoWU KimS KimPhase II enzyme
inducer sulphoraphane blocks matrix metalloproteinase production in
articular chondrocytesRheumatology
(Oxford)48932938200910.1093/rheumatology/kep132
|
|
5.
|
C LianxuJ HongtiY
ChanglongNF-kappaBp65-specific siRNA inhibits expression of genes
of COX-2, NOS-2 and MMP-9 in rat IL-1beta-induced and
TNF-alpha-induced chondrocytesOsteoarthritis
Cartilage14367376200610.1016/j.joca.2005.10.00916376111
|
|
6.
|
JP PelletierJ Martel-PelletierSB
AbramsonOsteoarthritis, an inflammatory disease: potential
implication for the selection of new therapeutic targetsArthritis
Rheum4412371247200110.1002/1529-0131(200106)44:6%3C1237::AID-ART214%3E3.0.CO;2-F11407681
|
|
7.
|
EB KoppS GhoshNF-kappa B and rel proteins
in innate immunityAdv
Immunol58127199510.1016/S0065-2776(08)60618-57741027
|
|
8.
|
AS Baldwin JrThe NF-kappa B and I kappa B
proteins: new discoveries and insightsAnnu Rev
Immunol14649683199610.1146/annurev.immunol.14.1.6498717528
|
|
9.
|
R NewtonLM KuitertM BergmannIM AdcockPJ
BarnesEvidence for involvement of NF-kappaB in the transcriptional
control of COX-2 gene expression by IL-1betaBiochem Biophys Res
Commun2372832199710.1006/bbrc.1997.70649266823
|
|
10.
|
M ShakibaeiC CsakiS NebrichA
MobasheriResveratrol suppresses interleukin-1beta-induced
inflammatory signaling and apoptosis in human articular
chondrocytes: potential for use as a novel nutraceutical for the
treatment of osteoarthritisBiochem
Pharmacol7614261439200810.1016/j.bcp.2008.05.029
|
|
11.
|
ED JohnstoneG ChanCP SibleyST DavidgeB
LowenLJ GuilbertSphingosine-1-phosphate inhibition of placental
trophoblast differentiation through a G(i)-coupled receptor
responseJ Lipid Res4618331839200510.1194/jlr.M500095-JLR200
|
|
12.
|
M SchuppelU KurschnerU KleuserM
Schafer-KortingB KleuserSphingosine 1-phosphate restrains
insulin-mediated keratinocyte proliferation via inhibition of Akt
through the S1P2 receptor subtypeJ Invest
Dermatol12817471756200810.1038/sj.jid.570125918219276
|
|
13.
|
S PyneNJ PyneSphingosine 1-phosphate
signalling in mammalian cellsBiochem
J349385402200010.1042/0264-6021:349038510880336
|
|
14.
|
MH StradnerJ HermannH
AngererSphingosine-1-phosphate stimulates proliferation and
counteracts interleukin-1 induced nitric oxide formation in
articular chondrocytesOsteoarthritis
Cartilage16305311200810.1016/j.joca.2007.06.018
|
|
15.
|
R OgawaM TakahashiS HiroseA novel
sphingosine-1-phosphate receptor agonist KRP-203 attenuates rat
autoimmune myocarditisBiochem Biophys Res
Commun361621628200710.1016/j.bbrc.2007.07.06117673173
|
|
16.
|
JE HughesS SrinivasanKR LynchRL ProiaP
FerdekCC HedrickSphingosine-1-phosphate induces an antiinflammatory
phenotype in macrophagesCirc
Res102950958200810.1161/CIRCRESAHA.107.17077918323526
|
|
17.
|
JW SeolHB LeeNS KimSY
ParkTartrate-resistant acid phosphatase as a diagnostic factor for
arthritisInt J Mol Med245762200919513535
|
|
18.
|
MB GoldringThe role of cytokines as
inflammatory mediators in osteoarthritis: lessons from animal
modelsConnect Tissue
Res40111199910.3109/0300820990900527310770646
|
|
19.
|
CM ThomasCJ FullerCE WhittlesM
SharifChondrocyte death by apoptosis is associated with cartilage
matrix degradationOsteoarthritis
Cartilage152734200710.1016/j.joca.2006.06.01216859932
|
|
20.
|
M DaveM AtturG PalmerThe antioxidant
resveratrol protects against chondrocyte apoptosis via effects on
mitochondrial polarization and ATP productionArthritis
Rheum5827862797200810.1002/art.2379918759268
|
|
21.
|
A LiaciniJ SylvesterWQ LiInduction of
matrix metalloproteinase-13 gene expression by TNF-alpha is
mediated by MAP kinases, AP-1, and NF-kappaB transcription factors
in articular chondrocytesExp Cell
Res288208217200310.1016/S0014-4827(03)00180-012878172
|
|
22.
|
DD WoodEJ IhrieCA DinarelloPL
CohenIsolation of an interleukin-1-like factor from human joint
effusionsArthritis
Rheum26975983198310.1002/art.17802608066603852
|
|
23.
|
R LargoMA Alvarez-SoriaI
Diez-OrtegoGlucosamine inhibits IL-1beta-induced NFkappaB
activation in human osteoarthritic chondrocytesOsteoarthritis
Cartilage11290298200310.1016/S1063-4584(03)00028-112681956
|
|
24.
|
AR AminM DaveM AtturSB AbramsonCOX-2, NO,
and cartilage damage and repairCurr Rheumatol
Rep2447453200010.1007/s11926-000-0019-511123096
|
|
25.
|
JY ReginsterR DeroisyLC RovatiLong-term
effects of glucosamine sulphate on osteoarthritis progression: a
randomised, placebo-controlled clinical
trialLancet357251256200110.1016/S0140-6736(00)03610-211214126
|
|
26.
|
R SinghS AhmedN IslamVM GoldbergTM
HaqqiEpigallocatechin-3-gallate inhibits interleukin-1beta–induced
expression of nitric oxide synthase and production of nitric oxide
in human chondrocytes: suppression of nuclear factor kappaB
activation by degradation of the inhibitor of nuclear factor
kappaBArthritis Rheum46207920862002
|
|
27.
|
PS BurrageKS MixCE BrinckerhoffMatrix
metalloproteinases: role in arthritisFront
Biosci11529543200610.2741/181716146751
|
|
28.
|
J Martel-PelletierDJ WelschJP
PelletierMetalloproteases and inhibitors in arthritic diseasesBest
Pract Res Clin
Rheumatol15805829200110.1053/berh.2001.019511812023
|
|
29.
|
M ShakibaeiT JohnG Schulze-TanzilI
LehmannA MobasheriSuppression of NF-kappaB activation by curcumin
leads to inhibition of expression of cyclo-oxygenase-2 and matrix
metalloproteinase-9 in human articular chondrocytes: implications
for the treatment of osteoarthritisBiochem
Pharmacol7314341445200710.1016/j.bcp.2007.01.005
|
|
30.
|
PJ BarnesM KarinNuclear factor-κB: A
pivotal transcription factor in chronic inflammatory diseasesN Engl
J Med336106610711997
|
|
31.
|
L SuomalainenV PentikäinenL
DunkelSphingosine-1-phosphate inhibits nuclear factor κB activation
and germ cell apoptosis in the human testis independently of its
receptorsAm J Pathol1667737812005
|
|
32.
|
K MasukoM MurataH NakamuraK YudohK
NishiokaT KatoSphingosine-1-phosphate attenuates proteoglycan
aggrecan expression via production of prostaglandin E2 from human
articular chondrocytesBMC Musculoskelet
Disord829200710.1186/1471-2474-8-2917374154
|