Introduction
Prion diseases or transmissible spongiform
encephalopathies (TSEs) are neurodegenerative disorders that are
characterized by loss of motor control, dementia, central nervous
system (CNS) spongiosis, and microglial activation (1,2).
TSEs are caused by an infectious agent, prion, whose
major component is a pathological form of the prion protein termed
the scrapie isoform (PrPSc) (3).
PrPsc acts as a template for the conversion of normal form of the
prion protein (the cellular isoform, PrPc) to PrPsc (4). In many cases this is also
accompanied by the accumulation of the PrPSc that leads to neuronal
apoptosis, extensive neuronal loss, and mitochondrial disruption
(5). Many pathogenic
characteristics of PrPSc have been confirmed in a peptide
corresponding to residues 106-126 of PrP [PrP (106-126)] (6). Moreover, PrP (106-126) was reported
to induce apoptotic cell death via dysregulation of mitochondrial
homeostasis in neuronal cells (7). Thus, PrP (106-126) has been used as
a model to study prion-induced neuronal cell death and has been
postulated to induce mitochondrial dysfunction (8).
Mitochondria are essential organelles found in
various cell types that play a principal role in cell survival and
apoptotic cell death (9).
Mitochondrial oxidative damage contributes to a range of
degenerative diseases (10).
Mitochondrial dysfunction caused by unnatural regulation of
mitochondrial dynamic proteins may lead to neuropathological
changes in prion disorders (11).
In addition, PrP (106-126)-induced neuronal cell damage that occurs
in neurodegenerative disorders causes mitochondrial disruption
(12). Furthermore, oxidative
stress is key in mitochondrial-mediated apoptotic cell death
(13).
Oxidative stress is a baneful condition caused by
reactive oxygen species (ROS) and/or a decrease in antioxidant
levels (14). In
neurodegenerative disorders, oxidative stress-induced
neurodegeneration is mediated by ROS production (15). In addition, mitochondrial
dysfunction is associated with ROS (16). PrP (106-126)-induced neuronal cell
damage occurs in neurodegenerative disorders via regulation of
cellular oxidation pathways (17).
Lactoferrin (LF) is an 80 kDa protein found in
colostrum, milk, and mucosal secretions such as blood, saliva, and
tears (18). It is a
multifunctional protein of the transferrin family, which is
involved in the regulation of immune responses, regulation of
neutrophil apoptosis, antioxidation, iron binding ability, and
antimicrobial activity (19). The
antioxidation capability of LF is due to the scavenging of ROS
(20). For example, LF inhibits
the subsequent production of ROS by neutrophils (21). However, the molecular mechanism of
LF-mediated neuronal survival is only beginning to be
understood.
We hypothesized that LF can prevent PrP
(106-126)-induced oxidative stress and neuronal cell death by
regulating ROS generation. To test this hypothesis, we investigated
the antioxidant effect of LF in PrP (106-126)-induced neuronal cell
death. In particular, we tested whether LF protects from neuronal
cell death by PrP (106-126) and assessed the therapeutic value of
LF in the treatment of neurodegenerative disorders.
Materials and methods
Cell culture
The SH-SY5Y human neuroblastoma cell line was
obtained from the American Type Culture Collection (ATCC,
Rockville, MD, USA). Cells were cultured in Minimum Essential
Medium (MEM; Invitrogen-Gibco, Grand Island, NY, USA) that
contained 10% fetal bovine serum (FBS; Invitrogen-Gibco) and
penicillin-streptomycin (both 100 U/ml) in a humidified incubator
maintained at 37°C and 5% CO2.
Reagents
LF from bovine colostrums was purchased from
Sigma-Aldrich (St. Louis, MO, USA). The antioxidant agents
glutathione (GSH) and N-acetylcysteine (NAC) were purchased from
Sigma-Aldrich.
PrP (106-126) treatment
Synthetic PrP (106-126) (sequence,
Lys-Thr-Asn-Met-Lys-His-Met-Ala-Gly-Ala-Ala-Ala-Ala-Gly-Ala-Val-Val-Gly-Gly-Leu-Gly)
was synthesized by Peptron (Seoul, Korea). The peptide was
dissolved in sterile dimethylsulfoxide (DMSO) at a concentration of
10 mM and stored at −80°C.
Western blot analysis
SH-SY5Y was lysed in a buffer containing 25 mM
HEPES; pH 7.4, 100 mM NaCl, 1 mM EDTA, 5 mM MgCl2, 0.1
mM dithiothreitol (DTT), and protease inhibitor mixture. Proteins
were electrophoretically resolved by 10–15% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and
immunoblotting was performed as previously described. Equal amounts
of lysate protein were similarly electrophoretically resolved and
electrophoretically transferred to a nitrocellulose membrane.
Immunoreactivity was detected through sequential incubation with
horseradish peroxidase-conjugated secondary antibody and enhanced
chemiluminescence reagents. The antibodies used for immunoblotting
were phospho-c-Jun, N-terminal kinase (p-JNK; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), cleaved caspase-3 (Cell
Signaling Technology, Danvers, MA, USA), and β-actin (Santa Cruz
Biotechnology, Inc.).
Cellular fractionation
SH-SY5Y cells were resuspended in mitochondrial
buffer (210 mM sucrose, 70 mM mannitol, 1 mM EDTA, 10 mM HEPES),
broken by a 26-gauge needle, and centrifuged at 700 × g for 10 min.
The postnuclear supernatant was centrifuged at 10,000 × g for 30
min. The pellet was used as the mitochondrial fraction and the
supernatant was used as the cytosolic fraction. Total proteins were
obtained and subjected to western blotting.
Annexin V assay
Apoptosis was assessed by a commercial Annexin V
assay (Santa Cruz Biotechnology, Inc.) according to the
manufacturer’s protocol. Annexin V content was determined by
measuring fluorescence at an excitation wavelength of 488 nm and
emission wavelengths of 525 and 530 using a Guava easyCyte HT
System (Millipore, Billerica, MA, USA).
Immunofluorescence
SH-SY5Y cells cultured on glass cover-slips were
treated with PrP (106-126). Cells were washed with
phosphate-buffered saline (PBS) and fixed with cold acetone for 90
sec. Cells were washed with PBS, blocked with 5% FBS in Tris buffer
saline containing Tween-20, and incubated with anti-caspase-3 (2
μg/ml) and anti-p-JNK (2 μg/ml) monoclonal antibodies
for 48 h at 20°C. Unbound antibody was removed by an additional PBS
wash, and cells were incubated with labeled anti-rabbit Alexa Fluor
546 (for anti-caspase-3) IgG antibody (4 μg/ml) and Alexa
Fluor 488 (for anti-p-JNK) IgG antibody (4 μg/ml) for 2 h at
20°C. Finally, cells were mounted with DakoCytomation fluorescent
medium and visualized via fluorescence microscopy.
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) assay
TUNEL analysis was performed to measure the degree
of cellular apoptosis using an in situ ApoBrdU DNA
fragmentation assay kit (BioVision, San Francisco, CA, USA)
following the manufacturer’s instructions.
DCFH-DA assay
SH-SY5Y cells were incubated in minimum essential
medium (Hyclone Laboratories, Logan, UT, USA) containing 10
μM 2′,7′-dichlorodihydrofluorescein diacetate (H2-DCFDA) at
37°C for 30 min. Cells were washed with PBS and lysed in the
aforementioned lysis buffer. Cells were transferred to a clear
96-well plate and fluorescent emission from the bottom of the plate
was measured at 515 nm with an excitation wavelength of 488 nm
using a SpectraMax M2 instrument (Molecular Devices, Sunnyvale, CA,
USA). SH-SY5Y cells were cultured on coverslips positioned in a
24-well plate. Cells were incubated in MEM (Hyclone Laboratories)
containing 10 μM H2-DCFDA) at 37°C for 30 min. Cells were
washed with PBS.
Mitochondrial transmembrane potential
(MTP) assay
The change in MTP was evaluated by the cationic
fluorescent indicator JC-1 (Molecular Probes, Eugene, OR, USA),
which aggregates in intact mitochondria (red fluorescence)
indicating high or normal MTP and low MTP when it remains in
monomeric form in the cytoplasm (green fluorescence). SH-SY5Y cells
were incubated in MEM containing 10 μM JC-1 at 37°C for 30
min, washed with PBS, and then transferred to a clear 96-well
plate. JC-1 aggregate fluorescent emission was measured at 583 nm
with an excitation wavelength of 526 nm, and JC-1 monomer
fluorescence intensity was measured with an excitation and emission
wavelength of 525 and 530 nm, respectively, using a Guava easyCyte
HT System (Millipore). SH-SY5Y cells were cultured on coverslips in
a 24-well plate, incubated in MEM containing 10 μm JC-1 at
37°C for 30 min, and then washed with PBS. Finally, cells were
mounted with DakoCytomation fluorescent medium and visualized via
fluorescence microscopy.
Statistical analysis
All data are expressed as the means ± standard
deviation (SD), and the data were compared using the Student’s
t-test and the ANOVA Duncan test with the SAS statistical package
(SAS, Cary, NC, USA). The results were considered to indicate
statistically significant differences at *P<0.05 or
**P<0.01.
Results
PrP (106-126)-induced neuronal cell death
is decreased by LF treatment in SH-SY5Y neuroblastoma cells
In a previous study, it was shown that LF inhibits
prion accumulation (22). Thus,
we presently examined whether LF protects against PrP
(106-126)-mediated neurotoxicity. To study the influence of LF on
PrP (106-126)-induced neuronal cell death, SH-SY5Y cells were
pretreated with various concentrations of LF (12 h) and then
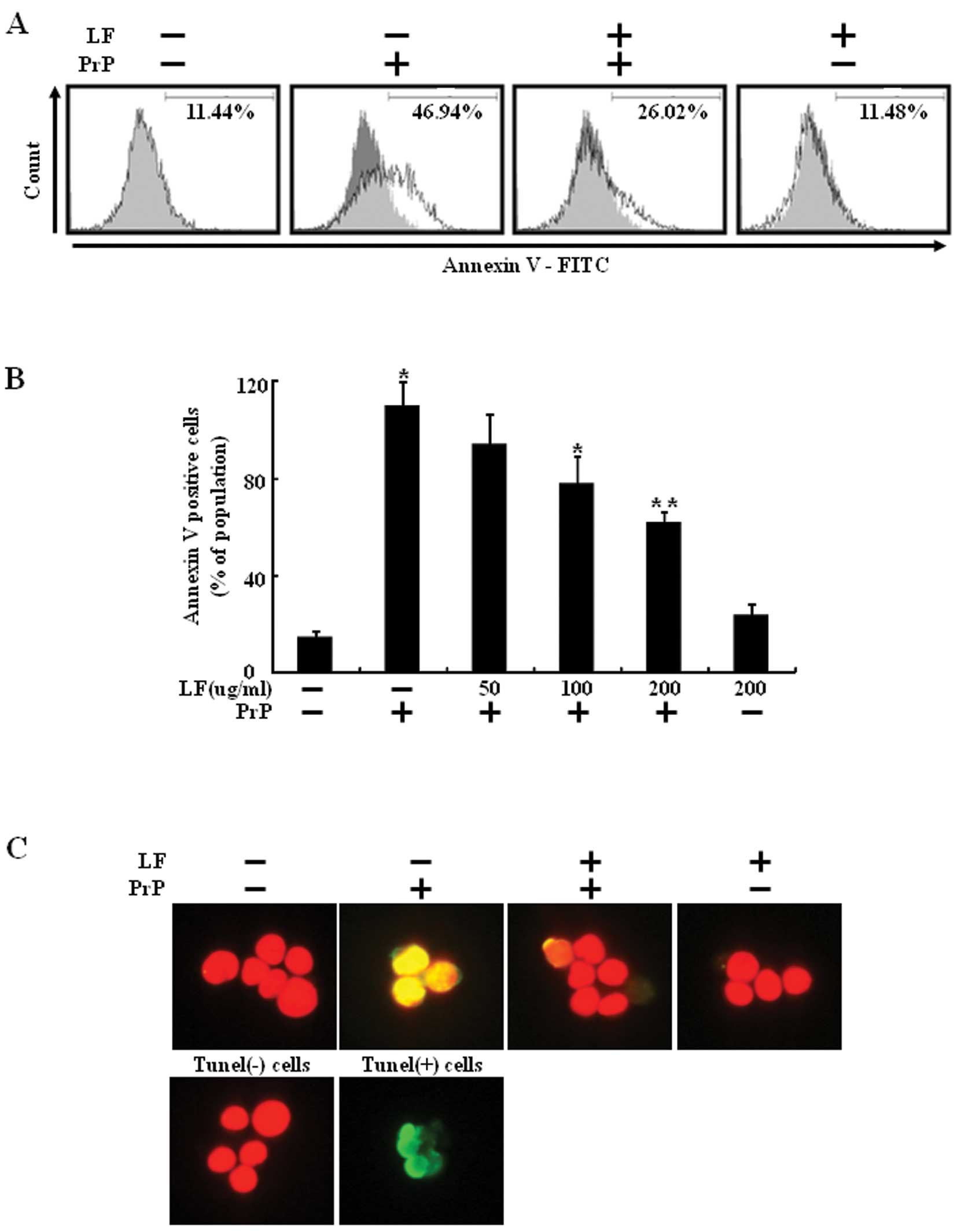
exposed to 100 μM PrP (106-126) for 8 h (Fig. 1B). The preventative effect of LF
was evaluated using the Annexin V assay of cell viability. As shown
in Fig. 1A, LF treatment
prevented PrP (106-126)-induced neuronal cell death. SH-SY5Y cells
were responsive to PrP (106-126) treatment (46.94% increase in
Annexin V-positive cells) and PrP (106-126)-induced neuronal cell
death was decreased by LF pretreatment (Fig. 1A). TUNEL assay revealed the
protective effect of LF on PrP (106-126)-induced apoptosis of
SH-SY5Y cells (Fig. 1C). These
results suggest that LF prevents PrP (106-126)-induced neuronal
cell death.
LF treatment suppresses PrP
(106-126)-mediated protein activation
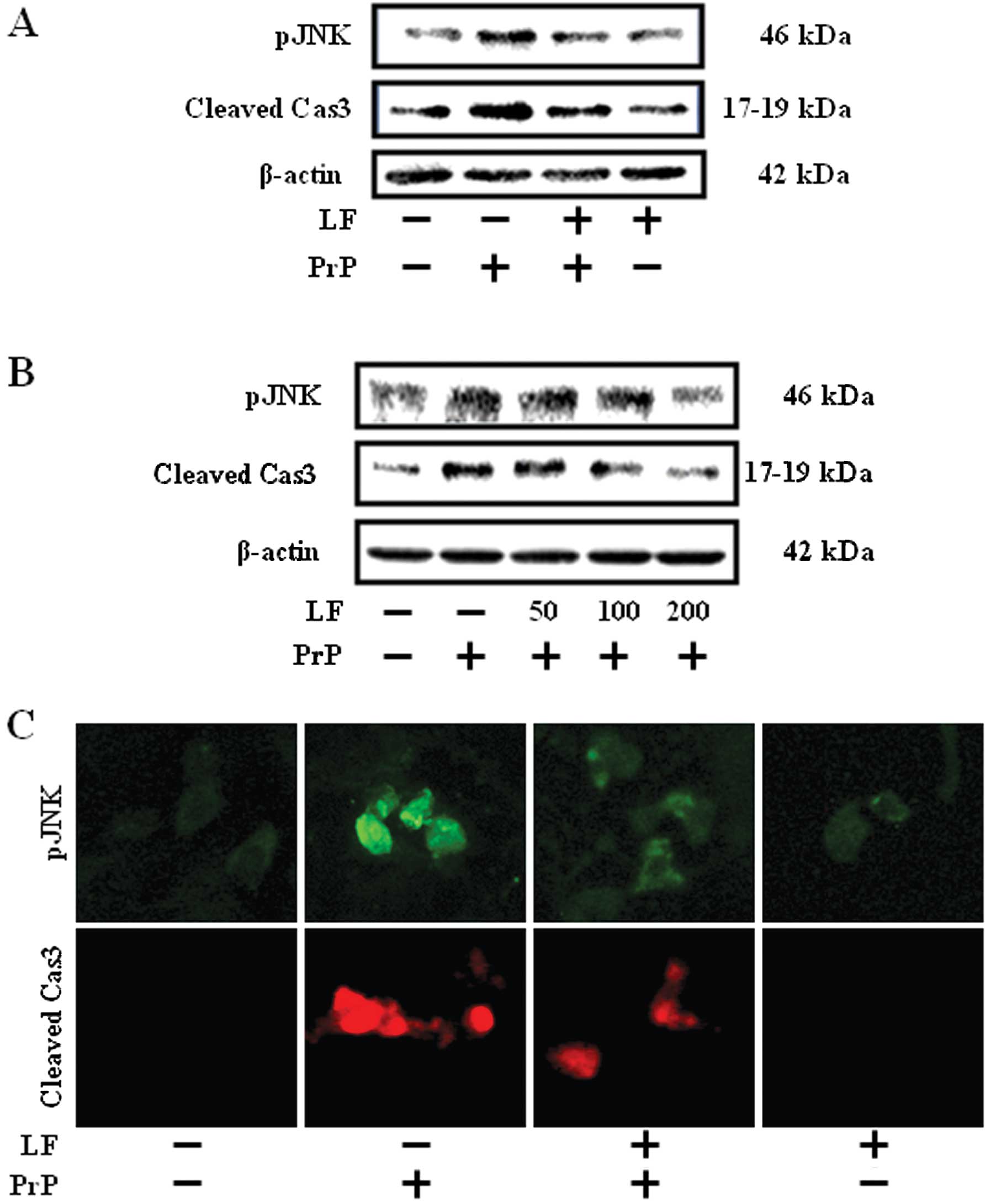
We examined the effects of LF treatment on the JNK
and caspase-3 activation. Western blot analyses revealed that
activation of JNK and caspase-3 increased expression in the 100
μM PrP (106-126)-treated group compared to the LF (200
μg/ml)-pretreated group and the control group (Fig. 2A). PrP (106-126) treatment induced
the activation of JNK and caspase-3 in SH-SY5Y cells. However, LF
treatment inhibited the activation of JNK and caspase-3 (Fig. 2A and B). Consistent with these
results, immunofluorescence monitoring also showed that LF
treatment completely inhibited PrP (106-126)-mediated protein
activation (Fig. 2C). These
results suggest that LF treatment suppresses PrP (106-126)-induced
protein activation.
LF treatment decreases PrP
(106-126)-induced oxidative stress via ROS scavenging
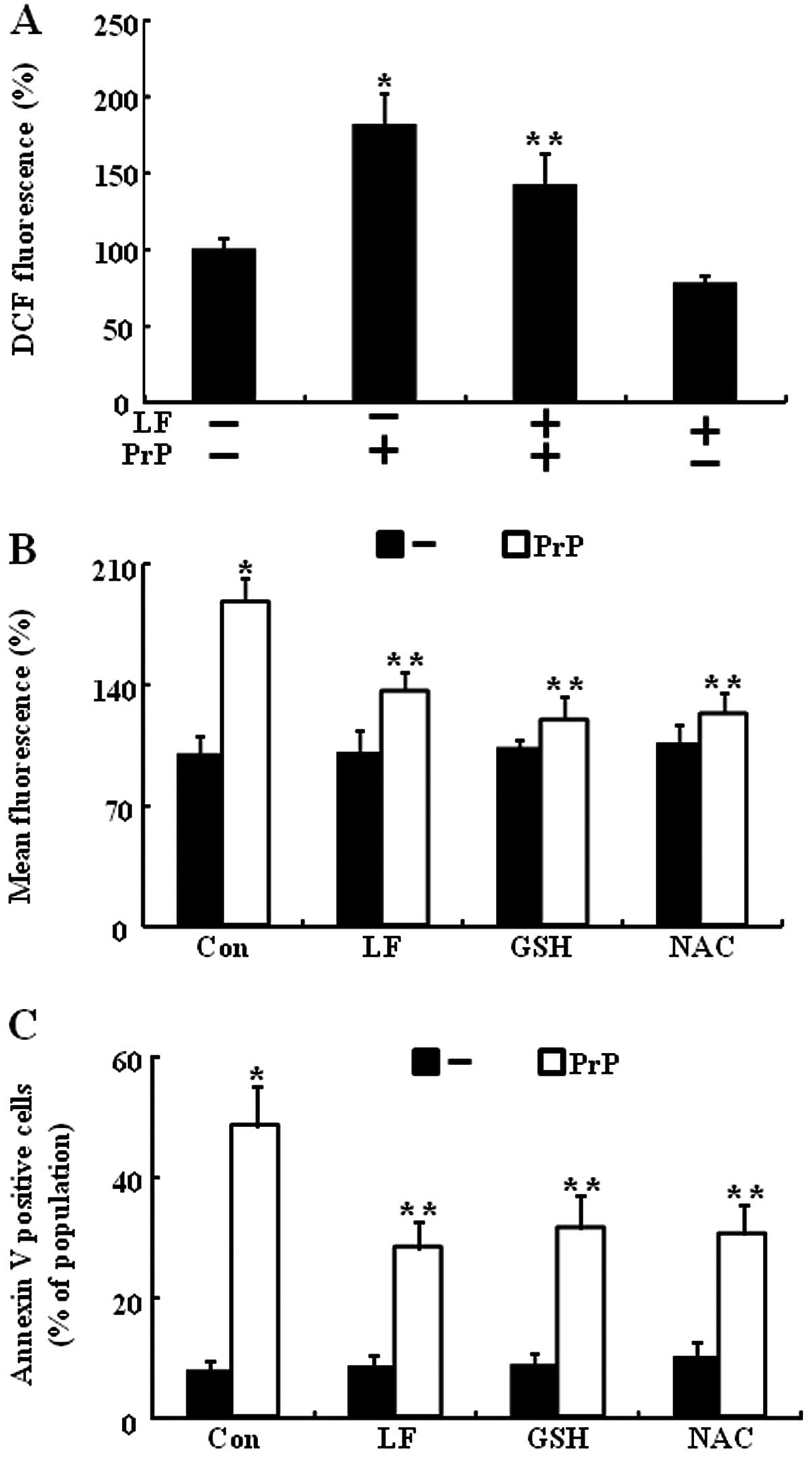
In a previous study, it was shown that LF is a
scavenger of ROS (20), and that
this protects against ROS-mediated cell death. PrP
(106-126)-induced neuronal cell death is mediated by ROS generation
(23). Thus, we next assessed
whether the protective effect of LF on PrP (106-126)-induced
neuronal cell death was related to ROS generation. SH-SY5Y cells
were preincubated 12 h with 200 μg/ml LF and then exposed to
100 μM PrP (106-126) for 12 h. LF treatment reduced PrP
(106-126)-induced ROS generation (Fig. 1A). How LF treatment might induce
PrP (106-126) resistance was studied by assessing the antioxidative
properties and generation of ROS after treatment. Intracellular ROS
production was spectrophotometrically measured by the DCFH-DA assay
(Fig. 3A). After exposure to 100
μM PrP (106-126), DCF fluorescence intensity in SH-SY5Y
cells increased significantly to 175% of the control value, whereas
LF (200 μg/ml) or anti-oxidants (800 μM GSH or 4 mM
NAC) led to a decrease in DCF fluorescence intensity (Fig. 3B). These results suggest that LF
protects PrP (106-126)-induced neuronal cell death via the
prevention of PrP (106-126)-induced ROS generation (Fig. 3C).
PrP (106-126)-induced mitochondrial
dysfunction is suppressed by LF treatment
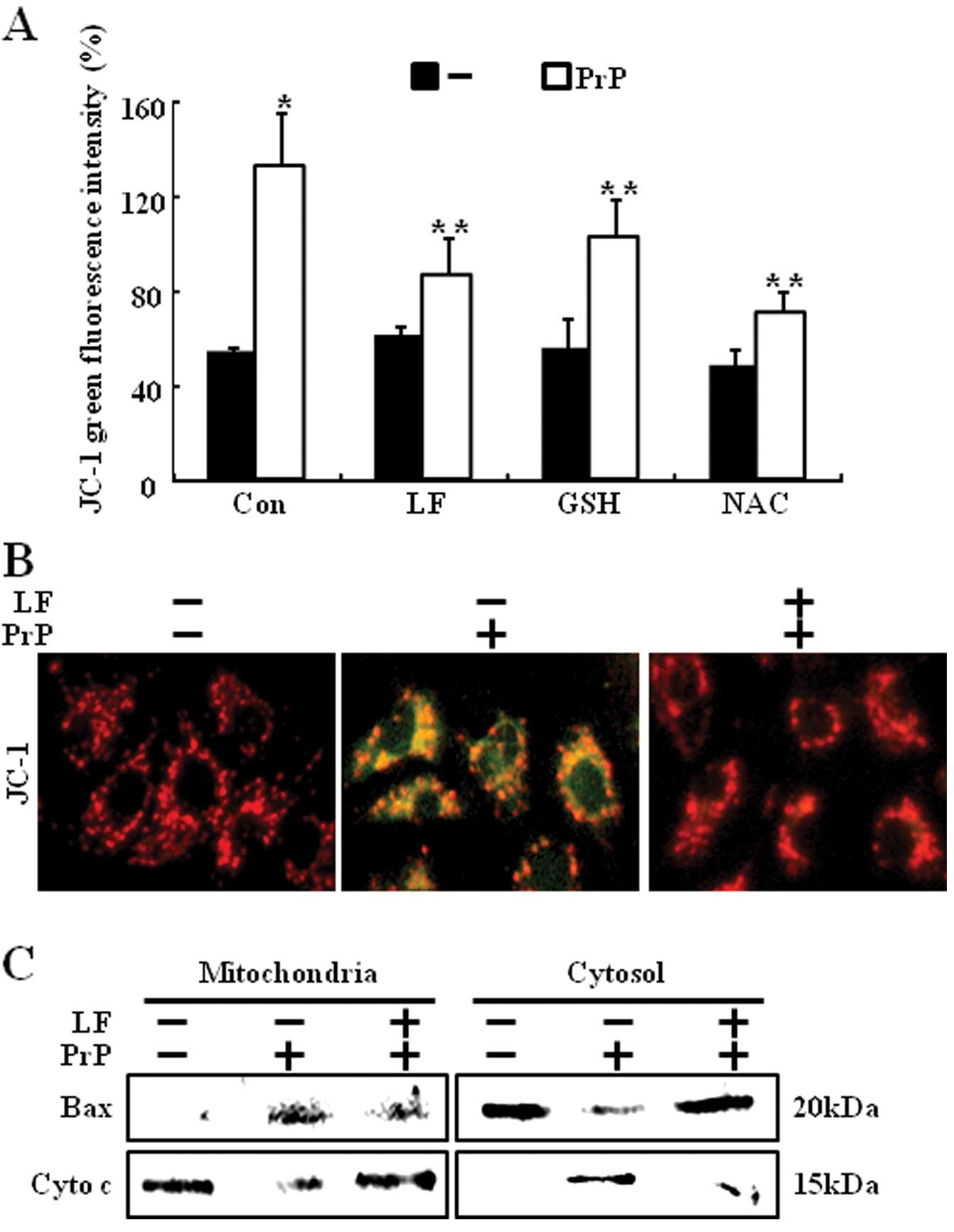
PrP (106-126)-induced apoptosis is mediated by
mitochondrial disruption (12).
Mitochondrial dysfunction occurs after apoptotic signals, including
loss of MTP and release of apoptotic factors into the cytosol
(24). We examined the effects of
LF or antioxidants on PrP (106-126)-induced mitochondrial
dysfunction. MTP was measured by flow cytometry. PrP
(106-126)-treated cells showed increased JC-1 monomers, while LF
pretreatment reduced PrP (106-126)-induced JC-1 monomers (Fig. 4A). Furthermore, pretreatment of
antioxidants also reduced PrP (106-126)-induced JC-1 monomers.
These results were confirmed by fluorescence microscopy images of
JC-1 stained cells (Fig. 4B).
Consistent with these results, LF-treatment cells prevented PrP
(106-126)-induced cytochrome c release and Bax translocation
(Fig. 4C).
Discussion
Prion diseases are fatal neurodegenerative disorders
(25). The main component of
prion disease is the abnormal isoform of prion protein (PrPsc)
(26). PrP (106-126) maintains
the neurotoxic characteristics of the entire pathological PrPSc and
is commonly used as a suitable model to study the mechanism of
prion disorders (5). However,
this peptide mechanism is not fully understood. In previous
studies, it has been shown that PrP (106-126) induces neurotoxicity
via mitochondrial disruption and ROS generation. LF is an 80 kDa
protein. It is a multifunctional protein of the transferrin family
and its functions include antimicrobial activity, antibacterial
activity, cell proliferation, and antioxidant ability (27). LF protects from programmed cell
death via antioxidant activity that is due to the scavenging of ROS
(20). Moreover, LF inhibits
PrPsc accumulation in scrapie-infected cells (22). However, the affirmative effect of
LF on PrP (106-126)-induced neuronal cell death is not completely
understood. In this study, LF treatment protected against PrP
(106-126)-induced neuronal cell death (Fig. 1). In addition, PrPc-deficient mice
were more sensitive to oxidative stress (28). Oxidative stress plays an important
role in neurodegenerative disorders (13). Thus, we considered whether LF
treatment could mediate ROS scavenger ability. Our results
demonstrate that LF protects against PrP (106-126)-induced ROS
generation in SH-SY5Y cells (Fig. 3A
and B). These results suggest that PrP (106-126) mediates
apoptotic cell death and ROS generation, and that these
consequences are decreased by LF treatment. ROS can activate JNK
protein. Indeed, PrP (106-126) induces neuronal cell damage by
activating JNK and caspase-3 proteins (Fig. 2). JNK activation has been
documented in neurodegenerative diseases (29). By contrast, LF treatment inhibits
PrP (106-126)-mediated protein activation including JNK and
caspase-3 (Fig. 2). These results
indicate that LF treatment inhibits PrP (106-126)-mediated JNK and
caspase-3 activation, and support the view that LF-mediated ROS
scavenging downregulates PrP (106-126)-mediated protein activation.
NAC protects cells against mitochondrial dysfunction (30). Furthermore, PrP (106-126)-induced
apoptotic cell death occurs through mitochondrial disruption in
neuronal cells (12). Our
findings additionally show that LF or antioxidants (GSH and NAC)
prevent neuronal cell death due to PrP (106-126)-mediated
mitochondrial dysfunction (Fig.
4). Collectively, these results indicate that LF treatment
protects from PrP (106-126)-induced neuronal cell death by ROS
scavenging associated antioxidant activity. Moreover, LF possesses
antioxidant activity and prevents PrP (106-126)-mediated
mitochondrial disruption. In addition, these findings also suggest
that LF may have clinical benefits when used for neurodegenerative
chemotherapy such as in patients with prion disorders.
Acknowledgements
This study was supported by the
Cooperative Research Program for Agriculture Science and Technology
Development (PJ907116) in Rural Development Administration and by
the National Research Foundation of the Korea Grant funded by the
Korean Government (2010-E00019).
References
|
1.
|
Beringue V, Couvreur P and Dormont D:
Involvement of macrophages in the pathogenesis of transmissible
spongiform encephalopathies. Dev Immunol. 9:19–27. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Ermolayev V, Cathomen T, Merk J, et al:
Impaired axonal transport in motor neurons correlates with clinical
prion disease. PLoS Pathog. 5:e10005582009. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Ogayar A and Sanchez-Perez M: Prions: an
evolutionary perspective. Int Microbiol. 1:183–190. 1998.PubMed/NCBI
|
|
4.
|
Bate C and Williams A: Monoacylated
cellular prion protein modifies cell membranes, inhibits cell
signaling, and reduces prion formation. J Biol Chem. 286:8752–8758.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Hur K, Kim JI, Choi SI, Choi EK, Carp RI
and Kim YS: The pathogenic mechanisms of prion diseases. Mech
Ageing Dev. 123:1637–1647. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Florio T, Paludi D, Villa V, et al:
Contribution of two conserved glycine residues to fibrillogenesis
of the 106-126 prion protein fragment. Evidence that a soluble
variant of the 106-126 peptide is neurotoxic. J Neurochem.
85:62–72. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Anantharam V, Kanthasamy A, Choi CJ, et
al: Opposing roles of prion protein in oxidative stress- and ER
stress-induced apoptotic signaling. Free Radic Biol Med.
45:1530–1541. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Jeong JK, Moon MH, Lee YJ, Seol JW and
Park SY: Autophagy induced by the class III histone deacetylase
Sirt1 prevents prion peptide neurotoxicity. Neurobiol Aging. May
8–2012.(Epub ahead of print).
|
|
9.
|
Nicholls DG and Budd SL: Mitochondria and
neuronal survival. Physiol Rev. 80:315–360. 2000.PubMed/NCBI
|
|
10.
|
Murphy MP and Smith RA: Targeting
antioxidants to mitochondria by conjugation to lipophilic cations.
Annu Rev Pharmacol Toxicol. 47:629–656. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Choi SI, Ju WK, Choi EK, et al:
Mitochondrial dysfunction induced by oxidative stress in the brains
of hamsters infected with the 263 K scrapie agent. Acta
Neuropathol. 96:279–286. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
O’Donovan CN, Tobin D and Cotter TG: Prion
protein fragment PrP-(106-126) induces apoptosis via mitochondrial
disruption in human neuronal SH-SY5Y cells. J Biol Chem.
276:43516–43523. 2001.PubMed/NCBI
|
|
13.
|
Kitazawa M, Wagner JR, Kirby ML,
Anantharam V and Kanthasamy AG: Oxidative stress and
mitochondrial-mediated apoptosis in dopaminergic cells exposed to
methylcyclopentadienyl manganese tricarbonyl. J Pharmacol Exp Ther.
302:26–35. 2002. View Article : Google Scholar
|
|
14.
|
Blokhina O, Virolainen E and Fagerstedt
KV: Antioxidants, oxidative damage and oxygen deprivation stress: a
review. Ann Bot. 91:179–194. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Park KW and Jin BK: Thrombin-induced
oxidative stress contributes to the death of hippocampal neurons:
role of neuronal NADPH oxidase. J Neurosci Res. 86:1053–1063. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Wang J, Yu Y, Hashimoto F, Sakata Y, Fujii
M and Hou DX: Baicalein induces apoptosis through ROS-mediated
mitochondrial dysfunction pathway in HL-60 cells. Int J Mol Med.
14:627–632. 2004.PubMed/NCBI
|
|
17.
|
Pietri M, Caprini A, Mouillet-Richard S,
et al: Overstimulation of PrPC signaling pathways by prion peptide
106-126 causes oxidative injury of bioaminergic neuronal cells. J
Biol Chem. 281:28470–28479. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Tuccari G and Barresi G: Lactoferrin in
human tumours: immunohistochemical investigations during more than
25 years. Biometals. 24:775–784. 2011.PubMed/NCBI
|
|
19.
|
Brock JH: The physiology of lactoferrin.
Biochem Cell Biol. 80:1–6. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Burrow H, Kanwar RK and Kanwar JR:
Antioxidant enzyme activities of iron-saturated bovine lactoferrin
(Fe-bLf) in human gut epithelial cells under oxidative stress. Med
Chem. 7:224–230. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Baveye S, Elass E, Mazurier J and Legrand
D: Lactoferrin inhibits the binding of lipopolysaccharides to
L-selectin and subsequent production of reactive oxygen species by
neutrophils. FEBS Lett. 469:5–8. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Iwamaru Y, Shimizu Y, Imamura M, et al:
Lactoferrin induces cell surface retention of prion protein and
inhibits prion accumulation. J Neurochem. 107:636–646. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Jeong JK, Seol JW, Moon MH, et al:
Cellular cholesterol enrichment prevents prion peptide-induced
neuron cell damages. Biochem Biophys Res Commun. 401:516–520. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Kroemer G, Galluzzi L and Brenner C:
Mitochondrial membrane permeabilization in cell death. Physiol Rev.
87:99–163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Harris DA: Cellular biology of prion
diseases. Clin Microbiol Rev. 12:429–444. 1999.PubMed/NCBI
|
|
26.
|
Sakudo A and Ikuta K: Prion protein
functions and dysfunction in prion diseases. Curr Med Chem.
16:380–389. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Hedlin P, Taschuk R, Potter A, Griebel P
and Napper S: Detection and control of prion diseases in food
animals. ISRN Veterinary Sci. 2012:242012. View Article : Google Scholar
|
|
28.
|
Brown DR and Besinger A: Prion protein
expression and super-oxide dismutase activity. Biochem J.
334:423–429. 1998.
|
|
29.
|
Tsirigotis M, Baldwin RM, Tang MY, Lorimer
IAJ and Gray DA: Activation of p38MAPK contributes to expanded
polyglutamine-induced cytotoxicity. PLoS One. 3:e21302008.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Mai S, Klinkenberg M, Auburger G,
Bereiter-Hahn J and Jendrach M: Decreased expression of Drp1 and
Fis1 mediates mitochondrial elongation in senescent cells and
enhances resistance to oxidative stress through PINK1. J Cell Sci.
123:917–926. 2010. View Article : Google Scholar
|