Introduction
Subarachnoid hemorrhage (SAH) frequently results in
complications including intracranial hypertension, rebleeding and
vasospasm. The extravasated blood is responsible for a cascade of
reactions involving release of various vasoactive and
pro-inflammatory factors from blood and vascular components in the
subarachnoid space. The communicating hydrocephalus following SAH
is one of the complex and multifactorial neurological disorders,
which arises from fibrosis in the subarachnoid space. Spinal
arachnoiditis and periradicular ‘inflammation’ of the arachnoid
membrane and adjacent peridural structures lead to fibrosis within
and around the lumbar dural sac and the spinal nerve roots
(1). Fibrosis causes cavitas
subarachnoidalis stenosis, which in turn blocks the cerebrospinal
fluid (CSF) circulation. The mechanism of fibrosis following SAH
has yet to be defined.
Thrombin (TH) is an Na+-activated,
allosteric serine protease that plays opposing functional roles in
blood coagulation. It is produced in the brain either immediately
after a cerebral hemorrhage or after the blood-brain barrier (BBB)
breakdown that occurs following several types of brain injury
(2). It was reported that TH
activity was increased in CSF after SAH and there was a significant
correlation between coagulation activity in the subarachnoid space
and clearance of SAH (3). Vesey
et al revealed that TH stimulates proinflammatory and
proliferative responses in primary cultures of human proximal
tubule cells (PTC), suggesting that the proinflammatory and
fibroproliferative actions of TH on human PTC may help explain the
extent of tubulointerstitial fibrosis observed in kidney diseases
where fibrin deposition is evident (4). Based on these reports, we
hypothesized that TH also plays an important role in fibrosis of
subarachnoid meninges after SAH.
TGF-β is key in tissue homeostasis and the
disruption of the TGF-β pathway has been implicated in numerous
human diseases, including cancer, autoimmune, fibrotic, and
cardiovascular diseases. TGF-β regulates cellular responses in a
positive or a negative way. For example, the anti-inflammatory
properties of TGF-β are beneficial in atherosclerosis, while the
profibrotic effects contribute to fibrosis in hypertension and
cardiac damage. TGF-β is believed to be the most important
extracellular matrix (ECM) regulator (5). In vascular smooth muscle cells
(VSMCs), endothelial cells, and fibroblasts, TGF-β1 increases the
synthesis of ECM proteins, such as fibronectin, collagens and
PAI-1. TGF-β reduces collagenase production and stimulates the
expression of tissue inhibitor of metalloproteinases (TIMPs),
resulting in an overall inhibition of ECM degradation and leading
to excessive matrix accumulation (5,6).
The mechanisms involved in TGF-β-mediated vascular fibrosis are
complex, including activation of Smad proteins, protein kinases,
production of mediators and crosstalk between pathways. Further
insights into the molecular mechanisms involved in ECM accumulation
may contribute to a better understanding of this pathological
process and may improve therapeutic strategies.
Materials and methods
Animals
Female Wistar rats (200–300 g) were housed in a room
at a temperature of 22±2°C under a 12-h light/dark schedule and
given water and food ad libitum. The procedures involving
experimental animals adhered to the law and notification of the
Chinese Government and were approved by the Laboratory Animal Care
and Use Committee of Xinxiang Medical College and Shantou
University.
Reagents
Rat α-thrombin and SB-431542, a specific inhibitor
of type 1 TGF-β receptor (TβR1), were purchased from Sigma (St.
Louis, MO, USA). TGF-β1 and TGF-β immunoassay kit was from R&D
Systems Inc., (Minneapolis, MN, USA). Anti-phospho-Smad1 (S465), 2
(S467), 3 (S423 + S425), anti-Smad1, 2 and 3, and anti-β-actin were
from Abcam (Cambridge, MA, USA).
Injection of TH into SAH and specimen
preparations
Seventy-eight female rats were randomly assigned
into 3 experimental groups (Group 1, TH group; Group 2, SB-431542
group; and Group 3, TH + SB-431542 group; n=21 in each group) and
the control group (n=15). All rats were anesthetized with 10%
chloral hydrate and were punctured at 5 mm below the margo
occipitalis with a 22-gauge needle into the cistema magna.
Subsequently, Groups 2 and 3 were injected with SB-431542 (0.1
μmol), which was dissolved in 10 μl of vehicle
(DMSO:water, 1:2), injected into the cistema magna through a 50
μl microsyringe and a polyethylene tube. One hour after
SB-431542 injection, 0.3 ml (10 U/ml) of TH (Groups 1 and 3) or
isotonic sodium chloride (control group) was infused into CSF at a
rate of 0.1 ml/minute. Animals were kept in the head down position
for 30 min after infusion before putting them back into the
cages.
At Day 0, 10 and 20 after infusion of TH, 7 rats
from the experimental groups (Groups 1, 2 and 3) and 5 rats from
the control group were randomly selected and sacrificed, and CSF
(100 μl/rat) was drawn respectively. CSF was centrifuged for
20 minutes at 2,000 rpm. The supernatant was collected and
maintained at −80°C for TGF-β1 measurement.
TGF-β1 assay
Concentrations of TGF-β1 in each CSF sample were
measured using an enzyme-linked immunosorbent assay (ELISA) kit
(Promega). The TGF-β1 levels were measured in 50 μl CSF
after 1N HCl was added at 1:3 and incubated for 10 min to activate
TGF-β1. Following neutralization with 1 ml 1N NaOH/0.5 M HEPES, the
samples were assayed by ELISA. No significant cross-reactivity with
other cytokines was observed.
Primary rat meningocyte culture
Rat subarachnoid meninges were removed from the
brains of rats. Cells were dissociated and plated in
collagen-coated 6-well plates with Eagle’s minimum essential medium
and 15% fetal bovine serum at 37°C in 95% air/5% CO2.
Culture of rat meningocytes was grown to 80% confluence and then
placed in serum free medium containing 0.1% bovine serum albumin
(BSA) for a minimum of 12 h prior to treatment. Specified
inhibitor, SB-431542 (10 μM), or vehicle was next added in
serum free medium for 30 min prior to the addition of TGF-β1 (2 and
4 ng/ml) or vehicle. TGF-β1 treatments were for either 6 h for RNA
analyses or 8 h for protein analyses.
Total RNA isolation and real-time
quantitative PCR (qRT-PCR)
Total RNA from control and treatments was isolated
using TRIzol according to the manufacturer’s instructions. RNA
obtained was analyzed using Agilent bioanalyzer and quantified via
a NinoDrop spectrophotometer (Thermo Scientific, Wilmington, DE,
USA). RNA (1 μg) per treatment was reverse transcripted
using random primers and the Applied Biosystems reverse
transcription kit. Five microliters of each reverse transcription
reaction was used for 50 μl real-time PCR using conditional
96-well format. TaqMan probes for CCN2/CTGF, β-actin and
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used in
real-time PCR analyses. Real-time PCRs were run in ABI 7700 system
at thermal cycler conditions of 50°C for 2 min, 95°C for 1 min, and
60°C for 1 min for 40 cycles. Data were analyzed using the
2−ΔΔCt method, and CCN2/CTGF mRNA levels were normalized
to β-actin and GAPDH mRNAs, and no treatment control.
Protein isolation and western
blotting
Cells were homogenized in a lysis buffer [25 mM
Tris-HCl (pH 7.4), 0.5 mM EDTA, 0.5 mM EGTA, 1 mM PMSF, 25
μg/ml leupeptin, 1 mM DTT, 135 mM NaCl, 25 mM NaF, 0.5%
Triton X-100] and centrifuged. The supernatant, each containing 100
ng of total protein, was separated on a sodium dodecyl sulphate
(SDS)-10% polyacrylamide gel coupled to a 4% polyacrylamide
stacking gel. After transferring the separated proteins to
polyvinylidene difluoride (PVDF) membranes, the membranes were
soaked in 20 ml of 4% BSA in Tris-buffered saline Tween-20 (TBST)
buffer for 1 h at room temperature, and were then probed 1 h with
either anti-CTGF, anti-phospho-Smad1, 2 and 3 primary antibody, and
then with HRP-conjugated secondary antibodies. Antigens were
detected using the Pierce chemiluminescent substrate system (ECL).
Following anti-phospho antibody analyses, the same membranes were
used with anti-Smad1, 2 or 3 analysis, i.e. after blotting with
anti-phospho-Smad1, 2 and 3, the membranes were stripped, and
re-detected using ECL for background check. These membranes were
then used for anti-Smad1, 2 and 3 analyses, respectively. The actin
protein was analyzed for all the blots at final stripping
steps.
Histological evaluation
After obtaining CSF at Day 10 and 20 post-infusion,
rat brains were fixed with 5% paraformaldehyde and then sectioned
transversely with a thickness of 5 mm at the frontal lobe, apical
lobe and end-lobe. Specimens were embedded in paraffin wax and
5-μm sections were stained with Masson’s method. The
sections were examined with microscopy. The thickness of the
meninges was analyzed with Fuji micro-image analysis system. The
section of each sample was taken from at least 3 separate
subarachnoid meninges individually.
Statistical analysis
Analysis of variance (ANOVA) was used to assess the
comparison (paired) of values. The LSD method was used for multiple
comparisons between groups. Linear correlation analysis was used to
compare the concentration of TGF-β1 in CSF with the average
thickness of subarachnoid meninges. P<0.05 was considered to
indicate a statistically significant difference.
Results
TH induces expression of the TGF-β1
A high level of TH was present in patient CSF after
SAH (7). Kasuya et al
reported that the TH activity in CSF after SAH was correlated with
the degree of SAH (8). To
elucidate the mechanism and cascade reaction of CSF after SAH, we
first infused TH into SAH. All animals treated with TH infusion
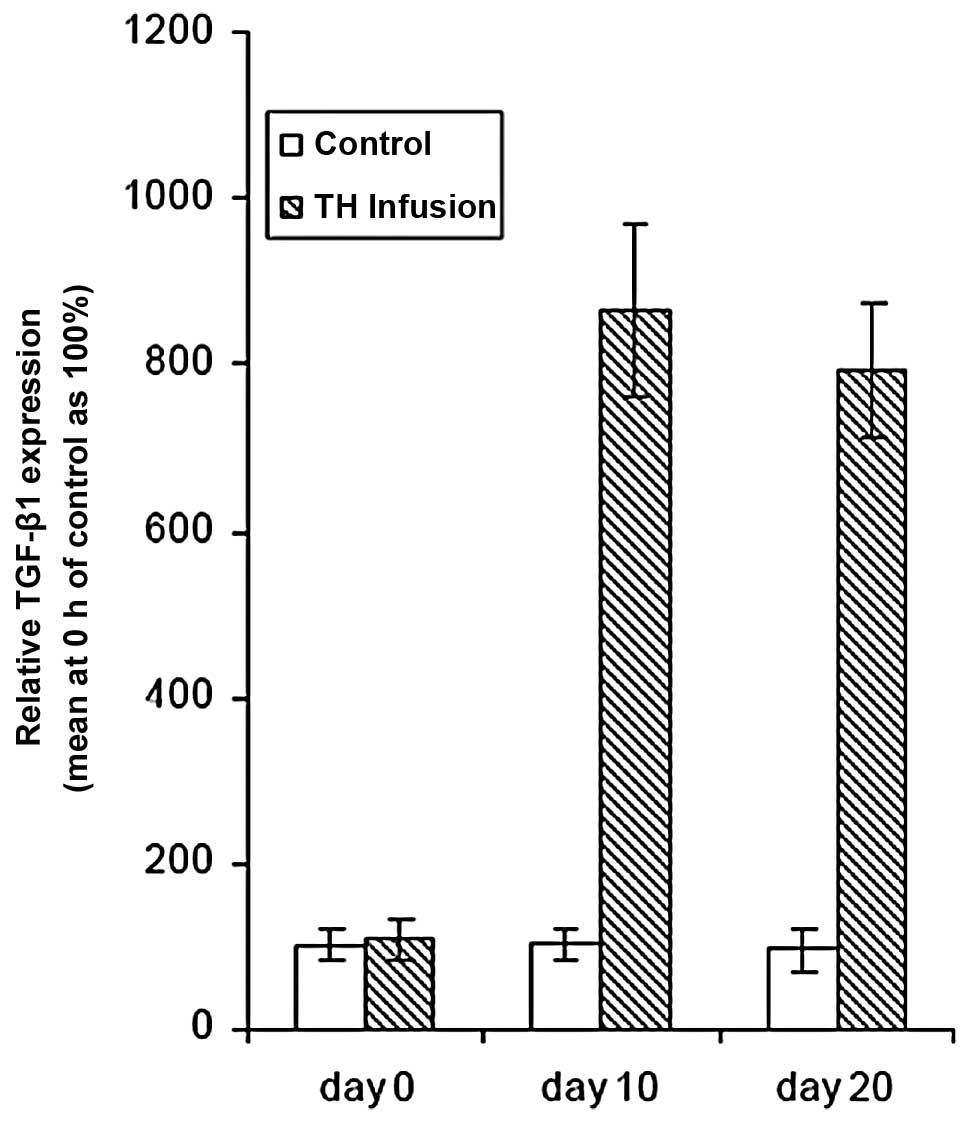
showed a significant increase in total TGF-β1 levels in the CSF.
Ten days after TH infusion, TGF-β1 expression increased 7.6-fold.
At Day 20 after TH infusion, TGF-β1 expression was slightly lower
than that at Day 10. There were no significant differences between
Day 10 and 20 (P>0.05) (Fig. 1
and Table I). The results
suggested that the TGF-β1 pathway may be evoked by accumulated TH
in CSF.
 | Table ITime course of TGF-β1 in rat CSF
induced by TH infusion. |
Table I
Time course of TGF-β1 in rat CSF
induced by TH infusion.
| Group | Mouse ID | Day 0 | Day 10 | Day 20 |
|---|
| Controls | 1 | 44.58 | 30.83 | 46.03 |
| 2 | 34.55 | 38.74 | 36.56 |
| 3 | 36.91 | 64.71 | 34.51 |
| 4 | 31.20 | 27.48 | 21.88 |
| 5 | 31.34 | 23.17 | 29.11 |
| Mean ± SD | 35.72±5.50 | 36.99±6.51 | 33.62±8.97 |
| TH (3 U/0.3 ml) | 11 | 36.44 | 426.58 | 286.42 |
| 12 | 39.75 | 264.71 | 276.91 |
| 13 | 41.34 | 339.63 | 406.99 |
| 14 | 36.42 | 286.42 | 191.87 |
| 15 | 41.86 | 291.20 | 281.13 |
| 16 | 31.00 | 286.42 | 302.69 |
| 17 | 41.34 | 268.55 | 236.14 |
| Mean ± SD | 38.31±3.62 | 309.1±57.31a | 283.2±66.22a |
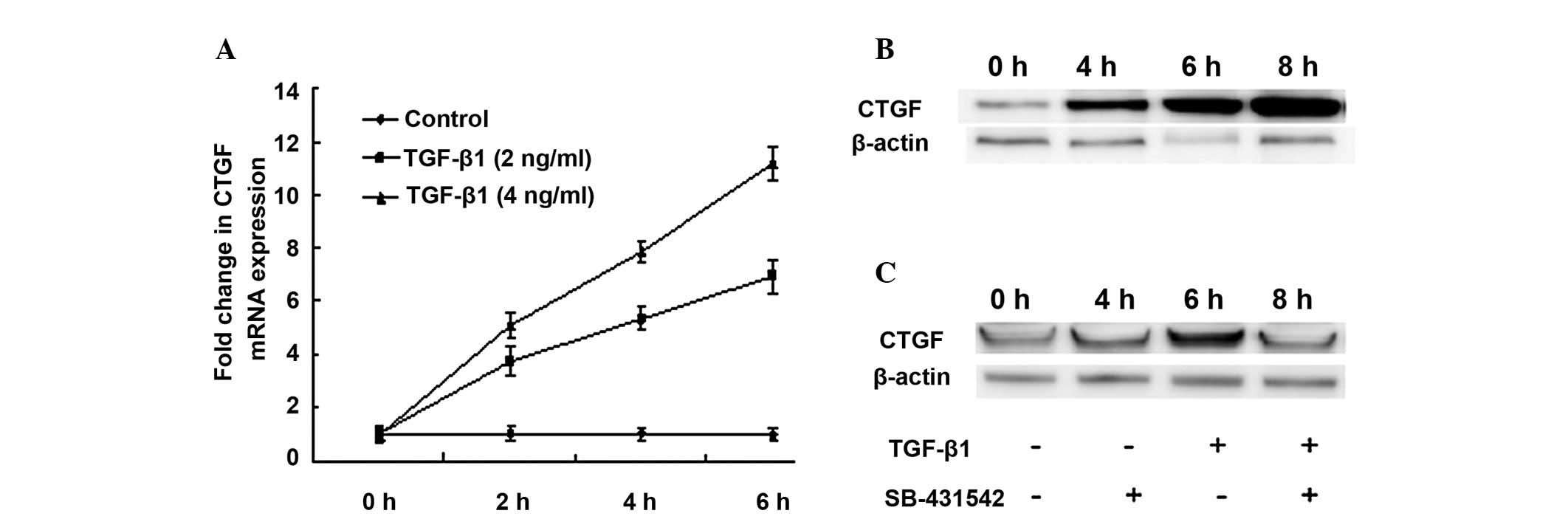
TGF-β1 induces expression of the CTGF in
primary meningocytes
Several studies have indicated that CTGF could be
selectively stimulated by TGF-β1. We investigated whether this
cascade reaction also occurs in the tissues or cells of the
subarachnoid space. We isolated meningocytes from subarachnoid
meninges for a primary culture. The expression of CTGF mRNA in the
culture of meningocytes was increased gradually in response of
TGF-β1 treatment. The responses were both dose and time dependent
(Table II and Fig. 2A). The expression of CTGF proteins
was also increased gradually (Fig.
2B). Pre-treatment of SB-431542 for 30 min blocked expression
of CTGF protein which was induced by TGF-β1 (Fig. 2C). Since CTGF was known for its
role as a mediator of the chronic fibrotic effects, our results
suggested that TH-induced TGF-β1-CTGF cascade may also play a role
in CSF after SAH.
| Table IITGF-β1 stimulated CTGF mRNA
expression. |
Table II
TGF-β1 stimulated CTGF mRNA
expression.
| Time course (h) | 0 | 2 | 4 | 6 |
|---|
| Controls | 82±16 | 84±14.7 | 83±15.5 | 85±16.1 |
| TGF-β1 (2 ng/ml) | 84±17.1 | 274±12.1 | 496±14.5 | 602±13.3 |
| TGF-β1 (4 ng/ml) | 86±18 | 416±12.5 | 661±13.2 | 945±15.3 |
| SB-431542 (10
μM) + TGF-β1 (2 ng/ml) | 88±10 | 90±9.7 | 93±5.9 | 89±11.1 |
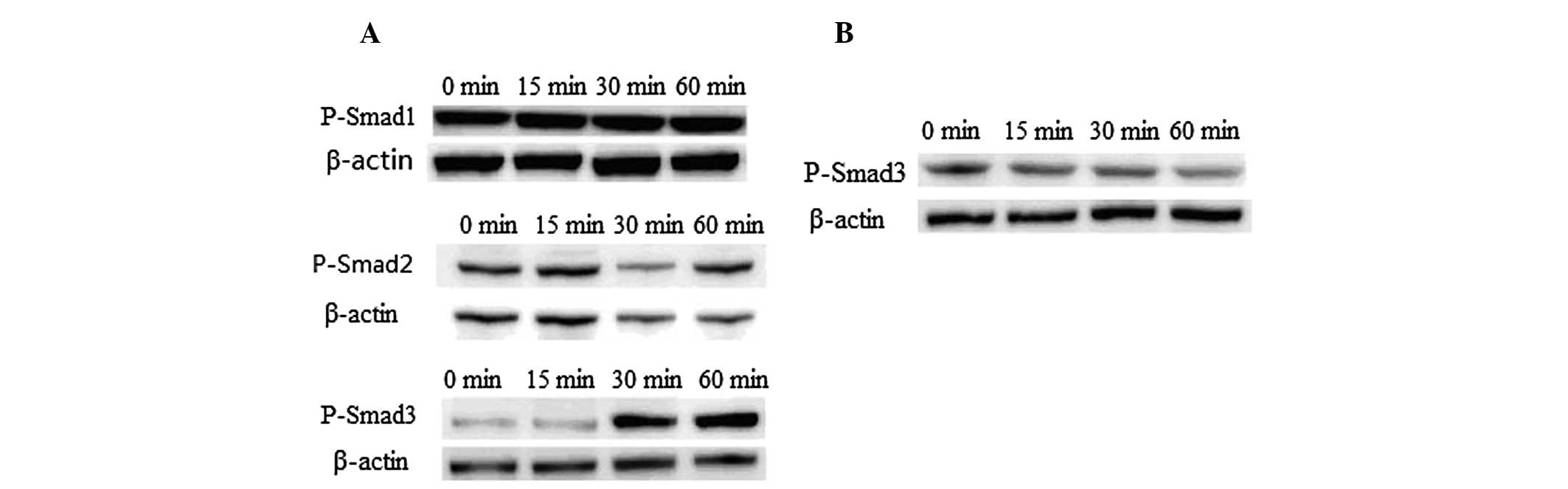
Smad3 is critical for CTGF expression
induced by TGF-β1 in primary meningocytes
TGF-β1 signaling was mediated by Smads in several
cell types. Increasing evidence shows that TGF-β1 acts by
activating its downstream mediators, Smad2/3, to mediate renal
fibrosis (9,10). To determine whether the Smads also
play a role in mediating meningocyte fibrosis, we assessed the
activation of Smad signaling by TGF-β1. Western blot analysis
revealed that phosphorylation of Smad3 was initiated by TGF-β1 when
primary meningocytes were treated with TGF-β1 at 15 min
post-treatment and persisted through 60 min. By contrast,
phosphorylation of Smad-1 and Smad-2 were not altered by TGF-β1
(Fig. 3A). Pre-treatment of
SB-431542 for 30 min blocked signaling of Smad3 completely
(Fig. 3B). We also determined
protein expressions of Smad1, 2 and 3 to ascertain whether they are
also altered by TGF-β1. Our results indicated that protein
expression levels were not affected by TGF-β1 in the period of
treatment (data not shown), suggesting that only activated Smad3 is
essential for CTGF induction by TGF-β1 in our experimental
conditions.
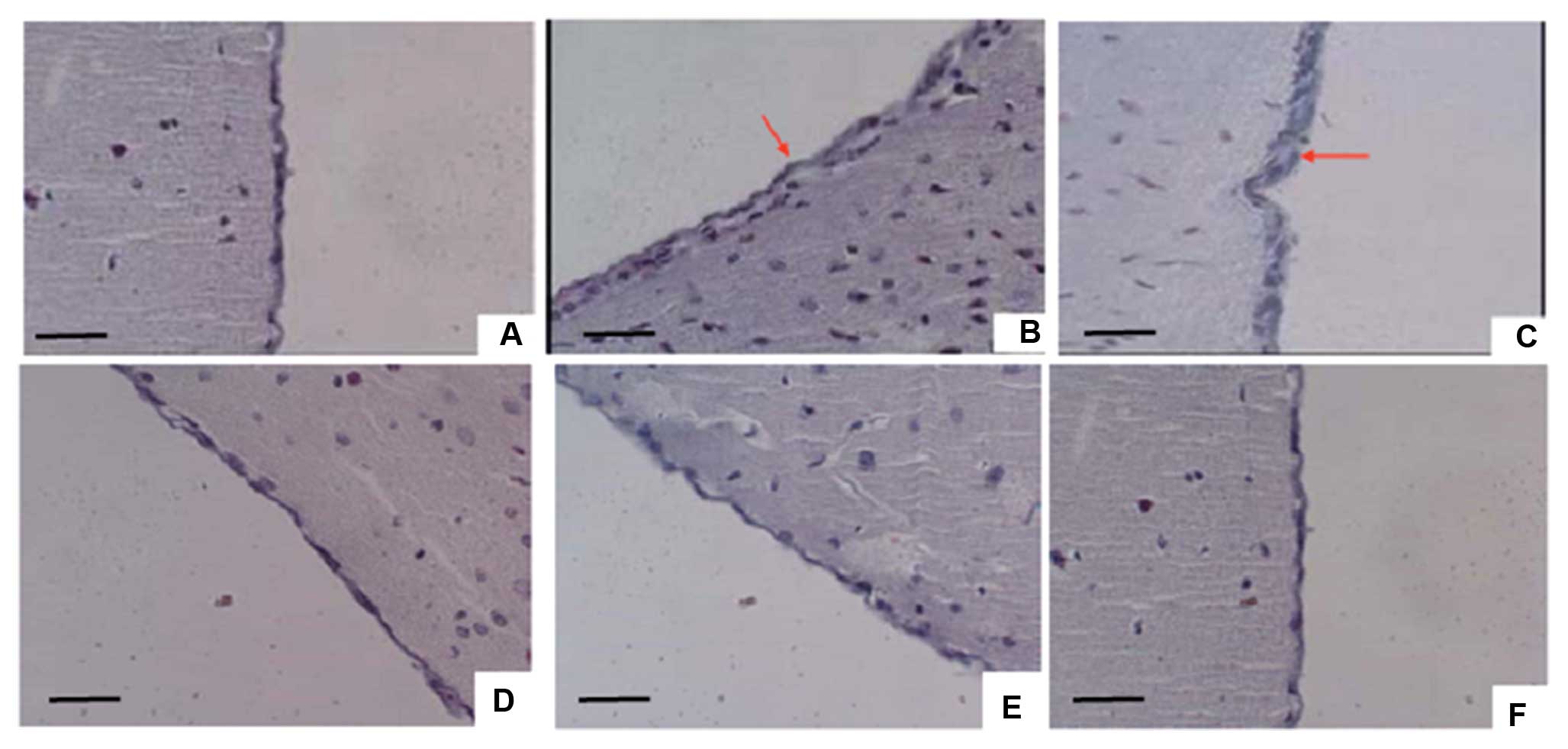
Pathological changes of subarachnoid
meninges by TH infusion
Rat subarachnoid meninges showed marked inflammatory
changes in the meningeal collagen fibers following TH infusion at
Day 10 and 20 of the experiment (Fig.
4, Tables III and IV), irrespective of the concomitant
application of isotonic sodium chloride (data not shown). Mice
exposed to TH demonstrated meningeal inflammation by Day 10, which
was resolved completely by Day 20. Masson’s trichrome staining
revealed slight collagen within inflammatory areas of subarachnoid
meninges in TH-treated rats at Day 10, whereas there was
significant collagen accumulation in the rats at Day 20. To
elucidate whether TH is a cause of proliferation in subarachnoid
meninges and its possible route, we pre-injected SB-431542 to block
the TGF-β1 signaling pathway. Our results indicated that there was
no obvious inflammation or collagen accumulation at Day 10 and 20
following TH infusion at pre-treated groups (Fig. 4). This strongly suggested that TH
could be one cause of subarachnoid meningeal fibrosis following
SAH, and the TGF-β1 signaling pathway could be critical for this
pathological process.
| Table IIITH induces a significant collagen
accumulation at rat subarachnoid meninges. |
Table III
TH induces a significant collagen
accumulation at rat subarachnoid meninges.
| Days after TH
infusion | 0 | 10 | 20 |
|---|
| Isotonic sodium
chloride | 3.43±1.10 | 3.13±0.17 | 3.53±0.25 |
| THa | 3.68±0.43 | 4.29±0.52c | 5.67±0.77d |
| SB-431542b | 3.57±0.64 | 3.61±1.01 | 3.59±0.75 |
| SB-431542b + THa | 3.45±1.23 | 3.47±0.47 | 3.61±0.39 |
| Table IVTime course of thickness of rat
subarachnoid meninges in different experimental groups. |
Table IV
Time course of thickness of rat
subarachnoid meninges in different experimental groups.
| Group | Mouse ID | Day 0 | Day 10 | Day 20 |
|---|
| TH | 1 | 4.40 | 3.69 | 6.20 |
| 2 | 3.23 | 4.16 | 4.96 |
| 3 | 4.11 | 4.58 | 6.63 |
| 4 | 3.67 | 4.88 | 5.46 |
| 5 | 3.47 | 3.54 | 6.79 |
| 6 | 3.61 | 3.27 | 6.40 |
| 7 | 3.61 | 5.41 | 6.23 |
| Mean ± SD | 3.73±0.4 | 4.22±0.77a | 6.11±0.67a |
| SB-431542 + TH | 11 | 3.32 | 3.18 | 3.81 |
| 12 | 3.72 | 3.06 | 3.33 |
| 13 | 2.82 | 3.40 | 3.40 |
| 14 | 3.84 | 3.04 | 3.24 |
| 15 | 3.43 | 4.96 | 3.53 |
| 16 | 2.96 | 3.02 | 3.28 |
| 17 | 3.25 | 3.36 | 3.45 |
| Mean ± SD | 3.34±0.42 | 3.43±0.69 | 3.48±0.22 |
Discussion
Alterations in the CSF circulation may follow SAH;
however, the frequency and extent to which this occurs is unknown.
A previous report revealed that leptomeningeal fibrosis is the
pathoanatomic basis of increased resistance to CSF outflow
(11). Fibrosis of the arachnoid
villi has been suggested as the cause for obstruction of CSF flow
(12). Accurate definition of
these abnormalities is important since in some instances they
result in symptomatic communicating hydrocephalus. The origin of
chronic communicating hydrocephalus following SAH is not well
understood. Genetic studies revealed that numerous cytokines,
growth factors or related molecules in the cellular signal pathways
play an important role in the development of hydrocephalus. To
establish the mechanism of the communicating hydrocephalus
following SAH, we infused CSF with TH, resulting in proinflammatory
and proliferative responses in rat meninges of SAH. The effect of
TH could be completely blocked by a TGF-β1 inhibitor, SB-431542,
suggesting that TH-stimulated proliferation of meninges is through
the TGF-β1 signaling pathway.
Shirato et al discovered that TH stimulates
kidney endothelial cells to secrete both cellulose protein and
TGF-β1 (13). TGF-β1 is an
important cytokine and growth-signaling molecule in the brain.
TGF-β1 expresses at a high level after SAH (7). In mouse models, severe hydrocephalus
has been observed in transgenic mouse overexpression of TGF-β1 in
astrocytes (14,15). Furthermore, several studies
indicated that TGF-β1 plays a central role in the response to brain
injury, and is involved in a variety of disorders of the central
nervous system such as stroke (16,17), ischemia (18,19) and abscess (20). Under various pathological
conditions, TGF-β1 expression is found in neurons, suggesting a
role as a neuronal crisis cytokine. The mechanism of how TGF-β1
promotes the collagen protein production is complex. At the level
of transcription and translation, TGF-β1 participates with nuclear
factor and activates type 1 collagen gene promoter, then stimulates
the expression of collagen junctura fibrosa protein gene by
reporter gene, and increases the composition of collagen protein.
However, it also upregulates the expression of proteinase inhibitor
through restraining the breakdown of proteins such as
metalloprotease, collagenase, stromelysin, and elastase.
To elucidate the cascade reactions of the TGF-β1
signaling pathway in subarachnoid meningeal fibrosis, we
investigated several downstream factors of TGF-β1. Our results
revealed that one downstream factor of TGF-β1, CTGF, was selected
to be stimulated in primary subarachnoid meningocytes treated by
TH. CTGF is well known for its role as a downstream mediator of the
chronic fibrotic effects of TGF-β1. Activated by TGF-β1, CTGF
induces fibroblasts to become myofibroblasts that deposit layers of
collagen, which occurs faster than the natural rate of collagen
breakdown. The affected organs become stiff and cannot perform
functions essential to life and health. FibroGen studies have
demonstrated that TGF-β1 alone only causes a transient increase in
fibrosis and that scar formation is persistent only when CTGF is
present (http://www.fibrogen.com/rd/ctgf/fibrosis.html).
Previous studies have demonstrated that the levels of CTGF
correlate with the degree and severity of fibrosis in several
diseases, including diabetic nephropathy, glomerulosclerosis, IgA
nephropathy, diabetic retinopathy, advanced macular degeneration,
cirrhosis, biliary atresia, congestive heart failure, lung fibrosis
and scleroderma. This prompted us to investigate the molecular
mechanisms that might link changes in subarachnoid meningeal
fibrosis, TGF-β1 and CTGF gene expressions. In this study, we
demonstrated that TH-induced overexpression of TGF β1-signaling is
one of the mechanisms of communicating hydrocephalus after SAH.
In general, TGF-β1 signals through a generic
Smad-mediated pathway. However, the signaling pathways that mediate
TGF-β1 induction of CTGF have been shown to vary depending on the
cell type being examined (21)
and, to date, there have been no studies on subarachnoid
meningocyte. The Smad family is divided into three subclasses:
receptor regulated Smads, activin/TGF-β receptor regulated (Smad2
and 3) or BMP receptor regulated (Smad1, 5, and 8). We selected
anti-phospho and non-phospho-Smad1, 2 and 3 in this study. Smad1
was used to eliminate the non-specific induction of TGF-β1. We
elucidated that activation of Smad3 is critical for stimulation of
CTGF expression. Both phosphorylation of Smad3 and CTGF expression
could be blocked by SB-431542. Our results strongly suggest that
the proliferative responses in rat meninges induced by TH are due
to TH-triggered activation of the TGF-β1 signaling pathway. This
cascade reaction might be a mechanism of communicating
hydrocephalus post SAH. To our knowledge, this is the first study
to explore the TH-TGF β1-Smad3-CTGF signaling pathway associated
with subarachnoid meningeal fibrosis and communicating
hydro-cephalus.
Abbreviations:
|
SAH
|
subarachnoid hemorrhage;
|
|
TH
|
thrombin;
|
|
CSF
|
cerebrospinal fluid;
|
|
CTGF
|
connective tissue growth factor;
|
|
ECM
|
extracellular matrix;
|
|
TGF-β
|
transforming growth factor β
|
Acknowledgements
This study was supported by the
National Natural Science Foundation of China (nos. 30772498 and
30872297 to P. Z.).
References
|
1
|
Hoyland JA, Freemont AJ, Denton J, Thomas
AM, McMillan JJ and Jayson MI: Retained surgical swab debris in
post-laminectomy arachnoiditis and peridural fibrosis. J Bone Joint
Surg Br. 70:659–662. 1988.PubMed/NCBI
|
|
2
|
Lee KR, Betz AL, Kim S, Keep RF and Hoff
JT: The role of the coagulation cascade in brain edema formation
after intracerebral hemorrhage. Acta Neurochir. 138:396–400. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kasuya H, Shimizu T, Okada T, Takahashi T,
Summerville T and Kitamura K: Activation of the coagulation system
in the subarachnoid space after subarachnoid haemorrhage: Serial
measurement of fibrinopeptide A and bradykinin of cerebrospinal
fluid and plasma in patients with subarachnoid haemorrhage. Acta
Neurochir. 91:120–125. 1988. View Article : Google Scholar
|
|
4
|
Vesey DA, Cheung CW, Kruger WA, Poronnik
P, Gobe G and Johnson DW: Thrombin stimulates proinflammatory and
proliferative responses in primary cultures of human proximal
tubule cells. Kidney Int. 67:1315–1329. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Leask A and Abraham DJ: TGF-β signaling
and the fibrotic response. FASEB J. 18:816–817. 2004.
|
|
6
|
Verrecchia F and Mauviel A: Transforming
growth factor-β signaling through the Smad pathway: role in
extracellular matrix gene expression and regulation. J Invest
Dermatol. 118:211–215. 2002.
|
|
7
|
Flood C, Akinwunmi J, Lagord C, Daniel M,
Berry M, Jackowski A and Logan A: Transforming growth factor-β1 in
the cerebrospinal fluid of patients with subarachnoid hemorrhage:
titers derived from exogenous and endogenous sources. J Cereb Blood
Flow Metab. 21:157–162. 2001.
|
|
8
|
Kasuya H, Shimizu T and Takakura K:
Thrombin activity in CSF after SAH is correlated with the degree of
SAH, the persistence of subarachnoid clot and the development of
vasospasm. Acta Neurochir. 140:579–584. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bottinger EP: TGF-β in renal injury and
disease. Semin Nephrol. 27:309–320. 2007.
|
|
10
|
Wang W, Koka V and Lan HY: Transforming
growth factor-β and Smad signaling in kidney diseases. Nephrology.
10:48–56. 2005.
|
|
11
|
Bech RA, Juhler M, Waldemar G, Klinken L
and Gjerris F: Frontal brain and leptomeningeal biopsy specimens
correlated with cerebrospinal fluid outflow resistance and B-wave
activity in patients suspected of normal-pressure hydrocephalus.
Neurosurgery. 40:497–502. 1997.
|
|
12
|
Massicotte EM and Del Bigio MR: Human
arachnoid villi response to subarachnoid hemorrhage: possible
relationship to chronic hydrocephalus. J Neurosurg. 91:80–84. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shirato K, Osawa H, Kaizuka M, Nakamura N,
Sugawara T, Nakamura M, Tamura M, Yamabe H and Okumura K: Thrombin
stimulates production of fibronectin by human proximal tubular
epithelial cells via a transforming growth factor-beta-dependent
mechanism. Nephrol Dial Transplant. 18:2248–2254. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Galbreath E, Kim SJ, Park K, Brenner M and
Messing A: Overexpression of TGF-β1 in the central nervous system
of transgenic mice results in hydrocephalus. J Neuropathol Exp
Neurol. 54:339–349. 1995.
|
|
15
|
Cohen AR, Leifer DW, Zechel M, Flaningan
DP, Lewin JS and Lust WD: Characterization of a model of
hydrocephalus in transgenic mice. J Neurosurg. 91:978–988. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim JS, Yoon SS, Kim YH and Ryu JS: Serial
measurement of interleukin-6, transforming growth factor-beta, and
S-100 protein in patients with acute stroke. Stroke. 27:1553–1557.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Knuckey NW, Finch P, Palm DE, Primiano MJ,
Johanson CE, Flanders KC and Thompson NdL: Differential neuronal
and astrocytic expression of transforming growth factor β isoforms
in rat hippocampus following transient forebrain ischemia. Brain
Res Mol Brain Res. 40:1–14. 1996.
|
|
18
|
Klempt ND, Sirimanne E, Gunn AJ, Klempt M,
Singh K, Williams C and Gluckman PD: Hypoxia-ischemia induces
transforming growth factor β1 mRNA in the infant rat brain. Brain
Res Mol Brain Res. 13:93–101. 1992.PubMed/NCBI
|
|
19
|
Henrich-Noack P, Prehn JH and Krieglstein
J: TGF-β1 protects hippocampal neurons against degeneration caused
by transient global ischemia. Dose-response relationship and
potential neuroprotective mechanisms. Stroke. 27:1609–1614.
1996.
|
|
20
|
Ata AK, Funa K and Olsson Y: Expression of
various TGF-β isoforms and type I receptor in necrotizing human
brain lesions. Acta Neuropathol. 93:326–333. 1997.
|
|
21
|
Blom IE, Goldschmeding R and Leask A: Gene
regulation of connective tissue growth factor: new targets for
antifibrotic therapy? Matrix Biol. 21:473–482. 2002. View Article : Google Scholar : PubMed/NCBI
|