Introduction
Parkinson’s disease (PD) is a common age-related,
progressive neurodegenerative disease, characterized by the loss of
dopaminergic neurons in the substantia nigra pars compacta
(SNpc) (1,2). Current treatments principally
ameliorate clinical manifestations of the disease rather than
prevent neuronal death, which reflects an incomplete understanding
of the basis of PD (3). However,
oxidative stress and mitochondrial dysfunction have been considered
to be responsible for the pathological features of PD, such as
neuronal death and apoptosis (4,5).
Reactive oxygen species (ROS) produced generated by oxidative
stress are widely believed to contribute to the loss of
dopaminergic neurons, mainly in the form of apoptosis (6). The mitochondria, the most important
sites of ROS production, are crucial in the process of apoptotic
regulation (7). The impairment of
mitochondrial activity increases ROS levels and causes oxidative
damage to proteins, lipids and DNA (8). Thus, the reduction of oxidative
stress may alleviate mitochondrial damage and appears to be a
promising strategy in the prevention and treatment of PD.
Rapamycin, a lipophilic, macrolide antibiotic, is
widely used in clinical practice as an immunosuppressant. It
induces autophagy by inactivating the mammalian target of rapamycin
(mTOR) (9). Rapamycin binds
intracellularly to the FK506 binding protein and targets mTOR to
block the calcium-dependent and non-dependent mTOR signaling
pathway (10). A number of
studies have previously claimed that rapamycin is able to provide
neuronal protection in a series of experimental models of
neurodegenerative diseases. Ravikumar et al (11,12) showed that rapamycin protects
against mutant Huntington-induced degeneration in cell, fly and
mouse models of Huntington’s disease. Pre-treatment with rapamycin
has been shown to prevent apoptosis and reduce ubiquitinated
protein aggregation in differentiated PC12 cells. C57/BL mice
post-treated with rapamycin significantly demonstrated an
attenuated loss of dopaminergic neurons (13). A recent study illustrated that
rapamycin but not FK506 protects neurons from death in both
cellular and animal models of PD by blocking the translation of
RTP801 (14). The mitochondrial
protective effects of rapamycin have previously been introduced in
Drosophila and this protection may be accounted for by the
reduced mitochondrial load and enhanced mitochondrial clearance by
autophagy (15). Certain studies
have shown that rapamycin reduces oxidative stress in
frataxin-deficient yeast cells (16) and restores the mitophagy inhibited
by monoamine oxidase B (Mao-B) in an inducible cell model (17). However, other studies have
indicated that rapamycin increases oxidative stress response in
adult stem cells (18) and have
shown that nucleolar disruption leads to oxidative damage and
Parkinsonism through mTOR repression (19). These studies suggest that the
protective role of rapamycin against oxidative stress in PD appears
not to be fully clarified, and the mitochondrial injuries and
apoptosis induced by oxidative stress remain to be elucidated.
In this study, we employed rapamycin in a rat model
of PD induced by 6-hydroxydopamine (6-OHDA). Rotational behaviors,
dopaminergic neuronal loss and mitochondrial injuries were
analyzed. By detecting markers of oxidative stress and apoptosis in
rapamycin-pre-treated PD rats, we demonstrate that rapamycin exerts
protective effects on the mitochondria, preventing oxidative stress
and apoptotic responses in a rat model of PD.
Materials and methods
Animals and induction of PD
Female Sprague-Dawley rats weighing 200–250 g were
obtained from the Experimental Animal Center of Soochow University
(Suzhou, China). All rats were housed in a temperature-controlled
environment at 22±1°C, with 12/12 h light/dark cycle and allowed
ad libitum access to food and water. All experimental
procedures were approved by the Animal Care and Use Committee of
Soochow University. For the induction of PD, the rats were
intraperitoneally (i.p.) anesthetized with chloral hydrate (400
mg/kg). For stereotactic surgery, 6-OHDA (16 μg dissolved in 4 μl
0.9% sodium chloride solution containing 0.2% ascorbic acid; Sigma)
was injected into 2 different sites within the right striatum.
Striatum injection coordinates were as follows: site 1:
anterior-posterior (AP) +0.7 mm, medial-lateral (ML) -3.0 mm,
dorso-ventral (DV) −4.5 mm; site 2: AP −0.2 mm, ML −2.6 mm, DV −6.0
mm. The sham-operated rats underwent the same surgical procedure in
the absence of 6-OHDA. After the surgery, animals were kept in a
temperature-controlled room for complete recovery.
Grouping, drug treatment and behavioral
testing
The rats were randomly divided into the following 5
groups and each group consisted of 24 animals: i) Sham-operated
rats treated as described above (group S); ii) PD model rats
pre-treated with the vehicle in the absence of rapamycin (group P);
iii) PD model rats pre-treated with a low dose of rapamycin (group
L-R) (0.05 mg/kg/day); iv) PD model rats pre-treated with a
moderate dose of rapamycin (group M-R) (0.5 mg/kg/day); v) PD model
rats pre-treated with a high dose of rapamycin (group H-R) (5
mg/kg/day). Rapamycin (Bomeibio, Hebei, China) was dissolved in
dimethyl sulfoxide (DMSO) and then diluted with 0.9% sodium
chloride solution with the final concentration of DMSO at 0.5%
(v/v). Different doses of rapamycin or vehicle (0.9% sodium
chloride solution containing 0.5% DMSO) were intragastrically
administered once daily to the rats from day 7 before the induction
of PD. Three weeks later, all animals underwent rotational testing
(day 25) to evaluate the motor asymmetry caused by the unilateral
nigrostriatal lesion. Each rat received R-(−)-Apomorphine
hydrochloride hemihydrate (APO; Sigma) (0.5 mg/kg i.p., dissolved
in 0.9% sodium chloride solution) and was placed in a transparent
cage immediately. Contralateral rotations (360°, in short axis) 30
min from the initiation of rotation were recorded.
Tyrosine hydroxylase (TH)
immunohistochemistry
On day 28, the rats were sacrificed by cardiac
perfusion with a solution of 4% paraformaldehyde (PFA). The brains
were removed, post-fixed in 10% formalin-PBS solution for 2 days,
and processed for paraffin embedding. To evaluate TH+
neurons in the substantia nigra, we examined serial sections
located in areas between −4.8 and −5.8 mm from the bregma in the
rostrocaudal direction, according to the Rat Brain Atlas by Paxinos
and Watson (20). The brain
sections were cut into 4 μm slices. Following deparaffinization and
rehydration, antigen retrieval was performed and endogenous
peroxidase activity was blocked. After the sections were blocked
with 3% normal goat serum, rabbit polyclonal antibody against TH
(1:1,000 dilution; Sigma) was added for 1 h of incubation at 37°C.
The slices were washed prior to incubation with a horseradish
peroxidase-conjugated secondary antibody (1:200 dilution; Dako,
Carpinteria, CA, USA) at 37°C for 30 min. The antibody-peroxidase
complex was revealed by incubating the slices with a
3,3-diaminobenzidine peroxidase substrate kit (Sigma). A total of 8
sections per rat were analyzed for TH+ neurons using an
Olympus X71 photomicroscope. The measurements were carried out by 2
observers who were blinded to the source of the images. The number
of ipsilateral dopaminergic neurons was presented as a percentage
of TH+ cells in the ipsilateral hemisphere vs. the
contralateral one.
Transmission electron microscope (TEM)
analysis
Following perfusion, the striatum were dissected and
fixed in 2.5% glutaraldehyde in 0.01 M PBS (PH 7.4) for 3 h at 4°C,
followed by post-fixation in 1% OsO4 for 2 h at 4°C, and
then washed with the same buffer. Before being embeded in epoxy
resin, the specimens were dehydrated in gradient series of ethanol
(20–100%) and acetone. Ultra-thin slices were cut using an
ultramicrotome, collected on copper grids, and stained with uranyl
acetate and lead citrate. The slices were observed using an Hitachi
H-600 TEM.
Assays of peroxide levels and antioxidant
activities
In order to determine peroxide levels of
malondialdehyde (MDA) and the activities of antioxidant enzymes,
including superoxide dismutase (SOD) and glutathione peroxidase
(GSH-PX), the midbrain tissues were quickly removed and homogenized
with 0.9% sodium chloride solution on day 28. After being
centrifuged, the supernatant were collected for bioassays using
relative commercial assay kits (Jiancheng Bioengineering Institute,
Nanjing, China) according to the manufacturer’s instructions. MDA
levels in the brain tissue were expressed as nanomole per milligram
of protein (nmol/mg protein). The activity of SOD and GSH-PX was
presented as units per milligram of protein (U/mg protein).
Reverse transcriptional-polymerase chain
reaction (RT-PCR)
The striatum tissues were immediately removed and
frozen in liquid nitrogen. Total RNA was extracted using TRIzol
reagent (Invitrogen, Carslbad, CA, USA) according to our
standardized laboratory protocol. The cDNA was synthesized with the
PrimeScript™ RT Master Mix (Takara Bio, Inc., Shiga, Japan)
according to the manufacturer’s instructions. PCR amplification was
performed to analyze the gene expression. The specific primers used
for PCR analyses are presented in Table I. Parallel amplification of rat
GAPDH was performed as the endogenous control, and the intensity of
each band was quantified using gel analysis software (SigmaScan Pro
Image version 5.0). The gene expressions were presented as the
ratio between the band intensity value for Bcl-2 or Bax and the
value for GAPDH from the same RNA sample.
 | Table ISpecific primers and their sequences
used for RT-PCR analysis in this study. |
Table I
Specific primers and their sequences
used for RT-PCR analysis in this study.
| Primer | Sequence |
|---|
| Bcl-2 | F:
5′-CCGGGAGATCGATGAAGTA-3′
R: 5′-CATATTTGTTTGGGGCATGTCT-3′ |
| Bax | F:
5′-GCAGGGAGGATGGCTGGGGAGA-3′
R: 5′-TCCAGACAAGCAGCCGCTCACG-3′ |
| GAPDH | F:
5′-GTCGTGGAGTCTACTGGCGTCTT-3′
R: 5′-CAGTCTTCTGAGTGGCAGTGATGG-3′ |
Western blot analysis
The rapidly removed striatum tissues were
homogenized in ice-cold lysis buffer (containing 1%
phenylmethanesulfonyl fluoride). Each sample (containing 50 μg
protein) was mixed with the same volume of sample buffer and boiled
for 5 min. The proteins with different molecular weights were
separated on 12–15% SDS-PAGE gels and then transferred onto
nitrocellulose membranes. The membranes were blocked with 5%
non-fat dry milk in 0.1% TBST for 1 h at room temperature.
Subsequently, the membranes were incubated with the following
antibodies at 4°C overnight: mouse anti-rat Bax and Bcl-2 (1:200
dilution; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA),
rabbit anti-rat cleaved caspase-9 (1:500 dilution; Cell Signaling
Technology, Inc., Danvers, MA, USA), mouse anti-rat cytochrome
c (1:1,000 dilution; Santa Cruz Biotechnology, Inc.), and
mouse anti-rat β-actin (1:1,000 dilution; Beyotime Biotechnology,
Haimen, China). Goat anti-rabbit or goat anti-mouse
fluorescence-conjugated secondary antibodies (1:20,000 dilution;
Beijing Boisynthesis Biotechnology Co., Ltd., Beijing, China) were
then applied for 1 h of incubation at room temperature and the
immunofluorescence were detected with an Odyssey Near-infrared
two-color laser imaging system (Li-Cor; Lincoln, NE). The optical
density of each protein band was quantified by BandScan software
version 5.0. The optical density of each protein band was
normalized to the corresponding density of the β-actin band.
Statistical analysis
All statistical analyses were performed using SPSS
13.0 for Windows (SPSS Inc., Chicago, IL, USA). All quantitative
data are presented as the means ± standard deviation (SD). A
Student’s t-test or non-parametric Mann-Whitney U test was used to
analyze the differences between independent samples. One-way ANOVA
was initially performed to determine whether an overall
statistically significant difference existed before using the
Student’s t-test. A value of p<0.05 was considered to indicate a
statistically significant difference.
Results
Rapamycin provides behavioral
improvements in a rat model of PD
The injection of apomorphine did not produce
rotational behaviors in the sham-operated group (group S, n=24),
whereas the PD model group (group P, n=24) displayed a significant
number of contralateral rotations (406.67±67.72/30 min).
Pre-treatment with various doses of rapamycin significantly reduced
the number of contralateral rotations in group L-R (336.63±57.97/30
min, n=24), M-R (348.75±55.82/30 min, n=24) and H-R
(317.13±52.61/30 min, n=24), as compared with those in group P
(p<0.05 in all cases). However, there were no statistical
differences in rotations observed between groups L-R, M-R and H-R
(p>0.05 in all cases).
Rapamycin protects against the loss of
dopaminergic neurons and mitochondrial ultrastructual injuries in a
rat model of PD
The loss of dopaminergic neurons in the SNpc was
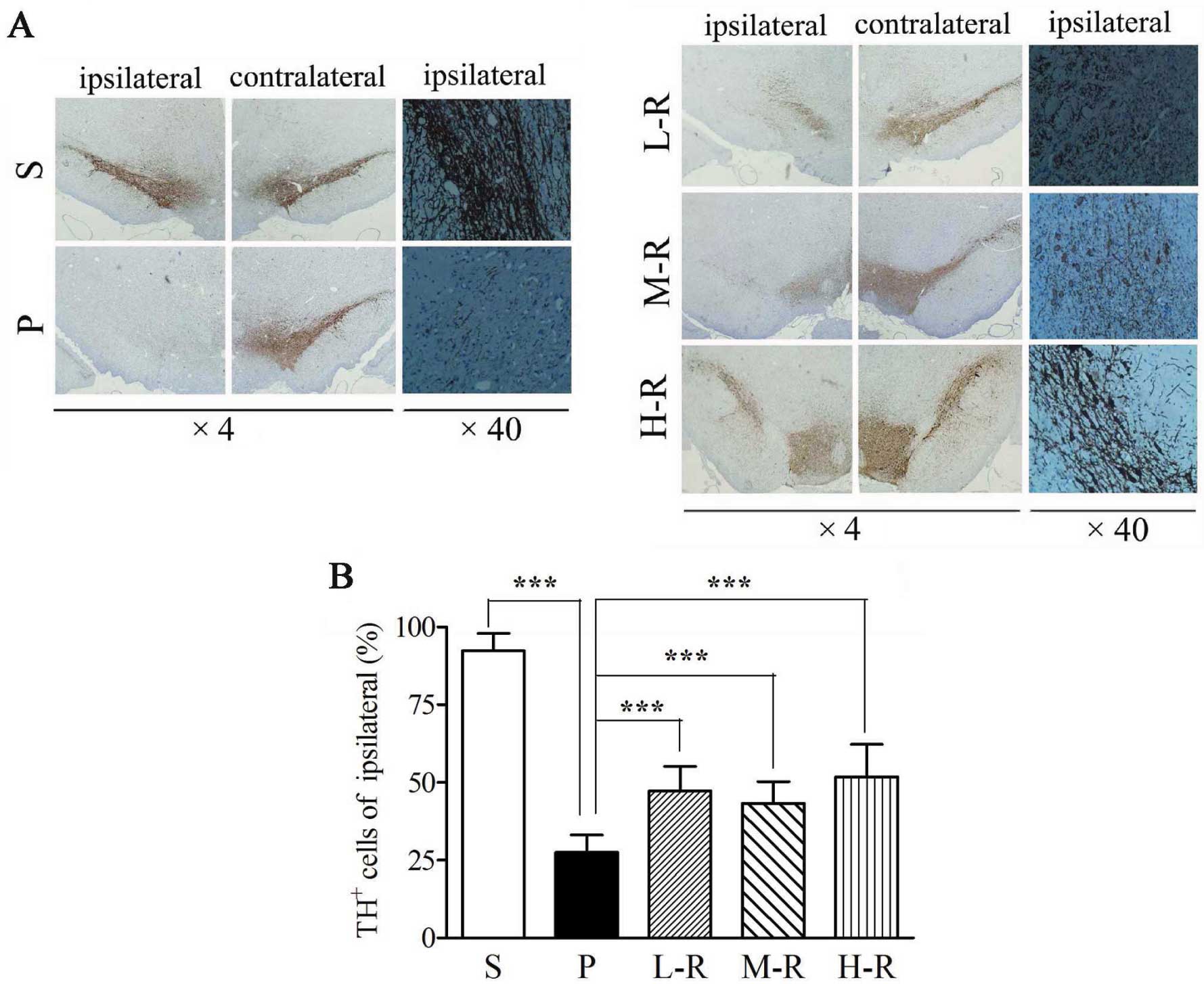
examined by TH immunohistochemistry (Fig. 1A). The number of remaining
TH+ neurons in the SNpc from the PD model rats (group P)
was significantly lower as compared with that in the SNpc from the
control rats (group S) (27.56±5.55 vs. 92.37±5.56%, p<0.001).
Pre-treatment with a low (L-R, 47.19±7.94%, p<0.001), moderate
(M-R, 43.17±7.09%, p<0.001) or high (H-R, 51.69±10.54%,
p<0.001) dose of rapamycin significantly increased the number of
TH+ neurons in the SNpc as compared to pre-treatment
with the vehicle (group P) (Fig.
1B). Although the number of TH+ neurons in group H-R
was higher than that in groups L-R and H-R, there was no
significant difference between them (p>0.05 in all cases)
(Fig. 1B). To analyze
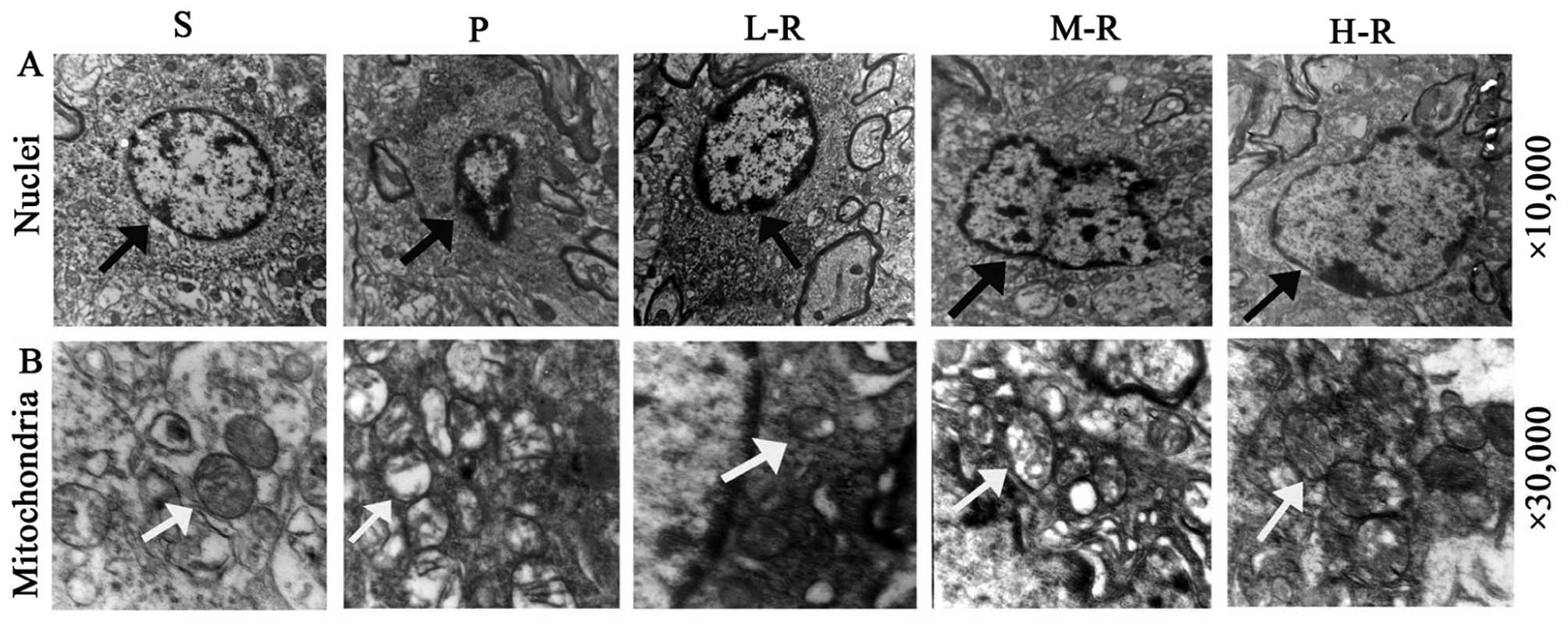
mitochondrial ultrastructural injuries, TEM analysis was performed.
In the striatum of the control rats (group S), healthy neurons
displayed normal nuclei with dispersed chromatin and mitochondria
with double membranes and clear cristae. In the PD model rats
(group P), the shrunken nuclei in the neurons demonstrated
condensed chromatin (Fig. 2A),
and the mitochondria were evidently swelled and vacuolated and the
cristae were lessened, distorted or had even disappeared (Fig. 2B). However, in the rats
pre-treated with rapamycin (groups L-R, M-R and H-R), the nuclei in
the neurons had less condensed chromatin and less swelled
mitochondria with distinct cristae and decreased vacuolations, as
compared with those in the rats pre-treated with the vehicle (group
P) (Fig. 2). Ultrastructural
manifestations of neurons appeared to be comparable between the
groups pre-treated with various doses of rapamycin.
Rapamycin reduces oxidative stress in a
rat model of PD
In the rats in group P, MDA levels were elevated as
compared with those in the rats in group S (30.62±4.55 vs.
13.87±3.32 nmol/mg protein, p<0.001) (Fig. 3A). However, the SOD (6.76±1.93 vs.
14.56±3.09 U/mg protein, p<0.001) and GSH-PX activity
(943.68±310.45 vs. 1797.12±313.53 U/mg protein, p<0.001) was
lower in group P compared to group S (Fig 3B and C). Following pre-treatment
with rapamycin, a reduction in MDA levels (24.63±4.56 nmol/mg
protein, p=0.003) and an increase in SOD (10.86±3.12 U/mg protein,
p=0.007) and GSH-PX (1324.65±251.63 U/mg protein, p=0.017) activity
were observed in the rats in group L-R as compared with those in
group P (Fig. 3). In the rats in
groups M-R and H-R, similar results were observed; the MDA levels
(M-R, 24.40±3.84 nmol/mg protein, p=0.002; H-R, 20.35±2.89 nmol/mg
protein, p<0.001) were decreased and SOD (M-R, 11.16±2.62 U/mg
protein, p=0.003; H-R, 12.01±3.25 U/mg protein, p<0.001) and
GSH-PX (M-R, 1302.45±236.38 U/mg protein, p=0.028; H-R,
1456.90±320.03 U/mg protein, p=0.001) activity was increased as
compared to group P (Fig. 3). No
statistically significant differences were observed in the levels
of MDA and SOD and GSH-PX activity between the PD rats pre-treated
with various doses of rapamycin (p>0.05 in all cases).
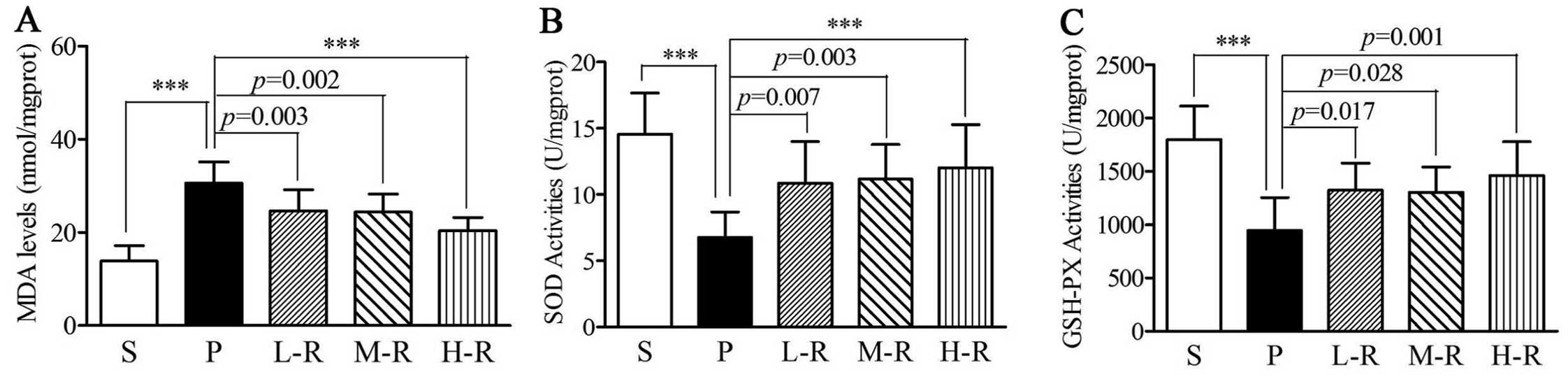 | Figure 3Levels of oxidative stress markers in
rats with Parkinson’s disease (PD). (A) Lower malondialdehyde (MDA)
levels were observed in the groups pre-treated with low (L-R,
p=0.003), moderate (M-R, p=0.002), and high (H-R, p<0.001) doses
of rapamycin compared to the group pre-treated with the vehicle
(P). (B) Increased superoxide dismutase (SOD) activity was observed
in the groups pre-treated with low (L-R, p=0.007), moderate (M-R,
p=0.003), and high (H-R, p<0.001) doses of rapamycin compared
the group pre-treated with the vehicle (P). (C) Increased
glutathione peroxidase (GSH-PX) activity was observed in the groups
pre-treated with low (L-R, p=0.017), moderate (M-R, p=0.028), and
high (H-R, p=0.001) doses of rapamycin compared to the group
pre-treated with the vehicle (P). Bars indicate the means ± SD of
the results. Each group consisted of 12 rats.
***p<0.001. |
Rapamycin alters the expression of Bcl-2
and Bax in a rat model of PD
In the rats in group P, RT-PCR analyses showed that
the Bcl-2 mRNA levels (33.40±5.20 vs. 61.19±17.24, p=0.001) were
reduced and Bax mRNA levels (57.86±12.97 vs. 28.10±9.11,
p<0.001) were elevated as compared with those in the rats in
group S (Fig. 4A-C).
Pre-treatment with rapamycin significantly increased Bcl-2
(47.19±9.97, p=0.005) and decreased Bax (44.65±7.78, p=0.008) mRNA
levels in group L-R as compared to pre-treatment with the vehicle
(group P). The mRNA levels of Bcl-2 (M-R, 45.59±8.53, p=0.005; H-R,
54.03±11.30, p<0.001) and Bax (M-R, 46.70±6.98, p=0.037; H-R,
41.85±8.49, p=0.001) in groups M-R and H-R were also significantly
different as compared with those in group P (Fig. 4A-C). Western blot analyses were
performed to evaluate the protein expression of Bcl-2 and Bax in
rapamycin-pre-treated and vehicle-pre-treated PD rats. Further
analyses revealed that pre-treatment with rapamycin significantly
upregulated Bcl-2 and downregulated Bax protein expression compared
to pre-treatment with the vehicle (p<0.05 in all cases); these
results were consistent with those from RT-PCR analyses (Fig. 4D-F). However, no statistically
significant differences were observed in the mRNA and protein
expression levels of Bcl-2 and Bax between the PD rats pre-treated
with various doses of rapamycin (p>0.05 in all cases).
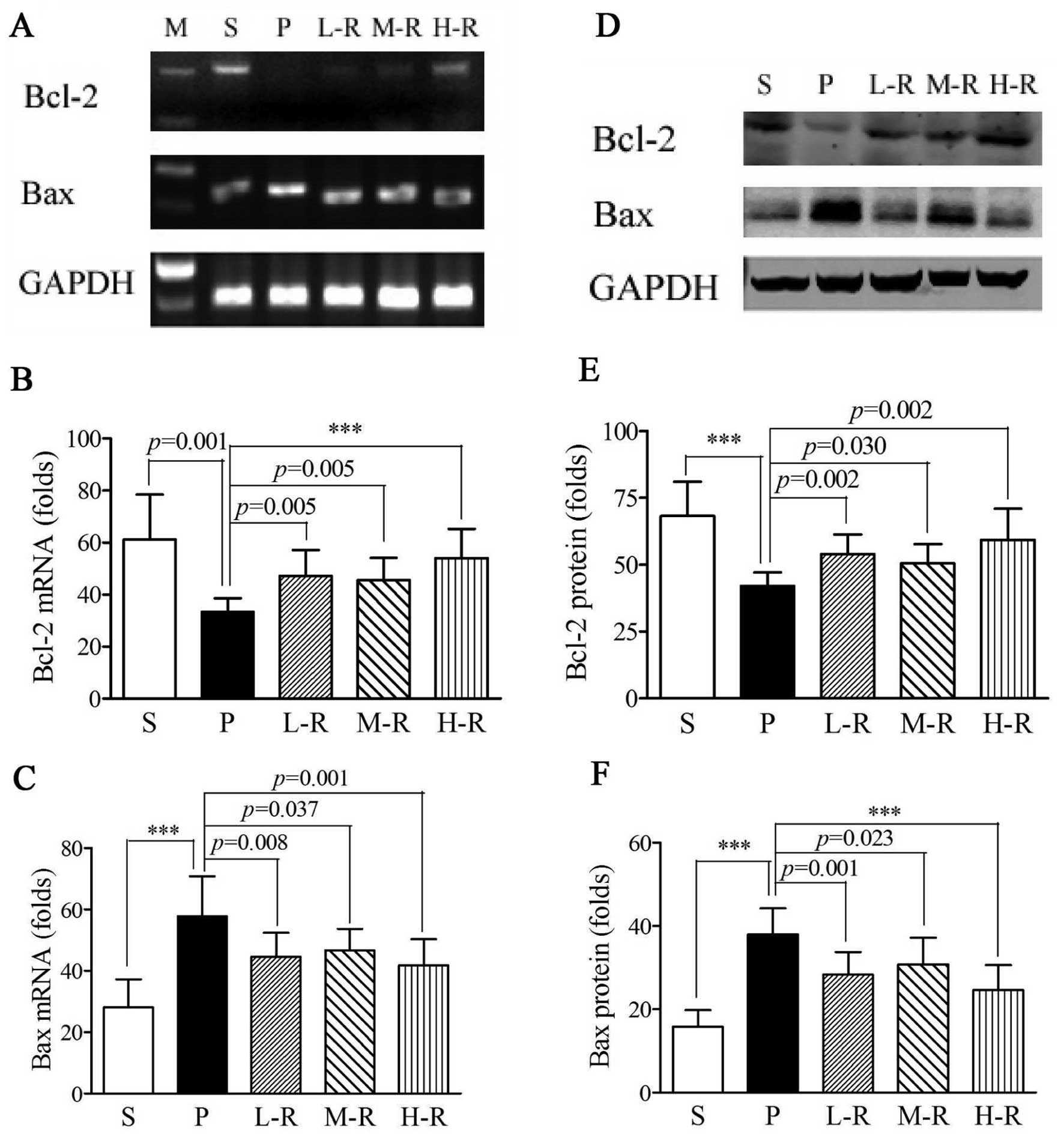 | Figure 4Expression of Bcl-2 and Bax in rats
with Parkinson’s disease (PD). (A-C) mRNA levels of Bcl-2 and Bax.
(A) Bcl-2 and Bax RT-PCR gel electrophoresis patterns with GAPDH as
the endogenous control. (B) Higher Bcl-2 (L-R, p=0.005; M-R,
p=0.005; H-R, p<0.001) and (C) lower Bax (L-R, p=0.008; M-R,
p=0.037; H-R, p=0.001) mRNA levels were observed in the groups
pre-treated with various doses of rapamycin compared to the group
pre-treated with the vehicle (P). (D-F) Protein expression of Bcl-2
and Bax. (D) Western blot analysis immunoreactive bands of GAPDH,
Bcl-2 and Bax are shown. (E) Increased Bcl-2 (L-R, p=0.002; M-R,
p=0.030; H-R, p=0.002) and (F) reduced Bax (L-R, p=0.001; M-R,
p=0.023; H-R, p<0.001) protein expression was observed in the
groups pre-treated with various doses of rapamycin compared to the
group pre-treated with the vehicle (P). Bars indicate the means ±
SD of the results. Each group consisted of 12 rats.
***p<0.001. L-R, low-dose rapamycin; M-R,
moderate-dose rapamycin; H-R, high-dose rapamycin. |
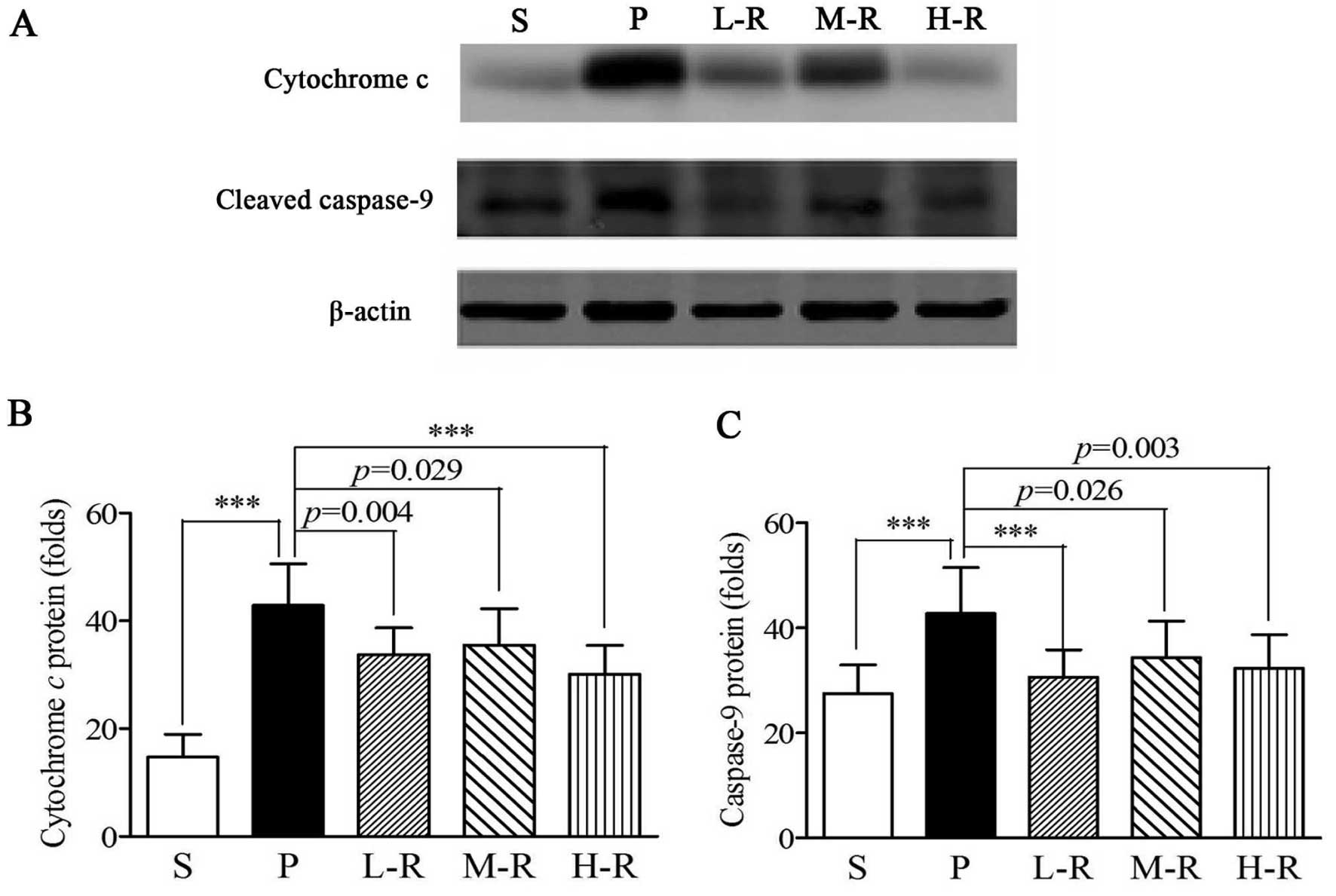
Rapamycin reduces cytochrome c release
and cleaved caspase-9 expression in a rat model of PD
Western blot analyses demonstrated that the release
of cytochrome c (42.86±7.73 vs. 14.74±4.17, p<0.001) and
the expression of cleaved caspase-9 (42.72±8.77 vs. 27.46±5.51,
p<0.001) were increased in the rats in group P, as compared with
the rats in group S (Fig. 5).
Pre-treatment with rapamycin significantly suppressed cytochrome
c release (33.80±4.86, p=0.004) and caspase-9 expression
(30.68±5.15, p<0.001) in group L-R, as compared with the group
pre-treated with the vehicle (group P) (Fig. 5). Pre-treatment with a moderate
and high dose of rapamycin also reduced the release of cytochrome
c (M-R, 35.53±6.73, p=0.029; H-R, 30.07±5.39, p<0.001)
and the expression of cleaved caspase-9 (M-R, 34.32±7.01, p=0.026;
H-R, 32.28±6.42, p=0.003), as compared to the group pre-treated
with the vehicle; no statistically significant differences were
observed between groups L-R, M-R and H-R (p>0.05 in all cases)
(Fig. 5).
Discussion
Oxidative stress and apoptosis play crucial roles in
the pathogenesis of PD (4).
Numerous efforts have been made to discover a therapeutic strategy
to prevent the degeneration of dopaminergic neurons. The mTOR
pathway regulates cell growth not only by regulating stress
responses (21), but also by
controlling mitochondrial energy metabolism (22). As a regulator of mTOR signaling,
rapamycin protects against neuronal death by RTP801 blockage or
through autophagy induction (13,14,23). Consistently, in this study, a
behavioral improvement and a reduced loss of TH+ neurons
were observed in the PD rats pre-treated with rapamycin. The aim of
this study was to investigate the mitochondrial protective role of
rapamycin in a rat model of PD by the analyses of oxidative stress
and apoptosis in vivo.
Oxidative stress is a common characteristic in a
number of current theories of the etiology of PD. Increases in
oxidative stress precede the signs of neuronal degeneration
(24), suggesting that oxidative
stress may be an early component of neuronal loss. Smith and Cass
(4,25) indicated that oxidative stress is
an early event in the course of dopamine depletion following 6-OHDA
administration and glial cell line-derived neurotrophic factor
(GDNF) reduced oxidative stress in a 6-OHDA model of PD. In
addition, nucleolar disruption leads to oxidative damage through
the mTOR pathway (19). To
investigate whether rapamycin suppresses oxidative stress in a rat
model of PD model, in this study, we detected the levels of MDA,
which has been shown to be associated with mitochondrial peroxide
activity (26). The levels of MDA
were higher in the PD rats compared to the sham controls, but were
significantly reduced following pre-treatment with rapamycin.
However, in the rapamycin-pre-treated PD rats, the levels of
antioxidant enzymes, such as SOD and GSH-PX were significantly
increased. The changes in peroxide activity and antioxidant enzyme
levels suggested that rapamycin reduced oxidative stress in
vivo, which may protect against dopamine neuronal death and
provide behavioral improvement in PD rats. Mitochondrial
dysfunction is considered critical to the pathogenesis of PD, while
oxidative stress and mitochondrial dysfunction reinforce each other
and constitute a vicious circle (27). To assess the therapeutic effects
on mitochondrial injuries, we performed TEM analyses and observed
nuclei and mitochondria ultrastructural injuries in the PD rats.
Pre-treatment with rapamycin led to a significant improvement in
6-OHDA-induced lesions, suggesting that rapamycin exerted a
protective effect against mitochondrial dysfunction, possibly
through the suppression of oxidative stress. Although the
therapeutic effects in the high-dose group exhibited optimization,
the results were not statistically different from those in the low-
and moderate-dose groups. This was consistent with the results of
behavioral analyses and the remaining TH+ neuronal
detections, suggesting that rapamycin protects against
6-OHDA-induced lesions, irrespective of the dose administered.
As is well known, apoptosis serves as a major
cellular mechanism in the pathogenesis of PD. Previous studies have
indicated that rapamycin protects against neuronal death or
apoptosis in in vitro and in vivo models of PD
(14). To evaluate the protective
effects against apoptosis, the expression of pro-apoptotic or
anti-apoptotic markers in rapamycin-pre-treated PD rats were
detected by RT-PCR and western blot analysis.
Mitochondrial-dependent apoptosis is finely regulated by a series
of pro- and anti-apoptotic proteins, such as Bcl-2 family proteins
that control the permeabilization of the mitochondrial outer
membrane (28). Increased Bax and
decreased Bcl-2 expression have been shown to reduce mitochondrial
membrane potential and increase ROS production in neurons (29). The increased Bcl-2 expression and
decreased Bax expression in rapamycin-pre-treated PD rats suggested
that rapamycin may play a beneficial role by preventing neurons
from subsequent pro-apoptotic insults. Cytochrome c is
usually found in the mitochondrial intermembrane space. The
translocation of Bax to the mitochondria is followed by the
permeabilization of the mitochondrial outer membrane, which results
in the release of cytochrome c and the activation of the
apoptotic program. Its release leads to caspase-9 cleavage and the
initiation of apoptosis (28,29). Thus, the reduced cytochrome
c release suggests that this protective effect of rapamycin
may be predominantly mitochondrial-dependent and decreased cleaved
caspase-9 may attenuate apoptotic responses to cellular oxidative
stress. However, it cannot be excluded that rapamycin may protect
against apoptosis initiated upstream of the mitochondria, such as
Fas/Fas-ligand-mediated apoptosis.
Additionally, the protective effects of rapamycin
against oxidative stress and apoptosis were not associated with the
dose of administration. It is worth noting that mTOR forms 2
distinct physical and functional complexes, termed mTOR complex 1
(mTORC1) and mTOR complex 2 (mTORC2). mTORC1, which is sensitive to
rapamycin, regulates translation and cell growth, whereas mTORC2 is
insensitive to rapamycin (30).
The L-3,4-dihydroxyphenylalanine (L-DOPA)-mediated activation of
mTORC1 has been shown to persist in mice that developed dyskinesia.
Moreover, the mTORC1 inhibitor, rapamycin, prevented the
development of dyskinesia without affecting the therapeutic
efficacy of L-DOPA (31). In this
study, the dissociation between the therapeutic effects and the
administered dose suggested that all doses of rapamycin may
selectively inhibit mTORC1 and have no interactions with mTORC2.
The specific inhibition of mTORC1 with a wide range of doses may
provide evidence supporting its clinical application for the
treatment of PD.
This study also raised the issue as to the mechanism
involved in the protective effects of rapamycin against oxidative
stress and apoptosis. Previous studies have provided evidence
linking mitochondrial dysfunction, oxidative stress and energy
depletion to neurodegenerative diseases, such as PD (27,32). Autophagy has been suggested to be
neuroprotective by enhancing the clearance of harmful protein
aggregates, and the dysfunction of autophagy may result in abnormal
mitochondrial function and oxidative stress (33). In addition, pre-treatment with
rapamycin has been shown to protect against apoptosis through the
induction of autophagy (13,15,23). Therefore, it can by hypothesized
that rapamycin reduces oxidative stress and protects against
apoptosis, possibly by enhancing autophagy, as shown in our
study.
In conclusion, this study demonstrates that
rapamycin provides behavioral improvement and reduces the loss of
dopaminergic neurons in PD. The neuroprotective properties of
rapamycin arise from its capacity to reduce oxidative stress and
mitochondrial injuries, which may consequently contribute to its
anti-apoptotic effects. Our findings provide evidence for the
employment of rapamycin as a therapeutic agent for the prevention
of neuronal degeneration in PD. The antioxidant and anti-apoptotic
mechanisms of rapamycin in PD remain to be elucidated in future
studies.
Acknowledgements
We thank Mr. Wen-Xuan Zhou, Mr. Zhong-Ji Zhou, Mr.
Ci-Yi Guo, Mr. Yu-Hai Chai and Mrs. Mei-Hua Ding for their
assistance.
Abbreviations:
|
PD
|
Parkinson’s disease
|
|
ROS
|
reactive oxygen species
|
|
mTOR
|
mammalian target of rapamycin
|
|
6-OHDA
|
6-hydroxydopamine
|
|
L-R
|
low dose of rapamycin
|
|
M-R
|
moderate dose of rapamycin
|
|
H-R
|
high dose of rapamycin
|
|
TH
|
tyrosine hydroxylase
|
|
TEM
|
transmission electron microscope
|
|
MDA
|
malondialdehyde
|
|
SOD
|
superoxide dismutase
|
|
GSH-PX
|
glutathione peroxidase
|
|
RT-PCR
|
reverse transcription-polymerase chain
reaction
|
References
|
1
|
Dauer W and Przedborski S: Parkinson’s
disease: mechanisms and models. Neuron. 39:889–909. 2003.
|
|
2
|
Marras C and Lang A: Invited article:
changing concepts in Parkinson disease: moving beyond the decade of
the brain. Neurology. 70:1996–2003. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Levy OA, Malagelada C and Greene LA: Cell
death pathways in Parkinson’s disease: proximal triggers, distal
effectors, and final steps. Apoptosis. 14:478–500. 2009.
|
|
4
|
Smith MP and Cass WA: Oxidative stress and
dopamine depletion in an intrastriatal 6-hydroxydopamine model of
Parkinson’s disease. Neuroscience. 144:1057–1066. 2007.PubMed/NCBI
|
|
5
|
Exner N, Lutz AK, Haass C and Winklhofer
KF: Mitochondrial dysfunction in Parkinson’s disease: molecular
mechanisms and pathophysiological consequences. EMBO J.
31:3038–3062. 2012.
|
|
6
|
Yamato M, Kudo W, Shiba T, Yamada KI,
Watanabe T and Utsumi H: Determination of reactive oxygen species
associated with the degeneration of dopaminergic neurons during
dopamine metabolism. Free Radic Res. 44:249–257. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee JE, Park JH, Shin IC and Koh HC:
Reactive oxygen species regulated mitochondria-mediated apoptosis
in PC12 cells exposed to chlorpyrifos. Toxicol Appl Pharmacol.
263:148–162. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hori A, Yoshida M, Shibata T and Ling F:
Reactive oxygen species regulate DNA copy number in isolated yeast
mitochondria by triggering recombination-mediated replication.
Nucleic Acids Res. 37:749–761. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Noda T and Ohsumi Y: Tor, a
phosphatidylinositol kinase homologue, controls autophagy in yeast.
J Biol Chem. 273:3963–3966. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sudarsanam S and Johnson DE: Functional
consequences of mTOR inhibition. Curr Opin Drug Discov Devel.
13:31–40. 2010.PubMed/NCBI
|
|
11
|
Ravikumar B, Duden R and Rubinsztein DC:
Aggregate-prone proteins with polyglutamine and polyalanine
expansions are degraded by autophagy. Hum Mol Genet. 11:1107–1117.
2002. View Article : Google Scholar
|
|
12
|
Ravikumar B, Vacher C, Berger Z, Davies
JE, Luo S, Oroz LG, et al: Inhibition of mTOR induces autophagy and
reduces toxicity of polyglutamine expansions in fly and mouse
models of Huntington disease. Nat Genet. 36:585–595. 2004.
View Article : Google Scholar
|
|
13
|
Pan T, Kondo S, Zhu W, Xie W, Jankovic J
and Le W: Neuroprotection of rapamycin in lactacystin-induced
neurodegeneration via autophagy enhancement. Neurobiol Dis.
32:16–25. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Malagelada C, Jin ZH, Jackson-Lewis V,
Przedborski S and Greene LA: Rapamycin protects against neuron
death in in vitro and in vivo models of Parkinson’s disease. J
Neurosci. 30:1166–1175. 2010.PubMed/NCBI
|
|
15
|
Ravikumar B, Berger Z, Vacher C, O’Kane CJ
and Rubinsztein DC: Rapamycin pre-treatment protects against
apoptosis. Hum Mol Genet. 15:1209–1216. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Marobbio CM, Pisano I, Porcelli V, Lasorsa
FM and Palmieri L: Rapamycin reduces oxidative stress in
frataxin-deficient yeast cells. Mitochondrion. 12:156–161. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Siddiqui A, Hanson I and Andersen JK:
Mao-B elevation decreases parkin’s ability to efficiently clear
damaged mitochondria: protective effects of rapamycin. Free Radic
Res. 46:1011–1018. 2012.PubMed/NCBI
|
|
18
|
Kofman AE, McGraw MR and Payne CJ:
Rapamycin increases oxidative stress response gene expression in
adult stem cells. Aging (Albany NY). 4:279–289. 2012.PubMed/NCBI
|
|
19
|
Rieker C, Engblom D, Kreiner G, Domanskyi
A, Schober A, Stotz S, et al: Nucleolar disruption in dopaminergic
neurons leads to oxidative damage and parkinsonism through
repression of mammalian target of rapamycin signaling. J Neurosci.
31:453–460. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Paxinos G and Watson C: The Rat Brain in
Stereotaxic Coordinates. 6th edition. Academic Press; London:
2007
|
|
21
|
Schieke SM, Phillips D, McCoy JP Jr,
Aponte AM, Shen RF, Balaban RS, et al: The mammalian target of
rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption
and oxidative capacity. J Biol Chem. 281:27643–27652. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wullschleger S, Loewith R and Hall MN: TOR
signaling in growth and metabolism. Cell. 124:471–484. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pan T, Rawal P, Wu Y, Xie W, Jankovic J
and Le W: Rapamycin protects against rotenone-induced apoptosis
through autophagy induction. Neuroscience. 164:541–551. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hall ED, Detloff MR, Johnson K and Kupina
NC: Peroxynitrite-mediated protein nitration and lipid peroxidation
in a mouse model of traumatic brain injury. J Neurotrauma. 21:9–20.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Smith MP and Cass WA: GDNF reduces
oxidative stress in a 6-hydroxydopamine model of Parkinson’s
disease. Neurosci Lett. 412:259–263. 2007.PubMed/NCBI
|
|
26
|
Long J, Liu C, Sun L, Gao H and Liu J:
Neuronal mitochondrial toxicity of malondialdehyde: inhibitory
effects on respiratory function and enzyme activities in rat brain
mitochondria. Neurochem Res. 34:786–794. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Onyango IG: Mitochondrial dysfunction and
oxidative stress in Parkinson’s disease. Neurochem Res. 33:589–597.
2008.
|
|
28
|
Martinou JC and Youle RJ: Mitochondria in
apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev
Cell. 21:92–101. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kirkland RA and Franklin JL: Bax, reactive
oxygen, and cytochrome c release in neuronal apoptosis. Antioxid
Redox Signal. 5:589–596. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bhagwat SV and Crew AP: Novel inhibitors
of mTORC1 and mTORC2. Curr Opin Investig Drugs. 11:638–645.
2010.PubMed/NCBI
|
|
31
|
Santini E, Heiman M, Greengard P, Valjent
E and Fisone G: Inhibition of mTOR signaling in Parkinson’s disease
prevents L-DOPA-induced dyskinesia. Sci Signal. 2:ra362009.
|
|
32
|
Lin MT and Beal MF: Mitochondrial
dysfunction and oxidative stress in neurodegenerative diseases.
Nature. 443:787–795. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee J, Giordano S and Zhang J: Autophagy,
mitochondria and oxidative stress: cross-talk and redox signalling.
Biochem J. 441:523–540. 2012. View Article : Google Scholar : PubMed/NCBI
|