Introduction
Cholestasis, which is characterized by an impairment
of bile formation and/or bile flow, is a commonly occurred
pathophysiological process in certain liver diseases (1). The persistence of cholestasis and
the retention of toxic bile salts in hepatocytes are associated
with acute and chronic liver failure, leading to biliary fibrosis,
cirrhosis and cancer (2,3). The accumulation of hydrophobic bile
acids (e.g., glycine conjugates of chenodeoxycholic acid) in the
liver is considered to be an important factor contributing to
hepatic injury (4,5). In healthy individuals, the portal
vein serum total bile acid concentration is approximately 20 μM; in
patients with cholestasis, concentrations can reach 300 μM
(6). Toxic bile salts induce
hepatocellular death through 2 mechanisms: necrosis at higher
concentrations (≥250 μM) (7,8)
and apoptosis at lower concentrations (≤100 μM) (9,10).
Both types of cell death seem to play a role in cholestatic liver
injury. The mechanisms involved in the toxicity of toxic bile salts
include disrupting cell membrane integrity through their detergent
action on lipid components (11)
and triggering the generation of reactive oxygen species (ROS),
which may eventually cause hepatocyte apoptosis or necrosis
(12). Glycochenodeoxycholic acid
(GCDC) is formed in the liver and acts as a detergent to solubilize
fats for absorption. It is the predominant human dihydroxy bile
salt under cholestatic conditions and is a likely candidate for
bile acid-mediated hepatocellular injury during cholestasis due to
its direct cytotoxic effects on hepatocytes (13). GCDC may cause hepatocyte apoptosis
by activating the death receptor or by inducing oxidative damage
that causes mitochondrial dysfunction, which subsequently triggers
apoptosis (12,14). Therefore, GCDC-induced hepatocyte
apoptosis is a reliable and reproducible model, which is widely
used to study the mechanisms involved in cholestatic liver
injury.
In traditional Chinese medicine, Hypericum
japonicum has been used for centuries as a raw material for
herbal tea and for the treatment of cholestasis, as well as acute
and chronic hepatitis (15).
Quercetin 7-rhamnoside (Q7R; molecular weight, 448.377) is one of
the main flavonoid components of Hypericum japonicum
(16). It has been reported that
Q7R possesses strong anti-porcine epidemic diarrhea virus activity,
which is not simply due to its general action as an antioxidant
(17,18). Amani et al reported that
Schouwia thebaica Webb extracts containing Q7R significantly
exert curative effects on carbon tetrachloride
(CCl4)-induced liver injury in rats (19). However, whether Q7R is one of the
active ingredients responsible for the hepatopreventive effects of
Hypericum japonicum has not yet been ascertained. Thus, the
aim of the present study was to determine the role of Q7R in
GCDC-induced apoptosis in L-02 cells and to elucidate the possible
mechanisms involved.
Materials and methods
Extraction and isolation of Q7R
Q7R (98.3% purity) was extracted and purified from
the herb, Hypericum japonicum. Briefly, a Hypericum
japonicum sample was pulverized to a powder. One kilogram of
powder was extracted by reflux with 10 l distilled water 3 times (2
h for each repetition). The mixture was filtered and collected. The
extract was then concentrated to 1.2 l and then 4.5 l aqueous
ethanol (95%) were added to the extract and stored at 4°C for 12 h.
The extract was then filtered and concentrated by rotary
vaporization at 60°C under reduced pressure and 400 ml residue was
obtained. The residue was then redissolved in water (total volume
1,000 ml), which was added into a glass column (6.0×80 cm,
containing 1.0 kg polyamide resin). In the polyamide resin column
chromatography, water and different concentrations of aqueous
ethanol (20 and 60%) were tested by a gradient elution program and
aqueous ethanol (60%) elution was collected. The elution fractions
were united and then concentrated by rotary vaporization at 60°C
under reduced pressure. The precipitate was filtered and washed
with water and absolute ethyl alcohol, respectively and was then
dried at 80°C for 1 h. Q7R was obtained, and identification was
performed by ultraviolet-visible spectroscopy (UV/Vis), electron
spray ionization-mass spectrometry, proton nuclear magnetic
resonance (1H-NMR) and carbon-13 nuclear magnetic
resonance (13C-NMR) spectroscopy. Based on spectral
analysis, the purified compound was verified as Q7R (19). The chemical structure of Q7R is
shown in Fig. 1. The purity
analysis was performed on a Waters 600 HPLC chromatography system
(Waters Corp., Milford, MA, USA) with an Agilent C18 column (5 μM,
250×4.0 mm ID) purchased from Agilent Technologies, Inc. (Santa
Clara, CA, USA). The mobile phase was prepared by mixing solvents,
methanol: 2.5% (v/v) acetic acid (36:64 v/v), and the pH was
adjusted to 2.5 with acetic acid. The detection wavelength and
column temperature were set at 255 nm and 30°C, respectively. The
flow rate was 0.4 ml/min. The loading volume was 3 μl. The purity
of Q7R was 98.3%. In this study, Q7R was dissolved in dimethyl
sulfoxide (DMSO; Sigma, St. Louis, MO, USA) for treatment.
Cell culture
The L-02 human normal liver cells were a kind gift
from professor Wen Chen (School of Public Health, Sun Yat-sen
University, Guangzhou, China). The cells were cultured at 37°C
(with 5% CO2 and 95% humidity) in Dulbecco’s modified
Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum
(FBS) (both from Gibco/Invitrogen, Carlsbad, CA, USA), 1%
penicillin and 1% streptomycin. The following day, the medium was
replaced with fresh DMEM and the cells were divided into 5 groups.
Untreated L-02 cells in the first group served as the controls.
Treated L-02 cells were stimulated with 100 μM GCDC (Sigma) in the
presence or absence of Q7R at the indicated concentrations and
periods or time. The stock solutions of Q7R were prepared in DMSO.
The final maximum concentration of DMSO in the experimental medium
did not exceed 0.1%.
Cell viability assay
Cell viability was evaluated by
methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay as
previously described (20). The
L-02 cells were seeded into 96-well plates at a density of 7,500
cells/well with 100 μl DMEM medium per well and incubated for 24 h.
In the present study, the cells were pre-treated with various
concentrations of Q7R (0, 50, 100 and 200 μM) for 30 min and
subsequently exposed to GCDC (100 μM) or the vehicle for an
additional 24 h. MTT (Sigma) was then added to each well followed
by incubation at 37°C for 4 h. Following incubation, the medium
containing MTT was removed and 150 μl DMSO were added. The optical
density of the homogenous purple solutions was measured using a
microplate reader (Mk3; Thermo Labsystems, Chicago, IL, USA) at 590
nm. The optical density of the formazan formed in the control
(medium only) cells was taken as 100% viability.
Detection of apoptosis assay by Hoechst
33258 staining
Following treatment as described above, the cells in
24-well plates were rinsed 3 times with phosphate-buffered saline
(PBS) and stained with Hoechst 33258. Subsequently, the L-02 cells
were viewed immediately by detecting fluorescence (excitation at
380 nm and emission at 460 nm) under a fluorescence microscope
(Nikon TE2000) and the images were recorded for later analysis. The
morphological features of apoptosis were evident, including
chromatin condensation, nuclear fragmentation and apoptotic body
formation.
Detection of apoptosis assay by flow
cytometry using Annexin V-FITC/PI double staining
The occurrence of apoptosis and/or necrosis was
evaluated by Annexin V binding and PI uptake as previously
described (21). The positioning
of the quadrants on the Annexin V/PI dot plots was performed and
living cells (Annexin V−/PI−), early
apoptotic/primary apoptotic cells (Annexin
V+/PI−), late apoptotic/secondary apoptotic
cells (Annexin V+/PI+) and necrotic cells
(Annexin V−/PI+) were distinguished.
Therefore, the total apoptotic cell population included the
percentage of cells with fluorescence Annexin
V+/PI− and Annexin
V+/PI+ staining. Annexin V binding was
performed using an Annexin V-FITC kit (Invitrogen) according to the
instructions of the manufacturer. Following treatment as described
in above, the cells were harvested and washed with PBS and
suspended in 100 μl Annexin V binding buffer. The cells were then
double stained with 10 μl Annexin V and 5 μl PI solution. The
samples were incubated for 20 min at room temperature and then
analyzed by flow cytometry (Gallios Flow Cytometer; Beckman
Coulter, Brea, CA, USA).
Analysis of intracellular ROS levels by
flow cytometry
The production of intracellular ROS was monitored by
flow cytometry using an oxidation sensitive fluorescent probe
(DCFH-DA; Beyotime, Shanghai, China). The cells were pre-treated
with various concentrations of Q7R (0, 50, 100 and 200 μM). Thirty
minutes later, GCDC (100 μM) was added to the medium followed by
incubation at 37°C for an additional 12 h. When the medium was
removed, the cells were harvested and washed twice with PBS. The
cells were then incubated with 10 μM DCFH-DA at 37°C for 30 min
according to the manufacturer’s instructions. The samples were
analyzed by flow cytometry (Gallios Flow Cytometer; Beckman
Coulter). Data were analyzed using FlowJo software (FlowJo Co.,
Ashland, OR, USA). For each sample, 10,000 events were
collected.
Analysis of intracellular glutathione
(GSH) levels
GSH plays a critical role in cellular defenses
against oxidative stress. The L-02 cells were cultured in a 6-well
plate for 24 h and then pre-treated with various concentrations of
Q7R (0, 50, 100 and 200 μM). After 30 min, GCDC (100 μM) was added
to the medium followed by and incubation at 37°C for 4 h. Following
incubation, the assay for GSH was performed using a GSH detection
kit (Beyotime) according to the manufacturer’s recommendations. The
GSH level was analyzed by microplate reader (Mk3; Thermo
Labsystems).
Mitochondrial membrane potential (Δψm)
assay
The Δψm was determined by the mean fluorescence
intensity of rhodamine 123 (Rh123; Sigma). The L-02 cells
(1×106 cells/well) were cultured in a 6-well plate for
24 h. Following treatment as described in above, the treated cells
were collected and resuspended at a concentration of
1×105 cells/ml in PBS. Rh123 (5 μg/ml) was added to the
cells, followed by an incubation for 30 min prior to analysis under
a fluorescence microscope (Nikon TE2000).
Analysis of the concentration of
intracellular Ca2+ by laser scanning confocal
microscopy
The L-02 cells (1×106 cells/well) were
added to each culture dish with coverslips for 24 h and the cells
were then pre-treated with various concentrations of Q7R (0, 50,
100 and 200 μM). After 30 min, GCDC (100 μM) was added to the
medium followed by incubation at 37°C for 24 h. The cultured L-02
cells were loaded with Ca2+ indicator dye by incubating
the culture dish in standard solution supplemented with 5 μM
Fluo-3-AM (Beyotime) for 50 min at room temperature. For measuring
alterations in the intracellular Ca2+ concentration, a
confocal laser-scanning microscope (Leica TCS SP5) was used. Fluo-3
was excited at the 488 nm line of an argon laser and the
fluorescence was measured at an emission wavelength >510 nm
selected with a longpass filter. An oil immersion objective (Zeiss
×16, NA 0.5) was used in all the experiments. Analysis of confocal
images was performed using analysis software.
Statistical analysis
All values were expressed as the means ± SD of 3
independent determinations. All experiments were carried out at
least 3 times, each time with 3 or more independent observations.
Statistical analyses were carried out using SPSS 16.0 software. The
statistical significance of differences between the mean values was
analyzed by one-way ANOVA followed by the Student-Newman-Keuls
test. P-values <0.05 were considered to indicate statistically
significant differences.
Results
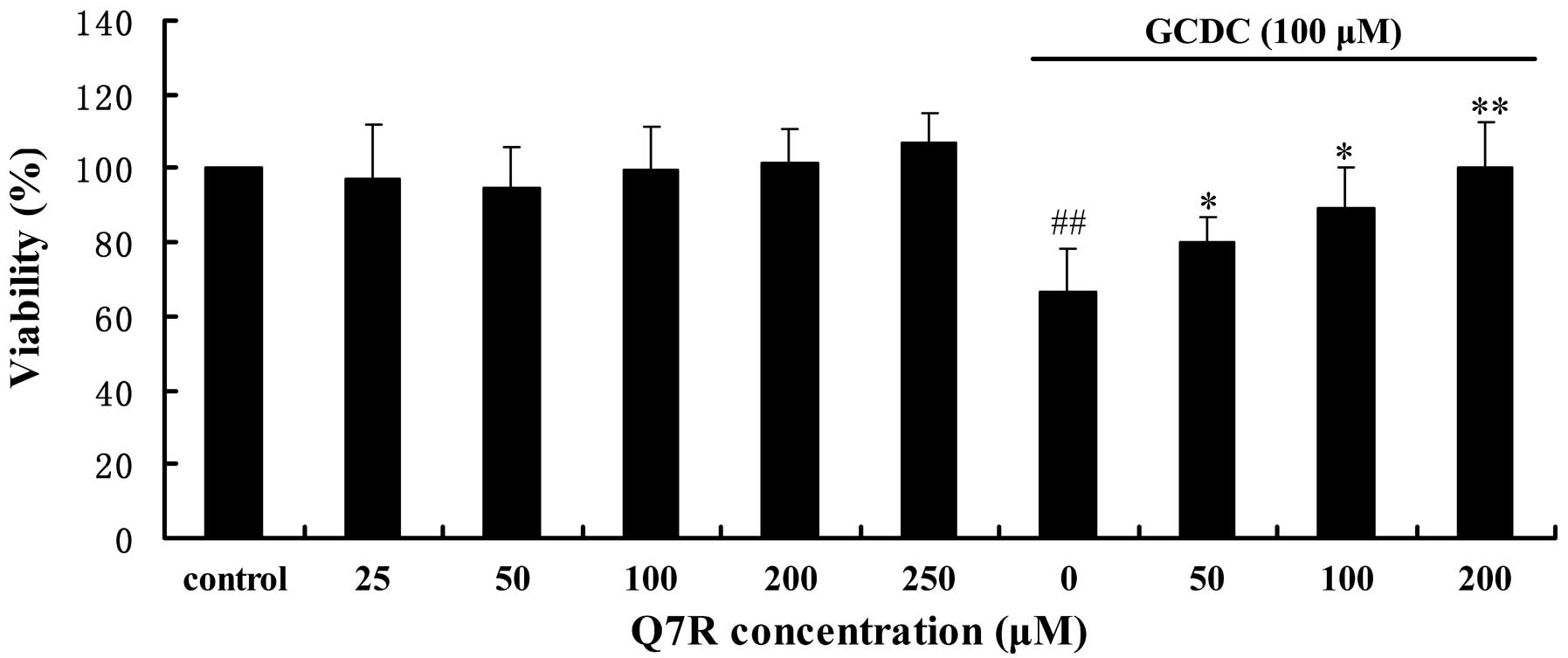
Cell viability
GCDC at 100 μM reduced the viability of L-02 cells,
whereas pre-treatment with various concentrations of Q7R (50, 100
and 200 μM) for 30 min significantly ameliorated the GCDC-induced
reduction in the viability of L-02 cells in a
concentration-dependent manner (Fig.
2).
Detection of apoptosis by Hoechst 33258
staining and Annexin V-FITC/PI double staining
The Hoechst 33258 dye stains the condensed chromatin
of apoptotic cells more brightly than the chromatin of normal
cells. A morphological observation of the L-02 cells indicated that
the control L-02 cells had regular and round-shaped nuclei,
revealed by nuclear staining with Hoechst 33258 (Fig. 3). By contrast, condensation and
fragmentation of the nuclei, characteristic of apoptotic cells were
evident in the L-02 cells treated with GCDC. On the other hand, the
number of apoptotic cells was significantly reduced in the cells
exposed to Q7R (50, 100 and 200 μM) in a concentration-dependent
manner.
The occurrence of apoptosis was further confirmed by
Annexin V/PI double staining. The apoptosis of the L-02 cells was
observed after 24 h of exposure to GCDC. Fig. 4 shows the extent of apoptosis of
the L-02 cells incubated with 100 μM GCDC with or without Q7R.
Shifts from no apoptosis (control) (bottom left quadrants) to early
(bottom right quadrants) and late apoptosis (upper right quadrants)
are clearly evident in the dot plots. The numbers of total (early +
late) apoptotic cells were markedly enhanced following treatment
with 100 μM GCDC after 24 h. On the other hand, pre-treatment with
various concentrations of Q7R (50, 100 and 200 μM) significantly
reduced the percentage of apoptotic L-02 cells.
Analysis of intracellular ROS levels by
flow cytometry
Treatment with 100 μM GCDC for 12 h increased ROS
production in the L-02 cells (Fig.
5). In addition, the results revealed a significant decrease in
intracellular ROS after 12 h of co-incubation with various
concentrations of Q7R (50, 100 and 200 μM).
Analysis of intracellular GSH levels
The intracellular concentration of GSH was
determined to elucidate the mechanisms through which Q7R exerts its
antioxidant effects. Fig. 6
demonstrates that the intracellular GSH content in the L-02 cells
was markedly reduced following exposure to 100 μM GCDC for 24 h,
whereas the reduction in GSH content was significantly inhibited
following pre-treatment with various concentrations of Q7R (50, 100
and 200 μM).
Δψm assay
Mitochondrial permeability transition (MPT) has been
shown to play a key role in bile acid-induced hepatocyte necrosis,
as well as other models of cellular apoptosis. As a consequence of
MPT, the Δψm collapses, thereby uncoupling the respiratory chain
and inhibiting adenosine triphosphate (ATP) biosynthesis during
cell apoptosis. The fluorescent dye, Rh123, is used to measure Δψm.
Treatment with 100 μM GCDC for 24 h significantly decreased the Δψm
in the L-02 cells when compared with the control cells, whereas the
reduction in Δψm was significantly inhibited following
pre-treatment with various concentrations of Q7R (50, 100 and 200
μM) in a concentration-dependent manner (Fig. 7).
Analysis of the concentration of
intracellular Ca2+ by laser scanning confocal
microscopy
It is well known that the increase in the
intracellular free Ca2+ concentration is associated with
the initiation of apoptosis. The ability of GCDC (100 μM) to induce
intracellular Ca2+ accumulation in the L-02 cells was
investigated in this experiment. To reveal the alterations in the
intracellular Ca2+ concentration, living cells were
observed under a confocal microscope using Fluo-3 AM as an
indicator. Fig. 8 illustrates
that the addition of GCDC (100 μM) to the cell medium induced a
significant increase in the intracellular Ca2+
concentration, whereas the addition of various concentrations of
Q7R (50, 100 and 200 μM) to the cell culture inhibited this
increase in a concentration-dependent manner.
Discussion
Hepatocyte damage by toxic bile acids is considered
to represent a major event in the progression of cholestatic liver
disease (22). Toxic bile acids,
particularly GCDC, which is the predominant dihydroxy bile acid in
cholestatic patients and has been held responsible for
cholestasis-associated liver injury, induce hepatocellular
apoptosis, thereby providing a cellular mechanism for bile
acid-mediated liver injury and fibrogenesis (23). A number of studies have attempted
to elucidate the intracellular mechanisms behind the GCDC-induced
apoptotic form of hepatocyte death (8,24).
GCDC has been reported to induce apoptosis by activating the death
receptor or extrinsic pathway, through the mitochondrial or
intrinsic pathway and by causing endoplasmic reticulum stress
(8,25). In this study, hepatocyte apoptosis
induced by GCDC (100 μM) was used to represent a model for human
cholestasis (26). The results
revealed that GCDC (100 μM) induced higher degrees of cell injury
and apoptosis in the L-02 cells, while Q7R reversed the
GCDC-induced reduction in cell viability. Furthermore, using
Hoechst 33258 and Annexin V-FITC/PI staining assays, to our
knowledge, this study showed for the first time that apoptosis
induced by GCDC (100 μM) was attenuated following treatment with
various effective concentrations of Q7R (Figs. 2 and 4). Thus, the mechanisms involved in this
anti-apoptotic effect were investigated.
Oxidative stress has been implicated in the
pathogenesis of hepatic injury during cholestasis in rats and
humans and can initiate hepatocyte apoptosis by inducing DNA damage
and upregulating the expression of transmembrane proteins, such as
FasL and Fas (4,8,27).
Hydrophobic bile acids are believed to cause hepatocellular
necrosis and apoptosis partly through the induction of MPT and the
generation of ROS in hepatic mitochondria and hepatocytes, leading
to the depletion of antioxidant defenses (13,28,29). Intracellular ROS generation by
mitochondria appears to be a primary factor or a mandatory signal
in GCDC-induced hepatocyte apoptosis (30). The results from the present study
demonstrated that Q7R effectively protected the L-02 cells against
oxidative damage caused by GCDC, as indicated by the suppression of
ROS formation. These data strongly suggest that Q7R plays an
important protective role against ROS overproduction induced by
GCDC in hepatocytes.
GSH is a tripeptide (γ-glutamylcysteinyl-glycine)
which directly plays a major antioxidant role in scavenging ROS
through the enzymes, GSH peroxidase and GSSG reductase, and
maintaining the intracellular redox state (31). GSH has been implicated in the
protection against the induction of apoptotic and necrotic cell
death in a variety of cell types (32,33). The results from the present study
indicated that Q7R markedly increased GSH levels in the presence of
GCDC. It is speculated that reduced hepatocyte damage with Q7R is
associated with diminished oxidative stress and reversed levels of
GSH.
Mitochondrial dysfunction can occur in
death-receptor-mediated apoptosis, particularly the so called ‘type
II cells’, such as hepatocytes (34). Mitochondrial membrane
perturbations are important features of bile acid-induced
apoptosis. Inhibitors of MPT suppress GCDC-induced rat hepatocyte
apoptosis (4). Hydrophobic bile
acids directly induce mitochondrial membrane potential
depolarization in hepatocytes (6). Our results revealed that 100 μM GCDC
induced damage to the mitochondrial membrane and decreased Δψm in
the L-02 cells, whereas Q7R attenuated mitochondrial membrane
perturbation.
Intracellular calcium is a universal messenger in
cells. It regulates physiological processes and is involved in
pathological processes, such as cell injury/death. Accumulating
evidence demonstrates the role of intracellular Ca2+
deregulation in the induction of apoptosis. Calcium toxicity is
considered to be an important factor contributing to bile
acid-induced cellular damage (35). An increase in the cytosolic free
calcium concentration induced by hydrophobic bile acids is
considered an important permissive factor that allows oxidave
stress to open the permeability pore (36). The cytosolic calcium-protein
kinase C (PKC) intracellular signaling pathway is an important
signaling pathway that regulates several key liver functions, while
abnormalities in this pathway have been implicated in liver cell
injury (37). The present study
demonstrated that the GCDC-induced increase in the intracellular
Ca2+ concentration was reduced following pre-treatment
with Q7R, suggesting that Q7R protects hepatocytes from apoptosis
by maintaining the intracellular free Ca2+
homeostasis.
On the basis of the results presented in this study,
to our knowledge, we demonstrate for the first time that Q7R exerts
protective effects against GCDC-induced apoptosis in L-02 cells at
least in part by decreasing intracellular ROS levels, ameliorating
GSH depletion, maintaining Δψm and intracellular free
Ca2+ homeostasis.
Acknowledgements
The authors gratefully acknowledge the financial
support provided by the Program for the Guangdong Province Science
and Technology Department (grant no. 2011B050300010).
References
|
1
|
Jones BA and Gores GJ: Physiology and
pathophysiology of apoptosis in epithelial cells of the liver,
pancreas, and intestine. Am J Physiol. 273:G1174–G1188.
1997.PubMed/NCBI
|
|
2
|
Oh SH, Yun KJ, Nan JX, Sohn DH and Lee BH:
Changes in expression and immunolocalization of protein associated
with toxic bile salts-induced apoptosis in rat hepatocytes. Arch
Toxicol. 77:110–115. 2003.PubMed/NCBI
|
|
3
|
Rodrigues CM and Steer CJ: Mitochondrial
membrane perturbations in cholestasis. J Hepatol. 32:135–141. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yerushalmi B, Dahl R, Devereaux MW,
Gumpricht E and Sokol RJ: Bile acid-induced rat hepatocyte
apoptosis is inhibited by antioxidants and blockers of the
mitochondrial permeability transition. Hepatology. 33:616–626.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guicciardi ME and Gores GJ: Apoptosis: a
mechanism of acute and chronic liver injury. Gut. 54:1024–1033.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Palmeira CM and Rolo AP:
Mitochondrially-mediated toxicity of bile acids. Toxicology.
203:1–15. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Galle PR, Theilmann L, Raedsch R, Otto G
and Stiehl A: Ursodeoxycholate reduces hepatotoxicity of bile salts
in primary human hepatocytes. Hepatology. 12:486–491. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Perez MJ and Briz O: Bile-acid-induced
cell injury and protection. World J Gastroenterol. 15:1677–1689.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Patel T, Bronk SF and Gores GJ: Increases
of intracellular magnesium promote glycodeoxycholate-induced
apoptosis in rat hepatocytes. J Clin Invest. 94:2183–2192. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kwo P, Patel T, Bronk SF and Gores GJ:
Nuclear serine protease activity contributes to bile acid-induced
apoptosis in hepatocytes. Am J Physiol. 268:G613–G621.
1995.PubMed/NCBI
|
|
11
|
Billington D, Evans CE, Godfrey PP and
Coleman R: Effects of bile salts on the plasma membranes of
isolated rat hepatocytes. Biochem J. 188:321–327. 1980.PubMed/NCBI
|
|
12
|
Sokol RJ, Straka MS, Dahl R, et al: Role
of oxidant stress in the permeability transition induced in rat
hepatic mitochondria by hydrophobic bile acids. Pediatr Res.
49:519–531. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maillette de Buy Wenniger L and Beuers U:
Bile salts and cholestasis. Dig Liver Dis. 42:409–418.
2010.PubMed/NCBI
|
|
14
|
Perez MJ, Macias RI and Marin JJ: Maternal
cholestasis induces placental oxidative stress and apoptosis.
Protective effect of ursodeoxycholic acid. Placenta. 27:34–41.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang N, Li P, Wang Y, et al:
Hepatoprotective effect of Hypericum japonicum extract and
its fractions. J Ethnopharmacol. 116:1–6. 2008.
|
|
16
|
Su J, Fu P, Shen Y, et al: Simultaneous
analysis of flavonoids from Hypericum japonicum Thunb. ex
Murray (Hypericaceae) by HPLC-DAD-ESI/MS. J Pharm Biomed Anal.
46:342–348. 2008.
|
|
17
|
Choi HJ, Kim JH, Lee CH, Ahn YJ, Song JH,
Baek SH and Kwon DH: Antiviral activity of quercetin 7-rhamnoside
against porcine epidemic diarrhea virus. Antiviral Res. 81:77–81.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Song JH, Shim JK and Choi HJ: Quercetin
7-rhamnoside reduces porcine epidemic diarrhea virus replication
via independent pathway of viral induced reactive oxygen species.
Virol J. 8:4602011. View Article : Google Scholar
|
|
19
|
Awaad AS, Maitland DJ and Soliman GA:
Hepatoprotective activity of Schouwia thebica webb. Bioorg
Med Chem Lett. 16:4624–4628. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rathee P, Rathee D, Rathee D and Rathee S:
In-vitro cytotoxic activity of β-Sitosterol triacontenate isolated
from Capparis decidua (Forsk.) Edgew. Asian Pac J Trop Med.
5:225–230. 2012.
|
|
21
|
Vermes I, Haanen C, Steffens-Nakken H and
Reutelingsperger C: A novel assay for apoptosis. Flow cytometric
detection of phosphatidylserine expression on early apoptotic cells
using fluorescein labelled Annexin V. J Immunol Methods. 184:39–51.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hofmann AF: Cholestatic liver disease:
pathophysiology and therapeutic options. Liver. 22:14–19. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rust C, Wild N, Bernt C, Vennegeerts T,
Wimmer R and Beuers U: Bile acid-induced apoptosis in hepatocytes
is caspase-6-dependent. J Biol Chem. 284:2908–2916. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee TY, Chen FY, Chang HH and Lin HC: The
effect of capillarisin on glycochenodeoxycholic acid-induced
apoptosis and heme oxygenase-1 in rat primary hepatocytes. Mol Cell
Biochem. 325:53–59. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang K, Brems JJ, Gamelli RL and Holterman
AX: Survivin signaling is regulated through nuclear factor-kappa B
pathway during glycochenodeoxycholate-induced hepatocyte apoptosis.
Biochim Biophys Acta. 1803:1368–1375. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Graf D, Kurz AK, Reinehr R, Fischer R,
Kircheis G and Häussinger D: Prevention of bile acid-induced
apoptosis by betaine in rat liver. Hepatology. 36:829–839. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kaplowitz N: Mechanisms of liver cell
injury. J Hepatol. 32(Suppl 1): S39–S47. 2000. View Article : Google Scholar
|
|
28
|
Kim JS, He L and Lemasters JJ:
Mitochondrial permeability transition: a common pathway to necrosis
and apoptosis. Biochem Biophys Res Commun. 304:463–470. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sokol RJ, Dahl R, Devereaux MW, Yerushalmi
B, Kobak GE and Gumpricht E: Human hepatic mitochondria generate
reactive oxygen species and undergo the permeability transition in
response to hydrophobic bile acids. J Pediatr Gastroenterol Nutr.
41:235–243. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sokol RJ, Winklhofer-Roob BM, Devereaux MW
and McKim JM Jr: Generation of hydroperoxides in isolated rat
hepatocytes and hepatic mitochondria exposed to hydrophobic bile
acids. Gastroenterology. 109:1249–1256. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Meister A: Glutathione metabolism and its
selective modification. J Biol Chem. 263:17205–17208.
1988.PubMed/NCBI
|
|
32
|
Fernandes RS and Cotter TG: Apoptosis and
necrosis: intracellular levels of glutathione influence mode of
cell death. Biochem Pharmacol. 48:675–681. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ben-Yoseph O, Boxer PA and Ross BD:
Assessment of the role of the glutathione and pentose phosphate
pathways in the protection of primary cerebrocortical cultures from
oxidative stress. J Neurochem. 66:2329–2337. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Scaffidi C, Fulda S, Srinivasan A, et al:
Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 17:1675–1687.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Venglovecz V, Rakonczay Z Jr, Ozsvári B,
et al: Effects of bile acids on pancreatic ductal bicarbonate
secretion in guinea pig. Gut. 57:1102–1112. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Anwer MS, Engelking LR, Nolan K, Sullivan
D, Zimniak P and Lester R: Hepatotoxic bile acids increase
cytosolic Ca2+ activity of isolated rat hepatocytes.
Hepatology. 8:887–891. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Berridge MJ: Inositol trisphosphate and
calcium signalling mechanisms. Biochim Biophys Acta. 1793:933–940.
2009. View Article : Google Scholar : PubMed/NCBI
|