Introduction
Cardiac hypertrophy, an increase in cardiomyocyte
size, has been recognized as an independent risk factor for future
cardiovascular morbidity and mortality (1). Angiotensin II (Ang II) has been
implicated in cardiomyocyte hypertrophy (2). The hypertrophic effects of Ang II
are mediated by several intracellular signaling pathways, including
the nuclear factor κ-light-chain-enhancer of activated B cells
(NF-κB) pathway (3). NF-κB has
been validated as a therapeutic target for the prevention of
cardiac hypertrophy and heart failure (4).
Tetramethylpyrazine (TMP, molecular structure shown
in Fig. 1C), a biologically
active ingredient isolated from the Chinese herb, Ligusticum
wallichii Franchat, has been widely used for the treatment of
ischemic cardiovascular diseases (5–7).
Its pharmacological functions include anti-ischemic (7), anti-inflammatory (8), antioxidant (9) and anti-arrhythmic properties
(7). The anti-inflammatory
properties of TMP have been reported to involve the suppression of
pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α)
and interleukin-1β (8). These
cytokines have been implicated in the pathogenesis of cardiac
hypertrophy (10,11). However, the effects of TMP on
cardiac hypertrophy and the expression of TNF-α in cardiomyocytes
remain unclear.
In light of these observations, we hypothesized that
TMP inhibits Ang II-induced cardiomyocyte hypertrophy, and that the
modulation of the NF-κB pathway, if present, is responsible for the
anti-hypertrophic effects of TMP. Furthermore, in this study, we
determined whether TMP regulates the Ang II-stimulated secretion
and expression of TNF-α in neonatal rat cardiomyocytes.
Materials and methods
Reagents
Dulbecco’s modified Eagle’s medium (DMEM), fetal
calf serum and tissue culture reagents were purchased from
Invitrogen Corp. (Carlsbad, CA, USA). TMP was obtained from the
National Institute for the Control of Pharmaceutical and Biological
Products, Beijing, China. [3H]leucine was obtained from
the China Institute of Atomic Energy, Beijing, China. Pyrrolidine
dithiocarbamate (PDTC; an NF-κB inhibitor) was obtained from
Sigma-Aldrich (St. Louis, MO, USA). All other chemicals were
obtained from Sigma-Aldrich, unless otherwise stated.
Cell culture of neonatal rat
cardiomyocytes
This study was carried out according to the National
Institutes of Health Guide for the Care and Use of Laboratory
Animals (NIH Publication no. 85–23, revised 1996). Approval was
provided by the Institutional Animal Care and Use Committee at the
Hubei University of Science and Technology, Xianning, China.
Primary cultures of neonatal rat cardiomyocytes were prepared
according to previously published protocols (12) with minor modifications. Briefly,
ventricles were excised from 1- to 3-day-old neonatal
Sprague-Dawley rats, which were decapitated to isolate the hearts.
Subsequently, the ventricular tissues were minced into ~1
mm3 sections and digested using 0.1% trypsin
(Sigma-Aldrich) and 0.1% collagenase type II (Sigma-Aldrich) in
D-Hanks solution and agitated for 7 min at 37°C to dissociate the
cardiomyocytes. The remaining tissues were transferred into a fresh
enzyme solution and allowed to dissociate for 7 min. This digestion
was repeated 5 times. Cell suspensions were collected, centrifuged
(5 min, 60 × g) and resuspended in DMEM supplemented with 5% fetal
bovine serum. Dissociated cells were pre-plated for 1 h at 37°C to
selectively remove non-myocytes. Non-adherent cardiomyocytes
(>90% purity) were plated on 6-well culture plates at a density
of 2×105 cells/ml DMEM containing 10% fetal bovine
serum, 100 U/ml penicillin and 100 μg/ml streptomycin. Following
incubation for 48 h at 37°C 5% CO2, the medium was
replaced by fetal bovine serum-free medium and the cells were
starved for 10 h prior to the addition of drugs. The cardiomyocytes
were subsequently incubated for a further 24 h with the vehicle
(control) or with 0.1 μM Ang II in the presence or absence of TMP
or NF-κB inhibitor at the indicated concentrations.
Protein synthesis
Protein synthesis was determined by measuring the
incorporation of [3H]leucine incorporation into
acid-insoluble protein, which is commonly used as evidence of
hypertrophy (13). The cells were
treated for 24 h in the presence or absence of 0.1 μM Ang II and
TMP (0.001–1.0 mM). [3H]leucine (1 μCi/ml) was added to
each culture dish during treatment with the drugs. Subsequently,
the cells were washed with PBS and treated with ice-cold 5%
trichloroacetic acid for 1 h to precipitate the protein. The
precipitates were then dissolved in 0.1 M NaOH solution. The
incorporation rate of [3H]leucine was quantified by
liquid scintillation spectrometry. Each experiment was performed in
triplicate.
RNA isolation and real-time PCR
Total RNA was isolated from the cultured neonatal
cardiomyocytes using TRIzol reagent (Invitrogen Corp.) according to
the manufacturer’s instructions. Subsequently, ~2 μg of total RNA
was reverse transcribed with ReverTra Ace reverse Transcriptase
(Toyobo Co., Ltd., Osaka, Japan). The RT reaction product was
heated at 95°C to terminate the reaction. The expression of β-MHC,
TNF-α and GAPDH mRNA was examined by real-time PCR using a
SYBR-Green dye. The primers used were as follows: rat β-MHC sense,
5′-TAACCCGAGGCAAGCTCACA-3′ and antisense,
5′-CACAATCATGCCGTGCTGAC-3′ (product size, 120 bp); rat TNF-α sense,
5′-GCCAATGGCATGGATC TCAAAG-3′ and antisense, 5′-CAGAGCAATGACTCCA
AAGT-3′ (product size, 357 bp); rat GAPDH sense, 5′-CTCAT
GACCACAGTCCATGCCATC-3′ and antisense, 5′-CGGAA GGCCATGCCAGTGAG-3′
(product size, 182 bp). Real-time PCR was performed on an iCycler
iQ Real-Time Detection System (Bio-Rad Laboratories, Hercules, CA,
USA). Amplification involved 1 cycle at 94°C for 3 min for initial
denaturation followed by 40 cycles of denaturation for 30 sec at
94°C, annealing for 30 sec at 60°C and extension for 45 sec at
72°C. All reactions were performed in triplicate and GAPDH served
as an internal control. The results were quantified as Ct values,
where Ct is defined as the threshold cycle of PCR at which the
amplified product is first detected and the values are expressed as
the ratio of the target gene to the control. The size of the PCR
products was confirmed by electrophoresis on 2% agarose gels with
ethidium bromide staining.
Measurement of TNF-α in the culture
medium
At the end of the drug treatment, the culture
supernatants were harvested, and the level of TNF-α protein was
measured using commercial ELISA kits (R&D Systems, Minneapolis,
MN, USA) according to manufacturer’s instructions.
Western blot analysis
To elucidate the mechanisms through which TMP exerts
its effects on hypertrophic cardiomyocytes, we examined the protein
expression of NF-κB (p-NF-κB p65).
At the end of the drug treatment, total protein was
extracted from the cardiomyocytes using the Protein Extraction kit
(Beyotime Institute of Biotechnology, Beijing, China). Protein
concentration was measured using the BCA Protein assay kit
(Beyotime Institute of Biotechnology). Equal amounts of protein
extract were analyzed by 10% SDS-PAGE and electrotransferred onto
an immobilon-P transfer membrane (Millipore, Bedford, MA, USA). The
membrane was probed overnight at 4°C with primary antibodies
against NF-κB p65 and the phosphorylated form of NF-κB p65 (p-NF-κB
p65) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) Following
incubation with HRP-linked rabbit IgG antibody, the signal was
visualized using the ECL Plus system (Beyotime Institute of
Biotechnology), according to the manufacturer’s instructions.
Immunoblotting signals were quantified using Image J software
[National Institutes of Health (NIH), Bethesda, MD, USA].
ELISA-based NF-κB transcription factor
activity assay
The cardiomyocytes were treated with the same
methods mentioned above, and then nuclear protein was extracted
using the Nuclear Protein Extraction kit (Beyotime Institute of
Biotechnology) and quantified using the BCA Protein assay kit
mentioned above according to the manufacturer’s instructions.
Nuclear extracts were frozen in liquid N2 and stored at
−70°C until use.
ELISA-based NF-κB transcription factor activity
assay was performed using a Trans-AM NF-κB p65 transcription factor
assay kit (Active Motif, Carlsbad, CA, USA) according to the
manufacturer’s instructions. The level of nuclear NF-κB p65 was
expressed as the absorbance at 450 nm (A450).
Statistical analysis
All values are expressed as the means ± SEM.
Statistical analyses were performed using unpaired Student’s
t-tests and one-way ANOVA where appropriate. A P-value <0.05 was
considered to indicate a statistically significant difference.
Results
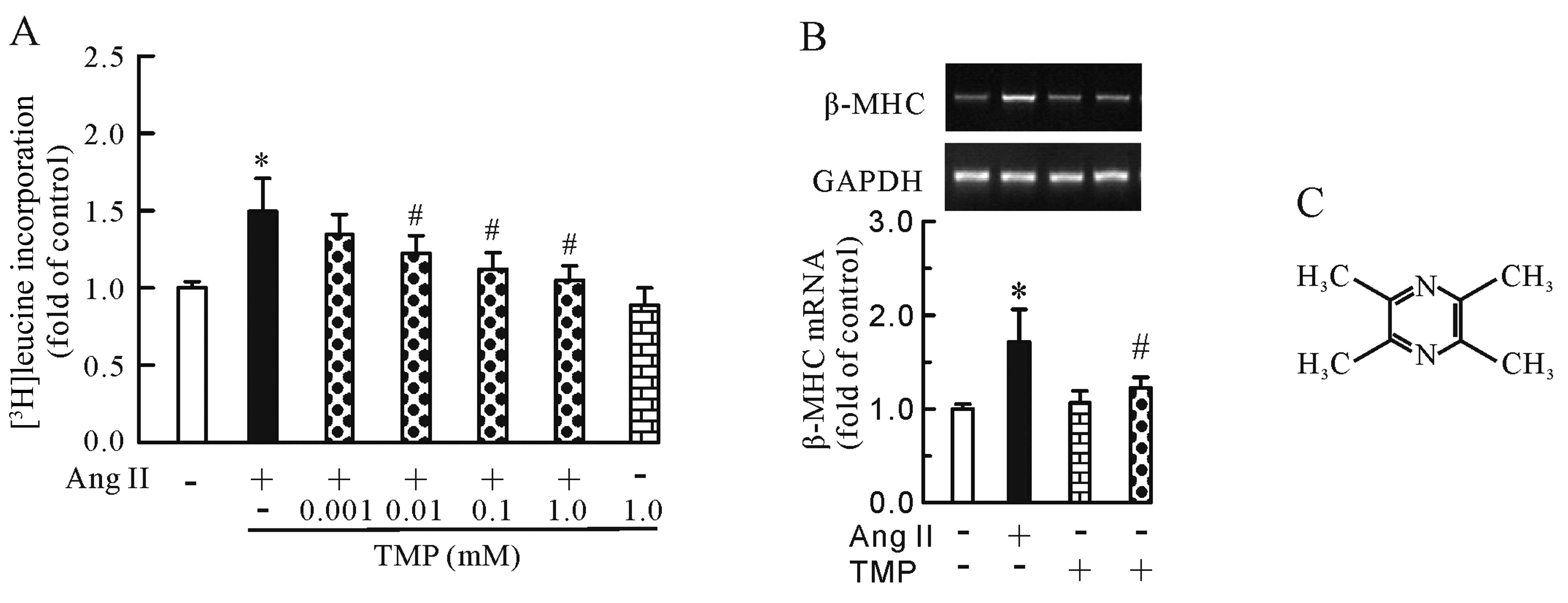
TMP attenuates the Ang II-induced
increase in protein synthesis and β-MHC mRNA expression
First, we measured the incorporation of
[3H]leucine and the mRNA expression of the hypertrophic
marker gene, β-MHC, in the cardiomyocytes to investigate the
anti-hypertrophic effects of TMP. As shown in Fig. 1, Ang II (0.1 μM) increased
[3H]leucine incorporation (P<0.05, Fig. 1A) and β-MHC mRNA expression
(P<0.05, Fig. 1B); however,
this increase was inhibited by TMP (0.001–1.0 mM) in a
dose-dependent manner. Of note, TMP (1.0 mM) alone had no effect on
[3H]leucine uptake or β-MHC mRNA expression.
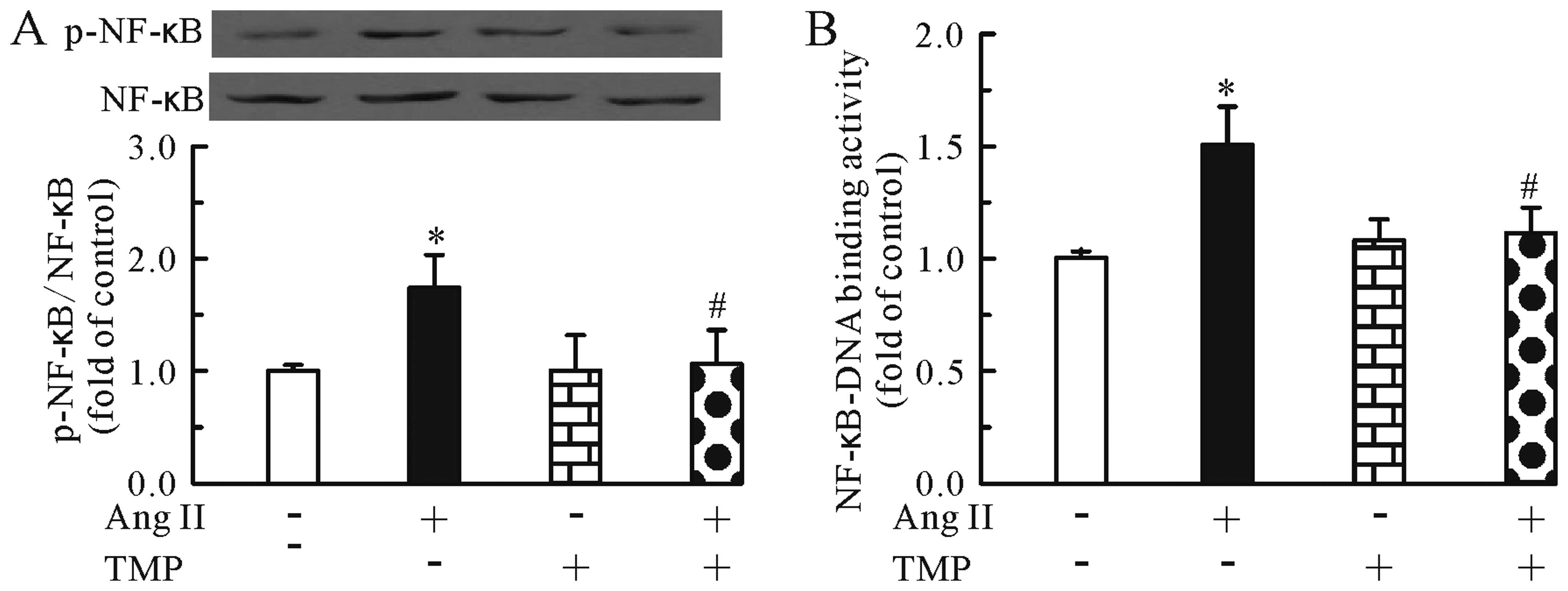
TMP prevents Ang II-induced NF-κB
activation and translocation
We then investigated whether TMP regulates the Ang
II-induced activation of the NF-κB pathway. As shown in Fig. 2, treatment with Ang II for 24 h
increased the protein levels of phosphorylated NF-κB p65 (Fig. 2A) and the NF-κB-DNA binding
activity (Fig. 2B) in the
cardiomyocytes. Conversely, TMP prevented these effects induced by
Ang II.
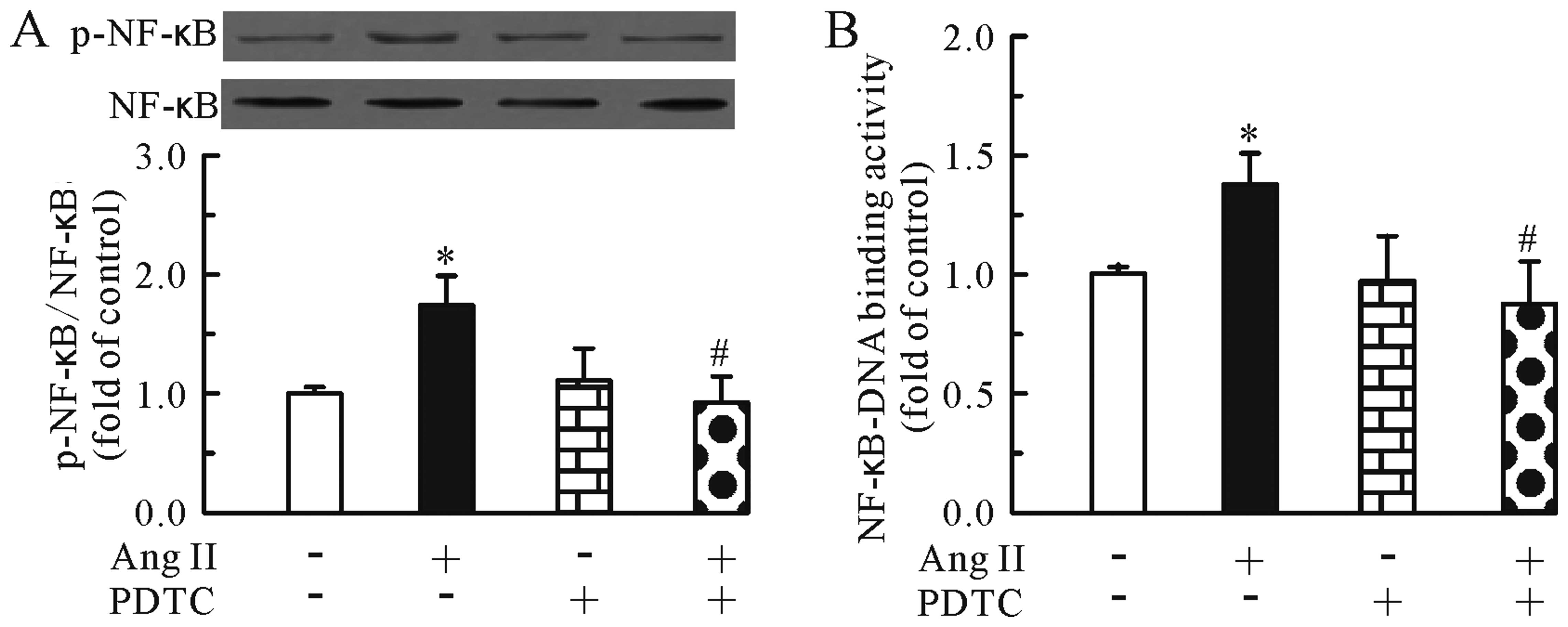
Anti-hypertrophic effects of TMP are
associated with inhibition of the NF-κB pathway
To investigate whether the modulation of NF-κB
pathway is responsible for the anti-hypertrophic effects of TMP,
the NF-κB inhibitor, PDTC (100 μM), was used in this study. The
cardiomyocytes were treated with Ang II (0.1 μM) for 24 h in the
presence or absence of PDTC (100 μM). First, we determined the
specificity of the inhibitor by western blot analysis. As shown in
Fig. 3, the NF-κB inhibitor
markedly inhibited the Ang II-induced upregulation of
phosphorylated NF-κB p65 (Fig.
3A) and the NF-κB-DNA binding activity (Fig. 3B) in the cardiomyocytes. In
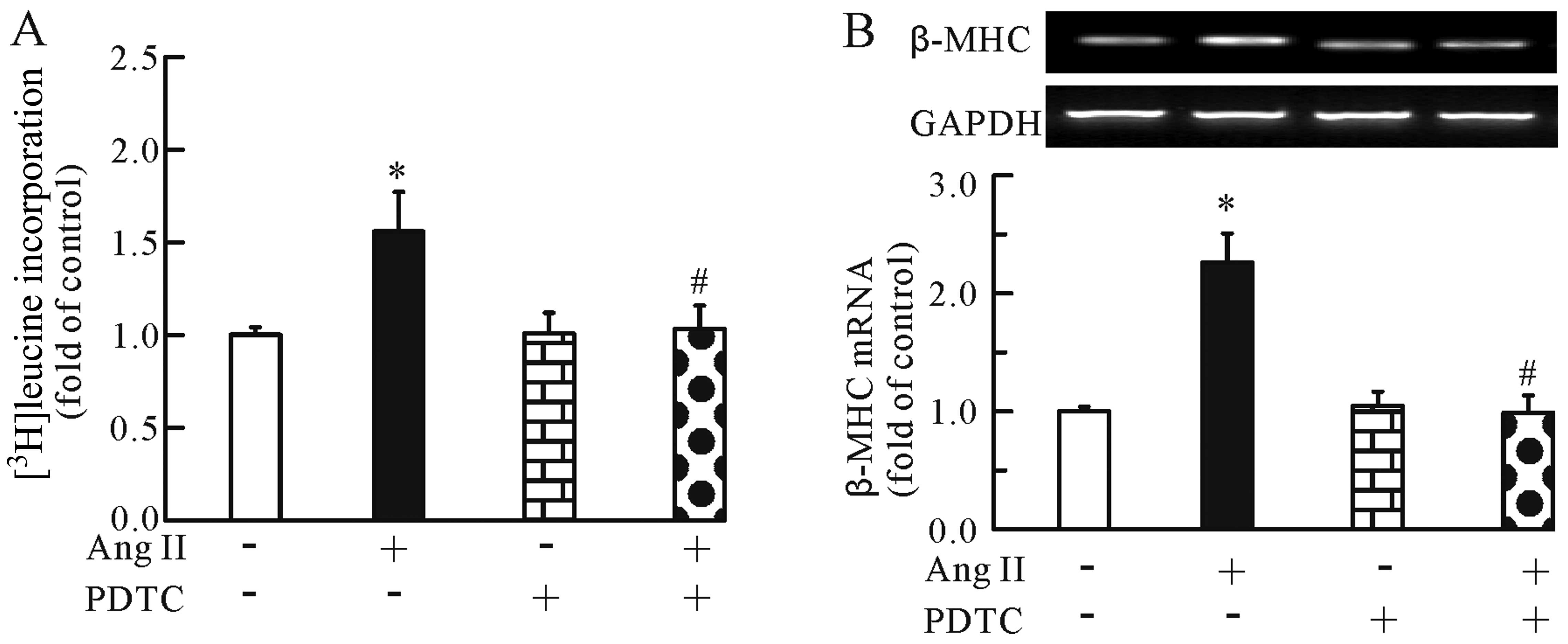
addition, the NF-κB inhibitor significantly inhibited the Ang
II-induced cardiomyocyte hypertrophy, as evidenced by the decrease
in [3H]leucine incorporation (Fig. 4A) and β-MHC mRNA expression
(Fig. 4B). Of note, PDTC alone
had no effect on [3H]leucine incorporation and β-MHC
mRNA expression. Thus, the modulation of the NF-κB pathway may be
one of the mechanisms involved in the anti-hypertrophic effects of
TMP.
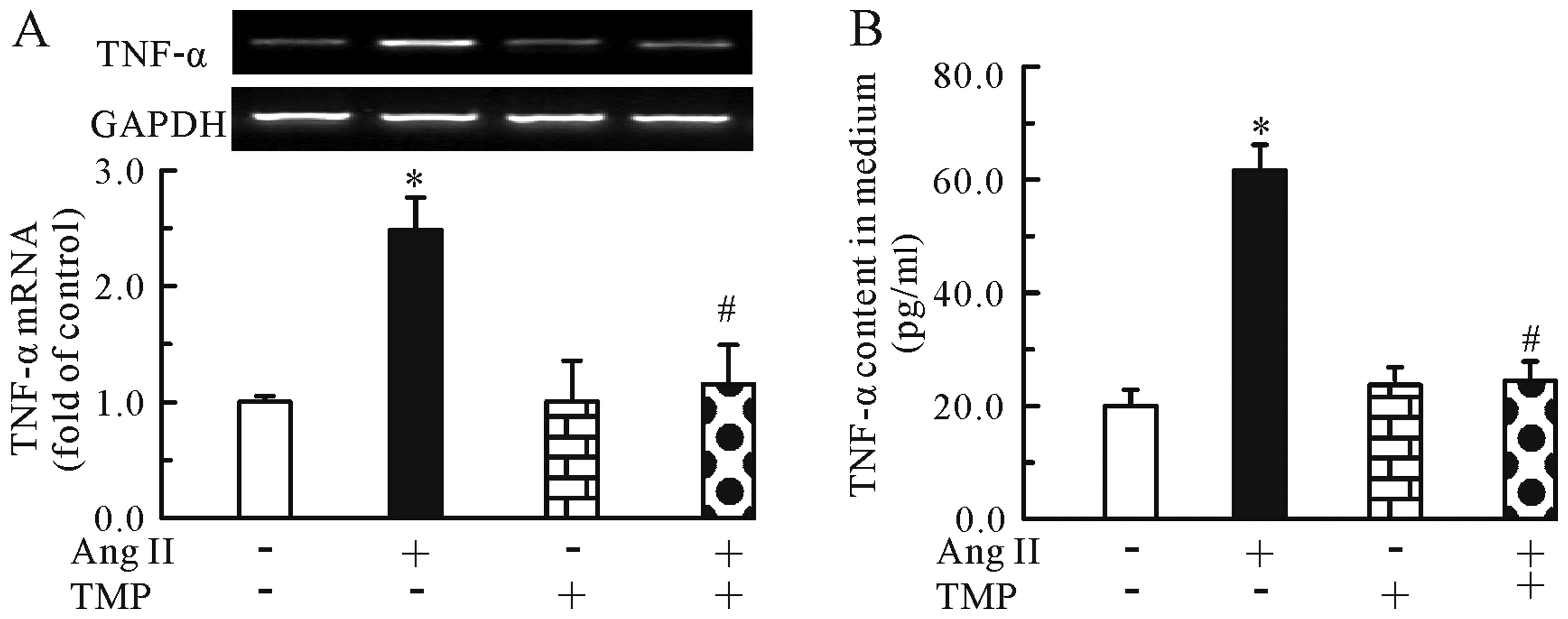
TMP inhibits the Ang II-induced
upregulation of TNF-α mRNA expression and protein secretion through
the suppression of the NF-κB pathway
Further experiments revealed the effects of TMP on
the mRNA expression and protein secretion of TNF-α. As shown in
Fig. 5, Ang II (0.1 μM) increased
the mRNA expression (Fig. 5A) and
protein secretion of TNF-α (Fig.
5B), whereas TMP (1.0 mM) markedly inhibited these effects
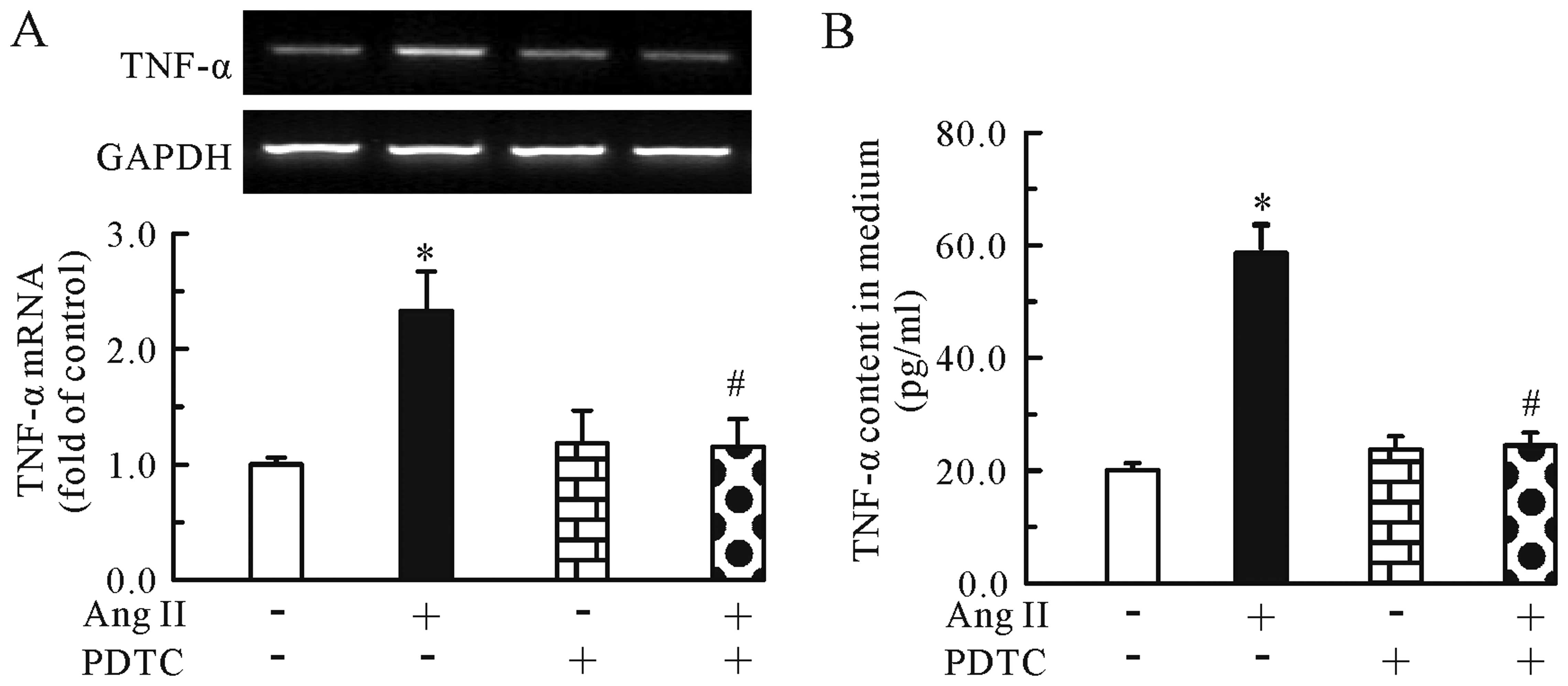
induced by Ang II. In addition, we found that compared to treatment
with Ang II (0.1 μM) alone, the combined treatment of Ang II and
PDTC (100 μM) significantly reduced the mRNA expression (Fig. 6A) and protein secretion (Fig. 6B) of TNF-α. Thus, TMP inhibited
the Ang II-induced upregulation of TNF-α mRNA expression and
protein secretion through the inhibition of the NF-κB pathway.
Discussion
The present study demonstrated that Ang II induced
hypertrophic growth in neonatal cardiomyocytes, as evidenced by the
increase in [3H]leucine incorporation and β-MHC mRNA
expression, which was significantly inhibited by treatment with
TMP. Ang II induced NF-κB activation in the cardiomyocytes, whereas
TMP decreased the levels of phosphorylated NF-κB. In addition, TMP
inhibited the Ang II-stimulated mRNA expression and protein
secretion of TNF-α in the cardiomyocytes, which was dependent on
NF-κB.
Previous in vivo and in vitro studies
support an important protective role of TMP (also known as
ligustrazine) in cardiac diseases. First, TMP exerts vasodilatory
effects by affecting the release of the vasoactive substances,
thromboxane A2 (TXA2) and prostacyclin
(PGI2), in isolated rat hearts, which may contribute to
its beneficial effects in myocardial hypoxia or ischemia (14). Second, TMP has been reported to
reduce ischemia-induced ventricular arrhythmias (7), and the possible ionic mechanism for
the anti-arrhythmic effect of TMP may involve its regulation of
cardiomyocyte ion channels, such as L-type Ca2+ channels
(15). Third, TMP has also been
suggested as a potent antioxidant with efficacy in lipid
peroxidation-induced heart toxicity (9). Furthermore, the protective role of
TMP in burn-induced myocardial injury has been suggested to be
associated with its inhibition of the release of TNF-α (16). Although a recent study
demonstrated that TMP exerts protective effects against dilated
cardiomyopathy in transgenic mice (17), the role of TMP in cardiac
hypertrophy and its possible mechanisms of action remain unknown.
In the present study, we first demonstrated that TMP inhibited Ang
II-induced cardiomyocyte hypertrophy in vitro and the
release of the pro-inflammatory cytokine, TNF-α, in
cardiomyocytes.
The underlying molecular mechanisms of cardiac
hypertrophy are extremely complex and involve intricate
interactions of multiple signaling pathways. Of these, the
involvement of the NF-κB pathway in the pathogenesis of cardiac
hypertrophy has been well established (4,18,19). Under pathological conditions, a
number of hypertrophic factors, such as Ang II (20) and isoproterenol (21) activate the NF-κB pathway in
cardiomyocytes. In unstimulated cells, the major form of NF-κB
complexes is an inactive heterodimer composed of the p50 and p65
subunits and is sequestered into the cytoplasm through its
interaction with specific inhibitory proteins, such as IκB
(22). Extracellular stimuli that
activate NF-κB induce the rapid phosphorylation of IκB and
subsequently, the dissociation of NF-κB from IκB. Once activated,
activated NF-κB translocates to the nucleus, where it acts as a
transcription factor by binding to regulatory DNA sequences,
triggering hypertrophic gene expression (23). The inhibition of NF-κB has been
demonstrated to exert anti-hypertrophic effects in cardiomyocytes
(4,24). In the present study, we therefore
hypothesized that the inhibition of the NF-κB pathway is the
molecular basis for the anti-hypertrophic effects of TMP. Our
results revealed that treatment with TMP markedly suppressed the
Ang II-induced secretion of phosphorylated NF-κB p65 in the
cardiomyocytes; the inhibition of NF-κB by the specific inhibitor,
PDTC, significantly inhibited Ang II-induced hypertrophy; thus, the
inhibition of NF-κB may be one of the mechanisms behind the
anti-hypertrophic effects of TMP. However, the mechanisms by which
TMP inhibits the NF-κB pathway remain unknown. Thus, further
studies are required to clarify this issue.
Although the direct pro-hypertrophic effects of
TNF-α have been well documented (10,25–27), there is also evidence to suggest
that TNF-α mediates the effects of other hypertrophic factors in an
autocrine or paracrine fashion. Previous studies have shown that
Ang II increases the expression of TNF-α in cardiomyocytes
(28). In the present study, we
also found that Ang II induced TNF-α secretion and expression in
neonatal cardiomyocytes. In TNF-α knockout mice, a previous study
demonstrated that TNF-α plays a role in mediating chronic Ang
II-induced effects on blood pressure and cardiac hypertrophy
(29). Taken together, these data
suggest that the autocrine release of TNF-α mediates the
hypertrophic effects of Ang II. In this study, treatment with TMP
attenuated the Ang II-induced secretion and expression of TNF-α,
which may also contribute to the anti-hypertrophic effects of
TMP.
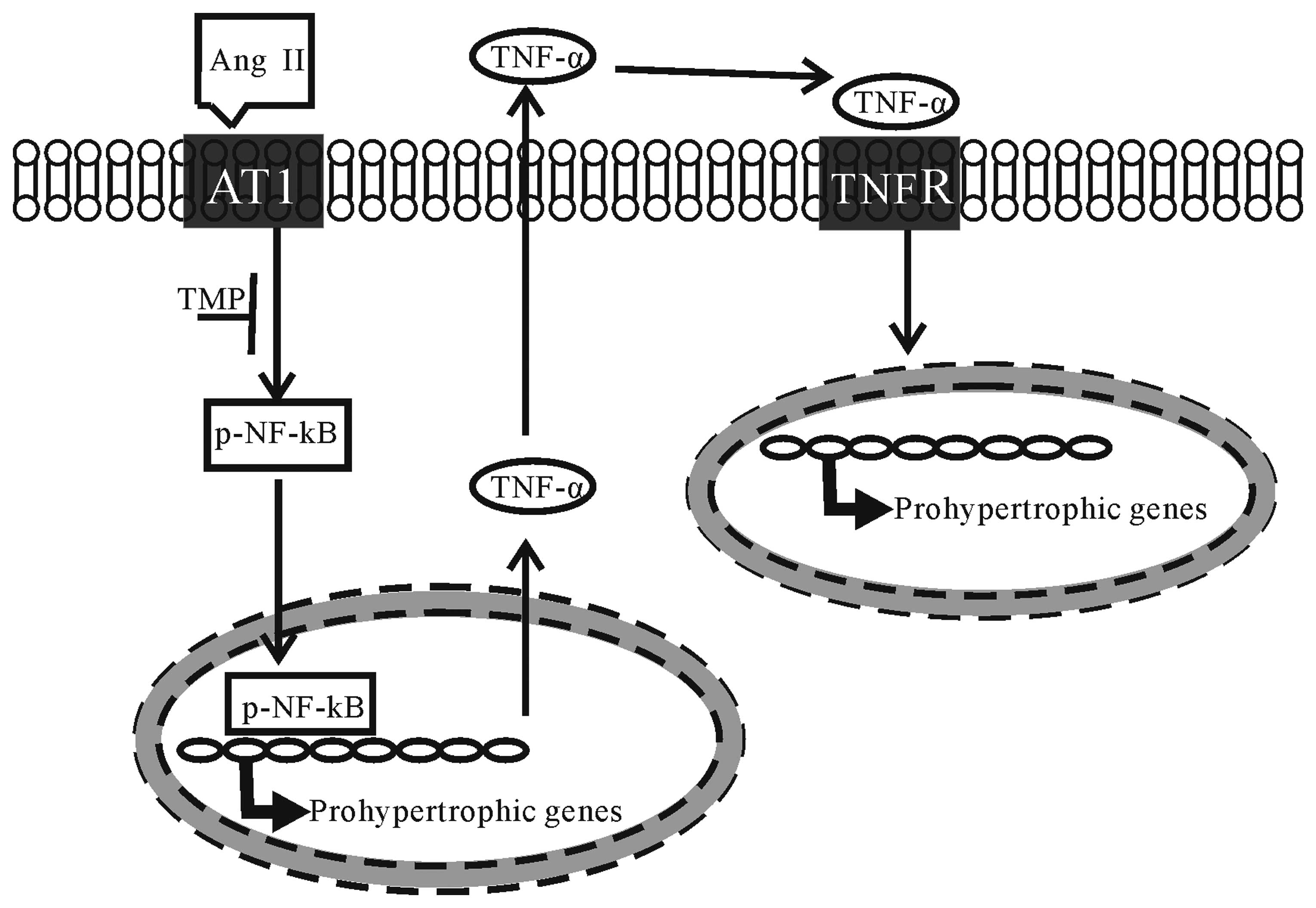
In conclusion, the present study investigated the
therapeutic effects of TMP on myocardial hypertrophy induced by Ang
II. To our knowledge, our results demonstrate for the first time
that TMP prevents Ang II-induced hypertrophy in neonatal
cardiomyocytes, which is attributed to its inhibition of the NF-κB
pathway and TNF-α secretion in cardiomyocytes (the proposed
mechanisms underlying the anti-hypertrophic effects of TMP are
illustrated in Fig. 7). These
findings raise the possibility of developing TMP as a therapeutic
drug for cardiac hypertrophy.
Acknowledgements
This study was supported by grants from the Science
and Technology Research Foundation of Hubei Provincial Educational
Department (no. B20122804) and the Science Fund of Hubei Science
and Technology University (nos. BK1104, KY0887 and ZX1201).
References
|
1
|
Levy D, Garrison RJ, Savage DD, Kannel WB
and Castelli WP: Prognostic implications of echocardiographically
determined left ventricular mass in the Framingham Heart Study. N
Engl J Med. 322:1561–1566. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sadoshima J and Izumo S: Molecular
characterization of angiotensin II - induced hypertrophy of cardiac
myocytes and hyperplasia of cardiac fibroblasts. Critical role of
the AT1 receptor subtype. Circ Res. 73:413–423. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee KH, Jang Y and Chung JH: Heat shock
protein 90 regulates IkappaB kinase complex and NF-kappaB
activation in angiotensin II-induced cardiac cell hypertrophy. Exp
Mol Med. 42:703–711. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gupta S, Young D, Maitra RK, et al:
Prevention of cardiac hypertrophy and heart failure by silencing of
NF-kappaB. J Mol Biol. 375:637–649. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen KJ and Chen K: Ischemic stroke
treated with Ligusticum chuanxiong. Chin Med J (Engl).
105:870–873. 1992.
|
|
6
|
Sutter MC and Wang YX: Recent
cardiovascular drugs from Chinese medicinal plants. Cardiovasc Res.
27:1891–1901. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Feng J, Wu G and Tang S: The effects of
tetramethylpyrazine on the incidence of arrhythmias and the release
of PGI2 and TXA2 in the ischemic rat heart. Planta Med. 65:268–270.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fan L, Wang K, Shi Z, Die J, Wang C and
Dang X: Tetramethylpyrazine protects spinal cord and reduces
inflammation in a rat model of spinal cord ischemia-reperfusion
injury. J Vasc Surg. 54:192–200. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu CF, Lin CH, Chen CF, Huang TC and Lin
SC: Antioxidative effects of tetramethylpyrazine on acute
ethanol-induced lipid peroxidation. Am J Chin Med. 33:981–988.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang GJ, Wang HX, Yao YS, Guo LY and Liu
P: The role of Ca2+/calmodulin-dependent protein kinase
II and calcineurin in TNF-alpha-induced myocardial hypertrophy.
Braz J Med Biol Res. 45:1045–1051. 2012.
|
|
11
|
Hu Y, Li T, Wang Y, et al: Tollip
attenuated the hypertrophic response of cardiomyocytes induced by
IL-1beta. Front Biosci. 14:2747–2756. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yokoyama T, Sekiguchi K, Tanaka T, et al:
Angiotensin II and mechanical stretch induce production of tumor
necrosis factor in cardiac fibroblasts. Am J Physiol.
276:H1968–H1976. 1999.PubMed/NCBI
|
|
13
|
Huang Y, Zhang H, Shao Z, et al:
Suppression of endothelin-1-induced cardiac myocyte hypertrophy by
PPAR agonists: role of diacylglycerol kinase zeta. Cardiovasc Res.
90:267–275. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Feng J, Liu R, Wu G and Tang S: Effects of
tetramethylpyrazine on the release of PGI2 and TXA2 in the hypoxic
isolated rat heart. Mol Cell Biochem. 167:153–158. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zou LY, Hao XM, Zhang GQ, Zhang M, Guo JH
and Liu TF: Effect of tetramethyl pyrazine on L-type calcium
channel in rat ventricular myocytes. Can J Physiol Pharmacol.
79:621–626. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gao S, Chen ZW, Zheng H and Chen XL:
Ligustrazine attenuates acute myocardium injury after thermal
trauma. Burns. 33:321–327. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao HP, Lu D, Zhang W, et al: Protective
action of tetramethylpyrazine phosphate against dilated
cardiomyopathy in cTnT(R141W) transgenic mice. Acta Pharmacol Sin.
31:281–288. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rajapurohitam V, Kilic A, Javadov S and
Karmazyn M: Role of NF-kappaB and p38 MAPK activation in mediating
angiotensin II and endothelin-1-induced stimulation in leptin
production and cardiomyocyte hypertrophy. Mol Cell Biochem.
366:287–297. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu Q, Chen Y, Auger-Messier M and
Molkentin JD: Interaction between NFkappaB and NFAT coordinates
cardiac hypertrophy and pathological remodeling. Circ Res.
110:1077–1086. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Valente AJ, Clark RA, Siddesha JM,
Siebenlist U and Chandrasekar B: CIKS (Act1 or TRAF3IP2) mediates
angiotensin-II-induced Interleukin-18 expression, and
Nox2-dependent cardiomyocyte hypertrophy. J Mol Cell Cardiol.
53:113–124. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Freund C, Schmidt-Ullrich R, Baurand A, et
al: Requirement of nuclear factor-kappaB in angiotensin II- and
isoproterenol-induced cardiac hypertrophy in vivo. Circulation.
111:2319–2325. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang D and Baldwin AS Jr: Activation of
nuclear factor-kappaB-dependent transcription by tumor necrosis
factor-alpha is mediated through phosphorylation of RelA/p65 on
serine 529. J Biol Chem. 273:29411–29416. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Baldwin AS Jr: The NF-kappa B and I kappa
B proteins: new discoveries and insights. Annu Rev Immunol.
14:649–683. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kawano S, Kubota T, Monden Y, et al:
Blockade of NF-kappaB ameliorates myocardial hypertrophy in
response to chronic infusion of angiotensin II. Cardiovasc Res.
67:689–698. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sun M, Chen M, Dawood F, et al: Tumor
necrosis factor-alpha mediates cardiac remodeling and ventricular
dysfunction after pressure overload state. Circulation.
115:1398–1407. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Janczewski AM, Kadokami T, Lemster B, Frye
CS, McTiernan CF and Feldman AM: Morphological and functional
changes in cardiac myocytes isolated from mice overexpressing
TNF-alpha. Am J Physiol Heart Circ Physiol. 284:H960–H969. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nakamura K, Fushimi K, Kouchi H, et al:
Inhibitory effects of antioxidants on neonatal rat cardiac myocyte
hypertrophy induced by tumor necrosis factor-alpha and angiotensin
II. Circulation. 98:794–799. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ock S, Ahn J, Lee SH, et al: Receptor
activator of nuclear factor-kappaB ligand is a novel inducer of
myocardial inflammation. Cardiovasc Res. 94:105–114. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sriramula S, Haque M, Majid DS and Francis
J: Involvement of tumor necrosis factor-alpha in angiotensin
II-mediated effects on salt appetite, hypertension, and cardiac
hypertrophy. Hypertension. 51:1345–1351. 2008. View Article : Google Scholar : PubMed/NCBI
|