Introduction
Non-alcoholic fatty liver disease (NAFLD) has gained
increasing attention worldwide due to its prevalence and its
association with insulin resistance and metabolic syndrome
(1,2). Hepatic steatosis is the basic
pathophysiological change occurring throughout the development of
NAFLD. Dietary effects on whole-body metabolism and its regulation
via the effects on lipid metabolic pathways are considered to be
crucial in the pathogenesis of hepatic steatosis (3,4).
The effect of high fructose intake on the pathogenesis of hepatic
steatosis due to an increase in daily fructose intake and its
harmful impact on hepatic lipid metabolism has gained much
attention (5–7). High-fructose feeding leads to
significant lipid accumulation in livers of rodents (7–9).
One important reason for this is that dietary fructose stimulates
endogenous de novo lipogenesis within the liver (10). However, the clear underlying
mechanisms by which fructose induces liver steatosis remain to be
clarified.
Previous studies demonstrated that endoplasmic
reticulum stress (ERS) and unfolded protein response (UPR), which
occurs following ERS, have a regulatory effect on lipid synthesis
in liver (11–13). On the other hand, ERS is
associated with the development of NAFLD. Specificallly, hepatic
ERS is accompanied by a fatty liver in genetically obese or chronic
high-fat-fed rodents (14,15).
An intervention study on ERS inhibitors, 4-phenylbutyric acid
(4-PBA) and tauroursodeoxycholic (TUDCA) found that resolved ERS is
able to ameliorate hepatic steatosis in ob/ob mice (15,16). In a short-term study, it was
suggested that ERS is involved in the development of lipid
accumulation in liver in mice fed a high fructose diet (17). Few studies have investigated the
role of ERS in hepatic steatosis induced by long-term high-fructose
feeding.
The aim of the present study was to clarify the role
of ERS in the development of fatty liver induced by long-term high
fructose intake. Fructose is a dietary factor that highly
stimulates lipogenesis, while ERS has a regulatory effect on
lipogenesis. We hypothesized that alleviation of ERS is able to
ameliorate hepatic steatosis in high-fructose-fed rats through
regulation of de novo lipogenesis. To prove this hypothesis,
4-phenylbutyric acid was used to inhibit the ERS induced by
high-fructose feeding in liver in Wistar rats. Lipid content, ERS
and lipogenic markers in liver were detected in order to
investigate the association between ERS and fructose-induced fatty
liver. Oxidative stress and hepatocyte apoptosis were also observed
to study the role of ERS in the development of NAFLD.
Materials and methods
Animals
Male Wistar rats supplied from the Experimental
Animal Center of Hebei Province (Shijiazhuang, China) were
conditioned in communal cages for 1 week at 22.0±0.5°C with a
12/12-h light/dark cycle (lights on 06.00 a.m.). Experimental
procedures were approved by the Animal Ethics Board of the Hebei
Research Institute for Endocrine and Metabolic Diseases and were in
accordance with China’s National Code of the Animal Care for
Scientific Experimentation. After an acclimatization period, the
rats (250–300 g) were divided into three groups. The control group
(Con) was fed a standard laboratory diet (18). The high-fructose (HFru) and
4-phenylbutyric acid (PBA) intervention (HFru-PBA) groups were fed
a high-fructose diet [35% calories from fructose, 35% calories from
starch, ~9% calories from fat and 21% calories from protein; based
on a recipe described in a study by Ren et al (17)]. PBA [dose: 0.35 g/kg.day, based on
a previous study (15)] was
administered to the HFru-PBA group by oral gavage subsequent to 4
weeks of high-fructose feeding. After 8-weeks, the rats were
sacrificed and liver tissues were collected.
Plasma glucose concentrations were determined using
a glucometer (Accu-Check Active, Roche Diagnostics GmbH, Mannheim,
Germany). Plasma insulin was measured using a radioimmunoassay kit
(Linco Research, St. Charles, MO, USA). The enzymatic activities of
ALT and AST were determined using a Biochemical Analyzer (Beckman
X20, Beckman Coulter, Brea, CA, USA). Plasma FFA concentration was
measured using an enzymatic colorimetric method (NEFA C test kit,
Jiancheng Biological Corporation, Jiangsu, China). Plasma
triglyceride concentrations and liver triglycerides (extracted from
homogenated liver tissues) were determined by a Peridochrom
Triglyceride GPO-PAP kit (Boehringer Mannheim, Mannheim,
Germany).
Histology staining
Small liver sections, fixed in 10% buffered
formalin, were processed for embedding in paraffin. Sections of 5–6
mm were cut for histopathological evaluation. Liver sections were
stained with hematoxylin and eosin (H&E staining) using a
standard protocol and then analyzed by light microscopy.
Hyperinsulinemic-euglycaemic clamp
The study was conducted between 09.00 and 12.00 a.m.
at week 8 in animals that had been fasted for 12 h. After being
anaesthetized with pentobarbitone (40 mg/kg, i.p.), the rat was
placed on a warm table to maintain rectal temperature at 37°C.
Catheters were inserted into the left femoral vein (for infusion of
glucose and insulin) and the femoral artery (for blood sampling).
After a basal period of 30 min, a hyperinsulinaemic-euglycaemic
clamp was performed, as previously described (19). In brief, human insulin (Actrapid;
Novo-Nordisk, Beijing, China) was infused at a constant rate of 4.1
mU/kg per min to achieve physiological hyperinsulinaemia (100–150
mU/l) for a period of 2 h. The blood glucose concentration was
clamped at the basal level by infusing glucose at variable rates.
Under these conditions, the glucose infusion rate (GIR) required to
maintain euglycaemia (usually calculated between 60 and 120 min)
reflects whole-body insulin sensitivity.
Western blot analysis
Liver samples were homogenized in ice-cold lysis
buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% Triton X-100, 10 mM NaP,
100 mM NaF, 2 mM Na orthovanadate, 1 mM EDTA, 1 mM EGTA, 10%
glycerol), supplemented with protease inhibitor cocktail tablets
(Roche) and DL-dithiothreitol, and solubilized for 30 min at 4°C.
Protein samples were then denatured in SDS sample buffer (125
mmol/l Tris-HCl, pH 6.8, 50% glycerol, 2% SDS, 5% β-mercaptoethanol
and 0.01% bromophenol blue). Equal amounts of tissue lysates were
resolved by SDS-PAGE and immunoblotted with antibodies against the
following proteins: ERS markers, phosphorylated pancreatic ER
kinase (p-PERK) (Thr980, Cell Signaling Technology, Danvers, MA,
USA), total- and phospho (Ser51)-eukaryotic translation initiation
factor 2α (eIF2α, Santa Cruz Biotechnology, Santa Cruz, CA, USA),
inositol-requiring kinase 1 (IRE1, Abcam, Cambridge, MA, USA),
phospho-IRE1 (Ser724, Abcam), X-box banding protein-1 (XBP1, Santa
Cruz Biotechnology), activation transcriptional factor 6 (ATF6,
Santa Cruz Biotechnology), C/EBP homologous protein (CHOP, Cell
Signaling Technology); upstream transcriptional factors of
lipogenesis: sterol regulatory element-binding protein-1c (SREBP1c,
Santa Cruz Biotechnology), carbohydrate responsive element binding
protein (ChREBP, Santa Cruz Biotechnology); downstream lipogenic
enzymes, acetyl-CoA carboxylase (ACC, Upstate, Lake Placid, NY,
USA), fatty acid synthase (FAS, Abcam) and stearoyl-CoA desaturase
1 (SCD1, Cell Signaling Technology). Antibodies against β-actin
(Santa Cruz Biotechnology) were blotted in each gel as the loading
control.
Determination of oxidative
parameters
Tissue homogenates were centrifuged for 15 min at
15,000 × g, and then the clear supernatants were removed for
analysis. Lipid peroxidation in the liver was measured by the
formation of malondialdehyde (MDA). The levels of MDA and the
activities of antioxidant enzymes including SOD, GSH-Px and CAT
were assayed using commercial assay kits according to the
manufacturer’s instructions. The MDA level was expressed as nmol/mg
protein. The activities of antioxidant enzymes were expressed as
U/mg protein.
Terminal deoxynucleotidyl
transferase-mediated dUTP nick end-labeling method (TUNEL)
assay
Paraffin sections (6-μm) were collected on
poly-L-lysine-coated glass slides, and the nuclear DNA
fragmentation of apoptotic cells was labeled in situ with
the ApopTag Peroxidase in situ Apoptosis Detection kit
(Intergen Co., Purchase, NY, USA). DNA fragmentation was determined
using a TUNEL assay as described by Boncompagni et al
(20). TUNEL-positive nuclei were
counted.
Statistical analyses
The results are presented as means ± SE. Differences
were considered significant when p<0.05 tested by one-way
analysis of variance (ANOVA). When significant variations were
found, the Tukey-Kramer multiple comparisons test was applied.
Results
Baseline characteristics and plasma
parameters in three groups of rats fed for 8 weeks
After 8 weeks of feeding, no difference was observed
in body mass for the three groups of rats. Visceral white adipose
tissue (WAT) was increased in the HFru group compared with the Con
group (p<0.05), while visceral WAT was significantly decreased
in the HFru-PBA group compared with the HFru group. Total
cholesterol (21), triglyceride
(TG) and free fatty acids in plasma were all increased in the HFru
group compared with the Con group, but improved in the HFru-PBA
group. No difference was observed in plasma ALT and AST for the
three groups. Basal plasma glucose and insulin concentrations were
both increased in the HFru group compared with the Con group, but
decreased in the HFru-PBA group compared with the HFru group
(Table I).
 | Table IBasal characteristics and plasma
parameters in three groups of rats at the end of the 8th week (mean
± SD, n=12). |
Table I
Basal characteristics and plasma
parameters in three groups of rats at the end of the 8th week (mean
± SD, n=12).
|
Characteristics | Con | HFru | HFru-PBA |
|---|
| Body mass (g) | 385±20 | 383±23 | 374±20 |
| Visceral WAT
(g) | 2.5±0.3 |
3.7±0.4b |
3.1±0.5a,d |
| TC (mmol/l) | 1.22±0.10 |
1.57±0.14b |
1.37±0.17a,d |
| TG (mmol/l) | 0.91±0.09 |
1.87±0.19b |
1.34±0.16b,d |
| FFAs (mmol/l | 0.53±0.15 |
1.09±0.19b |
0.69±0.11a,d |
| ALT (IUl/l) | 31±2 | 33±2 | 31±3 |
| AST (IUl/l) | 86±10 | 91±12 | 82±10 |
| FBG (mmol/l) | 4.53±0.20 |
5.73±0.39b |
5.07±0.29b,d |
| FINS (ng/ml) | 2.77±0.48 |
3.99±0.80b |
3.39±0.40c,d |
Systemic insulin resistance was induced
by high-fructose feeding but ameliorated by PBA intervention after
8 weeks of feeding
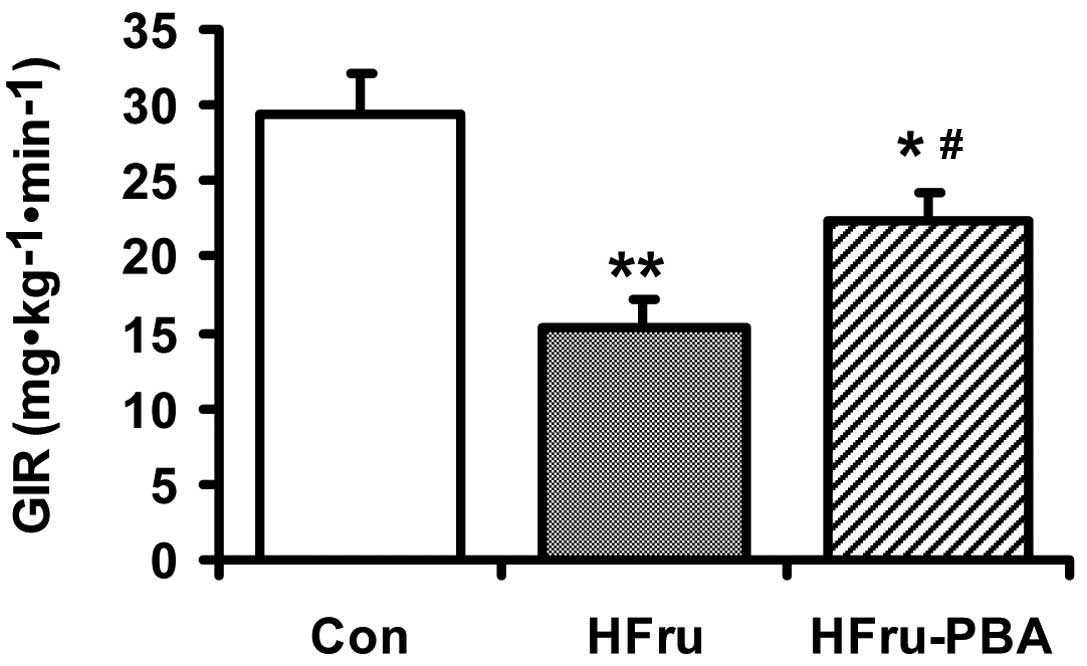
After 8 weeks of feeding, the GIR during the
hyperinsulinemic-euglycaemic clamp was reduced by 48% in the HFru
group compared with the Con group (p<0.01) (n=6 in each group).
In the HFru-PBA group, GIR was increased by 46% through PBA
intervention (p<0.01) (Fig.
1).
PBA intervention ameliorated
high-fructose feeding-induced hepatic steatosis after 8 weeks
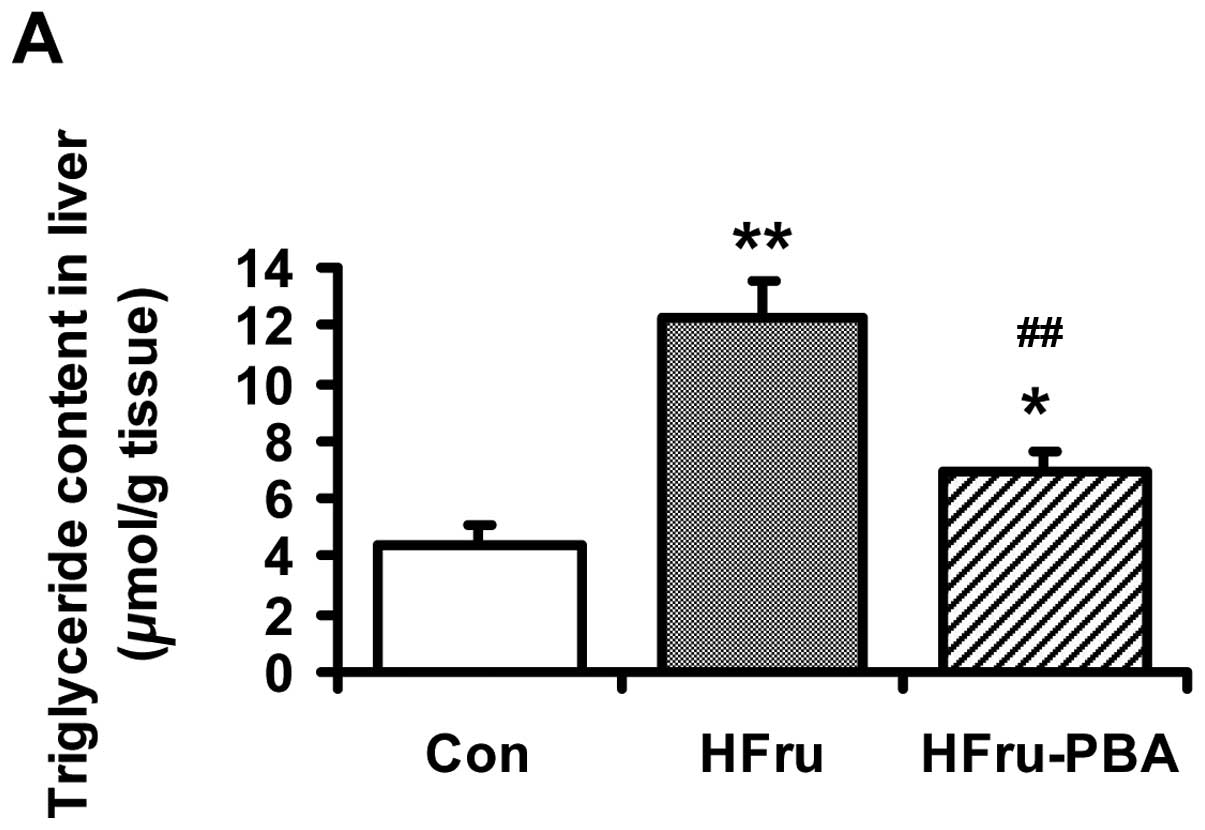
After rats were fed for 8 weeks, the liver
triglyceride content was increased by 1.8-fold (p<0.01) in the
HFru group compared with the Con group. PBA intervention decreased
liver triglyceride content by 43% (p<0.01) (Fig. 2A). H&E staining revealed
marked vacuolar degeneration in the liver in the HFru group at the
end of the 8th week, indicating fat accumulation in the liver.
Reduced liver lipid deposition was observed in the HFru-PBA group
(Fig. 2B).
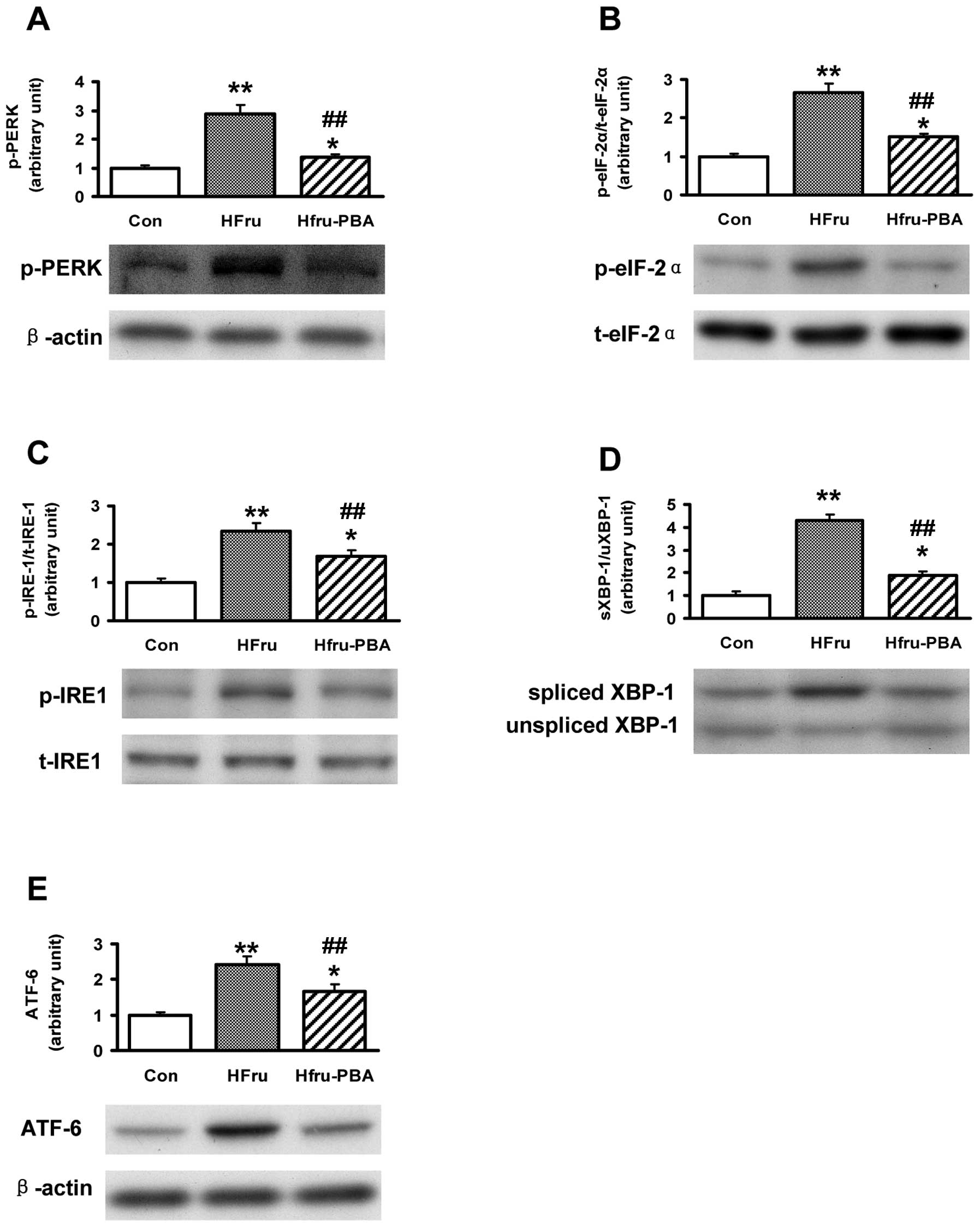
ERS markers were activated by
high-fructose feeding but normalized by PBA intervention after 8
weeks of feeding
After 8 weeks of feeding, the protein expression of
the activation forms of ERS markers, including phosphorylated PERK
(p-PERK), phosphorylated eIF2α (p-eIF2α), phosphorylated IRE-1
(p-IRE-1), spliced XBP1 (shown as the ratio of spliced XBP1 to
unspliced XBP1; spliced XBP1 is the activated form of XBP1) and
ATF6, were all upregulated in rat livers in the HFru group (all
p-values <0.01); while the protein expression of the above
markers in liver were significantly inhibited in the HFru-PBA group
(all p-values <0.01) (Fig.
3).
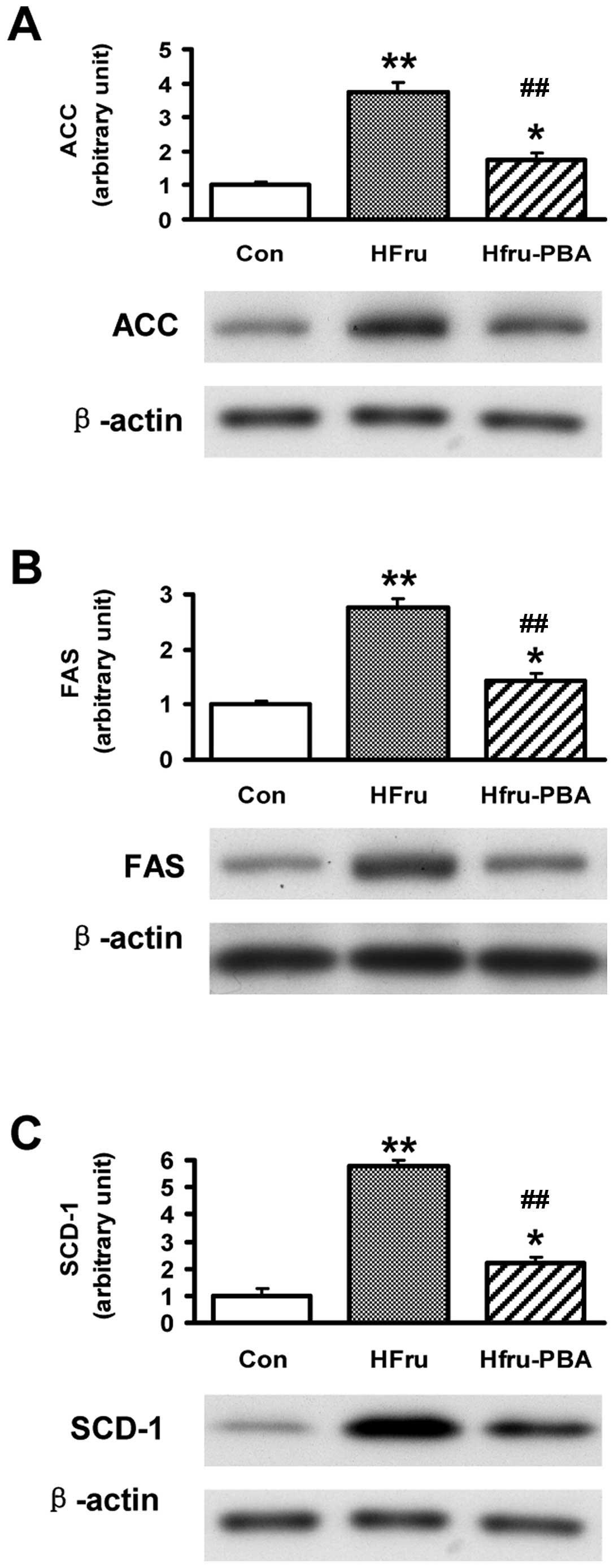
Protein expression of lipogenic enzymes
were stimulated by high-fructose feeding but were decreased by PBA
intervention after 8 weeks
Compared with the Con group, protein levels of the
key lipogenic enzymes including ACC, FAS and SCD1 were
significantly increased by 2.8, 5.7 and 3.8-fold, respectively, in
the liver tissues in rats after 8 weeks of high-fructose feeding
(all p-values <0.01). After PBA intervention, the proteins
levels of ACC, FAS and SCD1 were almost normalized in rat livers in
the HFru-PBA group (all p-values <0.01) (Fig. 4).
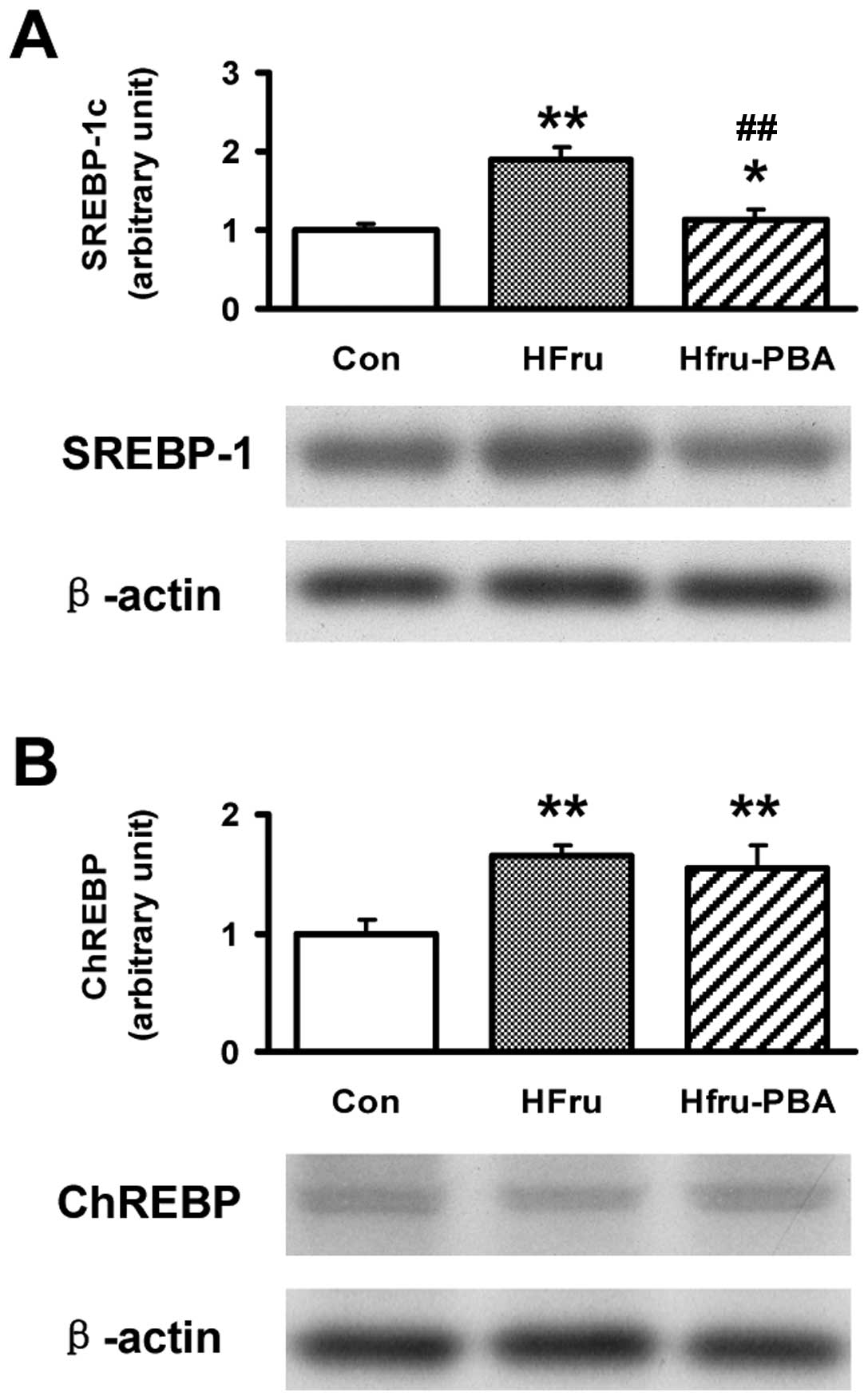
Upregulated protein expression of SREBP1c
induced by high-fructose feeding was decreased by PBA intervention
after 8 weeks
Protein contents of SREBP1c in rat livers in the
HFru group were upregulated by 1.9 and 4.3-fold, respectively, but
downregulated by 66 and 57%, respectively, with PBA intervention
(both p-values <0.01). Compared with the Con group, the protein
expression of ChREBP was increased by 66% in the HFru group
(p<0.01) but did not change with PBA intervention (Fig. 5).
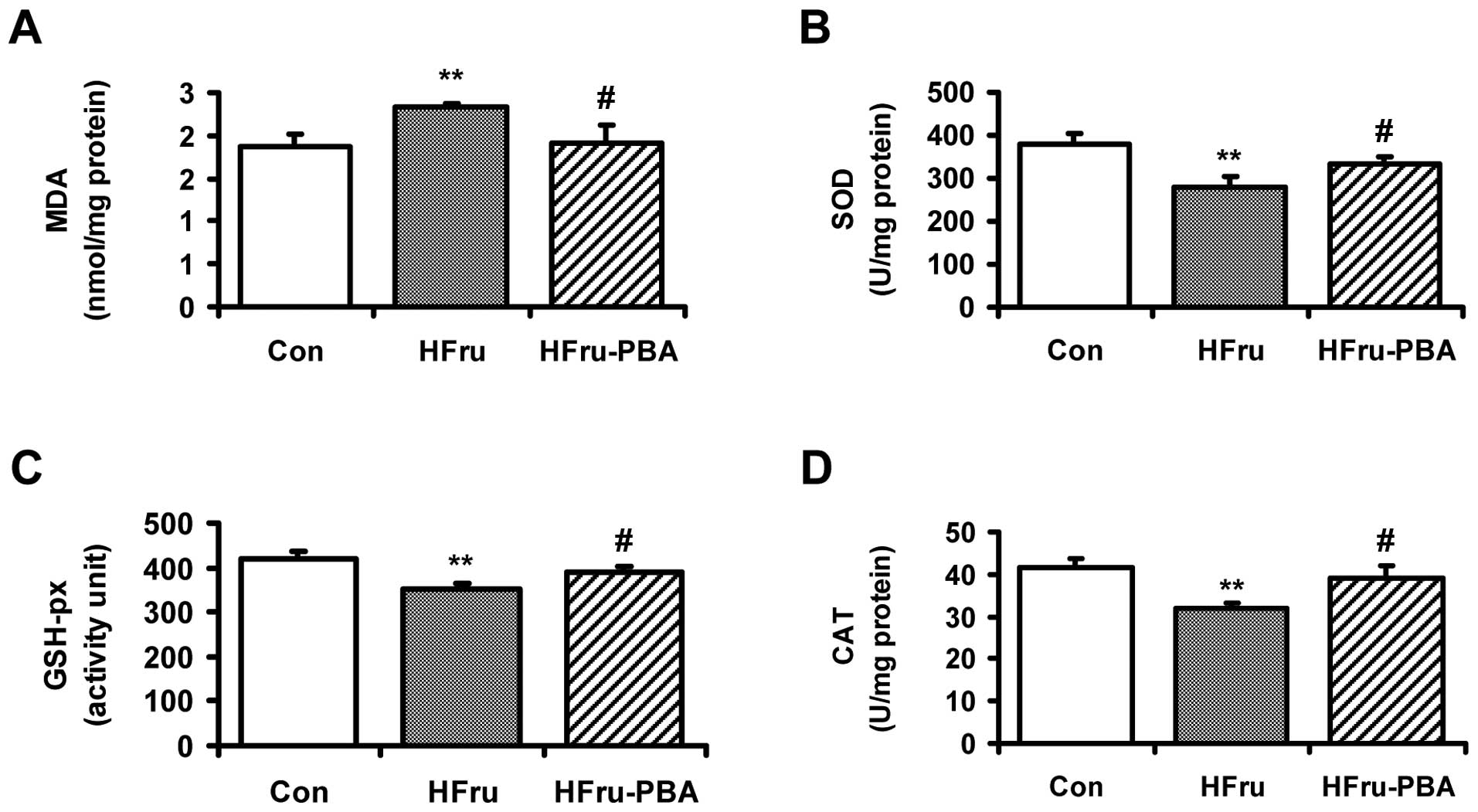
Oxidative stress in liver tissue in
high-fructose-fed rats was relieved by PBA intervention
The MDA level was significantly increased in the
HFru group compared with the Con group after 8 weeks of feeding. By
contrast, PBA intervention decreased the MDA level significantly in
the HFru-PBA group (both p-values <0.01) (Fig. 6A). Compared with the Con group,
the activities of SOD, GSH-px and CAT in in high-fructose-fed rat
livers were significantly decreased by 26.4, 16.4 and 23.1%,
respectively. In the HFru-PBA group, the activities of the
abovementioned parameters in livers were recovered by 18.2, 11.1
and 21.5%, respectively, following PBA intervention (all p-values
<0.05) (Fig. 6B–D).
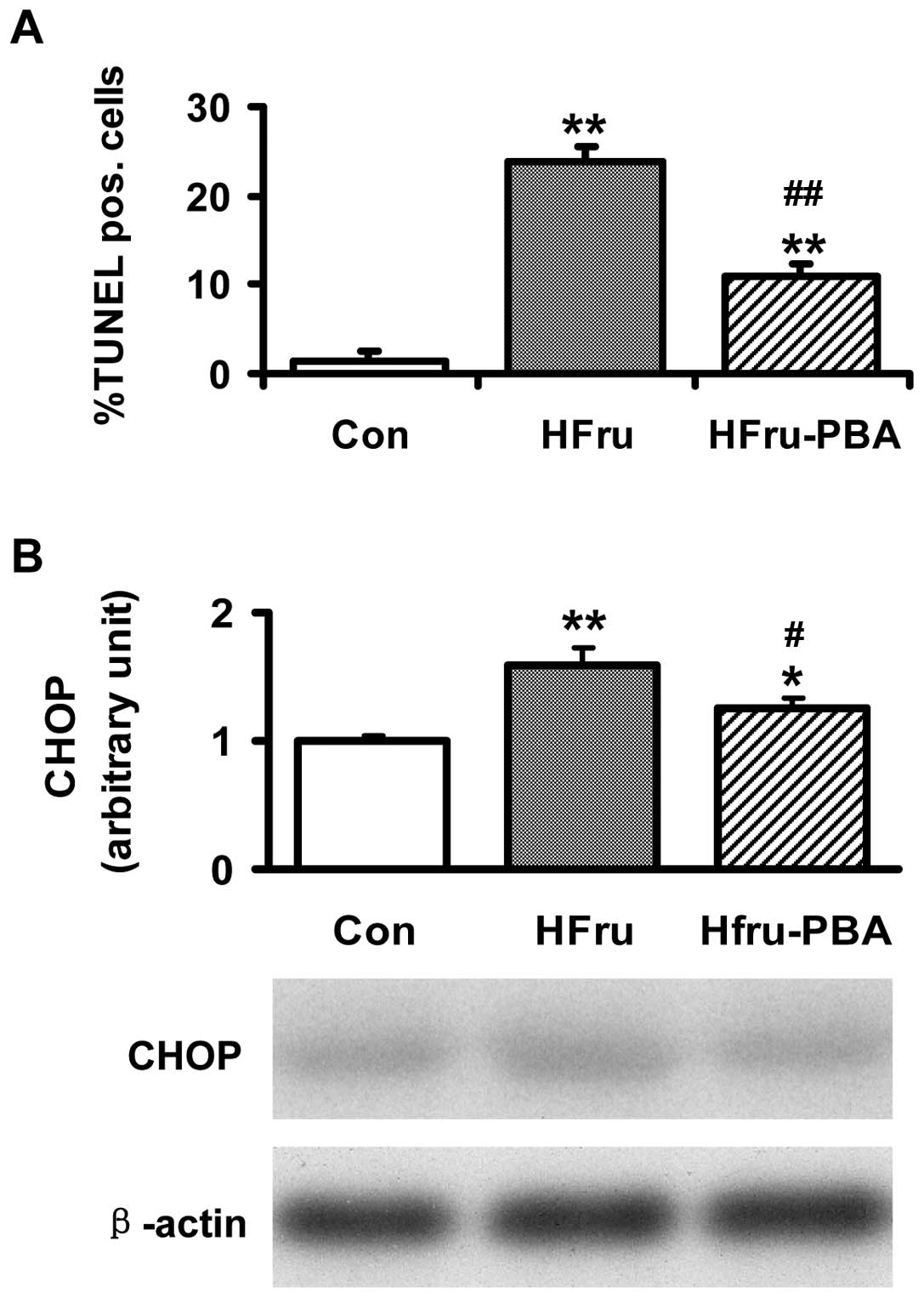
Changes in TUNEL assay and the protein
expression of CHOP indicate that apoptosis in hepatocytes was
increased in livers in high-fructose-fed rats while it was relieved
in livers in high-fructose-fed rats following PBA intervention
Compared with the Con group, the percentage of
TUNEL-positive cells in livers was significantly increased (1.4 vs.
23.8%, p<0.01) in the HFru group. In the HFru-PBA group, PBA
treatment decreased the percentage of TUNEL-positive cells in
livers to 11.0% (p<0.01) (Fig.
7A). Compared with the Con group, the protein contents of CHOP
in livers were increased by 59% (p<0.01) in the HFru group. PBA
treatment decreased the protein contents of CHOP by 21% in rat
livers in the HFru-PBA group (p<0.05) (Fig. 7B).
Discussion
Animal studies have shown that a high intake of
fructose leads to hepatic steatosis and whole-body insulin
resistance (8,9). In the present rat study, 8 weeks of
high-fructose feeding induced liver lipid accumulation, increased
glucose and insulin levels in plasma and decreased GIR during
hyperinsulinaemic-euglycaemic clamp study, which are consistent
with previous studies (9). The
mechanisms by which fructose induces hepatic steatosis remain to be
clarified. From previous studies (17), ERS is possibly involved in the
development of fatty liver induced by high-fructose feeding.
ERS involves the disruption of endoplasmic reticulum
homeostasis. Unfolded protein response (UPR) is the self-protective
mechanism in endoplasmic reticulum required to cope with ERS. Since
no direct marker of ERS is currently available, the transcription
factors of the three pathways of UPR are used as indirect ERS
markers, including the PERK-eIF2α, IRE-1-XBP1 and ATF6 pathways.
ERS has been shown to be involved in the development of NAFLD and
insulin resistance in genetically obese mice models or high-fat-fed
rodent models (14,15). Inhibition of ERS is able to
ameliorate hepatic steatosis in ob/ob mice (15,16). Additionally, ERS is induced in
mice fed on a short-term high-fructose diet in hepatic steatosis
(17). However, few studies have
been conducted on the role of ERS in hepatic steatosis induced by
long-term intake of fructose. Results of the present study show
that the hepatic lipid accumulation induced by long-term
high-fructose feeding was accompanied by ERS in liver in Wistar
rats, as reflected by the activation of 3′ UPR pathways. However,
resolved ERS by PBA intervention ameliorated hepatic steatosis,
indicating that ERS is involved in the pathogenesis of fatty liver
induced by high-fructose feeding. On the other hand, PBA
intervention improved the whole-body glucose metabolism and insulin
sensitivity as shown by the decreased glucose and insulin levels
and increased GIR in the hyperinsulinemic-euglycaemic clamp study.
The results confirmed that ERS is associated with insulin
resistance.
Fructose is known to be a highly lipogenic dietary
factor, and increased hepatic lipogenesis is an important mechanism
by which fructose induces hepatic steatosis (22). Studies (11–13) have shown that ERS and UPR have a
regulatory effect on the lipid synthesis in the liver. In obese
rodents, the ERS-induced dissociation of ERS chaperon GRP78 leads
to an upstream lipogenic transcriptional factor SREBP1c maturation
and result in hepatic steatosis, indicating ERS has a regulatory
effect on SREBP1c (12). On the
other hand, the ERS marker along the IRE-1-XBP1 pathway of UPR,
XBP1 has been established as a novel transcription factor governing
hepatic lipogenesis (13). Since
it has been proven that ERS and UPR have a regulatory effect on the
lipid synthesis in liver (11–13), we hypothesized that the
alleviation of ERS decrease lipid contents in liver by inhibiting
de novo lipogenesis. As shown in the results, the key
lipogenic enzymes including ACC, FAS and SCD1 were all upregulated
by high-fructose feeding, which reflects increased de novo
lipogenesis, while in the Fru-PBA group, the protein expression of
ACC, FAS and SCD1 was significantly decreased. This finding
confirms our hypothesis and suggests that resolved ERS by PBA is
capable of inhibiting de novo lipogenesis and thus
ameliorating hepatic steatosis. We then assessed the abovementioned
upstream transcriptional factors, including SREBP1c, ChREBP
[another well-known upstream transcriptional factor of de
novo lipogenesis in liver (23)] and spliced XBP1. The results
demonstrated that long-term fructose intake increased the protein
expression of SREBP1c, ChREBP and spliced XBP1, whereas PBA
intervention decreased the protein levels of SREBP1c and spliced
XBP1, but not ChREBP. Combined with the observation that ACC, FAS
and SCD1 in liver were downregulated following PBA intervention,
the results indicate that ERS has a regulatory effect on the
lipogenesis. Decreased expression of SREBP1c and XBP1 may
contribute to the decreased protein expression of key lipogenic
enzymes, resulting in improved hepatic steatosis. Of note, the
protein expression of ChREBP was not changed by PBA intervention.
However, the association between ChREBP and ERS remains to be
clarified. Our results show that ERS inhibition does not affect the
expression of ChREBP, suggesting that ERS does not have a
regulatory effect on ChREBP.
Another finding in our study is that ERS and
oxidative stress occur simultaneously in livers in
high-fructose-fed rats. Inhibition of ERS by PBA alleviated
oxidative stress in liver induced by high-fructose feeding, as
reflected by the increased activity of SOD, GSH-px and CAT
following PBA intervention compared with that in the high-fructose
group. These results suggest that ERS is connected to oxidative
stress in fatty liver in high-fructose-fed rats. Previous studies
have suggested that there is a complicated interaction between
endoplasmic reticulum and oxidative stress. Specifically, free
radicals, including reactive oxygen species (24), are one of the key messengers
between the two cell events (25). Evidence suggests that endoplasmic
reticulum is a potent source of ROS production. ROS produced in
endoplasmic reticulum under ERS has been estimated to account for
~25% of all ROS generated in cells. Sustained ERS can result in the
accumulation of reactive oxygen species and promotes oxidative
stress (26,27). In addition, C/EBP homologous
protein (CHOP) activation seems to enhance oxidative stress: as
previously shown, CHOP deletion reduces oxidative damage in mouse
models of diabetes (28). By
contrast, UPR in ERS has a protective effect against oxidative
stress response via the PERK and IRE-1-XBP1 pathways (29). The PERK pathway provides
protection from ROS via eIF2α. Mice lacking the ability to
phosphorylate eIF2α are characterized by a severe diabetic
phenotype that can be attenuated by a high antioxidant diet,
suggesting a role for eIF2α phosphorylation in the prevention of
oxidative stress (30). XBP1 may
also protect cells from oxidative damage. Mouse embryonic
fibroblasts deficient in XBP1 were more prone to cell death and
less able to activate antioxidant defenses following exposure to
hydrogen peroxide (31). The
results in the present study support that ERS and oxidative stress
are cross-linked in the development of fatty liver and that
inhibition of ERS can affect oxidative stress. However, the
interaction and the cause-effect relationship between the two
events remain unclear. Further investigations are needed to clarify
the relationship between the two stress responses in the
development of hepatic steatosis.
Besides relieving oxidative stress, apoptosis in
liver cells has been significantly decreased by PBA inhibition in
high-fructose-fed rats. Apoptosis is a cell event occurring in the
development of NAFLD and is associated with ERS and oxidative
stress (32–34). CHOP is best known as an important
mediator of ERS-induced cell death. Chronic ERS promotes apoptosis,
at least in part through the activation of CHOP (32). In addition to ERS, oxidative
stress is a key mechanism responsible for liver cell death and
liver damage (34). The
accumulation of ROS (24) also
contributes to the apoptosis of hepatocytes (35). Therefore, the decreased apoptotic
rate and decreased expression of CHOP in liver in the HFru-PBA
group may be the results of the improvement of ERS and oxidative
stress following PBA intervention.
Findings of this study have proven that long-term
high- fructose intake is capable of inducing ERS in liver with the
occurrence of hepatic steatosis. PBA intervention is able to
improve the lipid accumulation induced by high-fructose feeding
through inhibition of the upstream lipogenic transcriptional
factors SREBP1c and XBP1 and downregulation of the lipogenic key
enzymes. Besides ameliorating steatosis, oxidative stress and
apoptosis in liver were also alleviated when ERS was resolved by
PBA inhibition. The present study sheds new light on the role of
ERS in the development of fatty liver. ERS is therefore a potential
target for the prevention and treatment of NAFLD.
Acknowledgements
The authors thank Professor Jiming Ye (Health
Innovations Research Institute, RMIT University, Melbourne,
Victoria, Australia) for his advice on and interest in the study.
This study has been supported by fundings from the Chinese National
Science Foundation Grant (no. 81200639; allocation to L.-P.R.;
2012) and from the International Cooperation Foundation Grant in
Science and Technology Department of Hebei Province (no.
11396406-D; allocation to G.-Y.S.; 2011).
References
|
1
|
Malavolti M, Battistini NC, Miglioli L, et
al: Influence of lifestyle habits, nutritional status and insulin
resistance in NAFLD. Front Biosci (Elite Ed). 4:1015–1023. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cankurtaran M, Tayfur O, Yavuz BB, Geyik
S, Akhan O and Arslan S: Insulin resistance and metabolic syndrome
in patients with NAFLD but without diabetes: effect of a 6 month
regime intervention. Acta Gastroenterol Belg. 70:253–259.
2007.PubMed/NCBI
|
|
3
|
Moore JB: Non-alcoholic fatty liver
disease: the hepatic consequence of obesity and the metabolic
syndrome. Proc Nutr Soc. 69:211–220. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moore JB: Symposium 1: Overnutrition:
consequences and solutions Non-alcoholic fatty liver disease: the
hepatic consequence of obesity and the metabolic syndrome. Proc
Nutr Soc. 1–10. 2010.
|
|
5
|
Ouyang X, Cirillo P, Sautin Y, et al:
Fructose consumption as a risk factor for non-alcoholic fatty liver
disease. J Hepatol. 48:993–999. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nomura K and Yamanouchi T: The role of
fructose-enriched diets in mechanisms of nonalcoholic fatty liver
disease. J Nutr Biochem. 23:203–208. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yilmaz Y: Review article: fructose in
non-alcoholic fatty liver disease. Aliment Pharmacol Ther.
35:1135–1144. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kanuri G, Spruss A, Wagnerberger S,
Bischoff SC and Bergheim I: Role of tumor necrosis factor α (TNFα)
in the onset of fructose-induced nonalcoholic fatty liver disease
in mice. J Nutr Biochem. 22:527–534. 2011.
|
|
9
|
Hsieh PS: Inflammatory change of fatty
liver induced by intraportal low-dose lipopolysaccharide infusion
deteriorates pancreatic insulin secretion in fructose-induced
insulin-resistant rats. Liver Int. 28:1167–1175. 2008. View Article : Google Scholar
|
|
10
|
Aragno M, Tomasinelli CE, Vercellinatto I,
et al: SREBP-1c in nonalcoholic fatty liver disease induced by
Western-type high-fat diet plus fructose in rats. Free Radic Biol
Med. 47:1067–1074. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Oyadomari S, Harding HP, Zhang Y,
Oyadomari M and Ron D: Dephosphorylation of translation initiation
factor 2alpha enhances glucose tolerance and attenuates
hepatosteatosis in mice. Cell Metab. 7:520–532. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kammoun HL, Chabanon H, Hainault I, et al:
GRP78 expression inhibits insulin and ER stress-induced SREBP-1c
activation and reduces hepatic steatosis in mice. J Clin Invest.
119:1201–1215. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee AH, Scapa EF, Cohen DE and Glimcher
LH: Regulation of hepatic lipogenesis by the transcription factor
XBP1. Science. 320:1492–1496. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ozcan U, Cao Q, Yilmaz E, et al:
Endoplasmic reticulum stress links obesity, insulin action, and
type 2 diabetes. Science. 306:457–461. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ozcan U, Yilmaz E, Ozcan L, et al:
Chemical chaperones reduce ER stress and restore glucose
homeostasis in a mouse model of type 2 diabetes. Science.
313:1137–1140. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang JS, Kim JT, Jeon J, et al: Changes in
hepatic gene expression upon oral administration of
taurine-conjugated ursodeoxycholic acid in ob/ob mice. PLoS One.
5:e138582010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ren LP, Chan SM, Zeng XY, et al: Differing
endoplasmic reticulum stress response to excess lipogenesis versus
lipid oversupply in relation to hepatic steatosis and insulin
resistance. PLoS One. 7:e308162012. View Article : Google Scholar
|
|
18
|
Sewter C, Berger D, Considine RV, et al:
Human obesity and type 2 diabetes are associated with alterations
in SREBP1 isoform expression that are reproduced ex vivo by tumor
necrosis factor-alpha. Diabetes. 51:1035–1041. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ye JM, Doyle PJ, Iglesias MA, Watson DG,
Cooney GJ and Kraegen EW: Peroxisome proliferator-activated
receptor (PPAR)-alpha activation lowers muscle lipids and improves
insulin sensitivity in high fat-fed rats: comparison with
PPAR-gamma activation. Diabetes. 50:411–417. 2001. View Article : Google Scholar
|
|
20
|
Boncompagni E, Gini E, Ferrigno A, et al:
Decreased apoptosis in fatty livers submitted to subnormothermic
machine-perfusion respect to cold storage. Eur J Histochem.
55:e402011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu C, Chen Y, Cline GW, et al: Mechanism
by which fatty acids inhibit insulin activation of insulin receptor
substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase
activity in muscle. J Biol Chem. 277:50230–50236. 2002. View Article : Google Scholar
|
|
22
|
Samuel VT: Fructose induced lipogenesis:
from sugar to fat to insulin resistance. Trends Endocrinol Metab.
22:60–65. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Iizuka K, Bruick RK, Liang G, Horton JD
and Uyeda K: Deficiency of carbohydrate response element-binding
protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc
Natl Acad Sci USA. 101:7281–7286. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mosbah IB, Zaouali MA, Martel C, et al:
IGL-1 solution reduces endoplasmic reticulum stress and apoptosis
in rat liver transplantation. Cell Death Dis. 3:e2792012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Malhotra JD and Kaufman RJ: Endoplasmic
reticulum stress and oxidative stress: a vicious cycle or a
double-edged sword? Antioxid Redox Signal. 9:2277–2293. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shimizu Y and Hendershot LM: Oxidative
folding: cellular strategies for dealing with the resultant
equimolar production of reactive oxygen species. Antioxid Redox
Signal. 11:2317–2331. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cullinan SB and Diehl JA: Coordination of
ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway.
Int J Biochem Cell Biol. 38:317–332. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Song B, Scheuner D, Ron D, Pennathur S and
Kaufman RJ: Chop deletion reduces oxidative stress, improves beta
cell function, and promotes cell survival in multiple mouse models
of diabetes. J Clin Invest. 118:3378–3389. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gentile CL, Frye MA and Pagliassotti MJ:
Fatty acids and the endoplasmic reticulum in nonalcoholic fatty
liver disease. Biofactors. 37:8–16. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Back SH, Scheuner D, Han J, et al:
Translation attenuation through eIF2alpha phosphorylation prevents
oxidative stress and maintains the differentiated state in beta
cells. Cell Metab. 10:13–26. 2009. View Article : Google Scholar
|
|
31
|
Liu Y, Adachi M, Zhao S, et al: Preventing
oxidative stress: a new role for XBP1. Cell Death Differ.
16:847–857. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cao J, Dai DL, Yao L, et al: Saturated
fatty acid induction of endoplasmic reticulum stress and apoptosis
in human liver cells via the PERK/ATF4/CHOP signaling pathway. Mol
Cell Biochem. 364:115–129. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Alkhouri N, Carter-Kent C and Feldstein
AE: Apoptosis in nonalcoholic fatty liver disease: diagnostic and
therapeutic implications. Expert Rev Gastroenterol Hepatol.
5:201–212. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jin WP, Quan XQ, Meng FP, Cui XD and Piao
HJ: Relationship among hepatocyte apoptosis, P450 2E1 and oxidative
stress in alcoholic liver disease of rats. Zhongguo Wei Zhong Bing
Ji Jiu Yi Xue. 19:419–421. 2007.(In Chinese).
|
|
35
|
Haynes CM, Titus EA and Cooper AA:
Degradation of misfolded proteins prevents ER-derived oxidative
stress and cell death. Mol Cell. 15:767–776. 2004. View Article : Google Scholar : PubMed/NCBI
|