Introduction
Down syndrome (DS) is caused by the occurrence of
three copies of chromosome 21 and is associated with a number of
deleterious phenotypes, including cognitive impairment, childhood
leukemia and immune defects. These complicated and varied
phenotypes were generally thought to result from the interaction
between human chromosome 21 (Hsa21) genes with other disomic genes
(1). Recent bioinformatics
annotation has indicated that Hsa21 harbors >500 genes,
including 14 microRNA encoding genes (i.e., hsa-miR-99a, let-7c,
miR-125b, miR-155, miR-802, miR-3197, miR-3648, miR-3687, miR-4327,
miR-4759, miR-4760, miR-3118-5, miR-3156-3 and miR-548x) (2). Moreover, in previous studies, 5 of
14 Hsa21-derived miRNAs (i.e., hsa-miR-99a, let-7c, miR-125b-2,
miR-155 and miR-802) have been proven to be correlated with the
complex and variable phenotypes of DS and be overexpressed in the
heart, frontal cortex and hippocampus of DS fetuses (1,3–5).
However, the changes involved in the genome-wide miRNA expression
of DS fetuses under the influence of trisomy 21 have yet to be
determined. A miRNA can potentially regulate a large number of
protein-coding genes, while a target gene can also be regulated by
multiple miRNAs (6). Thus, the
molecular regulating mechanisms of the underlying progression of
complex and variable phenotypes of DS should be examined to
identify the genome-wide expression patterns of miRNAs.
MicroRNAs (miRNAs) are a class of endogenous, 18–25
nucleotides (nt) long, single-stranded RNAs that have emerged as
key post-transcriptional regulators of gene expression and play a
key role in various cell processes, such as cell proliferation,
differentiation, apoptosis, embryonic development and tissue
differentiation (2,7–9).
miRNAs are transcribed from intra- and inter-genetic regions of the
genome by RNA polymerase II (10). Following their processing, mature
miRNAs are assembled into ribonucleoprotein complexes known as
miRNA-induced silencing complexes (RISC). RISC subsequently
inhibits gene expression by perfect complementary binding, for mRNA
degradation, or imperfect binding at the 3′UTR region, to inhibit
translation (11). Its abnormal
expression has been proven to be involved in the occurrence and
development of diseases (11–14).
Hybridization-based methodologies, such as
microarray and PCR-based assays, have been used thus far to
identify and profile the association between the Hsa21-derived
miRNAs and the variable phenotypes of DS. Sethupathy et al
(5) first used quantitative PCR
(qPCR) to study the Hsa21-derived miR-155 in fibroblasts from
monozygotic twins discordant for trisomy 21 (one twin was
unaffected, whereas the other had trisomy 21) and confirmed miR-155
was overexpressed in the fibroblasts from the twin with DS. This
result may be relevant to the observed lower blood pressure in
individuals with DS. Subsequently, Kuhn et al (3) assessed the miRNA expression profiles
of DS hippocampus specimens using microarrays and verified that
five Hsa21-derived miRNAs were overexpressed in the DS brain.
Moreover, they confirmed that miR-155 and miR-802 are associated
with cognitive impairment in individuals with DS by regulating the
methyl-CpG-binding protein (MeCP2) gene (4,13).
Additionally, a number of studies indirectly
determined that Hsa21-derived miRNAs may partially contribute to
most of the observed DS phenotypes. For example, hsa-miR-125b and
miR-99a are associated with pediatric AML (non-DS) (15), 10 of 89 predicted miR-155 target
genes are involved in hematopoietic and myeloproliferative
disorders (14) and hsa-miR-99a,
let-7c and miR-125b may act as tumor suppressors that play an
important role in the reduced incidence of solid tumors in DS
individuals (16–18). Alternatively, previous studies
(1,15–18) mainly focused on Hsa21-derived
miRNA expression in tissues of human DS subjects, while few studies
have focused on determining the expression profiles of miRNAs
isolated from human blood samples. Thus, it would be clinically
relevant to investigate early gene regulatory mechanisms associated
with hemopoietic abnormalities and the immune defects in DS
patients. Furthermore, it is easier to obtain blood samples
compared with tissue samples.
In the present study, in order to investigate the
expression characteristic of miRNAs during the development of DS
fetus and to identify whether another miRNA gene is located on the
Hsa21 (in addition to the confirmed 14 Hsa21-derived miRNAs),
Illumina high-throughput sequencing technology was employed to
comprehensively characterize the miRNA expression profiles in the
cord blood of DS and control fetuses. Compared to previous
hybridization-based methods, high-throughput sequencing methods
have significant advantages. First, they can provide a more
integrated view of the miRNAs transcriptome and identify modest or
even low abundant miRNAs that previous methods are not able to
detect. Second, this method can successfully identify novel miRNAs
via the direct observation and validation of the folding potential
of the flanking genomic sequences (19). Using Illumina sequencing
technology, 395 known miRNAs and 181 novel candidate miRNAs were
identified in this study. Moreover, 149 known miRNAs were
significantly differentially expressed and two novel miRNAs were
located in the ‘DS critical region’ of chromosome 21
(chr21q22.2–22.3). The data obtained in this study provides
considerable insight into understanding the expression
characteristic of miRNAs in the DS fetal cord blood mononuclear
cells (CBMCs). Differentially expressed miRNAs may be associated
with the hemopoietic abnormalities and the immune defects of DS
fetuses and newborns.
Materials and methods
Clinical samples and ethics
statement
A total of six DS and six matched control fetal cord
blood samples (18–22 weeks of gestation) were obtained from the
Shenzhen People's Hospital, China. Three DS and three control cord
blood samples were combined to form pooled DS and control cord
blood samples, respectively, for small RNA library construction and
Illumina sequencing. The remaining three DS and three control
samples were used as the validation set to confirm the miRNA
differential expression patterns by qPCR. The cord blood samples
were obtained by puncture extraction with the help of a color
Doppler ultrasound as the prenatal women had been undergoing
prenatal diagnosis. The samples were then verified by protein
electrophoresis to exclude the contamination of maternal blood
cells. The characteristics of each case are provided in the
Table I. Written informed consent
for the biological studies was obtained from all the participants.
The study was approved by the Ethics Committee of the Shenzhen
People's Hospital.
 | Table IThe characteristics of each case. |
Table I
The characteristics of each case.
| Mother-ID | Age (years) | Gestational age
(weeks) | The result of
karyotype | Protein
electrophoresis |
|---|
|
|---|
| HBA (%) | HBF (%) |
|---|
| Patient 1a | 44 | 20 | 47,XX,+21 | 5.5 | 94.5 |
| Patient 2a | 36 | 20 | 47,XX,+21 | 6.7 | 93.3 |
| Patient 3a | 30 | 21 | 47,XY,+21 | 4.8 | 95.2 |
| Patient 4 | 34 | 20 | 47,XX,+21 | 6.2 | 93.8 |
| Patient 5 | 33 | 22 | 47,XY,+21 | 5.7 | 94.3 |
| Patient 6 | 32 | 19 | 47,XX,+21 | 6.4 | 93.6 |
| Control 1a | 37 | 21 | 46,XX | 4.8 | 95.2 |
| Control 2a | 36 | 20 | 46,XY | 6.4 | 93.6 |
| Control 3a | 30 | 20 | 46,XX | 5.8 | 94.2 |
| Control 4 | 34 | 18 | 46,XY | 6.8 | 93.2 |
| Control 5 | 33 | 22 | 46,XX | 6.6 | 93.4 |
| Control 6 | 32 | 21 | 46,XX | 5.2 | 94.8 |
CBMCs were separated by a Ficoll-Paque (Sigma, St.
Louis, MO, USA) density gradient centrifugation according to the
manufacturer's instructions. Briefly, 2 ml of blood (with EDTA as
an anticoagulant) was layered on 3 ml of Ficoll-Hypaque (Sigma) and
centrifuged for 25 min at 1,300 rpm at room temperature.
Mononuclear cells at the interface were aspirated with a Pasteur
pipette, washed twice in PBS by centrifugation for 10 min at 900
rpm at room temperature and dissolved in 1 ml of TRIzol®
reagent (Invitrogen, Carlsbad, CA, USA). The samples were then
stored at −80°C until further use.
Total RNA isolation and sequencing
Total RNA isolation was performed with CBMCs using
TRIzol reagent (Invitrogen) according to the manufacturer's
instructions. Small RNA library preparation and sequencing were
performed with Illumina sequencing technology (BGI-Shenzhen,
Shenzhen, China). Briefly, the small RNA (sRNA) population was
isolated by separating 10 μg of total RNA using denaturing
polyacrylamide gel electrophoresis (PAGE) and excising the portion
of the gel corresponding to the appropriate size (15–30 nt) based
on standard oligonucleotide markers. The sRNA was ligated with 3′
(5′-pUCGUAUGCCGUCUUCUGCUUGidT-3′) and 5′
(5′-GUUCAGAGUUCUACAGUCCGACGAUC-3′) adapters using T4 RNA ligase and
purified on a 15% Tris-Borate-EDTA (TBE) urea PAGE. The modified
sRNA was then reverse transcribed by Illumina′s small RNA RT-Primer
(5′-CAAGCAG AAGACGCATACGA-3′). The cDNA was used as the template
for PCR amplification (15 cycles) using Illumina′s small RNA primer
set (5′-CAAGCAGAAGACGGCATACGA-3′ and
5′-AATGATACGGCGACCACCGACAGGTTCAGAGTTCT ACAGTCCGA-3′). The amplified
cDNAs were then purified by 6% TBE PAGE and sequenced on the
Illumina Hi-Seq 2000 according to the manufacturer′s
instructions.
Read filter and small RNA annotation
Two sRNA sequencing data sets comprising the DS and
control CBMCs were obtained from Illumina fast track sequencing
services and were submitted to the NCBI's Gene Expression Omnibus
(http://www.ncbi.nlm.nih.gov/geo/) under
series the accession number GSE39436. The low quality reads were
filtered out to exclude those that were most likely to represent
the sequencing errors and 3′/5′ adaptor sequences were subsequently
trimmed to clean full-length reads and formatted into a
non-redundant FASTQ format. The frequencies of each unique small
RNA sequence reads were calculated as sequence tags (the number of
reads for each sequence reflects its relative abundance) and only
small RNA sequences ranging from 18 to 30 nt were retained for
subsequent analysis.
Unique sequencing reads that passed the filters were
mapped onto the reference human genome using the SOAP (version 2.0)
program with at most two mismatches (20). Subsequently, the unique sequence
reads that mapped to the human genome were aligned against the
miRBase version 17.0, Rfam 10.0 (http://rfam.sanger.ac.uk/), piRNABank (NCBI) and the
human gene UCSC annotation hg19 (http://genome.ucsc.edu/). Overlapping annotation
sequences from these data were classified as miRNA, tRNA, rRNA,
mRNA, snoRNA, snRNA, piRNA or other non-coding RNAs. The
unannotated sequences that did not overlap with any of these
annotations but could be coded by intergenic and intronic regions
of the human genome, served as a source of potential novel miRNAs
(11).
Normalization of the calculation of
transcript parts per million (TPM)
A normalization step was performed because the total
number of reads from different experiments was not the same and
variations in the number of reads of individual miRNA were possibly
due to sequencing depth. The number of reads from a unique sequence
(representing a miRNA) was divided by the total clone count of the
sample and multiplied by 106. The total clone count was
the sum of frequencies of all the residual unique sequences after
filtering.
Expression analysis of known miRNAs and
the prediction of novel miRNAs
Differentially expressed miRNAs between the two
libraries were identified by the fold-change method, as described
in a previous study (9). The
statistical significance (P) was inferred based on the Bayesian
method, which was developed for the analysis of digital gene
expression profiles and can account for the sampling variability of
tags with low counts (21).
Unless stated otherwise, the unique sequences with >10 TPM in
the two libraries was ignored and the most frequently observed
isomiR was used as the diagnostic sequence for comparison of miRNA
expression between libraries. A specific miRNA was deemed to be
significantly differentially regulated if the P determined by this
method was ≤0.001 and there was at least a 2-fold change in the
normalized sequence counts.
The unannotated sequences that could be aligned to
the human genome were considered for detecting novel candidate
miRNAs. Briefly, the 100 nucleotides of genomic sequences flanking
each side of the unannotated sequences were extracted. The RNA
secondary structures with a folding free energy of less than or
equal to −18 kcal/mol (Mfe ≤-18 kcal/mol) were predicted using
RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi). The
secondary structures were analyzed by MIREAP (default setting)
(http://sourceforge.net/projects/mireap/), which is a
sophisticated tool commonly used to identify novel miRNAs from
high-throughput sequencing data and has been widely used in related
studies (22,23). Concomitantly, the folded
structures identified were further tested by a publicly available
miRNA classifier (miRDeep) to reduce the number of false positives
(24). Additionally, the sRNAs
derived from the same precursors were grouped into a common family
and the most abundant member was selected as the representative of
the family.
qPCR
qPCR was performed as described previously (3), with minor modifications. Briefly,
total RNA was isolated from CBMCs using TRIzol reagent (Invitrogen)
as per the manufacturer's instructions. The RNAs were reverse
transcribed to cDNA with stem-loop-like RT primers (GenePharma,
Shanghai, China) that are specific for only the mature miRNA
species and then quantified on an Applied Biosystems 7500 Real-Time
PCR system (Applied Biosystems, Carlsbad, CA, USA) using SYBR-Green
PCR Master Mix (Toyobo, Osaka, Japan). The PCR reaction was
conducted at 95°C for 5 min, followed incubations at 95°C for 15
sec, 65°C for 15 sec and 72°C for 32 sec and repeated for 40
cycles. Each PCR was repeated at least three times. The relative
expression level of each miRNA was normalized against RNU6B
expression levels. The fold-change was calculated according to the
2−ΔΔCT method.
Target prediction and gene ontology (GO)
analysis
As previously described (9), the target genes for each
significantly differentially regulated miRNA were predicted by
miRecords (http://mirecords.biolead.org/), which integrates the
predicted targets from the following miRNA target prediction tools:
DIANA-microT, MicroInspector, miRanda, MirTarget2, miTarget,
NBmiRTar, PicTar, PITA, RNA22, RNAhybrid and
TargetScan/TargertScanS. Since miRNA target prediction often
suffers from a high false-positive rate, only the target genes
supported by at least 5 of 11 established target prediction
programs compiled by miRecords were taken into account.
The Gene Ontology (GO) terms of the predicted
targeted genes were annotated using the DAVID gene annotation tool
(http://david.abcc.ncifcrf.gov/summary.jsp). As
previously described (25), the
GeneMerge software was employed for identification of the
significant overrepresentation of particular GO terms. GO terms and
miRNAs were clustered using hierarchical clustering in R. A heatmap
was created in R using the heatmap function (default
parameters).
Results
Sequencing and annotation of sRNA
Using Illumina sequencing technology, a total of
24,026,800 and 21,448,373 raw sequence tags were generated in the
DS (P) and control (N) groups, respectively. Following removal of
the low-quality reads and masking of the adaptor sequences,
21,770,279 (P) and 17,482,100 (N) sequence tags were obtained
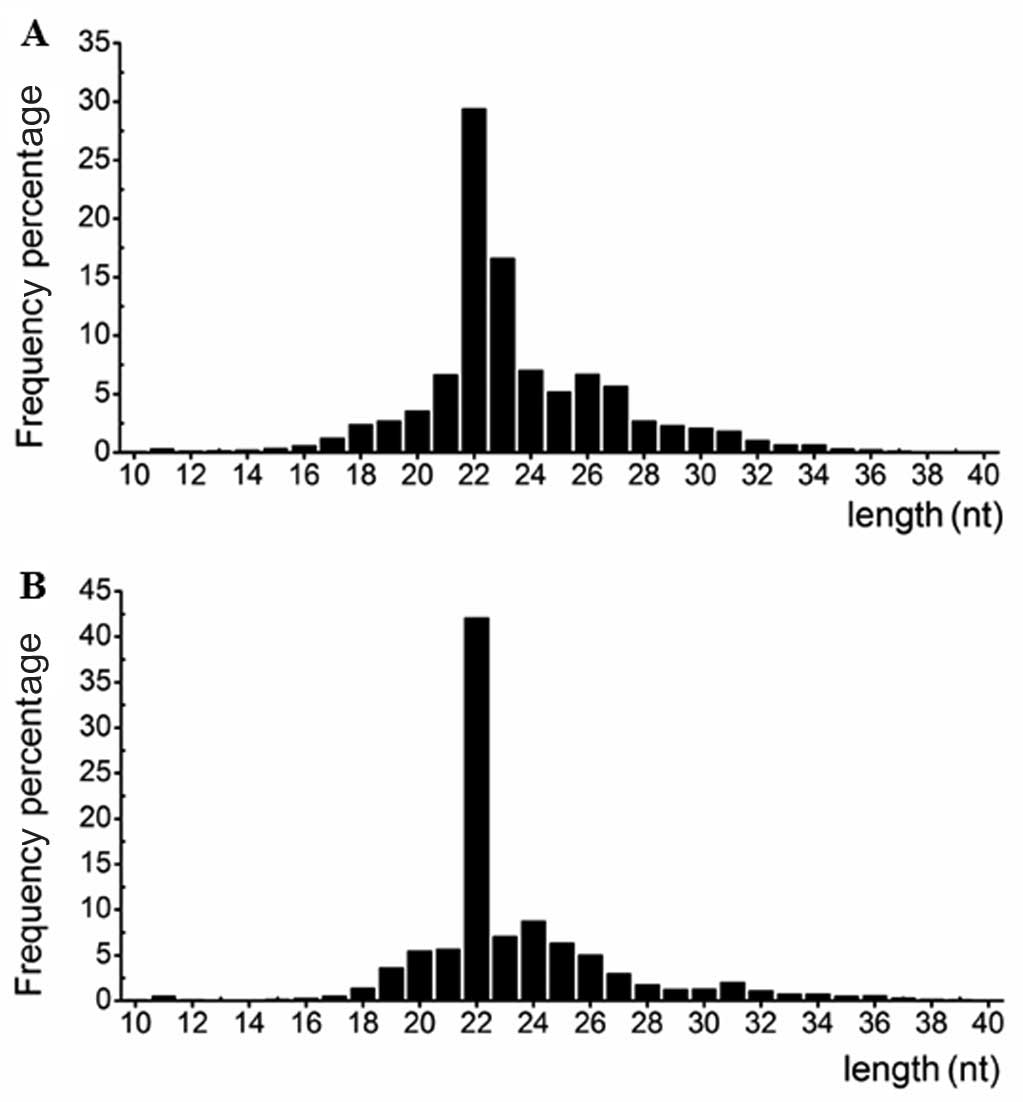
corresponding to 280,056 (P) and 232,814 (N) unique reads. Fig. 1 shows the length distribution of
the small RNAs. The majority of the small RNAs from the two
libraries were ~22 nt in length, which is consistent with the
typical size of miRNAs generated following Dicer activity. This
finding indicated that miRNAs were successively enriched from the
two libraries.
The 18–30 nt sequences were then mapped to the human
genome using SOAP (version 2.0), with a maximum of two mismatches.
In total, 8,087,250 (P) and 12,536,630 (N) sequence tags
represented by 58,523 (P) and 73,833 (N) unique reads were detected
to match the human genome. The sequences tags were annotated and
classified into different categories (miRNA, tRNA, rRNA, mRNA,
sn/snoRNA, piRNA, other non-coding RNAs and unannotated RNAs)
according to their overlap with the publicly available genome
annotations (data not shown). For miRNA, 2,208,096 (P) and
6,369,123 (N) sequence tags were annotated in (P) and (N),
respectively. The surplus unannotated sequence tags served as a
source of novel miRNAs.
Known miRNA expression patterns
In this study, a total of 395 miRNAs were detected
from the two libraries based on miRBase 17.0. To identify the
differentially regulated miRNAs in the two samples, we initially
normalized each miRNA count to TPM, as described in Materials and
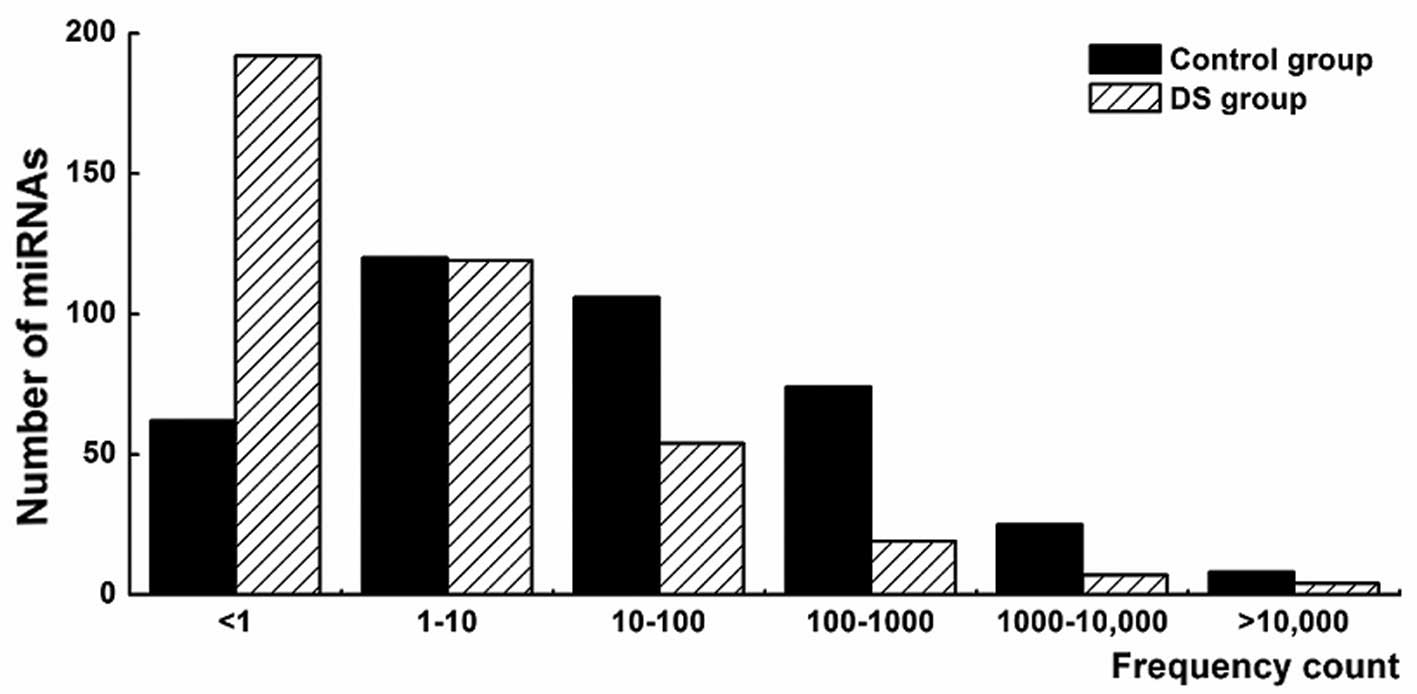
methods. The results showed that different miRNAs exhibited
significantly different expression levels ranging from <1 to
>10,000 TPM (Fig. 2). Several
members of the let-7 family, miR-103a, miR-107, miR-15b, miR-16,
miR-185 and miR-320a were observed as some of the highly expressed
miRNAs. While the members of the let-7 family, let-7a, c, d, e, f
and g represented ~43.7% of the total miRNA counts in the (N).
Notably, the let-7 family miRNAs were reported as highly abundant
miRNAs in various adult tissues and are crucial in embryogenesis
(26) and brain development
(27).
Based on the normalized number of reads per sample,
149 miRNAs were identified as significantly differentially
expressed with a fold change >2.0 and P<0.001. Of the 149
significantly differentially expressed miRNAs, 143 miRNAs were
downregulated and 6 miRNAs were upregulated in the DS group
(Table II). In addition, another
51 miRNAs were specifically expressed in the N group (data not
shown). These specifically enriched miRNAs can be selected as
candidate diagnostic biomarkers for DS fetuses. Of note, results of
this experiment demonstrated that 4 of 14 Hsa21-derived miRNAs
(hsa-miR-99a, let-7c, miR-125b-2 and miR-155) were downregulated in
the DS group. By contrast, other Hsa21-derived miRNAs were not
quantified in either of the two libraries by Illumina sequencing
technology, which might be associated with the lack of primers
specific for these miRNAs (3).
| Table IIThe six upregulated and 14
downregulated miRNAs that were differentially expressed in the DS
group (fold change >2.0 and P<0.001). |
Table II
The six upregulated and 14
downregulated miRNAs that were differentially expressed in the DS
group (fold change >2.0 and P<0.001).
| Relative count | | |
|---|
|
| | |
|---|
| miRNA-ID | Control group | Ds group | Fold change | P-value |
|---|
| hsa-miR-196b | 0.92 | 261.82 | 286.08 | 0 |
| hsa-miR-483-5p | 4.75 | 55.95 | 11.78 | 7.56E-209 |
|
hsa-miR-4732-5p | 21.85 | 241.01 | 11.03 | 0 |
| hsa-miR-320b | 61.72 | 159.71 | 2.59 | 3.11E-188 |
| hsa-miR-486-5p | 4357.89 | 11160.35 | 2.56 | 0 |
| hsa-miR-92b* | 55.43 | 129.35 | 2.33 | 2.38E-128 |
| hsa-miR-125b-2 | 22.88 | 8.13 | 2.81 | 3.26E-33 |
| hsa-miR-99a | 16.93 | 4.82 | 3.51 | 1.43E-32 |
| hsa-let-7c | 4134.17 | 986.14 | 4.19 | 0 |
| hsa-miR-16 | 3215.80 | 752.20 | 4.28 | 0 |
| hsa-miR-22 | 103.02 | 17.13 | 6.01 | 1.27E-297 |
| hsa-miR-17 | 172.75 | 22.05 | 7.84 | 0 |
| hsa-miR-98 | 20.19 | 1.88 | 10.72 | 3.14E-79 |
| hsa-miR-126 | 39.70 | 1.42 | 27.88 | 5.43E-198 |
| hsa-miR-181a | 880.39 | 24.53 | 35.89 | 0 |
| hsa-miR-181b | 369.98 | 4.18 | 88.51 | 0 |
| hsa-miR-21 | 950.34 | 7.30 | 130.12 | 0 |
| hsa-miR-223 | 862.20 | 5.42 | 159.07 | 0 |
| hsa-miR-27a | 35.87 | 0.18 | 195.24 | 3.13E-212 |
| hsa-miR-155 | 26.31 | 0.00 | #DIV/0! | 2.30E-162 |
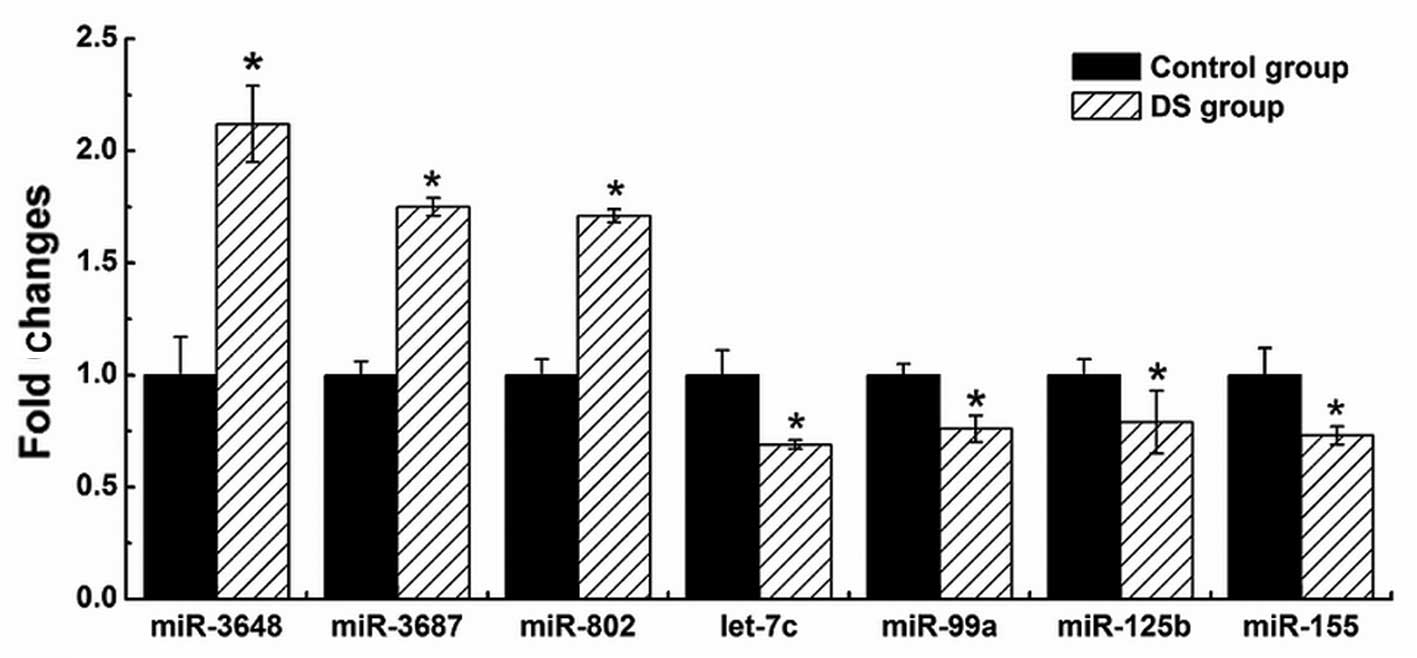
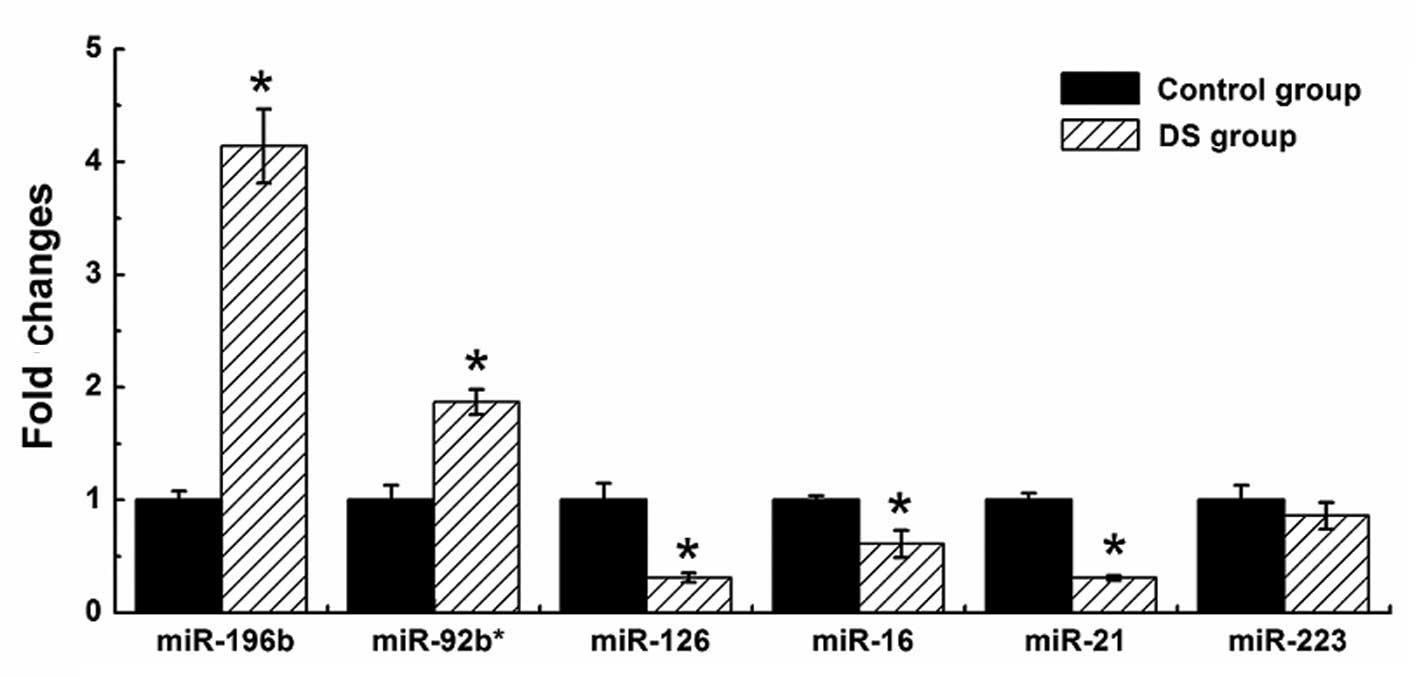
To validate the results of Illumina sequencing
studies, we performed qPCR assays with specific stem-loop RT
primers to examine the expression levels of the Hsa21-derived
mature miRNAs and randomly selected 6 significantly differentially
expressed miRNAs, including two upregulated miRNAs (hsa-miR-196b
and miR-92b*) and four downregulated miRNAs (hsa-miR-16, miR-126,
miR-21 and miR-223). The results demonstrated that four
Hsa21-derived mature miRNAs (hsa-miR-99a, let-7c, miR-125b-2 and
miR-155) were downregulated and three Hsa21-derived mature miRNAs
(hsa-miR-802, miR-3648 and miR-3687) were overexpressed by at least
50% in the DS fetal CBMCs (n=3) compared with matched control
specimens (Fig. 3), while a
further seven Hsa21-derived mature miRNAs remained undetected by
stem-loop quantity PCR in all the samples. The results of six
abnormally expressed non-Hsa21-derived miRNAs are shown in Fig 4. With the exception of miR-233, the
changes of five abnormally expressed non-Hsa21-derived miRNAs in
the DS group were consistent with the Illumina sequencing
results.
Identification of novel candidate
miRNAs
To identify novel candidate miRNAs from unmatched
sequences in the two libraries, we first removed the annotated
sequences, such as the known miRNAs, genomic repeats, coding
sequences and other small RNAs. In total, 59,098 (P) and 346,626
(N) detected sequence tags were derived from unannotated regions of
the human genome. As the predictions are based on identifying miRNA
precursors, genomic regions (100 nt) surrounding these unique
sequences were extracted and analyzed by MIREAP and miRDeep, which
are commonly used to identify novel candidate miRNAs from
high-throughput sequencing data (19,22). To reduce the false-positive rate,
only novel candidate miRNAs supported by these two independent
tools were considered. Using this method, 181 novel candidate
miRNAs were identified from the two libraries: 127 miRNAs from the
N group and 63 miRNAs from the DS group, but 9 miRNAs were shared
by both libraries.
We observed that the size of the 181 miRNAs ranged
from 20 to 24 nt and the minimum free energies of their precursors
varied from −18.6 to −71.5 kcal/mol with an average value of −36.1
kcal/mol. The length of the precursor hairpin structures ranged
from 61 to 101 nt. Of note, some novel candidate miRNAs in our
dataset had 1–7 isomiRs with varying frequencies, which can
strengthen the prediction of these molecules as novel miRNAs
(28). Additionally, two novel
candidate miRNAs (designated as hsa-miR-nov-1 and hsa-miR-nov-2)
genes were found to reside within the ‘DS critical region’ of human
chromosome 21 (chr21q22.2–q22.3) (Fig. 5), in which the genes have been
proven to be important in the pathologic process of DS (29). This observation suggest that the
two novel miRNA genes may also be crucial in the complex and
variable phenotypes of DS in the same manner as the five known
Hsa21-derived miRNAs (i.e., hsa-miR-99a, let-7c, miR-125b-2,
miR-155 and miR-802) (1). To
validate the novel miRNAs, we performed qPCR on four randomly
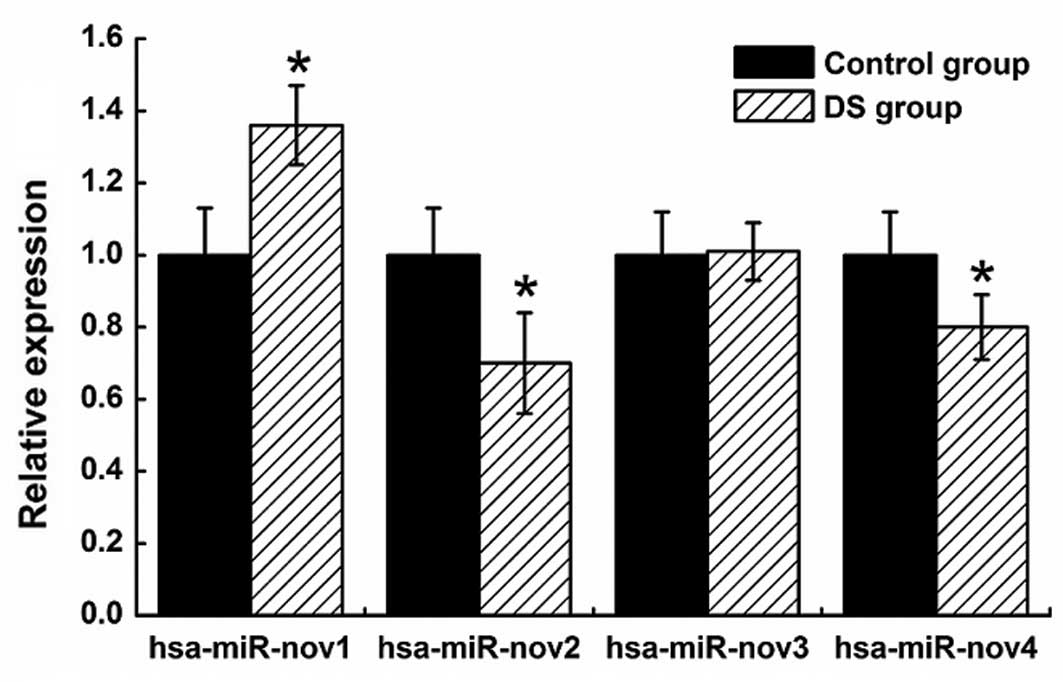
selected novel miRNAs (hsa-miR-nov1, hsa-miR-nov2, hsa-miR-nov3 and
hsa-miR-nov4), including the three aforementioned candidate miRNAs.
As a result, we have successfully validated the four novel
candidate miRNAs. Compared with the control samples, the
hsa-miR-nov1 was upregulated and the hsa-miR-nov2 and miR-nov4 were
downregulated in DS samples (Fig.
6).
In comparison with other miRNAs in the present
study, >92% of the putative novel miRNAs exhibited a lower
expression level with <10 TPM after normalizing for the
sequencing frequency and only 13 novel miRNAs were >10 TPM. This
finding suggested that the majority of putative novel miRNAs may
not have a significant role in DS. Nevertheless, the Hsa21-derived
novel miRNAs and the novel miRNAs that measured >10 TPM may be
involved in the development of the complex and variable phenotypes
of DS and deserve further functional investigation.
Target prediction and GO analysis
To explore the specific functions of the
differentially expressed miRNAs (fold change >2.0 and
P<0.001) in the developmental process of DS fetus, the mRNA
targets of each differentially expressed miRNA were identified by
miRecord tool (http://mirecords.biolead.org/) as described in
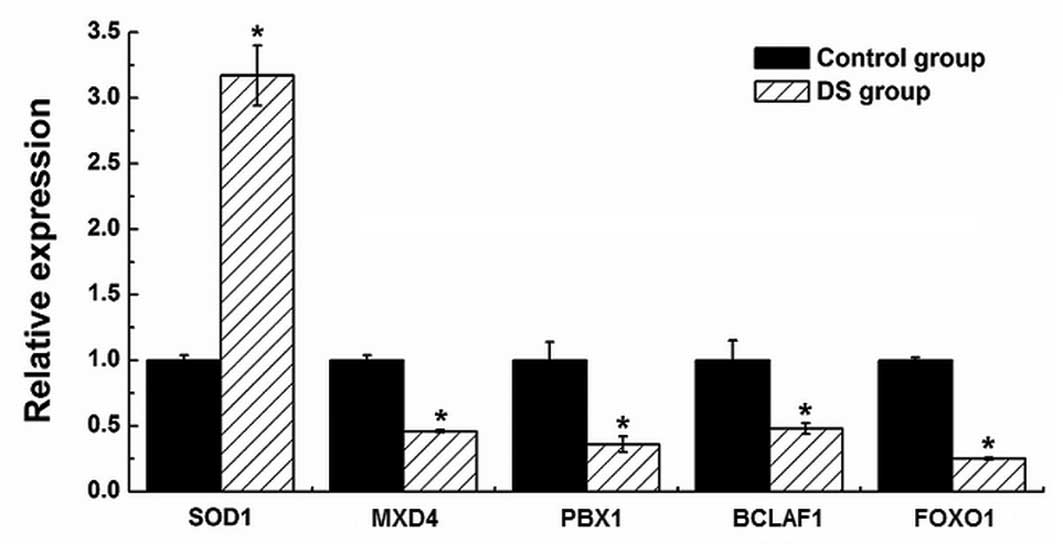
Materials and methods. Five mRNA targets (i.e., SOD1, MXD4,
PBX1, BCLAF1 and FOXO1) of differentially expressed miRNAs were
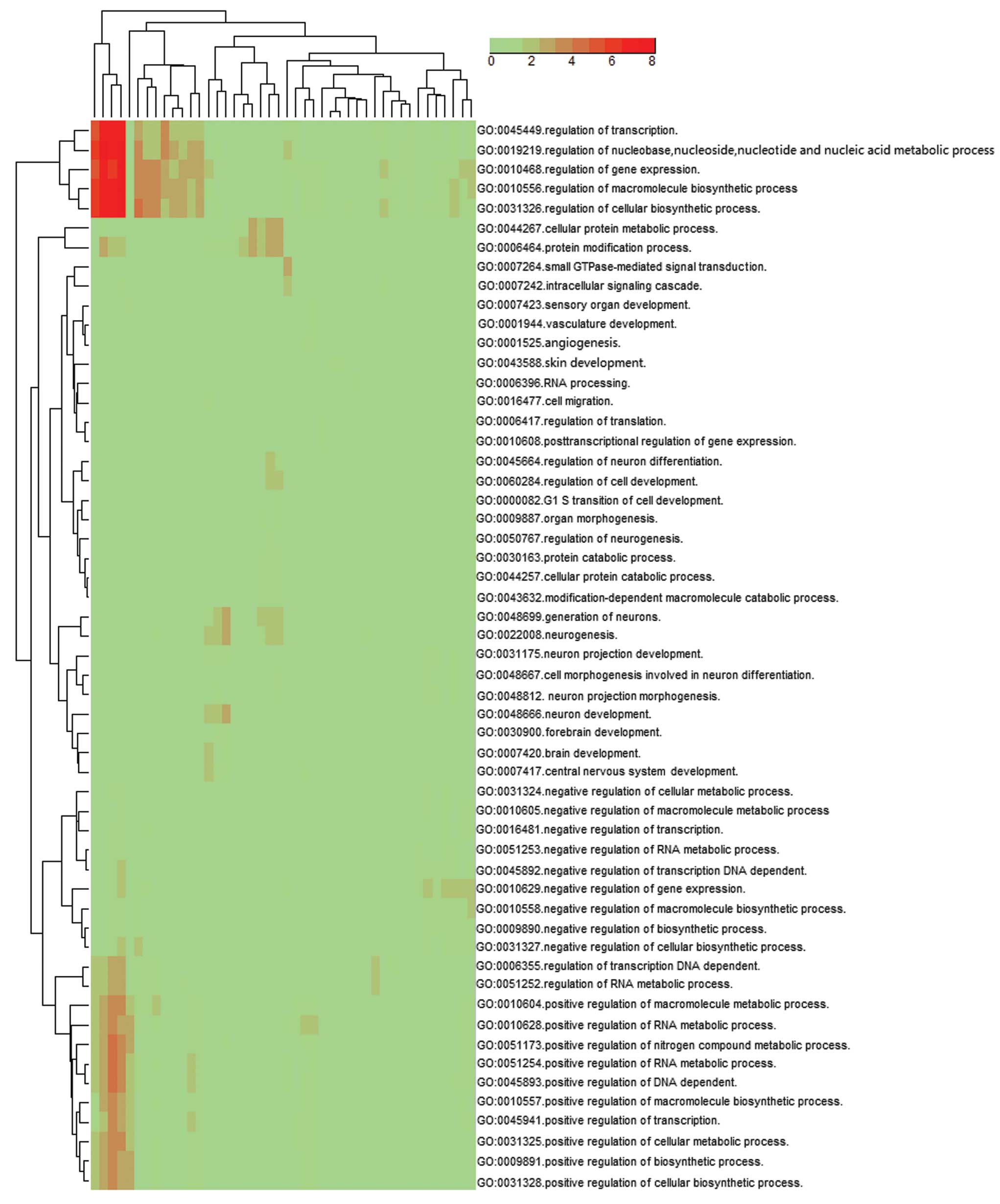
also quantified by qPCR in the (P) and (N) (Fig. 7). By examining the GO ‘biological
process’ classifications at level 5, results showed that the
cluster of overrepresented GO terms among the predicted target
genes of the up- and downregulated miRNAs in the DS fetus were
involved in the regulation of transcription (GO:0045449,
GO:0045941, GO:0045893), gene expression (GO:0010468, GO:0010628),
cellular biosynthetic process (GO:0031326, GO:0031328),
macromolecule biosynthetic process (GO:0010556, GO:0010604) and the
regulation of nucleobase, nucleoside, nucleotide and nucleic acid
metabolic process (GO:0019219) (P<0.001) (Fig. 8).
For the highly abundant (>1,000 TPM) and
significantly differentially expressed miRNAs, with the exception
of the aforementioned biological function, some clusters of
significant GO terms and their mRNA targets were also associated
with nervous system development, i.e., neurogenesis (GO:0022008),
generation of neurons (GO:0048699), neuron differentiation and
development (GO:0045664, GO:0048666), which might be associated
with the pathogenesis of cognitive impairment in DS patients. The
results also suggest that a set of highly abundant and
significantly differentially expressed miRNAs may promote the
progression of cognitive impairment in DS patients by regulating
genes in the pathway of nervous system development.
Discussion
To investigate the changes of genome-wide miRNA
expression of DS fetuses under the influence of trisomy 21 during
the development of DS fetus and to identify whether another miRNA
gene is located on the Hsa21, we have globally examined miRNA
expression profiles in the DS fetus and their normal counterparts
using Illumina deep sequencing technology. In total, 395 known and
181 novel miRNAs were identified in this experiment and two novel
candidate miRNAs were identified as residing within the ‘DS
critical region’ of chromosome 21 (chr21q22.2–22.3).
Compared with the miRNAs common to the two
libraries, the majority of differentially expressed miRNAs,
including four Hsa21-derived miRNAs (hsa-miR-99a, let-7c,
miR-125b-2 and miR-155) were downregulated in the DS group.
Notably, the aberrant expression of these Hsa21-derived miRNAs
including has-miR-802 has been proven to contribute to the complex
and variable phenotypes of DS (1). The miRNAs hsa-miR-155 and miR-802
are able to repress the expression of the MeCP2 gene leading
to cognitive impairment in individuals with DS (4). Additionally, hsa-miR-155 has also
been found to be important in the development of immunocytes during
inflammation through the regulation of Bach1, Sla, Cutl1, Csf1r,
Jarid2, Cebp-β, PU.1, Arntl, Hif1-α and Picalm, while
its sustained expression in the hematopoietic stem cells (HSC) is
likely to lead to a myeloproliferative disorder (14). The miRNAs hsa-miR-99a, let-7c and
miR-125b act as tumor suppressors and are capable of reducing the
incidence of solid tumors in DS patients (16–18). However, with the exception of
hsa-miR-802 being overexpressed by at least 50%, the remaining four
hsa21-derived miRNAs were downregulated in the DS fetal CBMCs in
the present study. These results are not consistent with previous
studies (3,5) that described these five miRNAs as
being overexpressed in DS fibroblast, brain and heart cells
(1). Concerning the discrepancies
between the data obtained in our study and previous studies, the
most important reasons may be due to the different samples (cord
blood cell and fetal tissues). Two groups extensively analyzed the
overexpressed Hsa21 genes in different trisomic cell/tissue types
(30,31). Prandini et al (31) found that, when the lymphoblastoid
cell lines were compared to the fibroblast cell lines derived from
individuals with DS and euploid control individuals, only 39% of
Hsa21 genes in lymphoblastoid cell lines and 62% in fibroblast cell
lines were overexpressed. Li et al (30) compared the difference of Hsa21
gene expression between the fibroblast and fetal hearts with
trisomy 21 and observed that the proportion of Hsa21 overexpressed
genes in fibroblast and fetal hearts was different (29 and 15%,
respectively). Additionally, the discrepancies between the relative
abundance of miRNA detected by Illumina sequencing technology and
stem-loop qPCR may be explained by the individual variation of
specimens and the application of different methods for miRNA
preparation and data analysis. Nonetheless, we may preliminarily
describe the miRNA expression profile of CMBC in the development of
DS fetus. Additionally, the data obtained in our study should be
validated using more DS cord blood samples.
Genome-wide miRNA expression profiling is likely to
enhance our understanding of miRNAs and their roles in the
development of the DS fetus. Numerous identified differentially
regulated miRNAs in this study have been reported to be involved in
various cell processes, such as cell proliferation,
differentiation, apoptosis, granulocyte maturation and activation.
For example, hsa-miR-233, a myeloid-specific miRNA, negatively
regulates myeloid progenitor proliferation, granulocyte
differentiation and activation and is involved in maintaining the
function of neutrophils. In the absence of hsa-miR-233,
granulocytes are hypermature and hypersensitive to activating
stimuli and exhibit increased fungicidal activity (12). Moreover, hsa-miR-196b and miR-21
have been found to be involved in granulopoiesis. Their
co-expression can completely block G-CSF-induced granulopoiesis
(32). Additionally, the
downregulation of hsa-miR-21 is able to restrain the generation of
mononuclear cells and the differentiation of dendritic cells (DC),
leading to the decrease of DC generation, and resulting in the
phagocytic function of DC inhibition (33). DC lacking hsa-miR-155 may further
impair its antigen presentation capacity, rendering it unable to
induce efficient T-cell activation in response to antigens
(34). Hsa-miR-155, a requirement
miRNA for normal immune function, is important in the function of B
and T lymphocytes and dendritic cells (35). Hsa-miR-155 deficiency results in
CD4+ T cells being more prone towards Th-2 differentiation as
compared with Th-1, leading to a reduced number of germinal center
B cells and extra-follicular B cells, with B cells failing to
produce high-affinity IgG antibodies (36). Furthermore, some miRNAs, including
hsa-miR-125a, miR-125b, miR-99b and let-7e are preferentially
expressed by the actively dividing centroblasts in germinal centers
and hsa-miR-125b overexpression is capable of inhibiting the
differentiation of primary B cells (37).
The GO ‘biological process’ classifications showed
that GO categories associated with the regulation of transcription,
gene expression, cellular biosynthetic process, macromolecule
biosynthetic process and the regulation of nucleobase, nucleoside,
nucleotide and nucleic acid metabolic process were enriched among
the target genes of significantly differentially expressed miRNAs
in the DS fetal CBMC. The majority of mRNA targets in these
categories have a well-documented association with immune
modulation, including SOD1, MXD4, PBX1, BCLAF1 and
FOXO1. These genes were targeted by hsa-miR-1, miR-320b,
miR-196b, miR-4732-5p and miR-486-5p, respectively. The SOD1
gene has been localized to the ‘DS critical region’ of chromosome
21. The overexpression of SOD1 may lead to a decrease in
lymphoproliferative responses, an increase in necrosis and
apoptosis of T-cells in vitro and play a crucial role in
T-cell functional deficits (38).
MXD4, a member of the Myc-Max-Mad network, has been proven
to be associated with the survival of Ag-specific T cells. The
downregulation of MXD4 may lead to an increase of T-cell
death (39). As a major global
developmental regulator, PBX1 is involved in the promotion
of progenitor cell proliferation in multiple tissues and has a
crucial role in maintaining the postnatal hematopoietic
compartment. The deletion of PBX1 in the thymus may lead to
T- and B-lineage cell reduction (40). Bcl-2 associated factor 1 (BCLAF1)
is a nuclear protein that is essential for proper homeostasis of T-
and B-cell lineages and the activation-induced proliferation of T
cells (41).
Bclaf1-deficient neonates exhibiting T cells were found to
have an activation-dependent proliferation defect ex vivo
(42). Furthermore, the
Forkhead-box transcription factor (FOXO1) is crucial in
maintaining the immune system homeostasis and was preferentially
expressed in mature peripheral T and B cells (43). The constitutive deletion of
Foxo1 in the T-cell lineage as well as acute deletion may
lead to T-cell homing defects to peripheral lymphoid organs
(44). FOXO1-deficient
mature B cells may fail to undergo class-switch recombination with
the resulting deficiency in IgG production (45). Combined with the decrease in the
number of immune cells in the blood circulatory system (46) and infection (47) presented in the DS patients, we
extrapolate that the differentially expressed miRNAs might be
involved in hemopoietic abnormalities and the immune defects of DS
fetuses and newborns.
Of note, with the exception of the aforementioned
results, target prediction of the miRNA pattern also revealed that
some highly abundant and significantly differentially expressed
miRNAs might be associated with the pathway of nervous system
development, i.e., neurogenesis, generation of neurons, neuron
differentiation and development. Notably, we found that the nervous
system development genes BDNF and CDK5R1 were the
targets of mir-103 family miRNAs (miR-103a/107). BDNF and
CDK5R1 are important for neuron proliferation,
differentiation, survival and neuronal migration (48,49), suggesting that mir-103 family
members might be associated with the pathogenesis of cognitive
impairment in DS patients. Cognitive impairment was the common
deleterious phenotype of all the DS patients. Additional
investigation of the differential expression patterns of miRNAs,
together with follow-up information in the clinic, may reveal a
miRNA signature as a biomarker for the detection of cognitive
impairment in DS patients.
Another aim of this study was to identify whether
another miRNA gene is located in the human chromosome 21. Notably,
2 of 181 novel candidate miRNAs (hsa-miR-nov1 and hsa-miR-nov2)
were identified in the ‘DS critical region’ of human chromosome 21
(Fig. 5). The lengths of
hsa-miR-nov1 and hsa-miR-nov2 are 23 and 21 nt, respectively. The
minimum free energy of both novel miRNAs is lower than −18
kcal/mol. By using the UCSC platform, we found that the
hsa-miR-nov1 gene resides in the anti-sense orientation within
intron 9 of the β-site APP cleaving enzyme 2 (BACE2) gene,
which is located at the chr21q22.2–22.3 and plays an important role
in the neurodegeneration and consequent dementia in AD and DS
(50,51). The hsa-miR-nov2 is located ~2.6
million base pairs downstream from the hsa-miR-nov1 gene in the
sense orientation within intron 11 of the human pyridoxal kinase
(PDXK) gene at Hsa21 genomic position q22.3. Significantly,
the expression mechanism of miRNA (10) suggests that the expression of
hsa-miR-nov1 and hsa-miR-nov2 might be associated with the
BACE2 and PDXK, respectively.
To speculate on the function of these two novel
candidate miRNAs, TargetScanS Custom (version 5.2) and GO were used
to predict their putative targets and analyze their biological
functions, respectively. The results showed that hsa-miR-nov1 has
211 conserved targets, including the MECP2 gene, which is
also the target of hsa-miR-155, miR-802, let-7c and miR-125b
(1) and may be associated with
cognitive impairment in DS individuals (4). The hsa-miR-nov2 miRNA regulates 101
conserved targets with a total of 105 conserved sites. The cluster
of overrepresented GO classes in the predicted targets of the two
novel miRNAs reveal that these miRNAs are mainly associated with
regionalization (GO:0003002), regulation of cell differentiation
(GO:0045595, GO:0045646 and GO:0045637), embryonic morphogenesis
and development (GO:0048598 and GO:0009790) and ion transport
(GO:0006811). However, although we have identified that
hsa-miR-nov1 and hsa-miR-nov2 were up- and downregulated in the DS
fetal CBMCs (Fig. 6),
respectively, the regulatory mechanisms of the two novel miRNAs
have yet to be confirmed.
In conclusion, the present study has provided
considerable insight into understanding the expression
characteristic of miRNAs in the DS fetal CBMCs and that the
differentially expressed miRNAs may be involved in hemopoietic
abnormalities and the immune defects observed in DS fetuses and
newborns. Additionally, certain highly abundant and differentially
expressed miRNAs might be involved in the pathway of cognitive
impairment of DS patients. To the best of our knowledge, this is
the first study to examine genome-wide miRNA expression profiling
in the DS fetus. Notably, the regulation mechanism of annotated-
and novel-hsa21-derived miRNAs in the development of DS blood cells
and the association between hsa21-derived miRNAs and the various
phenotypes of DS should be further investigated. Specifically, the
significantly differentially and specifically expressed miRNAs have
the potential to be selected as biomarkers in order to construct a
new antenatal diagnostic method for the DS fetus.
Acknowledgements
We are grateful to the patients for participating in
this study. We thank Dr Jianhua Yang (Sun Yat-sen University,
Guangzhou, P.R. China) and Wangmin Qiao (BGI, Shenzhen, P.R. China)
for their helpful comments regarding the data analysis. We would
also like to thank Zhaoyang Luo (Harbin Institute of Technology
Shenzhen Graduate School, Shenzhen, P.R. China) for his technical
assistance. This study was supported by the key project of Shenzhen
S&T program (no. 201001006) and Guangdong provincial S&T
program (no. 2012B032000008).
References
|
1
|
Elton TS, Sansom SE and Martin MM:
Trisomy-21 gene dosage over-expression of miRNAs results in the
haploinsufficiency of specific target proteins. RNA Biol.
7:540–547. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kozomara A and Griffiths-Jones S: miRBase:
integrating microRNA annotation and deep-sequencing data. Nucleic
Acids Res. 39:D152–157. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kuhn DE, Nuovo GJ, Martin MM, et al: Human
chromosome 21-derived miRNAs are overexpressed in down syndrome
brains and hearts. Biochem Biophys Res Commun. 370:473–477. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kuhn DE, Nuovo GJ, Terry AV Jr, et al:
Chromosome 21-derived microRNAs provide an etiological basis for
aberrant protein expression in human Down syndrome brains. J Biol
Chem. 285:1529–1543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sethupathy P, Borel C, Gagnebin M, et al:
Human microRNA-155 on chromosome 21 differentially interacts with
its polymorphic target in the AGTR1 3′ untranslated region: a
mechanism for functional single-nucleotide polymorphisms related to
phenotypes. Am J Hum Genet. 81:405–413. 2007.PubMed/NCBI
|
|
6
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cheng AM, Byrom MW, Shelton J and Ford LP:
Antisense inhibition of human miRNAs and indications for an
involvement of miRNA in cell growth and apoptosis. Nucleic Acids
Res. 33:1290–1297. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen CZ, Li L, Lodish HF and Bartel DP:
MicroRNAs modulate hematopoietic lineage differentiation. Science.
303:83–86. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vaz C, Ahmad HM, Sharma P, et al: Analysis
of microRNA transcriptome by deep sequencing of small RNA libraries
of peripheral blood. BMC Genomics. 11:2882010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bandiera S, Hatem E, Lyonnet S and
Henrion-Caude A: microRNAs in diseases: from candidate to modifier
genes. Clin Genet. 77:306–313. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Havelange V and Garzon R: MicroRNAs:
emerging key regulators of hematopoiesis. Am J Hematol. 85:935–942.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Szulwach KE, Jin P and Alisch RS:
Noncoding RNAs in mental retardation. Clin Genet. 75:209–219. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
O'Connell RM, Rao DS, Chaudhuri AA, et al:
Sustained expression of microRNA-155 in hematopoietic stem cells
causes a myeloproliferative disorder. J Exp Med. 205:585–594. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Malinge S, Izraeli S and Crispino JD:
Insights into the manifestations, outcomes, and mechanisms of
leukemogenesis in Down syndrome. Blood. 113:2619–2628. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ozen M, Creighton CJ, Ozdemir M and
Ittmann M: Widespread deregulation of microRNA expression in human
prostate cancer. Oncogene. 27:1788–1793. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nagayama K, Kohno T, Sato M, Arai Y, Minna
JD and Yokota J: Homozygous deletion scanning of the lung cancer
genome at a 100-kb resolution. Genes Chromosomes Cancer.
46:1000–1010. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Calin GA, Sevignani C, Dumitru CD, et al:
Human microRNA genes are frequently located at fragile sites and
genomic regions involved in cancers. Proc Natl Acad Sci U S A.
101:2999–3004. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Creighton CJ, Reid JG and Gunaratne PH:
Expression profiling of microRNAs by deep sequencing. Brief
Bioinform. 10:490–497. 2009. View Article : Google Scholar
|
|
20
|
Li R, Yu C, Li Y, et al: SOAP2: an
improved ultrafast tool for short read alignment. Bioinformatics.
25:1966–1967. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Anders S and Huber W: Differential
expression analysis for sequence count data. Genome Biol.
11:R1062010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu G, Wu J, Zhou L, et al:
Characterization of the small RNA transcriptomes of androgen
dependent and independent prostate cancer cell line by deep
sequencing. PLoS One. 5:e155192010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen X, Li Q, Wang J, et al:
Identification and characterization of novel amphioxus microRNAs by
Solexa sequencing. Genome Biol. 10:R782009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
An J, Lai J, Lehman ML and Nelson CC:
miRDeep*: an integrated application tool for miRNA identification
from RNA sequencing data. Nucleic Acids Res. 41:727–737. 2013.
|
|
25
|
Castillo-Davis CI and Hartl DL:
GeneMerge-post-genomic analysis, data mining, and hypothesis
testing. Bioinformatics. 19:891–892. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schulman BR, Esquela-Kerscher A and Slack
FJ: Reciprocal expression of lin-41 and the microRNAs let-7 and
mir-125 during mouse embryogenesis. Dev Dyn. 234:1046–1054. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wulczyn FG, Smirnova L, Rybak A, et al:
Post-transcriptional regulation of the let-7 microRNA during neural
cell specification. FASEB J. 21:415–426. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Morin RD, O'Connor MD, Griffith M, et al:
Application of massively parallel sequencing to microRNA profiling
and discovery in human embryonic stem cells. Genome Res.
18:610–621. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Patterson D: Molecular genetic analysis of
Down syndrome. Hum Genet. 126:195–214. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li CM, Guo M, Salas M, et al: Cell
type-specific over-expression of chromosome 21 genes in fibroblasts
and fetal hearts with trisomy 21. BMC Med Genet.
7:242006.PubMed/NCBI
|
|
31
|
Prandini P, Deutsch S, Lyle R, et al:
Natural gene-expression variation in Down syndrome modulates the
outcome of gene-dosage imbalance. Am J Hum Genet. 81:252–263. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Velu CS, Baktula AM and Grimes HL: Gfi1
regulates miR-21 and miR-196b to control myelopoiesis. Blood.
113:4720–4728. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gracias DT and Katsikis PD: MicroRNAs: key
components of immune regulation. Adv Exp Med Biol. 780:15–26. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lu LF and Liston A: MicroRNA in the immune
system, microRNA as an immune system. Immunology. 127:291–298.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rodriguez A, Vigorito E, Clare S, et al:
Requirement of bic/microRNA-155 for normal immune function.
Science. 316:608–611. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fernando TR, Rodriguez-Malave NI and Rao
DS: MicroRNAs in B cell development and malignancy. J Hematol
Oncol. 5:72012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gururajan M, Haga CL, Das S, et al:
MicroRNA 125b inhibition of B cell differentiation in germinal
centers. Int Immunol. 22:583–592. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Banerjee R, Mosley RL, Reynolds AD, et al:
Adaptive immune neuroprotection in G93A-SOD1 amyotrophic lateral
sclerosis mice. PLoS One. 3:e27402008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vasilevsky NA, Ruby CE, Hurlin PJ and
Weinberg AD: OX40 engagement stabilizes Mxd4 and Mnt protein levels
in antigen-stimulated T cells leading to an increase in cell
survival. Eur J Immunol. 41:1024–1034. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ficara F, Murphy MJ, Lin M and Cleary ML:
Pbx1 regulates self-renewal of long-term hematopoietic stem cells
by maintaining their quiescence. Cell Stem Cell. 2:484–496. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
McPherson JP, Sarras H, Lemmers B, et al:
Essential role for Bclaf1 in lung development and immune system
function. Cell Death Differ. 16:331–339. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sarras H, Alizadeh Azami S and McPherson
JP: In search of a function for BCLAF1. Scientific World Journal.
10:1450–1461. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dejean AS, Hedrick SM and Kerdiles YM:
Highly specialized role of Forkhead box O transcription factors in
the immune system. Antioxid Redox Signal. 14:663–674. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ouyang W, Beckett O, Flavell RA and Li MO:
An essential role of the Forkhead-box transcription factor Foxo1 in
control of T cell homeostasis and tolerance. Immunity. 30:358–371.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Dengler HS, Baracho GV, Omori SA, et al:
Distinct functions for the transcription factor Foxo1 at various
stages of B cell differentiation. Nat Immunol. 9:1388–1398. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
de Hingh YC, van der Vossen PW, Gemen EF,
et al: Intrinsic abnormalities of lymphocyte counts in children
with Down syndrome. J Pediat. 147:744–747. 2005.PubMed/NCBI
|
|
47
|
Ram G and Chinen J: Infections and
immunodeficiency in Down syndrome. Clin Exp Immunol. 164:9–16.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lee J, Duan W and Mattson MP: Evidence
that brain-derived neurotrophic factor is required for basal
neurogenesis and mediates, in part, the enhancement of neurogenesis
by dietary restriction in the hippocampus of adult mice. J
Neurochem. 82:1367–1375. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zuccotti P, Barbieri A, Colombrita C, et
al: Identification of post-transcriptional regulatory elements in
CDK5R1 3′UTR gene involved in CNS development and functioning. Eur
J Hum Genet. 19:3542011.
|
|
50
|
Holler CJ, Webb RL, Laux AL, et al: BACE2
expression increases in human neurodegenerative disease. Am J
Pathol. 180:337–350. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Webb RL and Murphy MP: β-Secretases,
Alzheimer's disease, and Down syndrome. Curr Gerontol Geriatr Res.
2012:3628392012.
|