1. Introduction
Gastric cancer (GC) is the fourth most commonly
diagnosed cancer and the second major cause of cancer-related
mortality worldwide (1,2). Despite improvements in surgery and
chemotherapy, the outcomes in patients with advanced gastric cancer
remain poor, with a five-year survival rate of <20% (3).
Over the past decade, targeted therapies have
greatly improved the outcome of a number of malignancies, including
breast, colorectal and lung cancer. However, less progress has been
made with regard to gastric cancer. The Trastuzumab for Gastric
Cancer (ToGA) study, investigating the effectiveness of trastuzumab
in human epidermal growth factor receptor 2 (HER2; ERBB2)-positive
advanced gastric or gastrooesophageal junction (GEJ) cancer
(4), represents a milestone in
the targeted therapy of gastric cancer. Moreover, a recent study
developed a genomic molecular map of gastric cancer and suggested
that collectively 37% of cases may be potentially treatable by
receptor tyrosine kinase (RTK)/RAS directed therapies (5).
Similar to HER2, cMET is another member of the RTK
family, and plays a key role in tumor survival, growth,
angiogenesis and metastasis (6–10).
A significant proportion of gastric cancers harbor cMET
overexpression and/or gene amplification (11,12), and the aberrant signaling of cMET
pathways in gastric cancer has been shown to correlate with a high
tumor stage and poor prognosis (11,13). The alternative activation of the
cMET pathway is considered to be an important mechanism responsible
for resistance therapeutics targeting HER family members, such as
HER2 and epidermal growth factor receptor (EGFR) (14,15). Recently, several cMET inhibitors
have been investigated in clinical trials, and the initial results
are encouraging (16,17). cMET is emerging as a promising
therapeutic target in gastric cancer, and may provide a potential
approach to overcoming resistance to other agents in targeted
therapy.
Although a number of review articles have focused on
the role of cMET in various malignancies, there is a lack of data
on its role in gastric cancer. Therefore, a greater understanding
of the role of cMET in gastric cancer is required.
In this review, we assess the role of cMET in
gastric cancer, summarize the preclinical and clinical trials of
cMET inhibitors, and discuss the challenges of cMET targeted
therapy. Finally, we present possible solutions, including the
exploration of biomarkers for population selection and drug
response assessment, and the establishment of patient-derived human
tumor tissue (PDTT) xenograft models for drug sensitivity
screening.
2. The cMET pathway
cMET was first identified in 1984 in a human
osteogenic sarcoma cell line treated with the carcinogen,
N-methyl-N′-nitronitrosoguanidine (18), by a genomic rearrangement that
fused the sequence from the translocated promoter region (TRP)
locus on chromosome 1 to a sequence from MET on chromosome 7
(19). A subsequent study
revealed that the encoded protein was an RTK (20).
Both hepatocyte growth factor (HGF) and scatter
factor (SF), are the ligands of cMET (21). HGF was originally identified as a
liver mitogen, while SF was recognized as a fibroblast-derived
modulators of epithelial cell mobility, then they were found to be
identical (22,23). Binding of HGF/SF to cMET leads to
receptor homodimerization and tyrosine residue phosphorylation,
recruitment of adaptor and effector proteins, which ultimately
triggers downstream activation of the RAS/mitogen-activated protein
kinase (MAPK), phosphoinositide 3-kinase (PI3K)/AKT, signal
transducer and activator of transcription (STAT), Ras-related C3
botulinum toxin substrate 1 (RAC1)-cell division cycle 42 (CDC42)
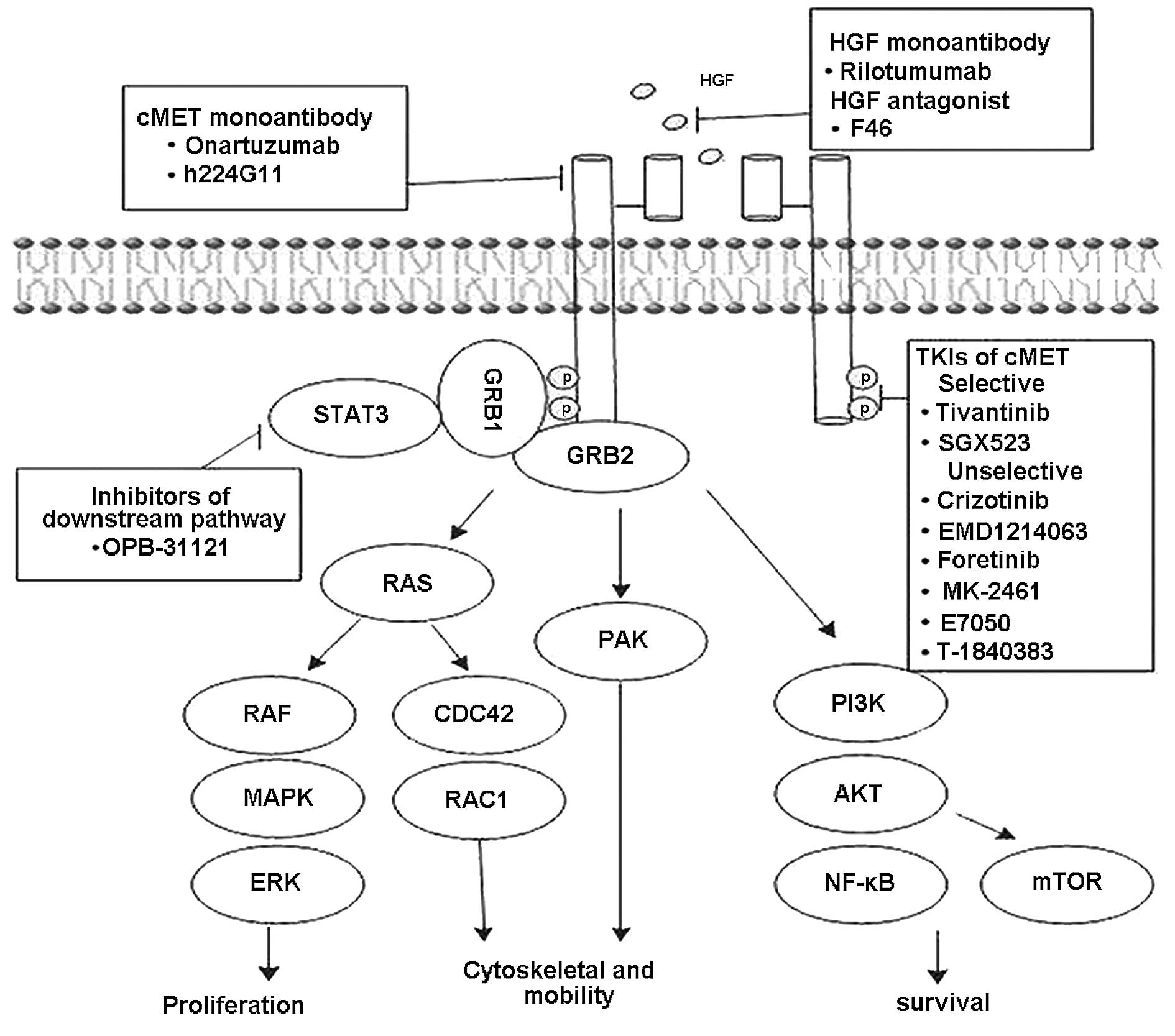
and p21 activated protein kinase (PAK) pathways (Fig. 1) (24–29). The cMET pathway can be modulated
by cell surface molecules, such as EGFR, ERBB2 and insulin-like
growth factor-1 (29–31). Under normal conditions, the cMET
signaling pathway is essential for a spectrum of physiological
processes, such as embryonic development, organ morphogenesis and
wound healing (32–36). However, the dysregulation of the
cMET pathway plays a causal role in tumor survival, growth,
angiogenesis and metastasis (6–10).
3. The role of cMET in gastric cancer
The cMET pathway can be oncogenic and is activated
by multiple mechanisms including, gene amplification, gene
mutation, protein overexpression and ligand-dependent autocrine and
paracrine, receptor crosstalk (10,28). The role of cMET in gastric
tumorigenesis was first identified in the human gastric tumor cell
line, GTL-16 (37). The
overexpression of TPR-MET RNA was detected in superficial gastritis
lesions with hyperplasia of glandular neck cells, suggesting the
possible involvement of this oncogene at an early stage of gastric
tumorigenesis (38). Similar
results were reported in another study (39).
cMET protein overexpression, as well as gene
amplification and mutation have been detected in gastric cancer
tissues and cell lines. Protein overexpression and gene
amplification can be determined by immunohistochemistry (ICH) and
RT-PCR/fluorescence in situ hybridization (FISH),
respectively. Among the retrospective studies (5,11–13,40–46) (Table
I), the increased expression of cMET was detected in
approximately 43% of patients with gastric cancer, while gene
amplification was detected in almost 12% of patients. Protein
overexpression and/or gene amplification significantly correlated
with the depth of tumor invasion and metastasis (11,13,45) and poor prognosis (5,11–13,40,43–46). Based on available evidence, it can
be inferred that gene amplification is likely to be more valuable
than protein overexpression as a prognostic marker. However, a lack
of consistent criteria on the determination of protein
overexpression and gene amplification limits the prognostic value
of these two markers. Consistent criterion that can evaluate cMET
expression and amplification is required.
 | Table IOverexpression and amplification
status of cMET in gastric cancer. |
Table I
Overexpression and amplification
status of cMET in gastric cancer.
|
Authors/(Refs.) | Year | No. of
Patients | OP (%) | Method | AP (%) | Method | Poor prognostic
marker |
|---|
| Tsugawa et
al (44) | 1998 | 70 | | | 10 | Slot blot
hybridization | AP |
| Nakajima et
al (11) | 2000 | 128 | 41.6 | ICH | 10.2 | Southern blot
hybridization | OP/AP |
| Park et al
(43) | 2000 | 43 | 67 | ICH | | | NR |
| Tang et al
(13) | 2004 | 232 | 68.8 | ICH | | | OP/AP |
| Retterspitz et
al (42) | 2010 | 94 | 50 | ICH | | | NR |
| Janjigian et
al (41) | 2011 | 38 | 63 | ICH | 0 | FISH | NR |
| Lee et al
(12) | 2011 | 482 | | | 21.2 | RT-PCR/FISH | AP |
| Graziano et
al (45) | 2011 | 230 | | | 10 | RT-PCR/FISH | AP |
| Lee et al
(40) | 2012 | 438 | 23.7 | ICH | 3.4 | SISH | AP |
| Deng et al
(5) | 2012 | 193 | | | 4 | SNP arrays | AP |
| Shi et al
(46) | 2012 | 128 | | | 30 | RT-PCR | AP |
| Total | | | 42.8 | | 12.1 | | |
In gastric cancer, cMET gene mutations appear to be
very rare; the majority of cMET mutations have been discovered in
papillary renal carcinoma (47,48). A germline missense cMET mutation
located at the juxtamembrane domain has been reported in a patient
with primary gastric cancer (49). Moreover, the Hs746T gastric cell
line harbors a splice site mutation of cMET, causing juxtamembrane
domain deletion (50). A large
proportion of gastric cancer patients harbor cMET overexpression
and/or gene alteration, providing evidence for the key role of cMET
in gastric cancer and a rationale for the development of cMET
inhibitors.
4. The development of cMET inhibitors in
gastric cancer
The increased understanding of the cMET pathway has
led to the development of cMET inhibitors, which focus on one of
the steps in the cMET pathway. Clinical trials investigating
monoclonal antibodies and small-molecule inhibitors directed at the
cMET axis are currently underway. The initial results of these
clinical trials are optimistic; thus, targeting the cMET pathway is
becoming a promising therapeutic strategy for gastric cancer. The
main strategies include, monoclonal antibodies or antagonists
against HGF or cMET, cMET selective or unselective tyrosine kinase
inhibitors (TKIs) and downstream pathway inhibitors (Fig. 1 and Table II).
| Table IIDevelopment of cMET inhibitors in
gastric cancer. |
Table II
Development of cMET inhibitors in
gastric cancer.
| Company | Compound | Type of agent | Development
phase | Initial
results |
|---|
| Amgen | Rilotumumab | HGF mAb | II and III | Rilotumumab + CT
vs. CT: median PFS 4.2 months vs. 5.6 months; OS 5.7 months vs.
11.1 months; suggest MET expression as predictive biomarker
(54). |
| Roche | MetMab | cMET mAb | III | MetMab: a patient
with chemo-refractory metastatic gastric cancer of the liver
achieved complete response lasting for 2 years by MetMAb
monotherapy (17). Suggesting
circulating HGF is a therapeutic response biomarker. |
| Daiichi Sankyo | Tivantinib | cMET selective
TKI | II | Tivantinib: Median
PFS 43 days, disease control rate 36.7%. No objective response
(64). |
| Exelixis | Cabozantinib | CMET unselective
TIK | II | Cabozantinib: 8/19
patients SD observed at 12 weeks, overall disease control rate 32%
at 12 weeks.
No objective response was observed (73). |
| Pfizer | Crizotinib | CMET unselective
TIK | I | Crizotinib: 2/4
patients with MET-amplified gastroesophageal cancer, tumor
shrinkage, (−30 and −16%) progression after 3.7 and 3.5 months;
MET, EGFR and HER2 amplification status may be evaluable (67). |
| Exelixis | Foretinib | CMET unselective
TIK | II | Foretinib: 15/73
patients SD (median 3.2 months); no response observed (70). |
| Otsuka | OPB-31121 | STAT3
inhibitor | I | OPB-31121: 1/5 SD
patients (>12 months) (75). |
| EMD Serono | EMD 1214063 | CMET unselective
TIK | I | |
| Merck | MK-2461 | CMET unselective
TIK | I | |
| Goetsch et
al | h224G11 | cMET mAb | Preclinical | |
| SGX | SGX523 | cMET selective
TKI | Preclinical | |
| Eliai | E-7050 | CMET unselective
TIK | Preclinical | |
| Takeda | T-1840383 | CMET unselective
TIK | Preclinical | |
| Samsung | F46 | HGF antagonist | Preclinical | |
5. Monoclonal antibodies to HGF
Rilotumumab (AMG 102) is a fully human monoclonal
antibody to HGF/SF. In vitro and in vivo studies have
confirmed the antitumor activity of rilotumumab (51,52). A phase 1 clinical study testing
the safety and pharmacokinetics of rilotumumab in 40 patients with
refractory advanced solid tumors, demonstrated that rilotumumab was
safe and well tolerated, and had a favorable pharmacokinetic
profile. A total of 16 of 23 (70%) evaluated patients had a best
response of stable disease (SD) with progression-free survival
(PFS) ranging from 7.9 to 40 weeks (53).
A multicenter, double-blind phase 1b/2 study,
assessed rilotumumab in combination with epirubicin, cisplatin and
capecitabine (ECX) in 121 advanced or metastatic gastric or
esophagogastric junction (EGJ) cancer patients (54). This study reported that the
addition of rilotumumab to the chemotherapeutic regimen improved
the median PFS from 4.2 to 5.6 months [hazard ratio (HR), 0.64; 80%
confidence interval (CI), 0.48–0.85], and the median and overall
survival (OS) from 8.9 to 11.1 months (HR, 0.73; 80% CI,
0.53–1.01). Further analysis of this study (54), revealed that the addition of
rilotumumab to the chemotherapeutic regimen in patients with
gastric tumors with high cMET expression improved median OS from
5.7 to 11.1 months (HR, 0.29; 95% CI, 0.11–0.76). Conversely, in
patients with low cMET expression, the addition of rilotumumab to
chemotherapy was associated with a trend towards an unfavorable OS
(HR, 1.84; 95% CI, 0.78–4.34). In the chemotherapy-only arm,
patients with a high cMET expression had a worse OS (HR, 3.22; 95%
CI, 1.08–9.63) than those with a low cMET expression; similar
trends were observed with PFS (16).
A phase III study to confirm the efficacy of
rilotumumab in advanced gastric and gastroesophageal cancer in
patients with high cMET expression is currently ongoing (55). Another phase II trial, assessing
[folinic acid (FOL, fluorouracil (F) and oxaliplatin (OX); FOLFOX]
alone or in combination with AMG 102 or panitumumab as first-line
therapy in patients with advanced gastroesophageal adenocarcinoma,
is also currently ongoing (56).
In addition to the typical outcome measures, such as PFS, OS,
objective response rate and safety, the study has been designed to
identify candidate predictive and prognostic biomarkers among
functional molecular alterations of the EGFR/RAS/RAF and HGF/cMET
pathways.
6. Monoclonal antibodies to cMET
MetMab (onartuzumab) is a monoclonal single-arm
humanized immunoglobulin (Ig) G1 antibody directed against cMET. In
an in vitro study, onartuzumab was first investigated in the
human glioblastoma cell line, U87, suggesting that the antibody may
exert tumor inhibitory effects, such as anti-proliferative,
anti-angiogenic and pro-apoptotic effects (57). MetMab has also been shown to be
effective against tumor xenografts (57).
In a phase I clinical trial, a patient with
chemo-refractory metastatic gastric cancer achieved a complete
response with MetMab monotherapy that lasted for two years. The
primary tumor had high cMET gene polysomy, as shown by FISH, and a
high cMET expression (2+), as observed by IHC. Intriguingly, HGF
serum levels were extremely high prior to treatment and declined
precipitously immediately after drug exposure, and remained low,
even at the time of widespread recurrence of the disease. This
observation suggests that circulating HGF is a biomarker for
therapeutic response (17).
Similar results have been reported in non-small cell lung cancer
(NSCLC); circulating HGF levels were measured as a pharmacodynamic
biomarker of onartuzumab activity (58). Other studies using PET with
(89)Zr-df-onartuzumab and (76)Br-onartuzumab in gastric carcinoma
xenografts showed that the uptake of both tracers significantly
correlated with tumor mass and cMET expression and was not affected
by the presence of plasma shed cMET (59).
Currently, a randomized, double-blind, phase II
study evaluating the efficacy and safety of onartuzumab in
combination with mFOLFOX6 in patients with metastatic HER2-negative
gastroesophageal cancer is ongoing (60).
Another currently ongoing phase III study introduced
an enrichment biomarker, enrolling patients with metastatic
HER2-negative, cMET-positive gastroesophageal cancer (61). The results of clinical trials on
potential biomarkers may provide recommendations on patient
selection and drug response assessment.
7. Tyrosine kinase inhibitors of cMET
Tivantinib is a selective, non-ATP competitive,
small-molecule inhibitor of cMET. In vitro and in
vivo studies have demonstrated that ARQ-197 inhibits cMET
activation in numerous human gastric cancer cell lines and
xenografts (62). Recent evidence
suggests that tivantinib inhibits microtubule polymerization, in
addition to inhibiting cMET; thus, tivantinib exerts its antitumor
activity in a manner independent of the cMET status (63). In a single-arm phase II study on
Asian patients with previously treated metastatic gastric cancer,
30 patients received tivantinib; cMET gene amplification (5
copies/cell) was observed in four patients (13.3%), and the disease
control rate was 36.7% (11/30). The median PFS was 43 days (95% CI,
29.0–92.0). No objective response was observed. Grade 3 or 4
adverse events (AEs) occurred in 13 patients (43.3%), in whom
neutropenia (n=4) and anemia (n=4) were recognized to be
drug-related. Only two patients discontinued treatment due to AEs.
There were no treatment-related deaths and no new reported AEs. No
obvious correlation was identified between treatment outcome and
specific biomarkers, including cMET gene amplification, cMET,
p-cMET and HGF expression in tumor and serum (64). Currently, a phase I/II trial is
recruiting patients to evaluate the response rate of the
combination of tivantinib plus FOLFOX as first-line therapy for
metastatic gastroesophageal cancer (65).
Crizotinib is an ATP competitive small-molecule
inhibitor for cMET and anaplastic lymphoma kinase (ALK), which has
shown marked antitumor activity in vitro and in vivo,
specifically in gastric cancer cells positive for MET amplification
(66). A recent study followed up
four patients as part of an expanded phase I cohort study; two of
four patients with MET-amplified gastroesophageal cancer treated
with crizotinib experienced tumor shrinkage (−30 and −16%) and
experienced progression after 3.7 and 3.5 months. The research
group also assessed MET, EGFR and HER2 amplification status using
FISH in 489 patients with gastroesophageal cancer. The gene
amplification rate of MET, EGFR and HER2 was 2, 4.7 and 8.9%,
respectively. The majority (84%) of the samples were wild-type for
all three genes. Survival analysis in patients with stages III and
IV disease revealed that the cMET amplified group had lower
survival rates (7.1 months; P<0.001) than the EGFR amplified
group (11.2 months; P=0.16) and the HER2 amplified group (16.9
months; P=0.89) when compared with the negative group (16.2 months)
(67).
Foretinib (GSK1363089 or XL880) is an
oral multikinase inhibitor that primarily targets cMET and vascular
endothelial growth factor receptor 2 (VEGFR2). It can prevent tumor
growth through a direct effect on tumor cell proliferation and
through the inhibition of invasion and angiogenesis mediated by HGF
and VEGF receptors (68). In an
in vitro study, foretinib appeared effective against gastric
cancer cells harboring not only cMET but also FGFR2 amplification
(69).
In a phase II study evaluating two dosing schedules
of oral foretinib (GSK1363089) in 74 patients with metastatic
gastric cancer, the best response was SD in ten (23%) patients
receiving intermittent dosing and five (20%) receiving daily
dosing. SD duration ranged from 1.9 to 7.2 months (median 3.2
months). Of 67 patients with tumor samples, three had cMET
amplification, one of whom had SD. Treatment-related AEs occurred
in 91% of patients; the rates of hypertension (35 vs. 15%) and
elevated aspartate aminotransferase levels (23 vs. 8%) were higher
with intermittent dosing. In both patients with high baseline tumor
phospho-MET (p-MET), the p-MET: total MET protein ratio decreased
following treatment with foretinib. However, no responses were
observed in this patient cohort; this may perhaps be due to the
evaluation of a non-molecularly selected population (70). The efficient development of
targeted therapies that may only benefit a fraction of patients
requires clinical trial designs that use biomarkers to identify
sensitive subpopulations (71).
Cabozantinib (XL184) is an orally bioavailable TKI
with activity against MET and VEGFR2, AXL, KIT, TIE2, FLT3 and RET
signaling. It showed antimetastatic, antitumor and antiangiogenic
activity in preclinical models (72). A phase II randomized
discontinuation trial of cabozantinib enrolled 397 patients with
advanced solid tumors. In the gastric cohort, a total of 21
patients were enroled, 19 patients had evaluable responses. The
best response was SD achieved by eight patients, and the overall
disease control rate was 32% at 12 weeks. No objective response was
observed (73).
With a better understanding of the role of the cMET
pathway in cancer, a number of other cMET inhibitors are currently
in development. Some molecules have already been investigated in
phase I/II clinical trials in patients with advanced solid tumors,
such as OPB-31121 (74,75) MK-2461 (76) and EMD 1214063 (77). Some, including SGX523 (78), T-1840383 (79), F46 (80), E7050 (81) and h224G11 (82), have been shown to exert effects on
gastric cell lines and xenografts in preclinical studies (Table II).
8. Resistance to cMET inhibitors
The clinical efficacy of targeted therapy is
hindered by the emergence of primary and acquired resistance. In
the ToGA trial, the addition of trastuzumab to the chemotherapeutic
regimen only led to an absolute increase in response rate of 12%
(4), indicating the existence of
de novo resistance. Moreover, a large proportion of those
patients initially responsive to trastuzumab developed acquired
resistance. With the introduction of cMET inhibitors into the
clinical setting, the same question cannot be avoided. To date,
little is known about the mechanisms responsible for resistance to
cMET inhibitors.
An in vitro and in vivo study
indicated that gastric cancer tumors bearing constitutive
activation of HER family members responded poorly to MET inhibition
(83). cMET activation may
mediate resistance to EGFR and HER2 in gastric cancer (14,15). Another study observed that the
acquisition of a mutation in the MET activation loop (Y1230),
destabilized the autoinhibitory conformation of MET and abrogated
an important aromatic stacking interaction with the inhibitor
(84). In a recent study, a
cMET-sensitive gastric cancer cell line was chronically exposed to
the cMET inhibitor, PF-04217903. As a result, a novel SND1-BRAF
fusion was observed and proven to be responsible for the resistance
(85).
The RTK family accounts for a high percentage of the
potential treatable genomic-targeted map of gastric cancer
(5); the crosstalk between RTKs
may also play an important role in drug resistance. Moreover, the
prolonged exposure of a gastric cancer cell line to TKIs has been
shown to lead to amplification and overexpression of wild-type Kras
and to overcome the inhibitory effects of cMET TKIs (86). These data suggest that targeting
cMET may be crucial to overcoming potential resistance to other
agents in targeted therapy. Thus, close attention should be paid to
this issue during the development of cMET inhibitors.
9. Conclusion
Increasing evidence suggests that cMET plays a key
role in the development of gastric cancer. A total of 12.1% of
gastric cancer patients harbor gene amplification and 42.8% have
protein overexpression (Table I).
cMET protein overexpression and/or gene amplification have been
shown to significantly correlate with the depth of tumor invasion
and metastasis and poor prognosis (11,13). cMET inhibitors have been
investigated in clinical trials, with encouraging initial results
(16,17). On the basis of these findings,
cMET is considered to be a promising therapeutic target in gastric
cancer.
However, with the rapid development of cMET
inhibitors, a number of trials have been published which show less
than favorable outcomes (70,73). These results can largely be
attributed to a lack of appropriate biomarkers for patient
selection and drug response assessment. Moreover, while a
proportion of gastric cancers harbor cMET overexpression and/or
amplification, it is unclear whether the cMET alteration is acting
as an oncogenic driver or a passenger. Recent clinical trials have
been designed with molecular alterations of cMET, EGFR/RAS/RAF as
biomarkers (56,61,84). Future clinical trials may also
assess molecular derangements such as cMET mutation, K-ras
amplification, EGFR and HER2 status as predictive markers (14,15,83,84,86).
Drug resistance is another critical issue in the
development of cMET inhibitors that needs to be addressed. Combined
therapies against different pathways and at different levels may be
a feasible approach to settle this issue. PDTT xenograft models,
which can reliably mimic disease response in humans, is an ideal
platform to study biomarker selection and drug resistance (87). PDTT can be used as a drug
sensitivity screening platform and may provide reliable information
for the treatment of patients. Several cMET-positive PDTT models
have been established to research biomarker selection and drug
resistance. Interestingly, alpha-fetoprotein producing gastric
cancer (AFPGC) with high cMET expression was found in our PDTT
models. Previous studies have also reported a higher frequency of
cMET expression in AFPGC compared with advanced gastric cancer
(88). We are currently using a
PDTT model to investigate whether AFPGC is a special subgroup for
cMET.
cMET is a promising target in gastric cancer, and it
is important to determine the specific subpopulations that are
likely to derive the greatest benefit from cMET inhibition.
Therefore, future studies should focus on the exploration of
biomarkers to optimize patient selection and drug response
assessment.
References
|
1
|
Garcia M, Jemal A, Ward EM, et al: Global
Cancer Facts and Figures 2007. GA: American Cancer Society;
Atlanta: 2007
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
3
|
Verdecchia A, Santaquilani M and Sant M:
Survival for cancer patients in Europe. Ann Ist Super Sanita.
45:315–324. 2009.PubMed/NCBI
|
|
4
|
Bang YJ, Van Cutsem E, Feyereislova A, et
al: Trastuzumab in combination with chemotherapy versus
chemotherapy alone for treatment of HER2-positive advanced gastric
or gastro-oesophageal junction cancer (ToGA): a phase 3,
open-label, randomised controlled trial. Lancet. 376:687–697. 2010.
View Article : Google Scholar
|
|
5
|
Deng N, Goh LK, Wang H, et al: A
comprehensive survey of genomic alterations in gastric cancer
reveals systematic patterns of molecular exclusivity and
co-occurrence among distinct therapeutic targets. Gut. 61:673–684.
2012. View Article : Google Scholar
|
|
6
|
Bussolino F, Di Renzo MF, Ziche M, et al:
Hepatocyte growth factor is a potent angiogenic factor which
stimulates endothelial cell motility and growth. J Cell Biol.
119:629–641. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang YW, Su Y, Volpert OV and Vande Woude
GF: Hepatocyte growth factor/scatter factor mediates angiogenesis
through positive VEGF and negative thrombospondin 1 regulation.
Proc Natl Acad Sci USA. 100:12718–12723. 2003. View Article : Google Scholar
|
|
8
|
Jeffers M, Fiscella M, Webb CP, Anver M,
Koochekpour S and Vande Woude GF: The mutationally activated Met
receptor mediates motility and metastasis. Proc Natl Acad Sci USA.
95:14417–14422. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Webb CP, Taylor GA, Jeffers M, et al:
Evidence for a role of Met-HGF/SF during Ras-mediated
tumorigenesis/metastasis. Oncogene. 17:2019–2025. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gherardi E, Birchmeier W, Birchmeier C and
Vande Woude G: Targeting MET in cancer: rationale and progress. Nat
Rev Cancer. 12:89–103. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nakajima M, Sawada H, Yamada Y, et al: The
prognostic significance of amplification and overexpression of cMET
and c-erb B-2 in human gastric carcinomas. Cancer. 85:1894–1902.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee J, Seo JW, Jun HJ, et al: Impact of
MET amplification on gastric cancer: Possible roles as a novel
prognostic marker and a potential therapeutic target. Oncol Rep.
25:1517–1524. 2011.PubMed/NCBI
|
|
13
|
Tang Z, Zhao M, Ji J, et al:
Overexpression of gastrin and cMET protein involved in human
gastric carcinomas and intestinal metaplasia. Oncol Rep.
11:333–339. 2004.PubMed/NCBI
|
|
14
|
Chen CT, Kim H, Liska D, Gao S,
Christensen JG and Weiser MR: MET activation mediates resistance to
lapatinib inhibition of HER2-amplified gastric cancer cells. Mol
Cancer Ther. 11:660–669. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kneissl J, Keller S, Lorber T, et al:
Association of amphiregulin with the cetuximab sensitivity of
gastric cancer cell lines. Int J Oncol. 41:733–744. 2012.PubMed/NCBI
|
|
16
|
Oliner KD, Tang R, Anderson A, et al:
Evaluation of MET pathway biomarkers in a phase II study of
rilotumumab (R, AMG 102) or placebo (P) in combination with
epirubicin, cisplatin, and capecitabine (ECX) in patients (pts)
with locally advanced or metastatic gastric (G) or esophagogastric
junction (EGJ) cancer. Proc ASCO abs. 4005. 2012.
|
|
17
|
Catenacci DV, Henderson L, Xiao SY, et al:
Durable complete response of metastatic gastric cancer with
anti-Met therapy followed by resistance at recurrence. Cancer
Discov. 1:573–579. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cooper CS, Park M, Blair DG, et al:
Molecular cloning of a new transforming gene from a chemically
transformed human cell line. Nature. 311:29–33. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Park M, Dean M, Cooper CS, et al:
Mechanism of met oncogene activation. Cell. 45:895–904. 1986.
View Article : Google Scholar
|
|
20
|
Park M, Dean M, Kaul K, Braun MJ, Gonda MA
and Vande Woude G: Sequence of MET protooncogene cDNA has features
characteristic of the tyrosine kinase family of growth-factor
receptors. Proc Natl Acad Sci USA. 84:6379–6383. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bottaro DP, Rubin JS, Faletto DL, et al:
Identification of the hepatocyte growth factor receptor as the cMET
proto-oncogene product. Science. 251:802–804. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nakamura T, Nishizawa T, Hagiya M, et al:
Molecular cloning and expression of human hepatocyte growth factor.
Nature. 342:440–443. 1989. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Stoker M, Gherardi E, Perryman M and Gray
J: Scatter factor is a fibroblast-derived modulator of epithelial
cell mobility. Nature. 327:239–242. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Peschard P and Park M: From Tpr-Met to
Met, tumorigenesis and tubes. Oncogene. 26:1276–1285. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ponzetto C, Bardelli A, Zhen Z, et al: A
multifunctional docking site mediates signaling and transformation
by the hepatocyte growth factor/scatter factor receptor family.
Cell. 77:261–271. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nguyen L, Holgado-Madruga M, Maroun C, et
al: Association of the multisubstrate docking protein Gab1 with the
hepatocyte growth factor receptor requires a functional Grb2
binding site involving tyrosine 1356. J Biol Chem. 272:20811–20819.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mughal A, Aslam HM, Khan AM, Saleem S,
Umah R and Saleem M: Bcr-Abl tyrosine kinase inhibitors-current
status. Infect Agent Cancer. 8:232013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Peters S and Adjei AA: MET: a promising
anticancer therapeutic target. Nat Rev Clin Oncol. 9:314–326. 2012.
View Article : Google Scholar
|
|
29
|
Yamamoto N, Mammadova G, Song RX, Fukami Y
and Sato K: Tyrosine phosphorylation of p145met mediated by EGFR
and Src is required for serum-independent survival of human bladder
carcinoma cells. J Cell Sci. 119:4623–4633. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Khoury H, Naujokas MA, Zuo D, et al: HGF
converts ErbB2/Neu epithelial morphogenesis to cell invasion. Mol
Biol Cell. 16:550–561. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bauer TW, Somcio RJ, Fan F, et al:
Regulatory role of cMET in insulin-like growth factor-I
receptor-mediated migration and invasion of human pancreatic
carcinoma cells. Mol Cancer Ther. 5:1676–1682. 2006. View Article : Google Scholar
|
|
32
|
Uehara Y, Minowa O, Mori C, et al:
Placental defect and embryonic lethality in mice lacking hepatocyte
growth factor/scatter factor. Nature. 373:702–705. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Woolf AS, Kolatsi-Joannou M, Hardman P, et
al: Roles of hepatocyte growth factor/scatter factor and the met
receptor in the early development of the metanephros. J Cell Biol.
128:171–184. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bladt F, Riethmacher D, Isenmann S, Aguzzi
A and Birchmeier C: Essential role for the cMET receptor in the
migration of myogenic precursor cells into the limb bud. Nature.
376:768–771. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang YW and Vande Woude GF: HGF/SF-met
signaling in the control of branching morphogenesis and invasion. J
Cell Biochem. 88:408–417. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chmielowiec J, Borowiak M, Morkel M, et
al: cMET is essential for wound healing in the skin. J Cell Biol.
177:151–162. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Giordano S, Ponzetto C, Di Renzo MF,
Cooper CS and Comoglio PM: Tyrosine kinase receptor
indistinguishable from the cMET protein. Nature. 339:155–156. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Soman NR, Correa P, Ruiz BA and Wogan GN:
The TPR-MET oncogenic rearrangement is present and expressed in
human gastric carcinoma and precursor lesions. Proc Natl Acad Sci
USA. 88:4892–4896. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yu J, Miehlke S, Ebert MP, et al:
Frequency of TPR-MET rearrangement in patients with gastric
carcinoma and in first-degree relatives. Cancer. 88:1801–1806.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee HE, Kim MA, Lee HS, et al: MET in
gastric carcinomas: comparison between protein expression and gene
copy number and impact on clinical outcome. Br J Cancer.
107:325–333. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Janjigian YY, Tang LH, Coit DG, et al: MET
expression and amplification in patients with localized gastric
cancer. Cancer Epidemiol Biomarkers Prev. 20:1021–1027. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Retterspitz MF, Monig SP, Schreckenberg S,
et al: Expression of {beta}-catenin, MUC1 and cMET in diffuse-type
gastric carcinomas: correlations with tumour progression and
prognosis. Anticancer Res. 30:4635–4641. 2010.
|
|
43
|
Park WS, Oh RR, Kim YS, et al: Absence of
mutations in the kinase domain of the Met gene and frequent
expression of Met and HGF/SF protein in primary gastric carcinomas.
APMIS. 108:195–200. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tsugawa K, Yonemura Y, Hirono Y, et al:
Amplification of the cMET, c-erbB-2 and epidermal growth factor
receptor gene in human gastric cancers: correlation to clinical
features. Oncology. 55:475–481. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Graziano F, Galluccio N, Lorenzini P, et
al: Genetic activation of the MET pathway and prognosis of patients
with high-risk, radically resected gastric cancer. J Clin Oncol.
29:4789–4795. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shi J, Yao D, Liu W, et al: Frequent gene
amplification predicts poor prognosis in gastric cancer. Int J Mol
Sci. 13:4714–4726. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Schmidt L, Duh FM, Chen F, et al: Germline
and somatic mutations in the tyrosine kinase domain of the MET
proto-oncogene in papillary renal carcinomas. Nat Genet. 16:68–73.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Olivero M, Valente G, Bardelli A, et al:
Novel mutation in the ATP-binding site of the MET oncogene tyrosine
kinase in a HPRCC family. Int J Cancer. 82:640–643. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lee JH, Han SU, Cho H, et al: A novel germ
line juxtamembrane Met mutation in human gastric cancer. Oncogene.
19:4947–4953. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Asaoka Y, Tada M, Ikenoue T, et al:
Gastric cancer cell line Hs746T harbors a splice site mutation of
cMET causing juxtamembrane domain deletion. Biochem Biophys Res
Commun. 394:1042–1046. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Burgess T, Coxon A, Meyer S, et al: Fully
human monoclonal antibodies to hepatocyte growth factor with
therapeutic potential against hepatocyte growth
factor/cMET-dependent human tumors. Cancer Res. 66:1721–1729. 2006.
View Article : Google Scholar
|
|
52
|
Jun HT, Sun J, Rex K, et al: AMG 102, a
fully human anti-hepatocyte growth factor/scatter factor
neutralizing antibody, enhances the efficacy of temozolomide or
docetaxel in U-87 MG cells and xenografts. Clin Cancer Res.
13:6735–6742. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gordon MS, Sweeney CS, Mendelson DS, et
al: Safety, pharmacokinetics, and pharmacodynamics of AMG 102, a
fully human hepatocyte growth factor-neutralizing monoclonal
antibody, in a first-in-human study of patients with advanced solid
tumors. Clin Cancer Res. 16:699–710. 2010. View Article : Google Scholar
|
|
54
|
Iveson T, Donehower RC, Davidenko I, et
al: 6504 ORAL safety and efficacy of epirubicin, cisplatin, and
capecitabine (ECX) plus rilotumumab (R) as first-line treatment for
unresectable locally advanced (LA) or metastatic (M) gastric or
esophagogastric junction (EGJ) adenocarcinoma. Eur J Cancer.
47:S4432011. View Article : Google Scholar
|
|
55
|
First-line treatment for locally advanced
or metastatic mesenchymal epithelial transition factor
(MET)-positive gastric, lower esophageal, or gastroesophageal
junction (GEJ) adenocarcinoma (RILOMET-1). http://www.clinicaltrials.gov/ct2/show/NCT01697072.
Accessed May 17, 2013
|
|
56
|
MEGA (Met or EGFR inhibition in
gastroesophageal adenocarcinoma). FOLFOX alone or in combination
with AMG 102 or Panitumumab as first-line treatment in patients
with advanced gastroesophageal adenocarcinoma. http://www.clinicaltrials.gov/ct2/show/NCT01443065.
Accessed May 17, 2013
|
|
57
|
Martens T, Schmidt NO, Eckerich C, et al:
A novel one-armed anti-cMET antibody inhibits glioblastoma growth
in vivo. Clin Cancer Res. 12:6144–6152. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Penuel E, Li C, Parab V, et al: HGF as a
Circulating biomarker of onartuzumab treatment in patients with
advanced solid tumors. Mol Cancer Ther. 12:1122–1130. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Jagoda EM, Lang L, Bhadrasetty V, et al:
Immuno-PET of the hepatocyte growth factor receptor Met using the
1-armed antibody onartuzumab. J Nucl Med. 53:1592–1600. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
A study of Onartuzumab (MetMAb) in
combination with mFOLFOX6 in patients with metastatic HER2-negative
gastroesophageal cancer. http://clinicaltrials.gov/ct2/show/NCT01590719.
Accessed May 15, 2013
|
|
61
|
A study of Onartuzumab (MetMAb) in
combination with mFOLFOX6 in patients with metastatic HER2-negative
and Met-positive gastroesophageal cancer (MetGastric). http://www.clinicaltrials.gov/ct2/show/NCT01662869.
Accessed May 15, 2013
|
|
62
|
Munshi N, Jeay S, Li Y, et al: ARQ 197, a
novel and selective inhibitor of the human cMET receptor tyrosine
kinase with antitumor activity. Mol Cancer Ther. 9:1544–1553. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Katayama R, Aoyama A, Yamori T, et al:
Cytotoxic activity of tivantinib (ARQ 197) is not due solely to
cMET inhibition. Cancer Res. 73:3087–3096. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Muro K, Ryu M-H, Yasui H, et al: A phase
II study of tivantinib monotherapy in patients with previously
treated advanced or recurrent gastric cancer. Proc ASCO abs.x 4082.
2012.
|
|
65
|
Phase I/II trial of Tivantinib with FOLFOX
for the treatment of advanced solid tumors and previously untreated
metastatic adenocarcinoma of the distal esophagus, gastroesophageal
junction or stomach. http://www.clinicaltrials.gov/ct2/show/NCT01611857.
Accessed May 15, 2013
|
|
66
|
Okamoto W, Okamoto I, Arao T, et al:
Antitumor action of the MET tyrosine kinase inhibitor crizotinib
(PF-02341066) in gastric cancer positive for MET amplification. Mol
Cancer Ther. 11:1557–1564. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lennerz JK, Kwak EL, Ackerman A, et al:
MET amplification identifies a small and aggressive subgroup of
esophagogastric adenocarcinoma with evidence of responsiveness to
crizotinib. J Clin Oncol. 29:4803–4810. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Qian F, Engst S, Yamaguchi K, et al:
Inhibition of tumor cell growth, invasion, and metastasis by
EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF
receptor tyrosine kinases. Cancer Res. 69:8009–8016. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kataoka Y, Mukohara T, Tomioka H, et al:
Foretinib (GSK1363089), a multi-kinase inhibitor of MET and VEGFRs,
inhibits growth of gastric cancer cell lines by blocking
inter-receptor tyrosine kinase networks. Invest New Drugs.
30:1352–1360. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Shah MA, Wainberg ZA, Catenacci DV, et al:
Phase II study evaluating 2 dosing schedules of oral foretinib
(GSK1363089), cMET/VEGFR2 inhibitor, in patients with metastatic
gastric cancer. PLoS One. 8:e540142013. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Freidlin B, McShane LM, Polley MY and Korn
EL: Randomized phase II trial designs with biomarkers. J Clin
Oncol. 30:3304–3309. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Yakes FM, Chen J, Tan J, et al:
Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor,
simultaneously suppresses metastasis, angiogenesis, and tumor
growth. Mol Cancer Ther. 10:2298–2308. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Schoffski P, Sgroi M, Burris HA, Lutzky J,
Rearden T, Sikic B, et al: Phase 2 randomized discontinuation trial
(RDT) of XL184 in patients (pts) with advanced solid tumors.
EORTC-NCI-AACR Symposium on Molecular Targets and Cancer
Therapeutics EJC Supplements. 8:1172010.
|
|
74
|
Kim MJ, Nam HJ, Kim HP, et al: OPB-31121,
a novel small molecular inhibitor, disrupts the JAK2/STAT3 pathway
and exhibits an antitumor activity in gastric cancer cells. Cancer
Lett. 335:145–152. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Oh D, Han S, Kim TM, et al: A phase I,
open-label, nonrandomized trial of OPB-31121, a STAT3 inhibitor, in
patients with advanced solid tumors. J Clin Oncol.
28:e130562010.
|
|
76
|
Camacho LH, Moulder SL, LoRusso PM, et al:
First in human phase I study of MK-2461, a small molecule inhibitor
of cMET, for patients with advanced solid tumors. J Clin Oncol.
26(Suppl 15): abs. 14657. 2008.
|
|
77
|
Falchook GS, Fu S, Amin HM, Piha-Paul SA,
Hong DS, Naing A, et al: 1245 Poster Phase I dose-escalation study
of the oral selective C-Met inhibitor EMD 1204831 in patients with
advanced solid tumours. Eur J Cancer. 47:S1582011.
|
|
78
|
Buchanan SG, Hendle J, Lee PS, et al:
SGX523 is an exquisitely selective, ATP-competitive inhibitor of
the MET receptor tyrosine kinase with antitumor activity in vivo.
Mol Cancer Ther. 8:3181–3190. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Awazu Y, Nakamura K, Mizutani A, et al: A
Novel Inhibitor of cMET and VEGF Receptor Tyrosine Kinases with a
Broad Spectrum of In Vivo Antitumor Activities. Mol Cancer Ther.
12:913–924. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Oh YM, Song YJ, Lee SB, et al: A new
anti-cMET antibody selected by a mechanism-based dual-screening
method: therapeutic potential in cancer. Mol Cells. 34:523–529.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Nakagawa T, Tohyama O, Yamaguchi A, et al:
E7050: a dual cMET and VEGFR-2 tyrosine kinase inhibitor promotes
tumor regression and prolongs survival in mouse xenograft models.
Cancer Sci. 101:210–215. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Goetsch L, Gonzalez A, Geronimi F,
Fabre-Lafay S, et al: Single or combined in vivo therapies of
cancer with h224G11, a humanized antibody targeting the cMET
receptor. Mol Cancer Ther. 8:B1272009. View Article : Google Scholar
|
|
83
|
Corso S, Ghiso E, Cepero V, et al:
Activation of HER family members in gastric carcinoma cells
mediates resistance to MET inhibition. Mol Cancer. 9:1212010.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Qi J, McTigue MA, Rogers A, et al:
Multiple mutations and bypass mechanisms can contribute to
development of acquired resistance to MET inhibitors. Cancer Res.
71:1081–1091. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Lee NV, Lira ME, Pavlicek A, et al: A
novel SND1-BRAF fusion confers resistance to cMET inhibitor
PF-04217903 in GTL16 cells through [corrected] MAPK activation.
PLoS One. 7:e396532012.PubMed/NCBI
|
|
86
|
Cepero V, Sierra JR, Corso S, et al: MET
and KRAS gene amplification mediates acquired resistance to MET
tyrosine kinase inhibitors. Cancer Res. 70:7580–7590. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Marangoni E, Vincent-Salomon A, Auger N,
et al: A new model of patient tumor-derived breast cancer
xenografts for preclinical assays. Clin Cancer Res. 13:3989–3998.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Amemiya H, Kono K, Mori Y, et al: High
frequency of cMET expression in gastric cancers producing
alpha-fetoprotein. Oncology. 59:145–151. 2000. View Article : Google Scholar : PubMed/NCBI
|