Introduction
Lysophosphatidic acid (LPA), which is released from
activated platelets (1) and the
hydrolysis of circulating lysophospholipids (2), is a simple phospholipid that is
involved in a number of cell processes including proliferation,
migration, platelet aggregation, neurite retraction and neuropathic
pain (1,3–6).
LPA is detected in serum, plasma, other biological fluids, and
tissues including brain (3). It
is produced in the course of blood coagulation in humans (7). Levels of LPA are increased after
brain injury, including intracerebral hemorrhage (ICH) (8) and cerebral ischemia (CI) (9). LPA is also produced by activated
platelets and stimulates platelet aggregation in turn, forming a
positive feedback and causing significantly elevated levels of LPA
in a local region of the brain, resulting in clotting hematoma in
ICH or thrombosis in CI.
Blood-brain barrier (BBB) disruption and brain edema
is an important pathophysiological event of brain injury after ICH
or CI. Proteolytic enzymes and inflammatory cytokines change the
BBB permeability properties, leading to brain edema formation.
Various proteolytic enzymes, including matrix metalloproteinase
(MMP)-9 and urokinase-type plasminogen activator (uPA) have been
suggested to be critical mediators for altering BBB permeability
(10). MMP-9 degrades the
basement membrane, and uPA stimulates the production of plasmin
that degrades various components of extracellular matrix. Previous
studies have indicated that LPA is involved in increasing
endothelial monolayer permeability (11), and that a high concentration of
LPA may disrupt the BBB (8).
However, the exact molecular mechanism that serves as a
contributory factor of LPA to BBB disruption remains to be
determined.
The Rho/Rho kinase (ROCK) signaling pathway is
important in the regulation of endothelial permeability. ROCK, the
downstream effector of Rho, is a serine/threonine kinase that is
activated by binding to the active GTP-bound form of Rho. Results
of recent studies have demonstrated that ROCK increases BBB
permeability via the upregulation of MMP-9 and disruption of tight
junction proteins (12,13). The ROCK inhibitor prevents injury
to brain endothelial cells by the reduction of MMP-9 activity
(14). Mounting evidence supports
that the Rho/ROCK signaling pathway is crucial in cancer cell
migration and invasion induced by LPA, which is due to the
upregulation of proteolytic enzyme secretion by Rho/ROCK signaling
pathway (15). Considering that
LPA is capable of increasing BBB permeability, and that the
Rho/ROCK pathway plays a role in LPA-induced proteolytic enzymes
secretion, including uPA and MMPs, we hypothesized that LPA-induced
ROCK activation is involved in proteolytic enzyme secretion,
leading to the disruption of BBB in vivo.
The aim of the present study was to examine whether
LPA induces the secretion of proteolytic enzymes MMP-9 and uPA, to
increase the permeability of BBB in vivo, and whether ROCK
is critical for LPA-induced proteolytic enzyme expression as well
as BBB disruption. The results obtained suggest mechanisms by which
LPA increases permeability of BBB through proteolytic enzyme
secretion mediated by a Rho/ROCK signaling pathway.
Materials and methods
Animals
Adult male Sprague-Dawley rats (weight, 250–280 g)
obtained from the Animal Center of Wuhan University were housed
under controlled temperature and lighting conditions with food and
water. All animal procedures were reviewed and approved by the
international guidelines for the ethical use of laboratory animals
and the National Institutes of Health Guide for the Care and Use of
Laboratory Animals.
Surgical procedure
Animals were anesthetized by intraperitoneal
injection of ketamine (60 mg/kg) and xylazine (10 mg/kg), and then
placed in a stereotaxic frame (Bilaney Consultants, Düsseldorf,
Germany). A scalpel was used to expose the skull, and a hole was
drilled at 3.0 mm laterally and 0.2 mm anterior to the bregma. A
microinjection needle was inserted stereotaxically into the right
caudate nucleus (coordinates: 0.2 mm anterior, 5.5 mm ventral and
3.0 mm lateral to the bregma). LPA (Sigma, St. Louis, MO, USA) was
dissolved in sterile phosphate-buffered saline (PBS) and a final
concentration of 100 μM was delivered in a final volume of 10 μl
over 5 min; the needle remained in place for an additional 5 min to
prevent reflux. ROCK inhibitor Y27632 (Sigma) was dissolved in
sterile PBS at a final concentration of 1 mM and co-injected with
LPA into the right caudate nucleus in some experiments to detect
whether Y27632 decreased BBB permeability by inhibiting ROCK. The
hole in the skull was sealed with bone wax and the scalp was
sutured. Control rats were infused with the same volume of PBS, and
the brains were removed 24 h after microinjection. Rectal
temperature was monitored and maintained between 36.5 and 37°C with
a heating pad throughout the procedure. The overall mortality rate
was <1%. Striatum tissues in each group were obtained as
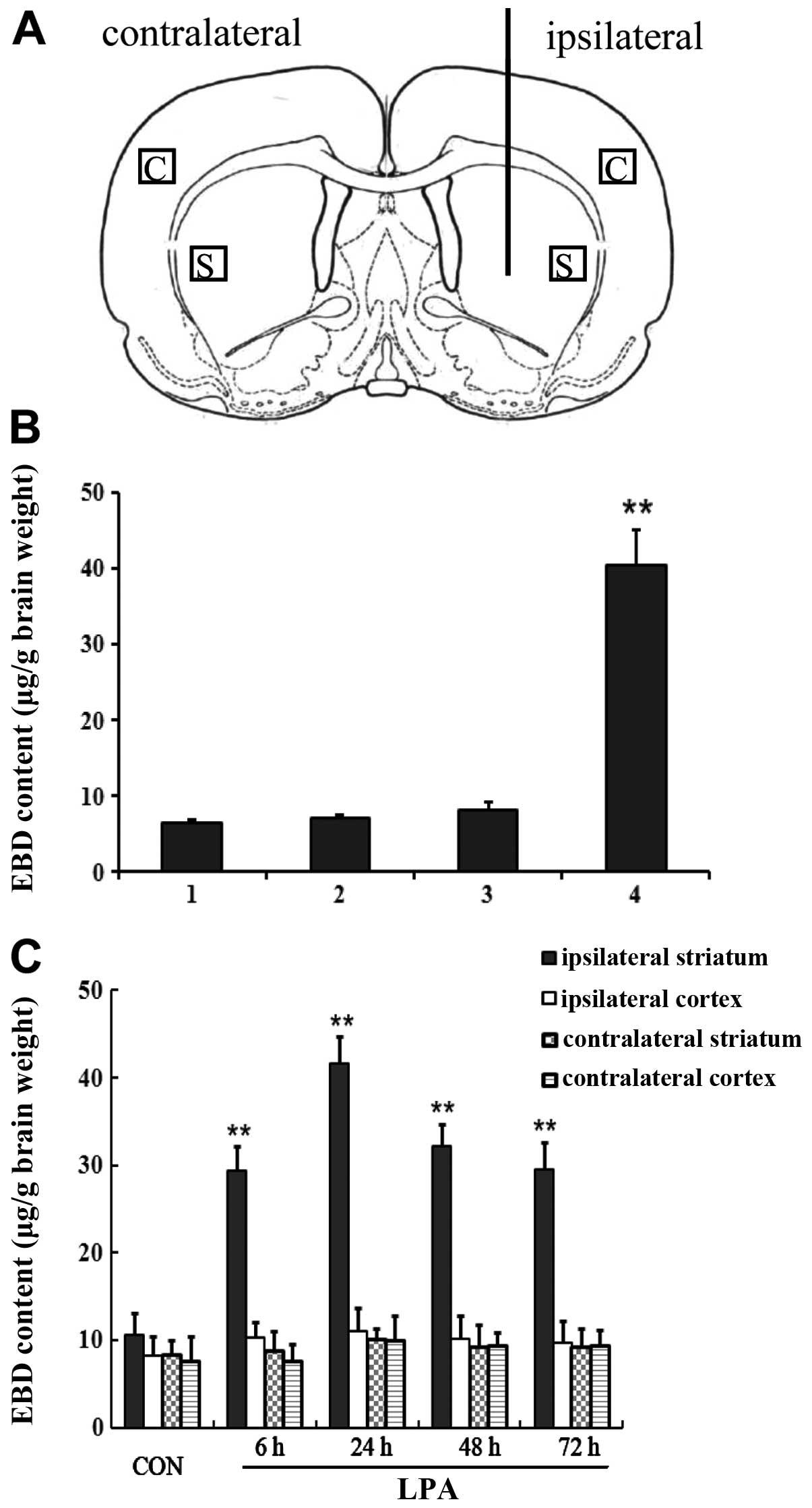
described in Fig. 1 for
subsequent detection.
Experimental groups
Animals (150 rats) were randomly assigned to each
group. Six rats used for the ultrastructural analysis of BBB were
sacrificed 24 h after surgery as well as controls (n=3 for each
group). Thirty-six rats used for determination of extravasated
Evans blue dye (EBD) for BBB integrity; 36 rats for quantitative
polymerase chain reaction (qPCR); 36 rats for western blot
detection; 36 rats for the immunohistochemistry evaluation (n=6 for
each time-point at 6, 24, 48 and 72 h after LPA was intracerebrally
injected and 24 h after LPA co-injected with Y27632 as well as for
controls).
Evans blue dye extravasation
BBB permeability was quantitatively evaluated by the
fluorescent detection of extravasated EBD at 6, 24, 48 and 72 h
after injection of LPA into the right caudate nucleus of rats
(16). Briefly, 2% EBD in saline
was injected from tail vein (4 ml/kg; Sigma) and allowed to
circulate for 60 min. To remove the intravascular dye, the animals
were perfused with saline through the left ventricle at 100 cm
H2O until colorless perfusion fluid was obtained from
the right atrium. After rats were decapitated, four sections of the
brains were removed: ipsilateral striatum and cortex, and
contralateral striatum and cortex (data not shown). Each tissue
sample was weighed, homogenized in 2 ml of 50% trichloroacetic acid
(wt/vol), and centrifuged at 18,000 × g for 20 min. EBD was
extracted from the tissue by using 50% trichloroacetic acid to
dissociate the dye from protein. Subsequent to centrifugation, the
supernatants containing EBD were diluted 4-fold with ethanol. For
fluorescence measurement, an aliquot was diluted with solvent (50%
trichloroacetic acid:ethanol =1:3). Tissue levels of EBD were
quantified by using a spectrofluorometer at an excitation
wavelength of 620 nm and an emission wavelength of 680 nm. Sample
values were compared with those of EBD standards mixed with the
solvent (100–1,000 μg/l). The EBD contents were expressed as μg/g
brain weight.
Ultrastructural analysis
Rats were perfused through the left ventricle with
saline followed by 2.5% glutaraldehyde and 2% paraformaldehyde in
0.1 M cacodylate buffer at pH 7.4. Tissue sections from the
striatum were additionally fixed in the same fixative for 2 h at
4°C. After washing, the slices were post-fixed in 1%
OsO4 for 1 h. The slices were subsequently dehydrated in
graded ethanol, and embedded in Epon 812. For electron microscopy,
ultrathin sections were processed by cutting with a diamond knife
on a Ultramicrotome (Leica Ultracut UCT; Leica Microsystems GmbH,
Wetzlar, Germany) and then collected on copper grids. The material
was air-dried, then stained for 10 min with 4.7% uranyl acetate and
for 2 min with lead citrate. The sections were examined and images
were captured on a Philips G212 electron microscope (FEI Tecnai;
FEI, Amsterdam, The Netherlands).
qPCR
Animals were sacrificed at 6, 24, 48 and 72 h
following LPA injection into the right caudate nucleus. Total RNA
was extracted from the right striatum tissues using TRIzol
(Invitrogen, Carlsbad, CA, USA) following the manufacturer’s
instructions. The concentration and purity were measured using the
NanoDrop™ method (3300 NanoDrop Analyzer; Thermo Scientific, Foster
City, CA, USA). Equal amounts (1 μg) of total RNA were reverse
transcribed into cDNA using a first-strand cDNA synthesis kit
(Transgen Biotech Co., Ltd., Beijing, China). PCR was performed
with primers for MMP-9, forward: 5′-ACGAGGACTCCCCTCTGCAT-3′ and
reverse: 5′-AGGCCTTGGGTCAGGTTTAGA-3′; uPA, forward:
5′-ACAGATTCCTGCTCGGGAGAT-3′ and reverse:
5′-CCAATGTGGGACTGAATCCAG-3′; ROCK2 forward:
5′-AACCTACTCCTGGAAGCCG-3′ and reverse: 5′-AGACAACGCTTCTGAGTTTCC-3′;
glyceraldehyde 3-phosphate dehydrogenase (GAPDH), forward: 5′-CAGT
GCCAGCCTCGTCTCAT-3′ and reverse: 5′-TGGTAACCA GGCGTCCGATA-3′).
Following an initial incubation for 15 min at 95°C, the reactions
were carried out for 40 cycles at 95°C for 15 sec and 60°C for 30
sec (florescence collection). The expression of MMP-9, uPA and
ROCK2 was normalized to GAPDH gene and compared with the
control.
Western blotting
The brains of rat were removed at 6, 24, 48 and 72 h
after LPA injection, and the right striatum was dissected,
immediately frozen and kept at −70°C. The frozen tissue was
homogenized in homogenization buffer (pH 7.4) with protease
inhibitors (Sigma) and centrifuged at 12,000 × g for 20 min at 4°C.
The supernatant was then collected and the total protein was
determined using a Micro BCA Protein Assay kit. Protein (50 μg) of
each sample was loaded onto the 8–10% SDS polyacrylamide gel
(SDS-PAGE), separated by electrophoresis and transferred to
nitrocellulose membranes (EMD Millipore Co., Billerica, MA, USA).
The membranes were incubated at 4°C overnight with primary
antibodies: anti-MMP-9 (goat polyclonal, 1:400), anti-uPA (rabbit
polyclonal, 1:400), anti-ROCK2 (goat polyclonal, 1:100) and
anti-β-actin (mouse monoclonal, 1:500), all from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). After being washed three
times for 10 min with washing solution, the membranes were
incubated with horseradish peroxidase-conjugated secondary
antibodies (1:1,000; ZSGB-Bio, Beijing, China). Immunoreactive
bands were visualized by an enhanced chemiluminescent substrate kit
and exposure to CL-XPosure film. The immunoreactivity of proteins
bands was quantitatively analyzed by Kodak Digital Science 1D
software (Eastman Kodak Co., Rochester, NY, USA) and expressed as a
mean optical density. Relative levels of MMP-9, uPA and ROCK2
protein were normalized to β-actin and compared with the
control.
Immunohistochemical staining
To assess the spatial distribution of MMP-9 and uPA
after LPA injection, immunohistochemistry was performed. Briefly,
the brain was dehydrated and embedded in paraffin. Then it was
sliced coronally into 4 μm sections from the rostral to the caudal
section of the injection site. The sections were de-waxed and
rehydrated, rinsed with distilled water and PBS, quenched with 3%
H2O2, blocked in 10% normal goat serum, and
incubated overnight at 4°C with primary antibodies: anti-MMP-9
(goat polyclonal, 1:100) or anti-uPA (rabbit polyclonal, 1:100)
both from Santa Cruz Biotechnology, Inc. The sections were then
incubated with biotinylated goat anti-mouse IgG or rabbit anti-goat
IgG (1:400; ZSGB-Bio) for 1 h at room temperature and incubated
with streptavidin-peroxidase for 30 min. The immunoreactions were
visualized with diaminobenzidine-H2O2
solution. Sections were washed, successively dehydrated in ethanol,
and defatted in xylenes. For the negative controls we used
non-specific IgG instead of the primary antibodies. To quantify the
number of immunoreactive cells labeled with MMP-9 or uPA, two
sections per rat were selected, and five randomly selected
non-overlapping high-power fields were examined from each
section.
Statistical analysis
Data were expressed as the means ± SEM. Statistical
analysis of quantified data was performed by analysis of variance
(ANOVA). P<0.05 was considered to indicate a statistically
significant difference.
Results
LPA increased the permeability of BBB in
ipsilateral striatum
To study the effect of LPA on the permeability of
BBB, it was injected directly into the rat brain. The extravasation
of EBD, as a marker of BBB breakdown, was quantified by
spectrofluorophotometric analysis. Since the striatum tissues for
the subsequent detection was obtained at a distance from the
injection site (Figs. 1A and
2A), the needle itself and the
volume of injected solution had no effect on the permeability of
BBB (Fig. 1B). No significant
difference on the extravasation of EBD in the ipsilateral cortex,
contralateral striatum and contralateral cortex between
LPA-injected rats and control rats was observed (Fig. 1C). Quantitative analysis of EBD in
another three regions demonstrated that leakage within the
ipsilateral striatum regions following LPA injection was highest
among the ipsilateral cortex and contralateral brain regions,
including the contralateral striatum and contralateral cortex
(Fig. 1C). We found that the
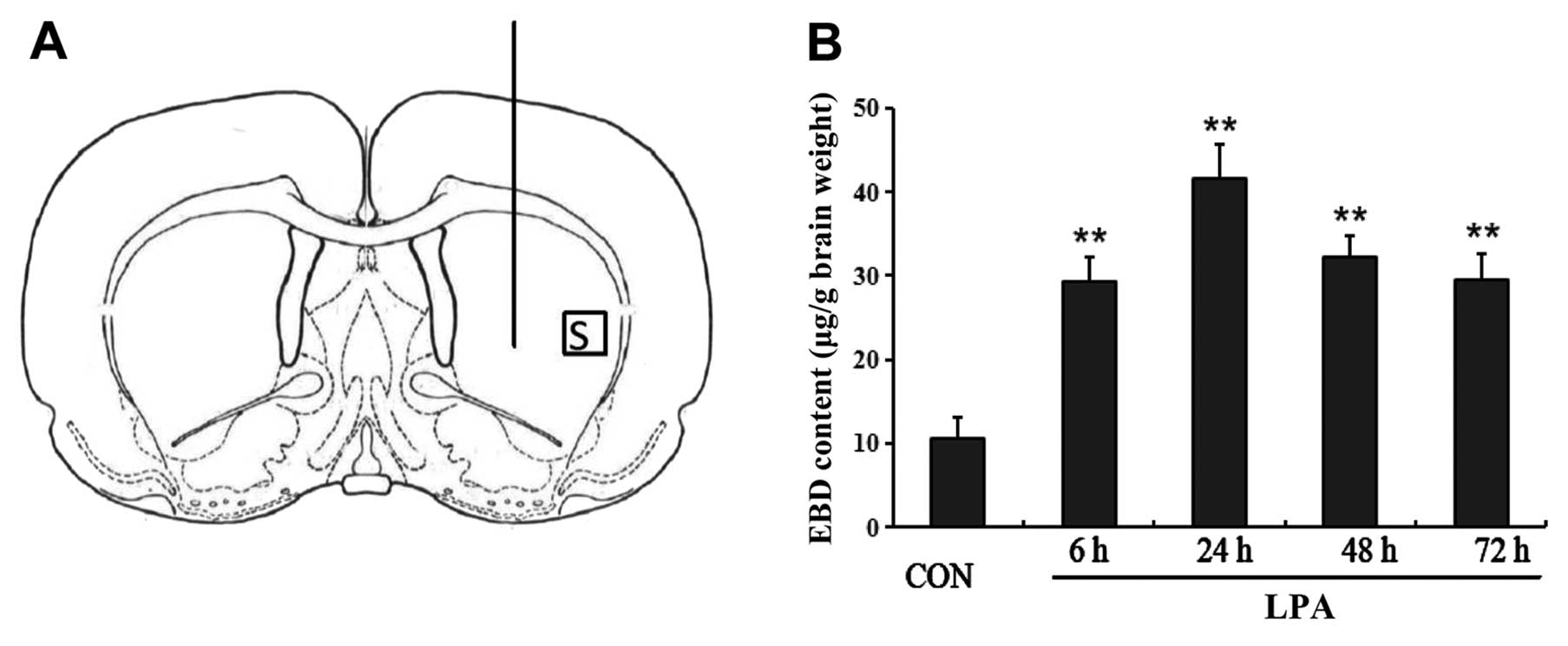
intracerebral injection of LPA resulted in an increase in the
permeability of BBB over time. LPA induced an increase in EBD
extravasation in the injected striatum, which began to increase 6 h
after LPA injection and peaked at 24 h, whereas it decreased at 48
and 72 h after LPA was injected intracerebtally (Fig. 2B). Significant differences were
noted in the extravasation of EBD in the ipsilateral striatum from
6 to 72 h after LPA injection compared with controls (P<0.01)
(Fig. 2B).
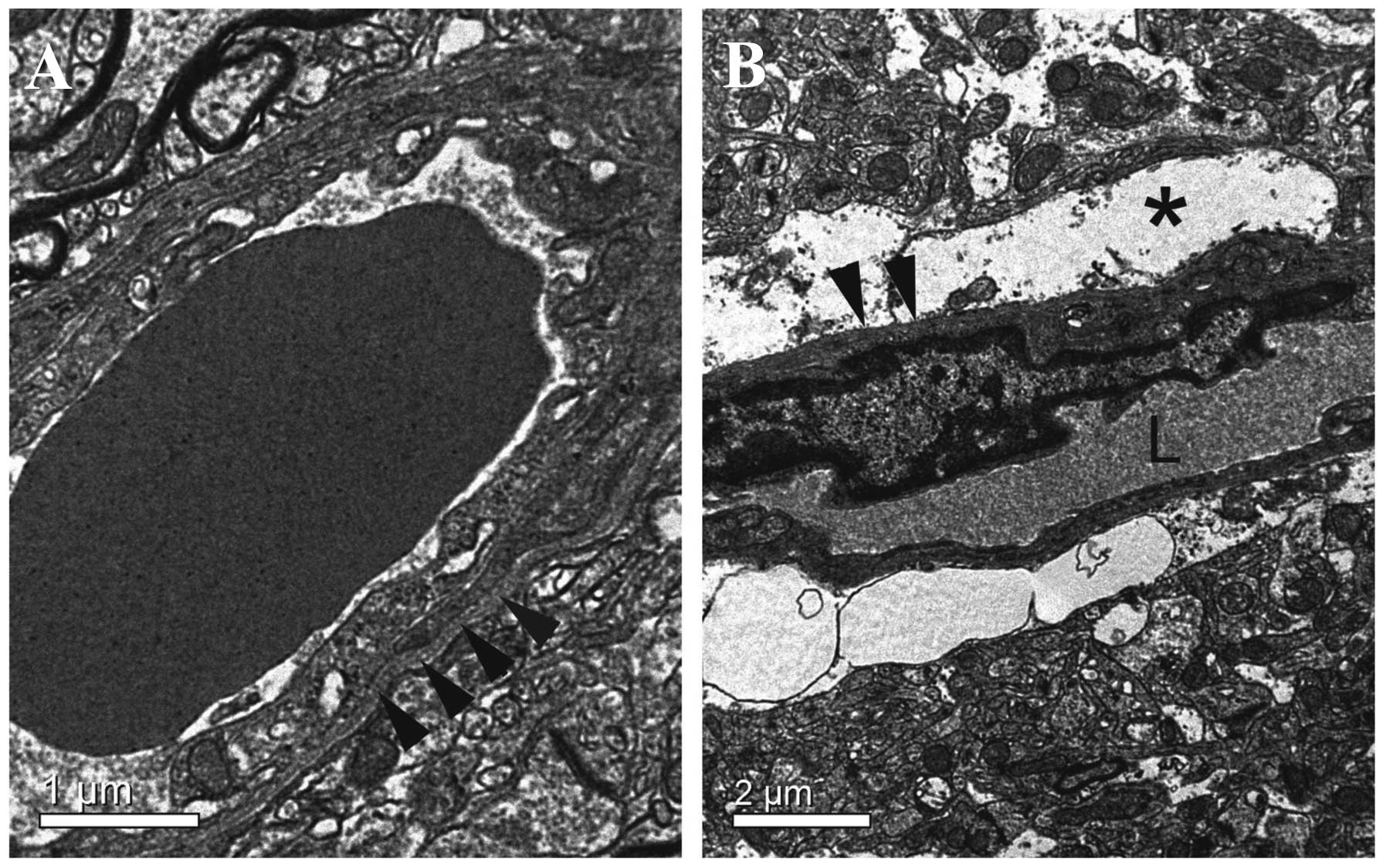
BBB ultrastructure alterations
Ultrastructural alterations of BBB were studied by
transmission electron microscopy in the ipsilateral striatum.
According to the result of EBD extravasation detection, the
detection time-point was 24 h after LPA injection when the BBB
permeability reached the peak point induced by LPA. Ultrastructural
analysis revealed no abnormalities in any structural or cellular
elements of the BBB in the control group (Fig. 3A). At 24 h after LPA was injected
intracerebrally, disruption of the BBB was evident. Characteristic
large spaces between capillaries and neuropil were observed. In
such capillaries, endothelial cells had little alteration in their
appearance and internal structure, although their pinocytic
activity was increased. The basement membrane was disrupted and
extremely coarse. Large swollen astrocytes or astrocytic end-feet
with scarce organelles and glycogen particles were observed, and
the extracellular space appeared considerably enlarged with the
presence of edema fluid, which narrowed capillary lumen (Fig. 3B).
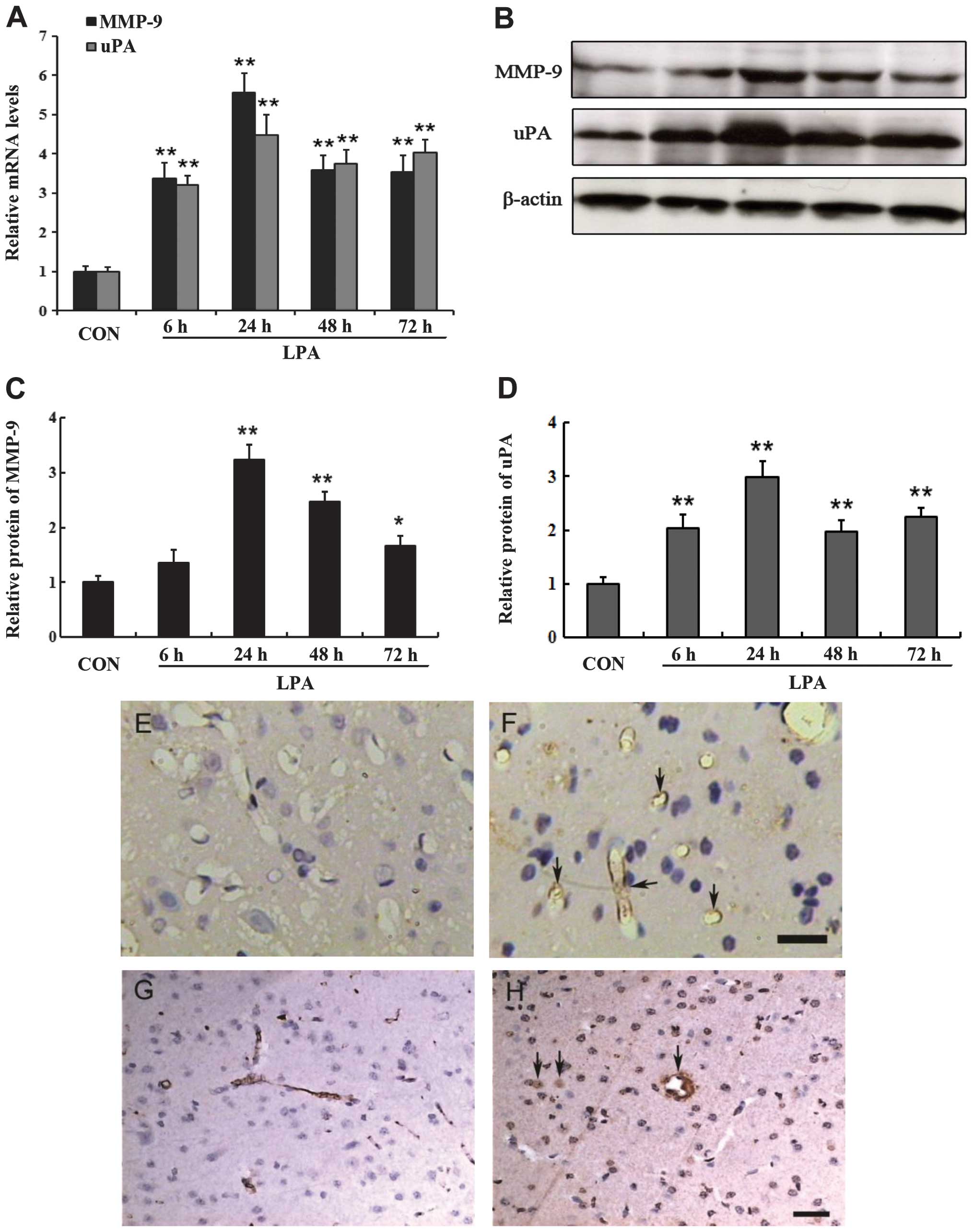
LPA induced the expression of proteolytic
enzymes
MMPs and uPA are important enzymes involved in BBB
damage, thus we determined the effects of LPA on the expression of
these proteolytic enzymes. According to the results of qPCR, LPA
significantly increased the mRNA expression of MMP-9 and uPA in the
ipsilateral striatum (Fig. 4A).
The mRNA expression of MMP-9 and uPA showed a significant
difference from 6 to 72 h following LPA injection as compared to
the controls (P<0.01) (Fig.
4A). The time course of MMP-9 and uPA protein expression
assessed by western blotting demonstrated that they were detectable
at 6 h following LPA injection and maximally expressed at 24 h
after LPA injection (Fig. 4B–D).
The protein expression of MMP-9 and uPA showed a statistical
difference from 24 to 72 h following LPA injection compared with
the controls (P<0.05) (Fig. 4C and
D). The spatial distribution of MMP-9 induced by LPA was
evaluated by immunohistochemical analysis. Diffuse MMP-9 expression
was observed in the right striatum where LPA was injected.
Immunoreactive MMP-9 appeared to stain with the endothelial cells
of microvessels (identified by morphometric criteria) at 6, 24, 48
and 72 h after LPA injection (Fig.
4F). By contrast, there was no similar labeling of endothelial
cells in the brain from rats that had been injected PBS as controls
(Fig. 4E). The MMP-9 expression
appeared to present with a significant number of endothelial cells
24 h after LPA injection. It was consistent with the results
obtained from the western blot analysis. The location of uPA
protein induced by LPA was also detected by immunohistochemistry.
In the LPA-treated groups, we observed that the staining for uPA
protein was predominantly associated with the microvessels and
parenchyma and diffusely within regions of the ipsilateral
striatum. Immunoreactive uPA was observed within endothelial cells
and fibrin deposition of microvessels, damaged neurons and glial
cells (identified by morphometric criteria), as well as scattered
accumulations within the extracellular space (Fig. 4H). In the control group, uPA was
slightly stained mainly on the endothelial cells of the
microvessels. No apparent staining of uPA was evident in the
neurons or glial cells in the ipsilateral striatum (Fig. 4G). Increased uPA immunoreactivity
was noted at 6, 24, 48 and 72 h following LPA injection, compared
with the PBS-injected controls. The expression of uPA protein was
initally detected in the ipsilateral striatum at 6 h after LPA
intracerebral injection, and became apparent at 24 h, then
decreased at 48 h, consistent with the results of uPA expression
detected by western blotting.
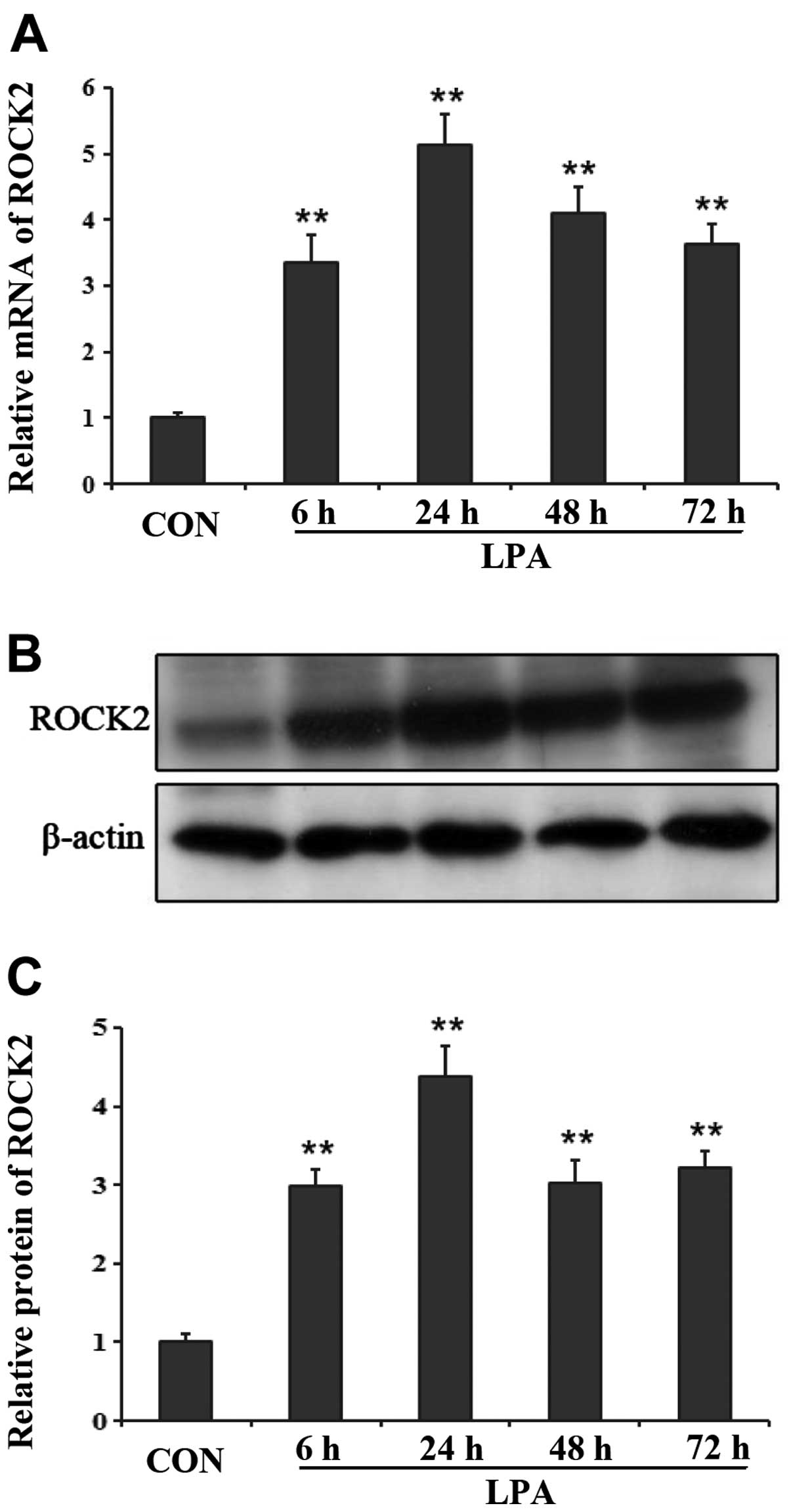
ROCK2 was involved in BBB disruption
induced by LPA
ROCK2 preferentially expressed in the brain and the
skeletal muscle (17), thus ROCK2
was examined in our experiment. By qPCR, we observed the changes of
the mRNA level of ROCK2 at different time-points following LPA
injection into the right striatum of rats. The expression level of
ROCK2 increased at 6 and 24 h following LPA injection, then
decreased gradually at 48 and 72 h after LPA injection (Fig. 5A). However, the different
time-point groups following LPA intracerebral injection all showed
a significant difference in the mRNA expression of ROCK2 compared
with the control group (P<0.01). Results of the western blot
analysis revealed similar changes in the protein expression of
ROCK2 at each time-point after LPA injection. At 24 h after LPA was
injected into the right striatum, the protein expression of ROCK2
reached a peak point and began to decrease at 48 h after LPA
injection (Fig. 5B and C). The
differences were significant between the LPA- and PBS-injected
groups (P<0.01).
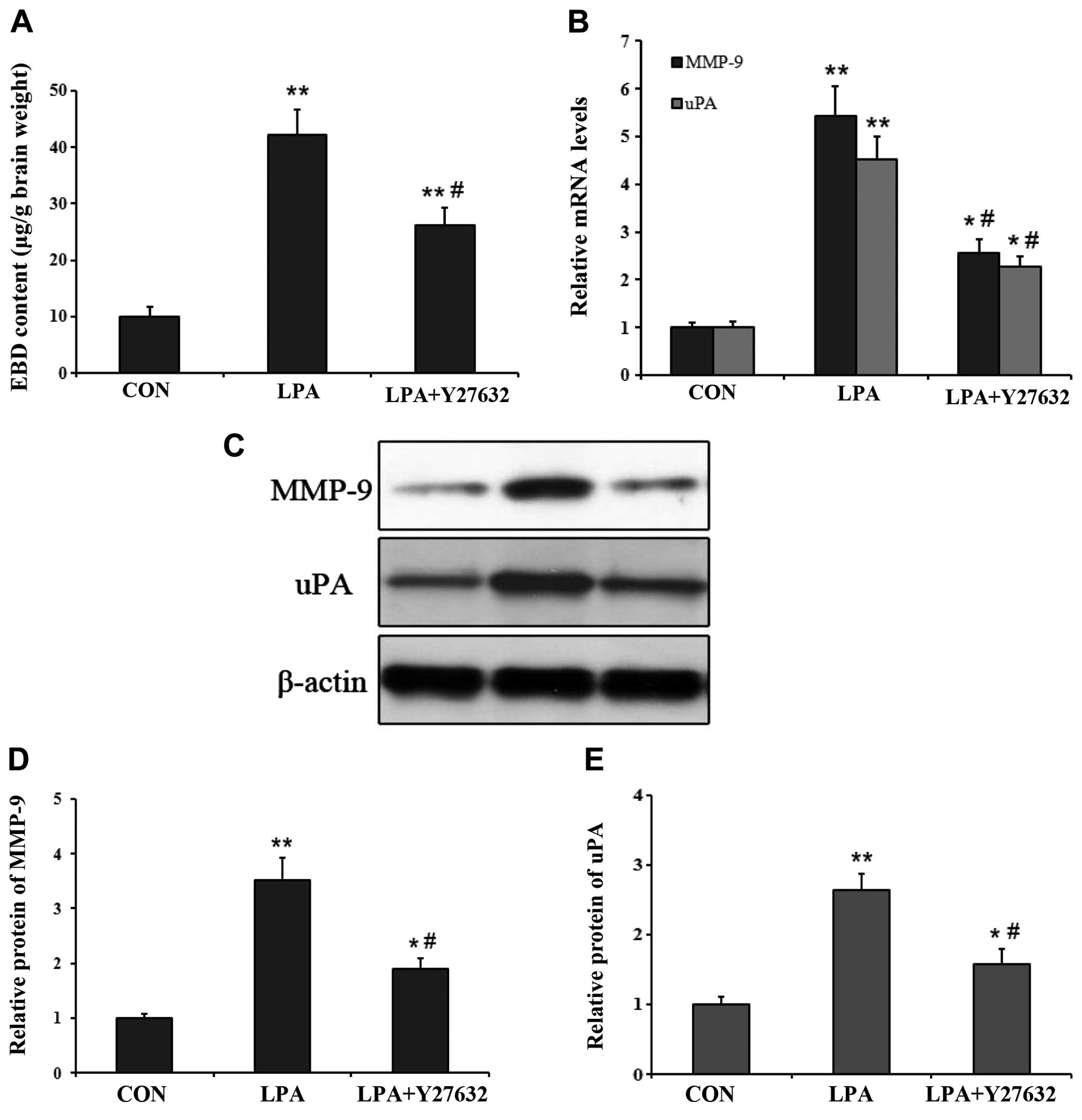
ROCK inhibitor decreased BBB permeability
and proteolytic enzyme expression
Since ROCK played a critical role in the
permeability of the rat BBB in certain pathological conditions
(13,18), the ROCK specific inhibitor Y27632
was utilized to evaluate its effects on the increased BBB
permeability induced by LPA. We examined the impact of Y27632 on
BBB permeability by measuring the EBD content in the ipsilateral
striatum at 24 h after LPA was co-injected with Y27632 into the
right caudate nucleus. Compared with the controls, EBD
extravasation in the ipsilateral striatum was significantly higher
in both 24 h after LPA injection and LPA co-injection with Y27632
(P<0.01) (Fig. 6A). However,
EBD extravasation was significantly lower at 24 h following LPA
co-injection with Y27632 than at the same time-point after LPA
injection (P<0.05) (Fig. 6A).
We also examined the effect of Y27632 on the mRNA and protein
expression of MMP-9 and uPA in the ipsilateral striatum after LPA
co-injection. The results of qPCR and western blot analysis
co-demonstrated that the mRNA and protein level of MMP-9 and uPA
were higher in both at 24 h after LPA injection and LPA
co-injection with Y27632, compared with the control group
(P<0.05) (Fig. 6B–E). However,
the mRNA and protein expression levels of MMP-9 and uPA were
significantly lower at 24 h after LPA co-injection with Y27632
compared with that at 24 h after LPA injection (P<0.05)
(Fig. 6B–E).
Discussion
In the present study, the underlying mechanisms by
which LPA increases the permeability of BBB have been demonstrated.
LPA was found to increase BBB permeability through the expression
of proteolytic enzymes, MMP-9 and uPA. Moreover, we have provided
evidence that ROCK is involved in LPA-induced proteolytic enzyme
expression and BBB disruption. These findings suggest a crucial
role of LPA in BBB damage, which may be due to activation of the
Rho/ROCK pathway and upregulation of the important proteolytic
enzymes MMP-9 and uPA.
The basement membrane is one of the basic components
of the BBB. Protein and collagen that compose the basement membrane
can be degraded by a variety of extracellular proteolytic enzymes,
including the MMP family. MMPs play a critical role in the
degradation of neurovascular integrity with BBB disruption. MMPs
comprise a large family of zinc-endopeptidases that increase the
permeability of BBB by degrading the extracellular matrix and tight
junction proteins in endothelial cells (19). MMPs, particularly MMP-9, have a
critical function in the proteolytic degradation of the basement
membrane of BBB in the pathological process (20,21). Early appearance of the activated
MMP-9 has been associated with an alteration of the BBB
permeability (22) and the
formation of vasogenic edema (23) subsequent to ischemic stroke
(24–26) and ICH (27,28). uPA is the serine protease that
converts plasminogen to the active protease plasmin. In addition to
playing a role in fibrinolysis, uPA may damage the basement
membrane of BBB via proteolysis and extracellular matrix breakdown.
The increased expression of uPA is detected in the ischemic
cerebral tissues and posttraumatic brain (29–31), which may be responsible for the
breakdown of the BBB (32). In
the present study, the results have shown that MMP-9 and uPA were
increased in the mRNA and protein level in the LPA-injected
striatum, which was parallel to EBD leakage, suggesting that LPA
induced the expression of proteolytic enzymes MMP-9 and uPA that
caused BBB dysfunction.
In studies on tumor cell progression and invasion,
LPA has been found to upregulate the expression of MMP-9 and uPA
through the Rho/ROCK signaling pathway (15,33). ROCK is a well-known downstream
effector of Rho and plays an important role in various
physiopathological processes. For example, ROCK is involved in
vascular smooth muscle cell contraction, actin cytoskeleton
organization, cell adhesion and motility. Accumulating data support
a critical role of the Rho/ROCK pathway in increasing permeability
of BBB (12,13). The possible mechanisms are the
disruption of tight junction proteins occluding and zonula
occludens-1 (13) and the
breakdown of basal membrane by the upregulation of MMP-9 and
downregulation of laminin expression (12).
Although recent findings suggest that Rho/ROCK
pathway is crucial in LPA-induced tumor progression (15,34), the definite mechanism by which
Rho/ROCK pathway mediates the breakdown of the BBB induced by LPA
remains unclear. In this study, we found a similar pattern of
changes in the ROCK2 expression, the brain EBD content that
indicated BBB permeability, and the expression of MMP-9 and uPA in
the ipsilateral striatum at different time-points following LPA
intracerebral injection. The present results support the hypothesis
that upregulation of the ROCK2 induced by LPA contributes to the
increase of proteolytic enzymes and subsequently BBB dysfunction.
On the one hand, LPA induced the expression of ROCK2 and stimulated
the synthesis of proteolytic enzymes leading to the increase of BBB
permeability. On the other hand, the inhibitor of ROCK2, Y27632,
blocked these subsequent actions induced by LPA in the right
striatum. Thus, we have demonstrated that the Rho/ROCK pathway may
be involved in LPA-induced proteolytic enzyme expression and BBB
disruption.
Jeong et al (15) reported that the Rho/ROCK pathway,
as a critical mediator, connected Ras to NF-κB in LPA-induced
proteolytic enzyme secretion in ovarian cancer cells, and that a
Gi-selective inhibitor pertussis toxin significantly inhibited
LPA-induced Rho activation, supporting the important role of Gi in
Ras and Rho activation in the LPA signaling axis. Although little
is known regarding the mechanism that the Rho/ROCK pathway mediates
BBB dysfunction induced by LPA, the importance of destructive
effects of LPA on the central nervous system in various
pathological conditions cannot be ignored. Therefore, the explicit
mechanism by which LPA induces BBB disruption through the Rho/ROCK
pathway remains to be confirmed in future studies by engaging the
LPA receptor antagonist and other antagonists in this signaling
cascade.
In conclusion, results of the present study have
shown that LPA increases BBB permeability by upregulation of
proteolytic enzymes MMP-9 and uPA, which may be suppressed by the
ROCK inhibitor. The present results suggest that LPA induces BBB
disruption through the Rho/ROCK signaling pathway and subsequent
production of proteolytic enzymes MMP-9 and uPA. However, future
studies are needed to confirm the explicit mechanism by which LPA
induces the BBB dysfunction through the Rho/ROCK pathway.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (81300984), the Natural Science
Foundation of Huber Province of China (no. 2010CHB02001) and the
Innovation Seed Fund of Wuhan University School of Medicine.
References
|
1
|
Moolenaar WH: Bioactive lysophospholipids
and their G protein-coupled receptors. Exp Cell Res. 253:230–238.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Van Meeteren LA, Frederiks F, Giepmans BN,
et al: Spider and bacterial sphingomyelinases D target cellular
lysophosphatidic acid receptors by hydrolyzing
lysophosphatidylcholine. J Biol Chem. 279:10833–10836.
2004.PubMed/NCBI
|
|
3
|
Tokumura A: Metabolic pathways and
physiological and pathological significances of lysolipid phosphate
mediators. J Cell Biochem. 92:869–881. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sorensen SD, Nicole O, Peavy RD, et al:
Common signaling pathways link activation of murine PAR-1, LPA, and
S1P receptors to proliferation of astrocytes. Mol Pharmacol.
64:1199–1209. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tigyi G and Parrill AL: Molecular
mechanisms of lysophosphatidic acid action. Prog Lipid Res.
42:498–526. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Inoue M, Xie W, Matsushita Y, Chun J, Aoki
J and Ueda H: Lysophosphatidylcholine induces neuropathic pain
through an action of autotaxin to generate lysophosphatidic acid.
Neuroscience. 152:296–298. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mauco G, Chap H, Simon MF and Douste-Blazy
L: Phosphatidic and lysophosphatidic acid production in
phospholipase C-and thrombin-treated platelets. Possible
involvement of a platelet lipase. Biochimie. 60:653–661. 1978.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tigyi G, Hong L, Yakubu M, Parfenova H,
Shibata M and Leffler CW: Lysophosphatidic acid alters
cerebrovascular reactivity in piglets. Am J Physiol.
268:H2048–H2055. 1995.PubMed/NCBI
|
|
9
|
Sun GY, Lu FL, Lin SE and Ko MR:
Decapitation ischemia-induced release of free fatty acids in mouse
brain. Relationship with diacylglycerols and lysophospholipids. Mol
Chem Neuropathol. 17:39–50. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen KM, Liu JY, Lai SC, Hsu LS and Lee
HH: Association of plasminogen activators and matrix
metalloproteinase-9 proteolytic cascade with blood-CNS barrier
damage of angiostrongyliasis. Int J Exp Pathol. 87:113–119. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schulze C, Smales C, Rubin LL and Staddon
JM: Lysophosphatidic acid increases tight junction permeability in
cultured brain endothelial cells. J Neurochem. 68:991–1000. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fujii M, Duris K, Altay O, Soejima Y,
Sherchan P and Zhang JH: Inhibition of Rho kinase by hydroxyfasudil
attenuates brain edema after subarachnoid hemorrhage in rats.
Neurochem Int. 60:327–333. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu K, Li Z, Wu T and Ding S: Role of rho
kinase in microvascular damage following cerebral ischemia
reperfusion in rats. Int J Mol Sci. 12:1222–1231. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ishiguro M, Kawasaki K, Suzuki Y, et al: A
Rho kinase (ROCK) inhibitor, fasudil, prevents matrix
metalloproteinase-9-related hemorrhagic transformation in mice
treated with tissue plasminogen activator. Neuroscience.
220:302–312. 2012. View Article : Google Scholar
|
|
15
|
Jeong KJ, Park SY, Cho KH, et al: The
Rho/ROCK pathway for lysophosphatidic acid-induced proteolytic
enzyme expression and ovarian cancer cell invasion. Oncogene.
31:4279–4289. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Baskaya MK, Dogan A, Rao AM and Dempsey
RJ: Neuroprotective effects of citicoline on brain edema and
blood-brain barrier breakdown after traumatic brain injury. J
Neurosurg. 92:448–452. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tawara S and Shimokawa H: Progress of the
study of rho-kinase and future perspective of the inhibitor.
Yakugaku Zasshi. 127:501–514. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang XN, Fu J and Wang WZ: The effects of
fasudil on the permeability of the rat blood-brain barrier and
blood-spinal cord barrier following experimental autoimmune
encephalomyelitis. J Neuroimmunol. 239:61–67. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rosenberg GA: Matrix metalloproteinases
and their multiple roles in neurodegenerative diseases. Lancet
Neurol. 8:205–216. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Asahi M, Asahi K, Jung JC, del Zoppo GJ,
Fini ME and Lo EH: Role for matrix metalloproteinase 9 after focal
cerebral ischemia: effects of gene knockout and enzyme inhibition
with BB-94. J Cereb Blood Flow Metab. 20:1681–1689. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Asahi M, Wang X, Mori T, et al: Effects of
matrix metalloproteinase-9 gene knock-out on the proteolysis of
blood-brain barrier and white matter components after cerebral
ischemia. J Neurosci. 21:7724–7732. 2001.PubMed/NCBI
|
|
22
|
Barr TL, Latour LL, Lee KY, et al:
Blood-brain barrier disruption in humans is independently
associated with increased matrix metalloproteinase-9. Stroke.
41:e123–e128. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rosell A, Cuadrado E, Ortega-Aznar A,
Hernandez-Guillamon M, Lo EH and Montaner J: MMP-9-positive
neutrophil infiltration is associated to blood-brain barrier
breakdown and basal lamina type IV collagen degradation during
hemorrhagic transformation after human ischemic stroke. Stroke.
39:1121–1126. 2008. View Article : Google Scholar
|
|
24
|
Romanic AM, White RF, Arleth AJ, Ohlstein
EH and Barone FC: Matrix metalloproteinase expression increases
after cerebral focal ischemia in rats: inhibition of matrix
metalloproteinase-9 reduces infarct size. Stroke. 29:1020–1030.
1998. View Article : Google Scholar
|
|
25
|
Gasche Y, Fujimura M, Morita-Fujimura Y,
Copin JC, Kawase M, Massengale J and Chan PH: Early appearance of
activated matrix metalloproteinase-9 after focal cerebral ischemia
in mice: a possible role in blood-brain barrier dysfunction. J
Cereb Blood Flow Metab. 19:1020–1028. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Aoki T, Sumii T, Mori T, Wang X and Lo EH:
Blood-brain barrier disruption and matrix metalloproteinase-9
expression during reperfusion injury: mechanical versus embolic
focal ischemia in spontaneously hypertensive rats. Stroke.
33:2711–2717. 2002. View Article : Google Scholar
|
|
27
|
Wang J and Tsirka SE: Neuroprotection by
inhibition of matrix metalloproteinases in a mouse model of
intracerebral haemorrhage. Brain. 128:1622–1633. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu H, Zhang Z, Li Y, et al: Time course of
upregulation of inflammatory mediators in the hemorrhagic brain in
rats: correlation with brain edema. Neurochem Int. 57:248–253.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dietzmann K, von Bossanyi P, Krause D,
Wittig H, Mawrin C and Kirches E: Expression of the plasminogen
activator system and the inhibitors PAI-1 and PAI-2 in
posttraumatic lesions of the CNS and brain injuries following
dramatic circulatory arrests: an immunohistochemical study. Pathol
Res Pract. 196:15–21. 2000. View Article : Google Scholar
|
|
30
|
Ito T, Takenaka K, Sakai H, Yoshimura S,
Hayashi K, Noda S and Sakai N: Elevation of mRNA levels of
tissue-type plasminogen activator and urokinase-type plasminogen
activator in hippocampus and cerebral cortex following middle
cerebral artery occlusion in rats. Neurol Res. 22:413–419.
2000.PubMed/NCBI
|
|
31
|
Armstead WM, Cines DB, Bdeir K,
Kulikovskaya I, Stein SC and Higazi AA: uPA impairs
cerebrovasodilation after hypoxia/ischemia through LRP and ERK
MAPK. Brain Res. 1231:121–131. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Patel TH, Sprague S, Lai Q, Jimenez DF,
Barone CM and Ding Y: Blood brain barrier (BBB) dysfunction
associated with increased expression of tissue and urokinase
plasminogen activators following peripheral thermal injury.
Neurosci Lett. 444:222–226. 2008. View Article : Google Scholar
|
|
33
|
Park SY, Jeong KJ, Panupinthu N, et al:
Lysophosphatidic acid augments human hepatocellular carcinoma cell
invasion through LPA1 receptor and MMP-9 expression. Oncogene.
30:1351–1359. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sawada K, Morishige K, Tahara M, Ikebuchi
Y, Kawagishi R, Tasaka K and Murata Y: Lysophosphatidic acid
induces focal adhesion assembly through Rho/Rho-associated kinase
pathway in human ovarian cancer cells. Gynecol Oncol. 87:252–259.
2002. View Article : Google Scholar : PubMed/NCBI
|