Introduction
The incidence of metastatic melanoma and mortality
rate has continually increased over the past decades (1,2).
The invasive and metastatic ability are the most fundamental
features of cancer cells which differ from normal cells, eventually
leading to tumor recurrence, progression and ultimately, death
(3). The median survival of
patients with metastatic melanoma is <1 year (4). Thus, an improvement in therapy for
metastatic melanoma is mandatory.
A number of studies have revealed that microRNAs
(miRNAs or miRs) are associated with the metastasis of colon
cancer, breast cancer and lung cancer cells (5–7).
Over the past few years, the association between miRNAs and tumors
has become a research hotspot. Certain miRNAs have been proven to
be potential biomarkers in clinical diagnosis (8,9),
which have the most extensive gene regulation functions on several
levels (10) and are involved in
a series of important biological processes, including early
embryonic development, cell proliferation, apoptosis, cell death
and fat metabolism, and even the differentiation of stem cells
(11). Due to the advances in
miRNA research, an increasing number of studies have demonstrated
that miRNAs regulate important tumor-related genes, and are thus
closely related to the development and occurrence of tumors. For
example, miR-7 and miR-331–3p have been shown to regulate the
expression of epidermal growth factor receptor (EGFR) and human
EGFR, which are biomarkers of human tumors (12). Jiang et al demonstrated
that miRNA-155 acts as a suppressor of the cytokine signaling 1
gene that is a tumor suppressor in breast cancer (13). However, a number of
tumor-associated miRNAs remain unexploited, and the mechanisms
responsible for the regulatory functions of miRNAs in metastatic
melanoma are not yet fully understood. Therefore, miRNAs may
provide a novel treatment method for cancers.
Given the detrimental effects of metastatic tumors
and the growing development of sequencing technology, small RNA
sequencing was adopted in our study to identify the differentially
expressed miRNAs in metastatic melanoma compared with primary
cutaneous melanoma samples. The target genes were predicted, and
functional and pathway analyses were performed to reveal their
roles in the progression of metastasis. Our findings may provide
new insight into the diagnosis and treatment of metastatic
tumors.
Materials and methods
Sequencing data
The GSE3623 sequencing data, including a total of 31
samples, was downloaded from the Gene Expression Omnibus (GEO)
database. A total of 7 melanoma samples were selected, including 4
primary cutaneous melanoma samples (used as controls) and 3
metastatic melanoma samples. Raw data were obtained in SRA format,
as previously described (14).
The platform used was GPL9052 Illumina Genome Analyzer (Homo
sapiens). Deep sequencing was performed on various tissues and
sequencing data were obtained. We transformed the data from SRA
format to FASTQ format, as previously described (15) using the SRA Toolkit (16). Sequence alignments were then
carried out with Bowtie alignments (17), allowing as many as 2 mismatches.
Bowtie is a rapid and memory-saving tool for short sequence
assembly. It aligns short sequences to reference genomes and can
read base pairs as long as 1024 bp, which renders it extremely
suitable for next-generation sequencing technologies (18). The expression level of miRNAs was
determined using miRDeep, as previously described (19), which has been proven to be of high
sensitivity and accuracy (20).
Screening of differentially expressed
miRNAs
After the expression levels were determined, the
differentially expressed miRNAs were screened out between the
metastatic melanoma and primary cutaneous melanoma (controls)
samples with limma package in R language (21); a p-value <0.05 and |logFC|>1
were set as the cut-off values.
Prediction of target genes of miRNAs
Target genes were predicted using TargetScanHuman
6.2, which is a software package developed by Lewis et al
(22). This software package was
used for predicting target genes in mammalian species based on
conservations across species and thermodynamic features of
miRNA-target gene complexes (23). Target genes with a prediction
score >0.9 were regarded as of high confidence.
Interaction network construction of
target genes
Genes usually interact with each other to exert
certain biological functions (24). Therefore, genes which interact
with target genes were identified by the String database (25) and the interaction network of these
genes was then established to broaden the understanding of their
functions. The String database can rate the interactions from the
aspects of homology, experiment and text mining.
Functional and pathway analyses
Functional and pathway analyses were performed for
the genes in the network with Expressing Analysis Systematic
Explorer (EASE) (26) (p<0.05)
which can identify the significant Gene Ontology (GO) terms and the
genes that exist in the GO categories (27).
Results
Sequencing data
After the raw data were transformed into FASTQ
format, the sequence read length was 36 bp. The number of original
reads, miRNA alignments and miRNA types for each sample are listed
in Table I. The expression levels
of the miRNAs in each sample were then determined (data not
shown).
 | Table IStatistical information of the
sequencing data. |
Table I
Statistical information of the
sequencing data.
| Group | Accession no. | No. of original
reads | No. of miRNA
alignments | No. of miRNA
types |
|---|
| Metastatic
melanoma | GSM893568 | 402556 | 17847 | 259 |
| GSM893576 | 1226137 | 18005 | 123 |
| GSM893577 | 708653 | 16493 | 262 |
| GSM893578 | 395686 | 31687 | 279 |
| Primary melanoma | GSM893564 | 97403 | 85049 | 338 |
| GSM893566 | 464804 | 40816 | 251 |
| GSM893573 | 1279059 | 40267 | |
Differentially expressed miRNAs
A total of 4 differentially expressed miRNAs were
identified in the metastatic melanoma samples as compared with the
primary cutaneous melanoma (control) samples according to the
criteria (p<0.05 and |logFC|>1). These 4 miRNAs were
hsa-miR-146, hsa-miR-27, hsa-miR-877 and hsa-miR-186. The
information for these 4 miRNAs is presented in Table II.
| Table IIDetails for the 4 differentially
expressed miRNAs. |
Table II
Details for the 4 differentially
expressed miRNAs.
| ID | p-value | |logFC| |
|---|
| hsa-miR-146 | 0.005384 | 1.91716394 |
| hsa-miR-27 | 0.035685 | 1.49264032 |
| hsa-miR-877 | 0.039706 | 2.73696559 |
| hsa-miR-186 | 0.041463 | 1.1356551 |
Target genes and the interaction
network
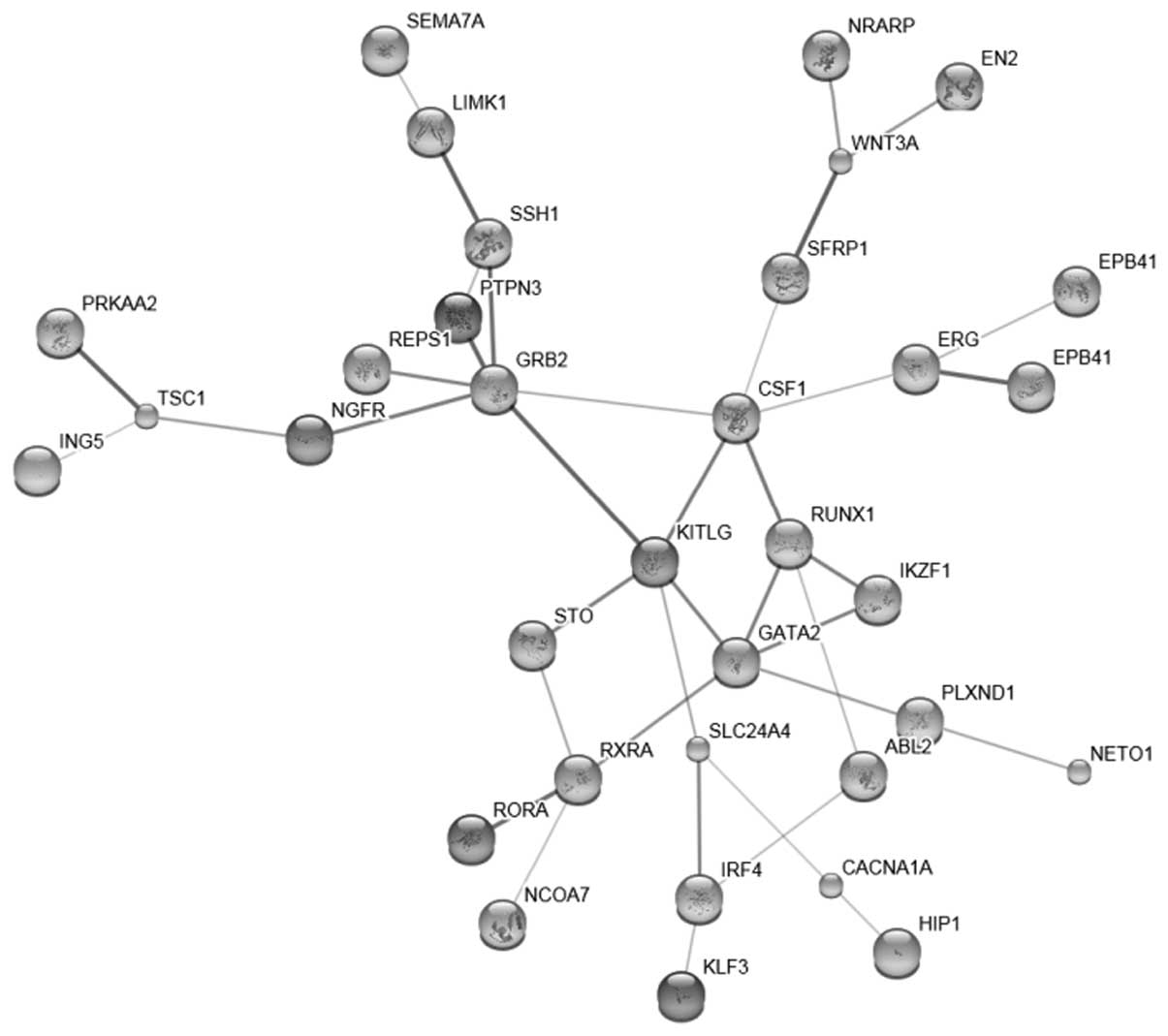
A total of 101 target genes with high confidence
were found for hsa-miR-27 (data not shown). A total of 41
interactions (data not shown) were disclosed and the network was
then constructed (Fig. 1).
Functional and pathway analyses of genes
in the network
A total of 4 significant functional terms and 1
pathway were identified (Table
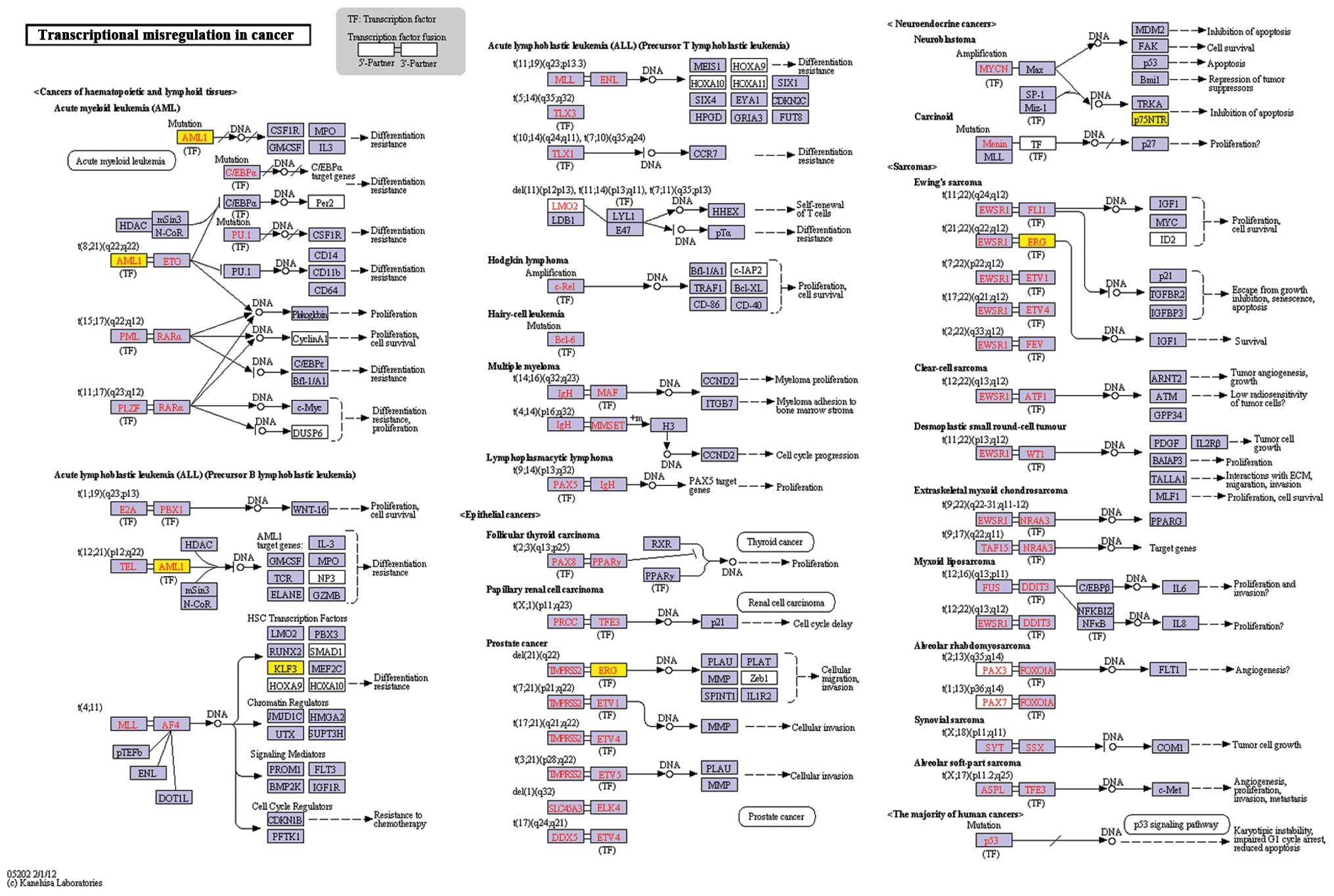
III) for the genes in the interaction network. The most
significant function was transcription, while the pathway of
transcriptional misregulation in cancer was also revealed (Fig. 2). The genes which were enriched at
the transcriptional level, included inhibitor of growth family,
member 5 (ING5), GATA binding protein 2 (GATA2) and erythroblast
transformation-specific (ETS)-related gene (ERG). Other genes, such
as Ikaros family zinc finger 1 (IKZF1), interferon regulatory
factor 4 (IRF4) and runt-related transcription factor (RUNX), as
well as retinoid X receptor, alpha (RXRA) and RAR-related orphan
receptor A (RORA) were also included in the list (Table II).
| Table IIIFunctional and pathway analyses of
the genes in the network. |
Table III
Functional and pathway analyses of
the genes in the network.
| Term - pathway | p-value | Genes |
|---|
| GO:0006350 -
Transcription | 0.001954 | ING5, GATA2, ERG,
IKZF1, RXRA, NCOA7, IRF4, RORA, RUNX1, HIP1, KLF3 |
| GO:0045449 -
Regulation of transcription | 0.003148 | ING5, GATA2, ERG,
IKZF1, RXRA, NCOA7, IRF4, EN2, RORA, RUNX1, HIP1, KLF3 |
| GO:0043228 -
Non-membrane bounded organelle | 0.016522 | GATA2, SSH1, PTPN3,
EPB41, TSC1, IKZF1, EPB41L4A, KITLG, IRF4, FGD6, ABL2, HIP1 |
| GO:0043232 -
Intracellular non-membrane bounded organelle | 0.016522 | GATA2, SSH1, PTPN3,
EPB41, TSC1, IKZF1, EPB41L4A, KITLG, IRF4, FGD6, ABL2, HIP1 |
| KO:05202 -
Transcriptional misregulation in cancer | 0.000498 | ERG, KLF3,
AML1 |
Discussion
We identified 4 differentially expressed miRNAs by
next-generation sequencing technology. In order to determine the
functions of these miRNAs, the target genes of hsa-miR-27 were
predicted and the interaction network of the genes was constructed.
Finally, the functions of and pathways associated with these genes
were analyzed in order to further elucidate the mechanisms
responsible for metastatic melanoma.
The 4 differentially expressed miRNAs were
hsa-miR-146, hsa-miR-27, hsa-miR-877 and hsa-miR-186. Rabinowits
et al found the overexpression of hsa-miR-146 in lung cancer
(28). Katakowski et al
demonstrated that the expression of miR-146b-5p inversely
correlated with glioma invasiveness in the brain (29). The study by Mertens-Talcott et
al indicated that miR-27a is overexpressed in breast cancer
cells, and that cell proliferation is decreased by the inhibition
of this miRNA using antisense molecules in MDA-MB-231 cells
(30). Scott et al
reported that in SKBr3 breast cancer cells, the inhibitor of
histone deacetylases, LAQ824, rapidly decreased miR-27a levels
(31). miR-27a has also been
shown to be upregulated in renal cell carcinoma as compared with
normal kidneys (32) and acts as
an oncogene in gastric adenocarcinoma (33). Guttilla and White further
indicated that in breast cancer cells, miR-27a regulated the
expression of FOXO1 (34), a
putative tumor suppressor. The overexpression of miR-877 has been
reported in endometrial serous adenocarcinomas (35). In a previous study, when
glyceollins were applied for the treatment of triple-negative
breast cancer, the downregulation of miR-877 was observed (36). Baffa et al indicated that
hsa-miR-186 was differentially expressed in paired primary and
metastatic cancers (37).
Leidinger et al demonstrated that hsa-miR-186 combined with
other miRNAs was able to distinguish melanoma patients from healthy
individuals (38). Therefore, we
consider that these 4 miRNAs are potential targets for modulating
the metastasis of melanoma cells.
To further elucidate the regulatory mechanisms, the
target genes of the 4 miRNAs were predicted by TargetScan. A total
of 101 target genes were revealed for hsa-miR-27. Functional
enrichment analysis revealed that the most significantly enriched
term was transcription, indicating that a number of gene
expressions are regulated, which may lead to the acquisition of
metastatic tumor cells. In addition, several target genes have been
disclosed and are worthy of further investigation. ING5 is
predicted to be one of the target genes which is linked to
tumorigenesis in human head and neck carcinoma (39). GATA2 is a member of the GATA
family of zinc-finger transcription factors. Bohm et al
found that the high expression of GATA2 is associated with
metastatic progression and biochemical recurrence in prostate
cancer through the regulation of key androgen-regulated genes
(40). ERG is a member of the ETS
family and a number of studies have investigated its role in
prostate cancer (41,42).
Overall, next-generation sequencing technology was
adopted in this study to identify the differentially expressed
miRNAs in metastatic melanoma as compared with primary cutaneous
melanoma samples. Moreover, the functions and pathways of target
genes were revealed, which may provide insight into the regulatory
mechanism. The data presented in our study may prove beneficial in
the diagnosis and treatment of metastatic melanoma and may aid in
the development of novel clinical applications using miRNAs.
However, further sutdies are required to confirm our results.
Acknowledgements
This study was supported by a grant from the
National Natural Science Foundation of China (no. 30900767 to
Jianglin Zhang). The authors thank Professor Ming Zhou for
providing insightful comments.
References
|
1
|
Hernandez BY, Green MD, Cassel KD,
Pobutsky AM, Vu V and Wilkens LR: Preview of Hawaii cancer facts
and figures 2010. Hawaii Med J. 69:223–224. 2010.PubMed/NCBI
|
|
2
|
Gray-Schopfer V, Wellbrock C and Marais R:
Melanoma biology and new targeted therapy. Nature. 445:851–857.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hatfield S and Ruohola-Baker H: microRNA
and stem cell function. Cell Tissue Res. 331:57–66. 2008.
View Article : Google Scholar
|
|
4
|
Agarwala SS: Current systemic therapy for
metastatic melanoma. Expert Rev Anticancer Ther. 9:587–595. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Calin GA and Croce CM: MicroRNA-cancer
connection: the beginning of a new tale. Cancer Res. 66:7390–7394.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Iorio MV, Ferracin M, Liu CG, et al:
MicroRNA gene expression deregulation in human breast cancer.
Cancer Res. 65:7065–7070. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ma L, Teruya-Feldstein J and Weinberg RA:
Tumour invasion and metastasis initiated by microRNA-10b in breast
cancer. Nature. 449:682–688. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yanaihara N, Caplen N, Bowman E, et al:
Unique microRNA molecular profiles in lung cancer diagnosis and
prognosis. Cancer Cell. 9:189–198. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhou J, Yu L, Gao X, et al: Plasma
microRNA panel to diagnose hepatitis B virus-related hepatocellular
carcinoma. J Clin Oncol. 29:4781–4788. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen CZ: MicroRNAs as oncogenes and tumor
suppressors. N Engl J Med. 353:1768–1771. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hatfield SD, Shcherbata HR, Fischer KA,
Nakahara K, Carthew RW and Ruohola-Baker H: Stem cell division is
regulated by the microRNA pathway. Nature. 435:974–978. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Giles KM, Barker A, Zhang PM, Epis MR and
Leedman PJ: MicroRNA regulation of growth factor receptor signaling
in human cancer cells. Methods Mol Biol. 676:147–163. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang S, Zhang HW, Lu MH, et al:
MicroRNA-155 functions as an OncomiR in breast cancer by targeting
the suppressor of cytokine signaling 1 gene. Cancer Res.
70:3119–3127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shumway M, Cochrane G and Sugawara H:
Archiving next generation sequencing data. Nucleic Acids Res.
38:D870–D871. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cock PJ, Fields CJ, Goto N, Heuer ML and
Rice PM: The Sanger FASTQ file format for sequences with quality
scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res.
38:1767–1771. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kaminuma E, Mashima J, Kodama Y, et al:
DDBJ launches a new archive database with analytical tools for
next-generation sequence data. Nucleic Acids Res. 38:D33–D38. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Langmead B, Trapnell C, Pop M and Salzberg
SL: Ultrafast and memory-efficient alignment of short DNA sequences
to the human genome. Genome Biol. 10:R252009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Trapnell C and Salzberg SL: How to map
billions of short reads onto genomes. Nat Biotechnol. 27:455–457.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Friedlander MR, Chen W, Adamidi C, et al:
Discovering microRNAs from deep sequencing data using miRDeep. Nat
Biotechnol. 26:407–415. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Buermans HP, Ariyurek Y, van Ommen G, den
Dunnen JT and ‘t Hoen PA: New methods for next generation
sequencing based microRNA expression profiling. BMC Genomics.
11:7162010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Smyth GK: Limma: linear models for
microarray data. Bioinformatics and Computational Biology Solutions
using R and Bioconductor. Gentleman R, Carey V, Dudoit S, Irizarry
R and Huber W: Springer; New York: pp. 397–420. 2005, View Article : Google Scholar
|
|
22
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Friedman RC, Farh KK, Burge CB and Bartel
DP: Most mammalian mRNAs are conserved targets of microRNAs. Genome
Res. 19:92–105. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tong AH, Drees B, Nardelli G, et al: A
combined experimental and computational strategy to define protein
interaction networks for peptide recognition modules. Science.
295:321–324. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Szklarczyk D, Franceschini A, Kuhn M, et
al: The STRING database in 2011: functional interaction networks of
proteins, globally integrated and scored. Nucleic Acids Res.
39:D561–D568. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hosack DA, Dennis G Jr, Sherman BT, Lane
HC and Lempicki RA: Identifying biological themes within lists of
genes with EASE. Genome Biol. 4:R702003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Steeg PS: Tumor metastasis: mechanistic
insights and clinical challenges. Nat Med. 12:895–904. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rabinowits G, Gercel-Taylor C, Day JM,
Taylor DD and Kloecker GH: Exosomal microRNA: a diagnostic marker
for lung cancer. Clin Lung Cancer. 10:42–46. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Katakowski M, Zheng X, Jiang F, Rogers T,
Szalad A and Chopp M: MiR-146b-5p suppresses EGFR expression and
reduces in vitro migration and invasion of glioma. Cancer Invest.
28:1024–1030. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mertens-Talcott SU, Chintharlapalli S, Li
X and Safe S: The oncogenic microRNA-27a targets genes that
regulate specificity protein transcription factors and the G2-M
checkpoint in MDA-MB-231 breast cancer cells. Cancer Res.
67:11001–11011. 2007. View Article : Google Scholar
|
|
31
|
Scott GK, Mattie MD, Berger CE, Benz SC
and Benz CC: Rapid alteration of microRNA levels by histone
deacetylase inhibition. Cancer Res. 66:1277–1281. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gottardo F, Liu CG, Ferracin M, et al:
Micro-RNA profiling in kidney and bladder cancers. Urol Oncol.
25:387–392. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu T, Tang H, Lang Y, Liu M and Li X:
MicroRNA-27a functions as an oncogene in gastric adenocarcinoma by
targeting prohibitin. Cancer Lett. 273:233–242. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Guttilla IK and White BA: Coordinate
regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast
cancer cells. J Biol Chem. 284:23204–23216. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hiroki E, Akahira J, Suzuki F, et al:
Changes in microRNA expression levels correlate with
clinicopathological features and prognoses in endometrial serous
adenocarcinomas. Cancer Sci. 101:241–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rhodes LV, Tilghman SL, Boue SM, et al:
Glyceollins as novel targeted therapeutic for the treatment of
triple-negative breast cancer. Oncol Lett. 3:163–171.
2012.PubMed/NCBI
|
|
37
|
Baffa R, Fassan M, Volinia S, et al:
MicroRNA expression profiling of human metastatic cancers
identifies cancer gene targets. J Pathol. 219:214–221. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Leidinger P, Keller A, Borries A, et al:
High-throughput miRNA profiling of human melanoma blood samples.
BMC Cancer. 10:2622010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li X, Nishida T, Noguchi A, et al:
Decreased nuclear expression and increased cytoplasmic expression
of ING5 may be linked to tumorigenesis and progression in human
head and neck squamous cell carcinoma. J Cancer Res Clin Oncol.
136:1573–1583. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bohm M, Locke WJ, Sutherland RL, Kench JG
and Henshall SM: A role for GATA-2 in transition to an aggressive
phenotype in prostate cancer through modulation of key
androgen-regulated genes. Oncogene. 28:3847–3856. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang J, Cai Y, Ren C and Ittmann M:
Expression of variant TMPRSS2/ERG fusion messenger RNAs is
associated with aggressive prostate cancer. Cancer Res.
66:8347–8351. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Carver BS, Tran J, Gopalan A, et al:
Aberrant ERG expression cooperates with loss of PTEN to promote
cancer progression in the prostate. Nat Genet. 41:619–624. 2009.
View Article : Google Scholar : PubMed/NCBI
|