Introduction
Gallbladder cancer is a relatively rare but highly
lethal disease and is the most common cancer of the biliary tract
and the seventh most common gastrointestinal carcinoma (1). Gallbladder cancer is a highly
invasive and aggressive disease with a dismal prognosis and the
5-year survival rate for all stages of gallbladder cancer is
approximately 5% (2,3). Gallbladder cancer has a very poor
prognosis due to its invasive and aggressive characteristics which
are determined by various factors. One of these factors is the
tumor microenviroment. Numerous studies have confirmed that tumor
necrosis factor-α (TNF-α) is a key cytokine amongst all cytokines
of the tumor microenvironment which promote tumor cell
proliferation and invasion. In a previous study, in an orthotopic
mouse model of pancreatic cancer, treatment with anti-TNF-α
antibody resulted the reduction of tumor growth and metastasis
(4). In studies on animal
thoracic neoplasms, dermatoma and gastrointestinal cancer, the
association between tumor cell growth and metastasis and the amount
of TNF-α in the tumor microenvironment has been demonstrated
(5–7). In patients with malignant tumors of
the prostate, it has also been shown that there is a correlation
the amount of TNF-α and the degree of malignancy, recurrence,
metastasis and prognosis (8).
Studies have investigated the effects of TNF-α on tumor cell
metastasis, demonstrating that TNF-α enhances the invasive capacity
of cancer cells (9,10).
However, the specific mechanisms responsible for
TNF-α promoting the progression of malignant tumors have not been
elucidated. Certain studies have found that TNF-α promotes a
variety of inflammatory cytokines and chemokines, thus affecting
the formation and development of tumor blood vessels, promoting
tumor invasion and metastasis (11). TNF-α activates associated cell
signaling pathways through the activation of transcription factors
and related genes, consequently affecting the activity of tumor
cells, promoting tumor cell proliferation (12). TNF-α directly leads to gene
damage, mutation, amplification of DNA, consequently affecting
tumor development (13–15). In addition, TNF changes the
function of immune cells, promoting tumor progression (16).
However, there are few reports on the function of
tumor-derived TNF-α. Studies on colon and ovarian cancer have found
that tumor-derived TNF-α plays an important role in tumor
progression (5,11).
However, the specific mechanisms responsible for
TNF-α promoting cancer cell growth and invasion are largely
unknown. Studies have demonstrated that TNF-α is a key cytokine
amongst all cytokines of the tumor microenvironment. Hagemann et
al (17) demonstrated that in
epithelial tumors, TNF-α stimulates matrix metalloproteinase (MMP)
secretion, thereby promoting tumor cell invasion. Kulbe et
al (18) found that in
ovarian cancer cells, TNF-α stimulates IL-8, monocyte chemotactic
protein-1 (MCP-1) and chemokine receptor expression, thus enhancing
tumor cell invasion and metastasis. Chua et al (19) demonstrated that TNF-α enhances
epithelial-mesenchymal transition in mammary epithelial cells.
Another study also found that TNF-α induces the expression of
vascular endothelial growth factor (VEGF), thus promoting
microvascularization (20).
Tumor cell-derived TNF-α is a important factor
produced by tumor cells and plays a key role in the tumor
microenviroment (21). Moreover,
TNF-α may even promote tumor growth at lower levels (22). Colon cancer cell-derived TNF-α
plays an important role in promoting proliferation through
autocrine mechanisms found in the tumor microenvironment (5). In ovarian cancer, it has been shown
that tumor-derived TNF-α plays an important role in promoting
invasion and metastasis (11,23,24).
However, whether gallbladder cancer cells produce
autocrine TNF-α, and whether gallbladder cancer cell-derived TNF-α
affects the biological behavior of the cells, remain unresolved
issues. Thus, in the present study, we examined various gallbladder
cancer cell lines expressing different levels of TNF-α in order to
determine the effects of TNF-α on gallbladder cancer proliferation,
invasion, metastasis and apoptosis, as well as the underlying
mechanisms involved.
Materials and methods
Cell culture
The gallbladder cancer cell line, SGC-996, was
provided by the Tumor Cytology Research Unit, Medical College,
Tongji University, Shanghai, China. NOZ cells were obtained from
the Health Science Research Resources Bank in Japan, and they were
isolated from ascites derived from a 48-year-old female patient
with gallbladder cancer (25).
Both the cell lines were cutured in Dulbecco’s modified Eagle’s
medium (DMEM) supplemented with 10% fetal bovine serum (FBS). All
the cells were incubated at 37°C under 95% air and 5%
CO2.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was extracted from the gallbladder cells
grown in 6-well plates using TRIzol reagent (Invitrogen, Carlsbad,
CA, USA) according to the manufacturer’s instructions. cDNA was
synthesized using the AVM First Strand cDNA synthesis kit
(Invitrogen). The primers for TNF-α and β-actin were synthesized
according to primer design principles. TNF-α yielded a 443 bp
product, and the sequences of the primers were as follows: forward,
5′-AGTGACAAGCCTGTAGCCC-3′ and reverse, 5′-GCAATGATCCCAAAGTAGACC-3′;
TFN receptor 1 (TNFR1) yielded a 223 bp product, and the sequences
of the primers were as follows: forward, 5′-TGCCA GGAGAAACAGAACA-3′
and reverse, 5′-AACCAA TGAAGAGGAGGGAT-3′. β-actin yielded a 254 bp
product, and the sequences of the primers were as follows: forward,
5′-CTGTCTGGCGGCACCACCAT-3′ and reverse, 5′-GCAA
CTAAGTCATAGTCCGC-3′. RT-PCR was performed under the following
conditions: 30 cycles of denaturation at 94°C for 30 sec, annealing
at 55°C for 30 sec, and extension at 72°C for 1 min fllowed by 10
min for final extension at 72°C. The data of TNF-α were normalized
relative to the expression of β-actin mRNA expression in the
respective samples.
Western blot analysis
The cells were washed twice with cold
phosphate-buffered saline (PBS) and then incubated on ice with 250
μl of RIPA buffer with 2.5 μl phenylmethylsulfonyl fluoride (PMSF)
for 20 min. The cells were collected and centrifuged at 13,000 rpm
for 10 min at 4°C. The protein concentrations of the cell lysates
were measured in duplicate using a BCA Protein assay kit (Beyotime
Institute of Biotechnology, Shanghai, China). The proportion of
protein lysates and 6X loading buffer according to the ratio of 4:1
were mixed and then boiled for 5 min at 100°C. Equal amounts of
total protein were resolved by sodium dodecyl sulfate (SDS
10%)-polyacrylamide gel electrophoresis and transferred onto
polyvinylidene fluoride (PVDF) membranes. The PVDF membranes were
then blocked with 5% non-fat milk in Tris-buffered saline with
Tween-20 (TBST) for 2 h. The diluted primary antibodies, including
polyclonal goat anti-human TNFR1 antibody (1:1,000), monoclonal
mouse anti-human TNF-α (1:500) (both from Santa Cruz Biotechnology,
Inc. Santa Cruz, CA, USA), monoclonal mouse anti-human AKT (1:500),
monoclonal mouse anti-human p-AKT (1:500), monoclonal mouse
anti-human nuclear factor-κB (NF-κB) (p65) (1:500), monoclonal
mouse anti-human p-NF-κB (p-p65) (1:500) (all from Cell Signaling
Technology, Danvers, MA, USA), monoclonal mouse anti-human Bcl-2
(1:500), monoclonal mouse anti-human Bax (1:500) and β-actin
(1:1,500) (all from Santa Cruz Biotechnology, Inc.) were then
incubated with the membranes overnight at 4°C. The appropriate
secondary antibody conjugated with horseradish peroxidase diluted
in TBST was added for 2 h at room temperature. Using a
chemiluminescence western blot immunodetection kit (Invitrogen), we
tested immunoreactivity according to the manufacturer’s
instructions and recorded the data on hyperfine-ECL detection film.
The amounts of TNF-α and TNFR1 protein were semiquantified as
ratios to β-actin suggested on each gel.
TNF-α siRNA plasmid construction and
transfection
Suitable siRNA target sequences were found in the
human TNF-α sequence. According to the design guidelines of siRNA
and the literature (5), DNA
template oligonucleotides corresponding to siRNA sequences were
synthesized as follows: 5′-GCGTGGAGCTGAGAGATAA-3′. A small hairpin
RNA (shRNA) of human TNF-α in a pGPU-GFP-neo gene transfer vector
encoding a green fluorescent protein (GFP) sequence was constructed
by GenePharma Co., Ltd. (Shanghai, China). The plasmids were
verified by DNA sequencing. Corresponding sequences (C-N) for the
negative contols (NC) were also provided by GenePharma Co., Ltd.
The SGC-996 cells were cultured in DMEM medium supplemented with
10% FBS. When the cells were at approximately 90% confluency, we
transfected the plasmids into the cells. The cells were transfected
using Lipofectamine 2000 (Invitrogen) according to the
manufacturer’s instructions. The transfection efficiency was
quantified by determining the percentage of cells that were
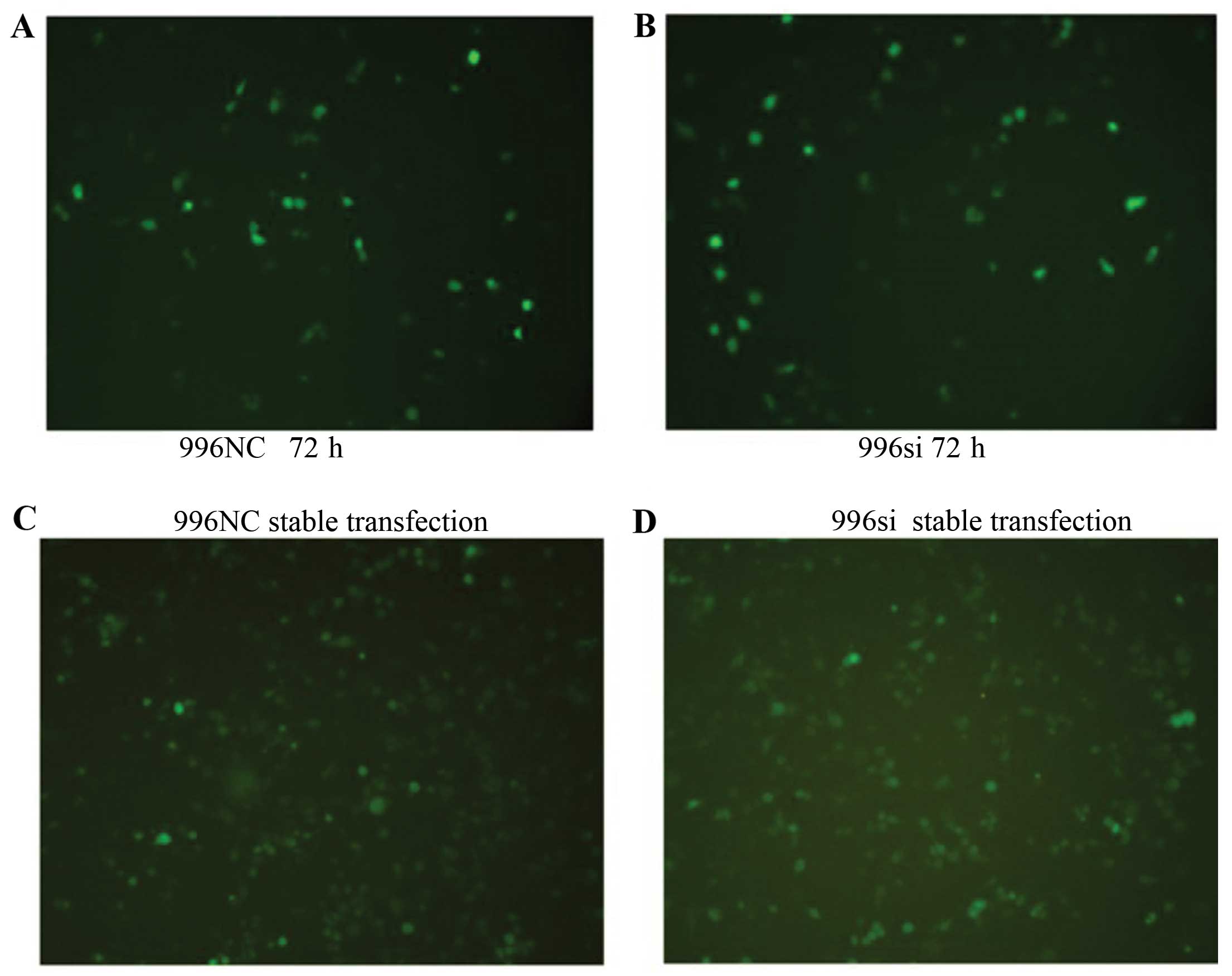
GFP-positive using a microscope (Fig.
3A and B). The culture medium was replaced with a selection
medium containing G418 at concentrations of 400 μg/ml (Alexis
Biochemicals, San Diego, CA, USA) 72 h later. When we obtained the
stably transfected cells, the cells were continuously maintained in
200 μg/ml of G418 (Fig. 3C and
D).
Ezyme-linked immunosorbent assay
(ELISA)
To analyze autocrine TNF-α in the untransfected
SGC-996 cells, TNF-α small interfering (siRNA)-transfected SGC-996
cells and the SGC-996 negative control (NC)-transfected cells,
these cells were seeded into plates at a density of
3×106 cells/well with 4 ml of DMEM medium supplemented
with 10% FBS. The amount of autocrine TNF-α in the cells was
determined after 1, 2, 3, 4 and 5 days by ELISA. ELISA was
performed to detect TNF-α in the cell culture supernatants of the
untransfected SGC-996 cells, the TNF-α siRNA-transfected SGC-996
cells and the SGC-996NC-transfected cells using a TNF-α (H) ELISA
kit (Wuhan Boster Biological Technology, Ltd., Wuhan, China). Two
hundred microliters of supernatant were added to each well. ELISA
was perforemd according to the manufacturer’s instructions. The
sensitivity of the assays was 7.8 pg/ml. The absorbance was
detected at 450 nm. Each plate test was repeated 3 times.
Cell proliferation assay
To analyze cell proliferation, the untransfected
SGC-996, TNF-α siRNA-transfected SGC-996 and SGC-996NC-transfected
cells were seeded into 96-well plates at a density of
103 cells/well with 100 μl of DMEM medium supplemented
with 10% FBS. The proliferative activity was determined after 1, 2,
3, 4 and 5 days by the addition of 10 μl of sterile
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
(5 mg/ml; Sigma, St. Louis, MO, USA) to each well. The reaction was
terminated after 4 h of incubation at 37°C by the addition of 100
μl of dimethyl sulfoxide (DMSO; Sigma). The optical density (OD)
value was obtained by measuring absorbance at a wavelength of 570
nm. Each well test was repeated 6 times.
In vitro cell migration assay
Cell motility was assayed using Transwells (24-well
format) with 8 μm pore polycarbonate membranes (BD Biosciences, San
Jose, CA, USA). The lower side of the membranes was covered with 5
μg fibronectin (BD Biosciences). To test cell motility induced by
TNF-α siRNA, the treated or untreated SGC-996 cells
(2×105) in 200 μl of DMEM medium with 2.5% FBS were
placed in the upper chamber. The lower chamber was filled with 700
μl DMEM medium with 10% FBS as the chemoattractant. The migration
chamber was incubated for 8 h at 37°C and 5% CO2. The
cells on the upper surface of the membrane were removed by gentle
scrubbing with a cotton swab. Membranes were fixed in a stationary
liquid of 95% ethanol and 5% acetic acid for 30 min and stained
with hematoxylin and eosin (H&E). The number of cells on the
lower surface of the membrane in 5 random visual fields (×400) was
then counted using a bright field light microscope. Each assay was
repeated in triplicate.
In vitro cell invasion assay
For invasion assays, Transwells (24-well format)
with 8 μm polycarbonate membranes (BD Biosciences) were used.
Briefly, the upper side of the membranes was coated with Matrigel
matrix (20 μg/well) and the membranes were then air-dried for 1 h
of incubation 37°C. The lower side of the membranes was coated with
5 μg fibronectin (BD Biosciences). Other experimental procedures
were the same as those for the migration assay.
Flow cytometric analysis
To determine the apoptosis induced by TNF-α siRNA,
the treated or untreated SGC-996 cells were seeded
(5×105/well) in 6-well plates in DMEM medium with 10%
FBS for 48 h to collect the cells and stained using the Annexin
V-PE/7-aminoactinomycin D kit (KeyGen Biotech, Nanjing, China)
according the manufacturer’s instructions, and analyzed using a
Becton-Dickinson FACSCalibur.
Ststistical analysis
Data were analyzed using GraphPad Prism 5 software.
Analysis of variance was conducted followed by one-way ANOVA or an
unpaired t-test. The data are expressed as the means ± standard
deviation (SD). A P-value <0.05 was considered to indicate a
statistically significant difference.
Results
mRNA expression of TNF-α and TNFR1 in the
NOZ and SGC-996 cells
We analyzed the mRNA expression of TNF-α and TNFR1
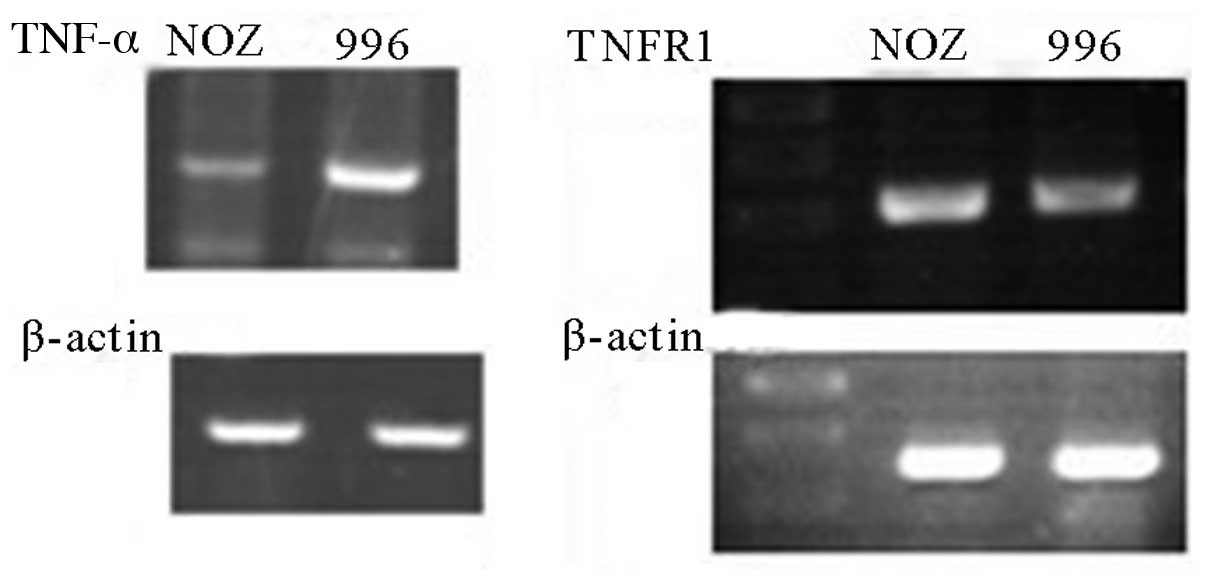
in the NOZ and SGC-996 cells. Using RT-PCR, we detected the mRNA
expression of TNF-α and TNFR1 in both cell lines (Fig. 1). The TNF-α mRNA expression level
in the NOZ cells was lower than that in the SGC-996 cells (Fig. 1). However, the mRNA levels of
TNFR1 were similar between the NOZ and SGC-996 cells (Fig. 1). Thus, we used the SGC-996 cells
to further examine the role of autocrine TNF-α.
Protein expression of TNFR1 and TNF-α in
the NOZ and SGC-996 cells
We then determined the TNFR1 and TNF-α protein
expression in the NOZ and SGC-996 cells by western blot analysis.
As expected, the protein expression of TNFR1 and TNF-α was detected
in both cell lines. We observed no difference in the TNFR1 protein
expression levels in the 2 cell lines by western blot analysis
(Fig. 2); however, the TNF-α
protein expression level in the NOZ cells was lower than that in
the SGC-996 cells.
mRNA and protein expression of TNF-α
after obtaining stably transfected SGC-996 (SGC-996si) cells
We used an RNAi-mediated method to silence TNF-α in
order to characterize the biological effects of TNF-α in the
SGC-996 cells. The DNA sequencing results verified that TNF-α siRNA
plasmid construction was successful. We obtained stably transfected
cells using G418 after 2 weeks, as shown by a screening test.
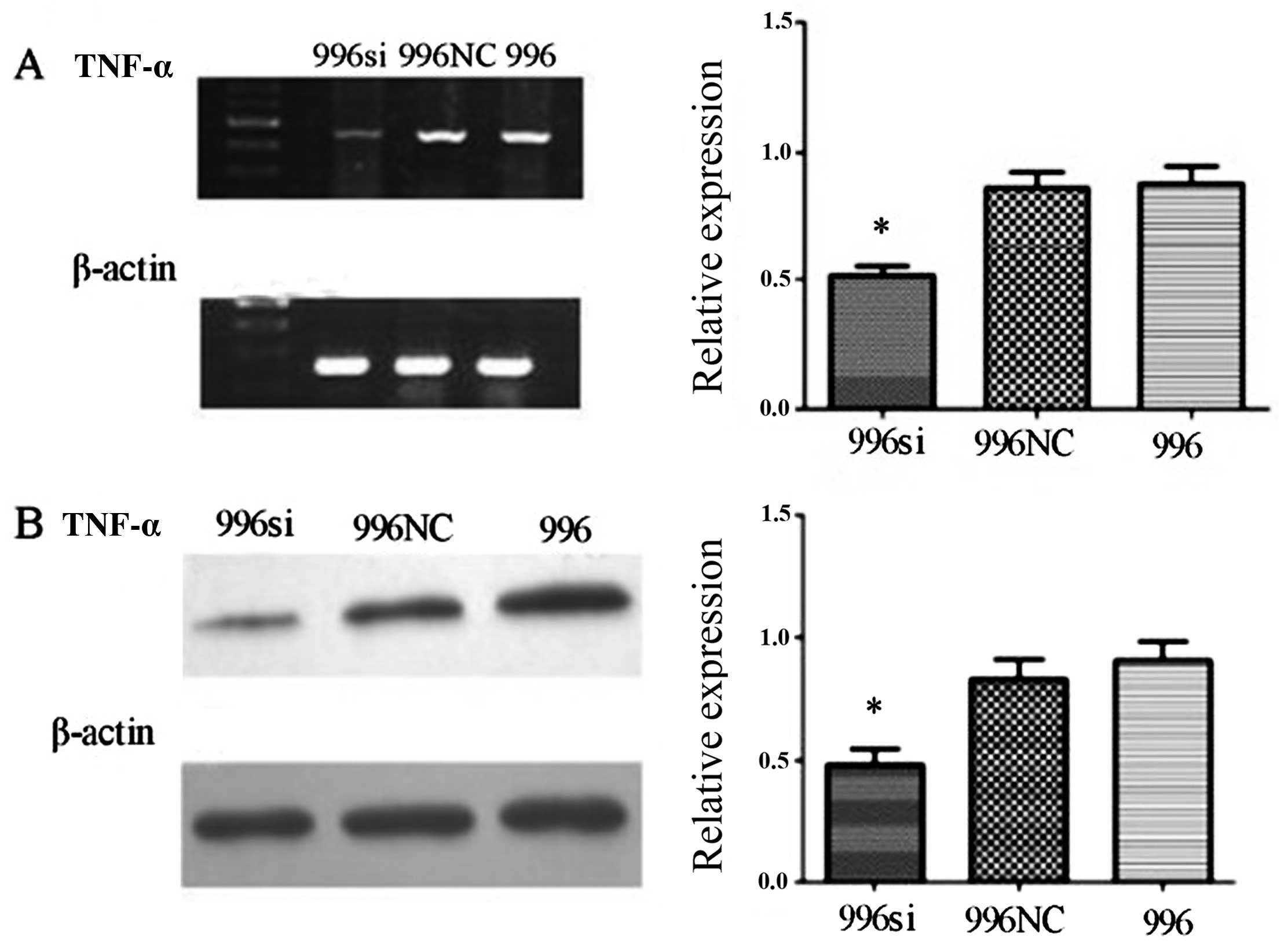
RT-PCR revealed a decrease in TNF-α mRNA levels in the
siRNA-transfected cells, while the levels of the β-actin gene
maintained relatively unaltered and mock transfection or
transfection with the C-N/siRNA vector had no effect on TNF-α mRNA
expression (Fig. 4A). We obtained
stably transfected SGC-996 cells by siRNA targeting TNF-α using
G418 after 2 weeks, as shown by a screening test; these stably
transfected cells were used for the for following experiments.
RT-PCR and western blot analysis indicated that autocrine TNF-α
mRNA and protein levels were markedly inhibited in the
siRNA-transfected SGC-996 (SGC-996si) cells. Semiquantitative
analysis revealed that the TNF-α mRNA and protein expression in the
SGC-996si group was markedly suppressed (Fig. 4).
Autocrine TNF-α protein levels in the
untransfected SGC-996, TNF-α siRNA-transfected SGC-996 and
SGC-996NC cell culture supernatants
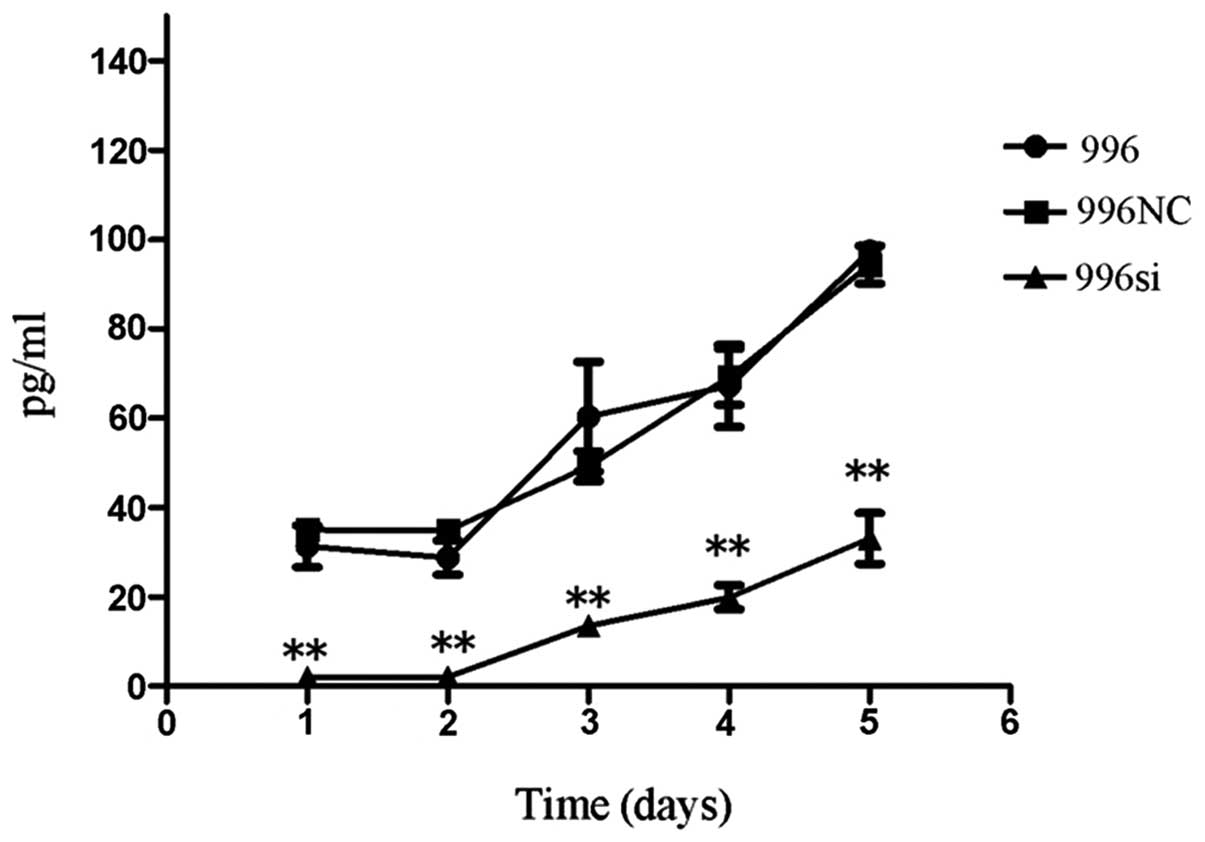
We then analyzed the autocrine TNF-α protein levels
in all cell culture supernatants of the untransfected SGC-996,
TNF-α siRNA-transfected SGC-996 and SGC-996NC-transfected cells
(Fig. 5). Consistent with the
mRNA levels, ELISA analysis revealed that when compared to the
untransfected SGC-996 and SGC-996NC-transfected cells, the TNF-α
siRNA-transfected SGC-996 cells showed a marked inhibition in the
production of autocrine TNF-α protein. By contrast, no transfection
(SGC-996) or transfection with the negative control (SGC-996NC) had
no effect on autocrine TNF-α protein levels (P<0.05).
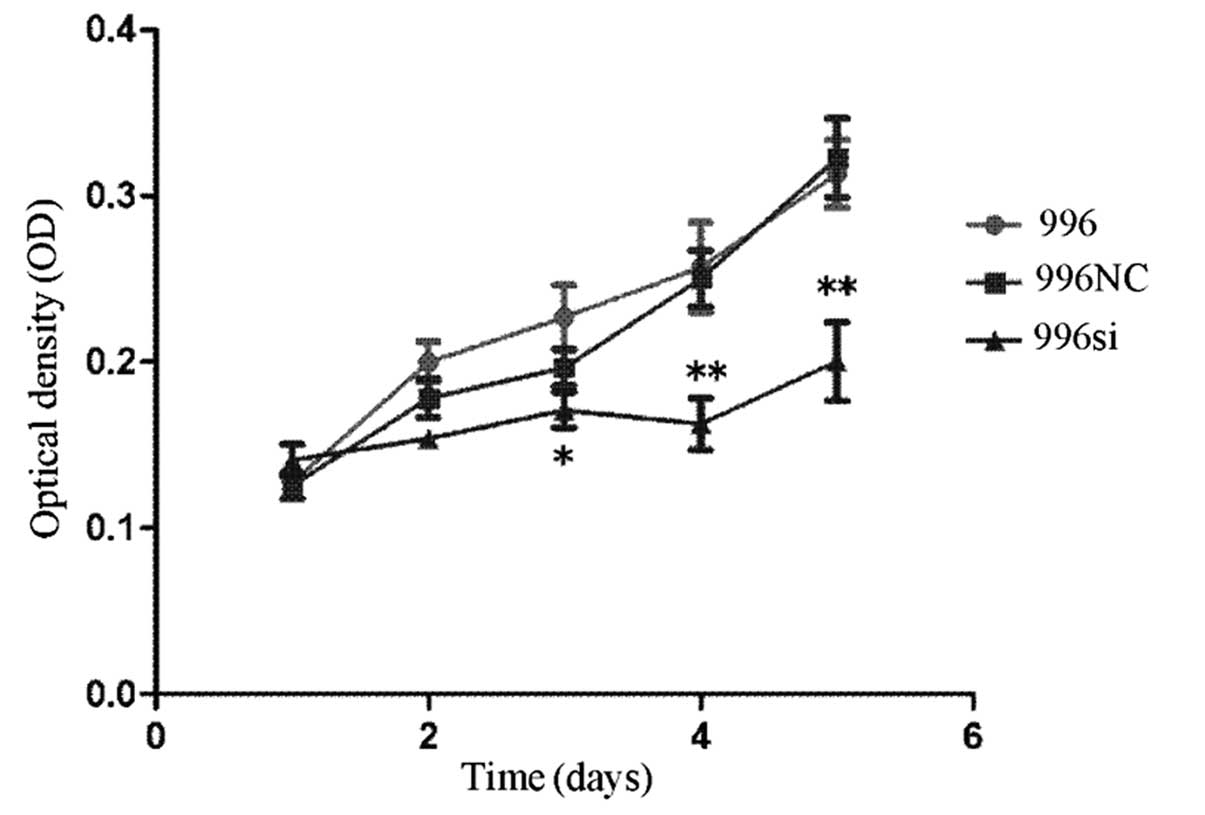
Knockdown of TNF-α decreases the
proliferation of SGC-996 cells in vitro
To determine whether the endogenous TNF-α promotes
cancer cell proliferation, we first treated the human SGC-996 cells
with siRNA directed against TNF-α. We transfected the SGC-996 cells
with TNF-α siRNA to induce the downregulation of TNF-α gene
expression with C-N/siRNA and mock-treated groups were used as
controls. We examined cell proliferation at 1, 2, 3, 4 and 5 days
by MTT assay. Compared to the C-N/siRNA-transfected cells and the
untransfected cells, cell proliferation in the TNF-α
siRNA-transfected group was slower (P<0.05) (Fig. 6). The data from the in
vitro cell proliferation assay indicated that the growth of the
cells in the TNF-α siRNA-transfected group was markedly reduced
compared with the untreated group, which was the same as the growth
of the C-N/siRNA-transfected group. These data support the
autocrine role of TNF-α in affecting the proliferaton of
gallbladder cancer cells.
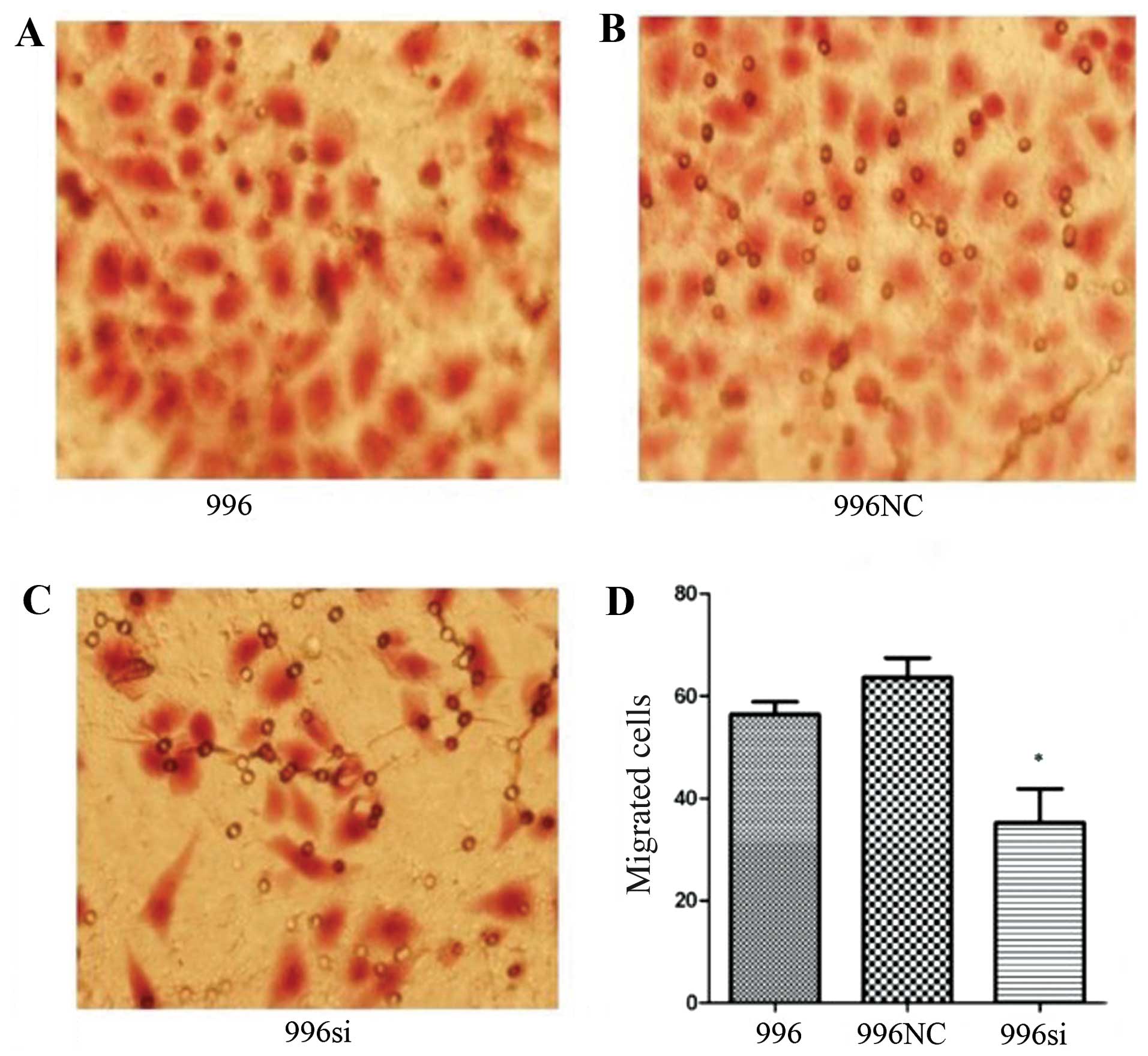
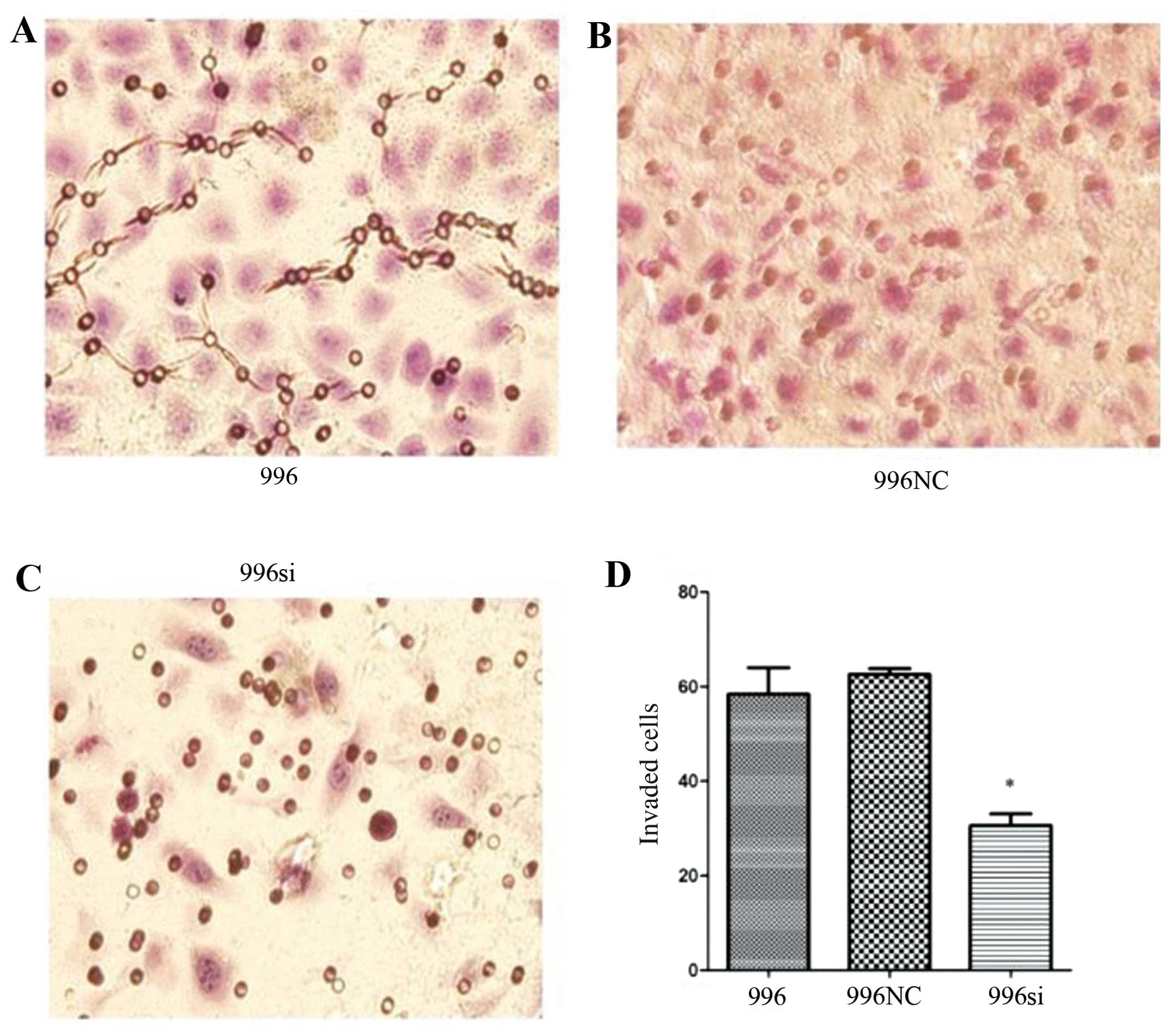
TNF-α knockdown infuences gallbladder
cancer cell migration and invasiveness
To determine whether the migration and invasiveness
of the SGC-996 cells depends on endogenously secreted TNF-α, we
used Transwell assay to examine the effects of the knockdown of the
TNF-α gene in the SGC-996 cells. Following staining with H&E, 5
different fields (×400, magnification) were counted to test the
numbers of migrated and invaded cells. The total number of cells in
the TNF-α siRNA group that migrated and invaded through the
Transwell polycarbonate filter was significantly lower than that of
the cells in the SGC-996 (untreated) group (Figs. 7 and 8) (P<0.05), which was similar to the
number of cells in the C-N/siRNA group. These data suggest that the
function of gallbladder cancer cell-derived TNF-α plays an
important role in the migration and invasion of gallbladder cancer
cells.
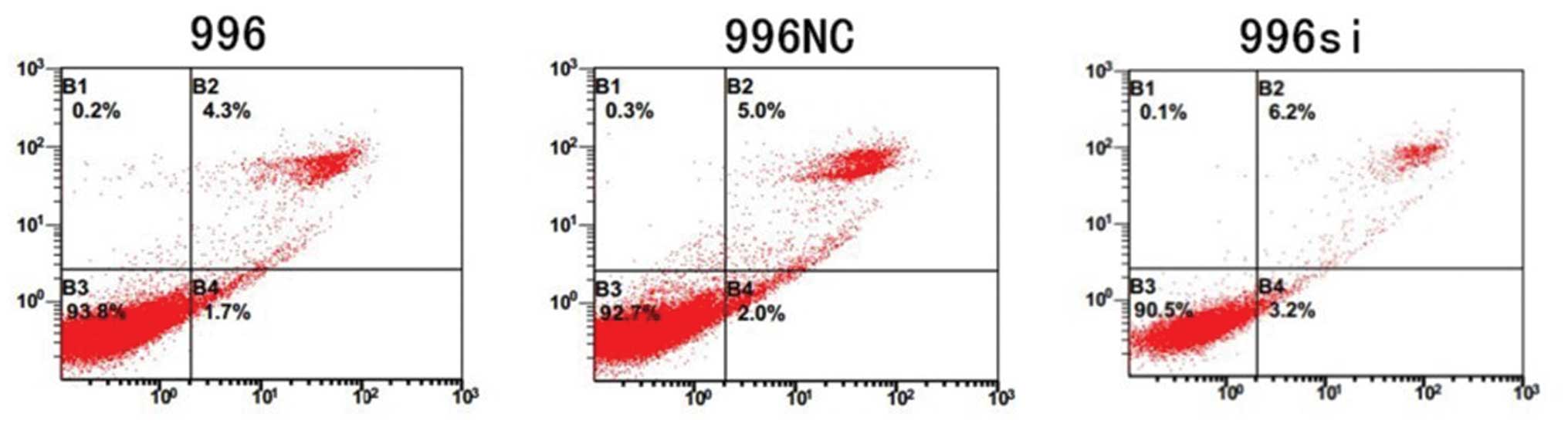
TNF-α knockdown does not increase the
apoptosis of SGC-996 cells
To investigate whether the induced effects of TNF-α
gene silencing on cell viability were due to apoptosis, we employed
flow cytometry (FCM) after the cells were stained with Annexin
V-PE/7-aminoactinomycin D. The untransfected SGC-996, TNF-α
siRNA-transfected SGC-996 cells and the SGC-996NC-transfected cells
exhibited a similar rate of apoptosis (Fig. 9).
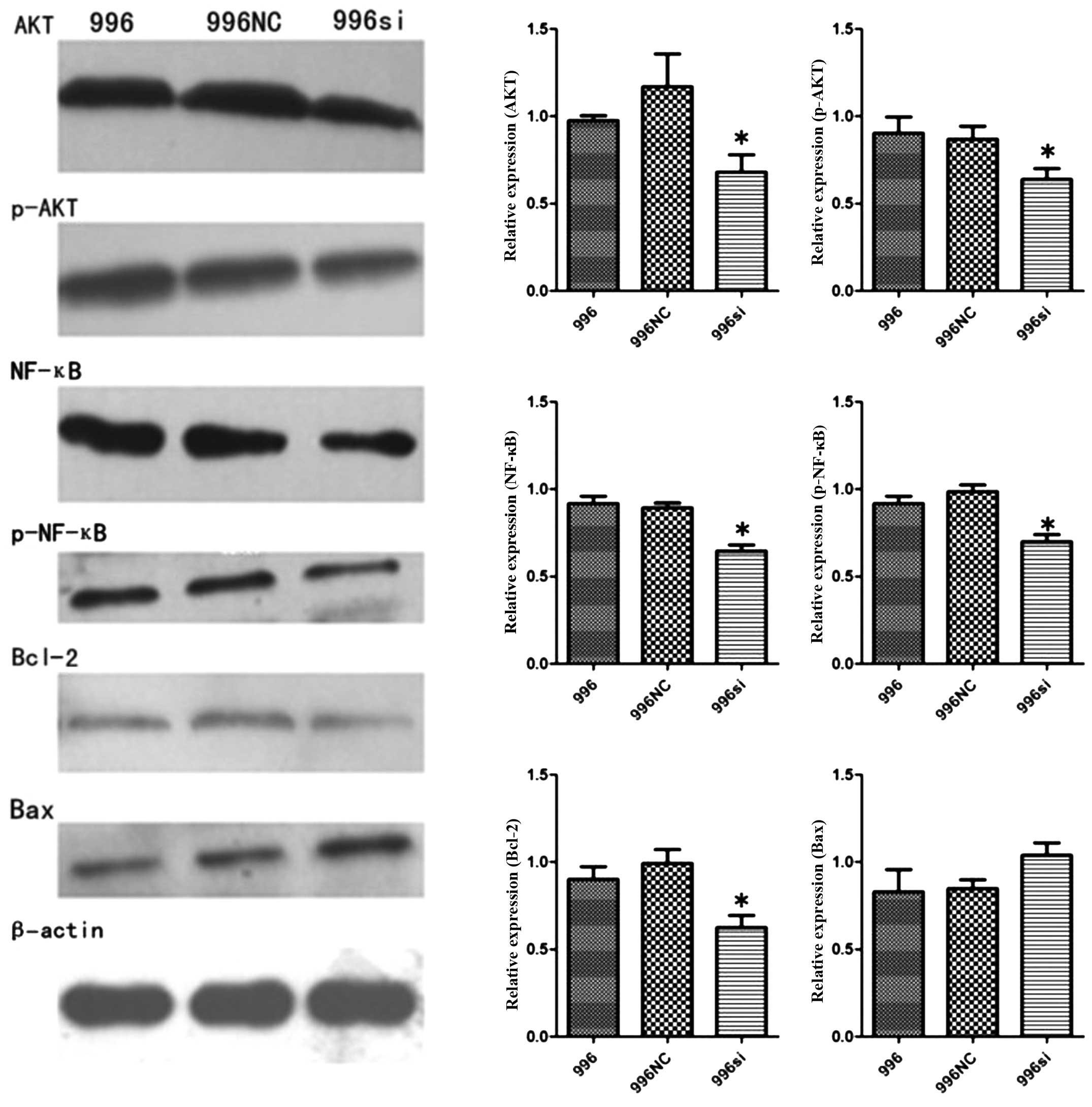
TNF-α knockdown decreases the activity of
the TNF-α-AKT-NF-κB-Bcl-2 pathway, but does not promote the
occurrence of apoptosis
To investigate the mechanisms of TNF-α silencing
responsible for the decrease in growth and invasion, we assessed
the changes in AKT, p-AKT, NF-κB (p65), p-NF-κB (p-p65), Bcl-2 and
Bax protein levels. These proteins are vital to the survival of
gallbladder cancer cells. The western blot analysis results
indicated that the AKT, p-AKT, NF-κB (p65), p-NF-κB (p-p65) and
Bcl-2 protein levels in the SGC-996si cells decreased (P<0.05)
in comparison to the SGC-996NC-transfected and untransfected
SGC-996 cells. We also examined the expression of the Bax gene,
which can promote the apoptosis of gallbladder cancer cells, and
found that TNF-α silencing did not significantly increase the
expression of the Bax gene P>0.05 (Fig. 10).
Discussion
In 1975, Carswell found a factor that can rapidly
cause hemorrhaging and tumor necrosis, named TNF. Thus, TNF was
initially identified and named as such, as it can cause tumor
necrosis (26). TNF-α, a very
important inflammatory cytokine with diverse biological
acitivities, such as the maintenance and homeostasis of host
defence and the immune system, has been shown to be involved in
malignant disease. TNF-α can promote cancer cell proliferation and
invasion and it is mainly produced by macrophages. Cellular
responses to TNF-α are mediated through its receptors, TNFR1 and
TNFR2. The expression of each receptor is independently regulated
on the surface of cells. The ability of TNF-α receptors to interact
with both identical and different downstream signaling pathways
explains their respective functions. TNF-α activates pathways
leading to different cell functions (27), such as cell survival and
proliferation, expression of inflammatory genes and cell death.
TNFR1 can signal each of these biological effects and plays a
crucial role in cell survival and proliferation through the
pathways of NF-κB and AP-1 (28,29).
To our knowledge, this study is the first to
demonstrate the role of tumor-derived TNF-α in promoting the
invasion and proliferation of human gallbladder cancer cells. We
further investigated the possible mechanisms underlying this
process. We determined whether TNF-α was expressed in two different
gallbladder cancer cell lines (NOZ and SGC-996). We found that the
mRNA and protein expression levels of TNF-α in the SGC-996 cells
were higher compared to the levels in the NOZ cells. Thus, on the
basis of our findings and those of previous studies (5,11,23,24), tumor-derived TNF-α may play an
important role in gallbladder tumor cell proliferation and
invasion.
We examined cell proliferation t 1, 2, 3, 4 and 5
days by MTT assay. Compared with the C-N/siRNA and untreated
groups, cell proliferation in the TNF-α siRNA group was slower. The
data from the in vitro cell proliferation assay indicated
that the growth of the cells in the TNF-α siRNA group was markedly
reduced compared with the untreated group and the C-N/siRNA group.
This result is different from that of a previous study on ovarian
cancer, in which the authors concluded that TNF-α RNAi cells grew
at a similar rate to normal cells (11). We repeated our experiment and
found that the results were consistent in the gallbladder cancer
cells. The reasons for this phenomenon require further
investigation.
A number of studies have reported that TNF-α can
induce the expression of MMPs, interleukin (IL)-8, CXC chemokine
receptor type 4 (CXCR), VEGF and MCP-1, thus enhancing tumor cell
invasion and metastasis (17,18,20). Our research team also found that
CXCR and VEGF-C/D promote gallbladder cancer cell proliferation and
invasion (30–32). In this study, we found that the
migration and invasion ability was inhibited when the TNF-α gene
was silenced in vitro. This suggests that tumor-derived
TNF-α exerts a profound effect on migration and invasion. As we
used the RNAi technology to silence the TNF-α gene in SGC-996
cells, the expressoin of the cytokines, MMPs, IL-8, CXCR, VEGF and
MCP-1, was decreased in the gallbladder cells (data not shown). In
the present study, to determine whether the suppressive effects of
TNF-α gene silencing on cell viability were due to apoptosis, we
employed flow cytometry after the cells were stained with red
fluorescence Annexin V-PE/7-aminoactinomycin D. The untransfected
SGC-996, TNF-α siRNA-transfected SGC-996 and SGC-996NC-transfected
cells exhibited a similar rate of apoptosis. Further studies are
required to elucidate the specific mechanisms responsible for this
phenomenon.
In this study, we assessed the changes in AKT,
p-AKT, NF-κB (p65), p-NF-κB (p-p65), Bcl-2 and Bax protein
expression. These proteins are vital to the survival of gallbladder
cancer cells. The western blot analysis results indicated that AKT,
p-AKT, NF-κB (p65), p-NF-κB (p-p65) and Bcl-2 protein levels in the
SGC-996si cells were decreased compared with those in the
SGC-996NC-transfected and untransfected SGC-996 cells. We also
examined the expression of Bax, which can promote the apoptosis of
gallbladder cancer cells, and found that TNF-α silencing did not
significantly increase the expression of the Bax gene.
In conclusion, in the present study, we verify the
biological behavior of gallbladder cancer cell-derived TNF-α. We
provide evidence that the reduction of autocrine TNF-α in
gallbladder cancer cells can exert inhibitory effects on the
ability of the cells to grow and migrate in vitro. This
provides further evidence that targeting TNF-α and its
intracellular pathways may prove useful in the treatment of
gallbladder cancer.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (no. 81272373), the Key Project
of Science and Technology Research Program in Fujian Province (no.
2009Y0024) and the Key Project of Science Research in Fujian
Medical University (no. 09ZD017), and the National Clinical Key
Specialty Construction Project (General Surgery) of China.
References
|
1
|
Donohue JH, Stewart AK and Menck HR: The
National Cancer Data Base report on carcinoma of the gallbladder,
1989–1995. Cancer. 83:2618–2628. 1998.PubMed/NCBI
|
|
2
|
Cubertafond P, Gainant A and Cucchiaro G:
Surgical treatment of 724 carcinomas of the gallbladder. Results of
the French Surgical Association Survey. Ann Surg. 219:275–280.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bartlett DL, Fong Y, Fortner JG, Brennan
MF and Blumgart LH: Long-term results after resection for
gallbladder cancer. Implications for staging and management. Ann
Surg. 224:639–646. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Egberts JH, Cloosters V, Noack A, et al:
Anti-tumor necrosis factor therapy inhibits pancreatic tumor growth
and metastasis. Cancer Res. 68:1443–1450. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zins K, Abraham D, Sioud M and Aharinejad
S: Colon cancer cell-derived tumor necrosis factor-alpha mediates
the tumor growth-promoting response in macrophages by up-regulating
the colony-stimulating factor-1 pathway. Cancer Res. 67:1038–1045.
2007. View Article : Google Scholar
|
|
6
|
Stathopoulos GT, Kollintza A, Moschos C,
et al: Tumor necrosis factor-alpha promotes malignant pleural
effusion. Cancer Res. 67:9825–9834. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Scott KA, Moore RJ, Arnott CH, et al: An
anti-tumor necrosis factor-alpha antibody inhibits the development
of experimental skin tumors. Mol Cancer Ther. 2:445–451.
2003.PubMed/NCBI
|
|
8
|
Michalaki V, Syrigos K, Charles P and
Waxman J: Serum levels of IL-6 and TNF-alpha correlate with
clinicopathological features and patient survival in patients with
prostate cancer. Br J Cancer. 90:2312–2316. 2004.PubMed/NCBI
|
|
9
|
Soria G, Ofri-Shahak M, Haas I, et al:
Inflammatory mediators in breast cancer: coordinated expression of
TNFα and IL-1β with CCL2 & CCL5 and effects on
epithelial-to-mesenchymal transition. BMC Cancer.
11:1302011.PubMed/NCBI
|
|
10
|
Radhakrishnan P, Chachadi V, Lin MF, Singh
R, Kannagi R and Cheng PW: TNFα enhances the motility and
invasiveness of prostatic cancer cells by stimulating the
expression of selective glycosyl- and sulfotransferase genes
involved in the synthesis of selectin ligands. Biochem Biophys Res
Commun. 409:436–441. 2011.
|
|
11
|
Kulbe H, Thompson R, Wilson JL, et al: The
inflammatory cytokine tumor necrosis factor-alpha generates an
autocrine tumor-promoting network in epithelial ovarian cancer
cells. Cancer Res. 67:585–592. 2007. View Article : Google Scholar
|
|
12
|
Akiyama M, Hideshima T, Hayashi T, et al:
Nuclear factor-kappaB p65 mediates tumor necrosis factor
alpha-induced nuclear translocation of telomerase reverse
transcriptase protein. Cancer Res. 63:18–21. 2003.
|
|
13
|
Li J, Sejas DP, Zhang X, et al: TNF-alpha
induces leukemic clonal evolution ex vivo in Fanconi anemia group C
murine stem cells. J Clin Invest. 117:3283–3295. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yan B, Wang H, Rabbani ZN, et al: Tumor
necrosis factor-alpha is a potent endogenous mutagen that promotes
cellular transformation. Cancer Res. 66:11565–11570. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Komori J, Marusawa H, Machimoto T, et al:
Activation-induced cytidine deaminase links bile duct inflammation
to human cholangiocarcinoma. Hepatology. 47:888–896. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li B, Vincent A, Cates J, Brantley-Sieders
DM, Polk DB and Young PP: Low levels of tumor necrosis factor alpha
increase tumor growth by inducing an endothelial phenotype of
monocytes recruited to the tumor site. Cancer Res. 69:338–348.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hagemann T, Wilson J, Kulbe H, et al:
Macrophages induce invasiveness of epithelial cancer cells via
NF-kappa B and JNK. J Immunol. 175:1197–1205. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kulbe H, Hagemann T, Szlosarek PW,
Balkwill FR and Wilson JL: The inflammatory cytokine tumor necrosis
factor-alpha regulates chemokine receptor expression on ovarian
cancer cells. Cancer Res. 65:10355–10362. 2005. View Article : Google Scholar
|
|
19
|
Chua HL, Bhat-Nakshatri P, Clare SE,
Morimiya A, Badve S and Nakshatri H: NF-kappaB represses E-cadherin
expression and enhances epithelial to mesenchymal transition of
mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2.
Oncogene. 26:711–724. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Johnston DA, Dong B and Hughes CC: TNF
induction of jagged-1 in endothelial cells is NFkappaB-dependent.
Gene. 435:36–44. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cox GW, Melillo G, Chattopadhyay U, Mullet
D, Fertel RH and Varesio L: Tumor necrosis factor-alpha-dependent
production of reactive nitrogen intermediates mediates IFN-gamma
plus IL-2-induced murine macrophage tumoricidal activity. J
Immunol. 149:3290–3296. 1992.
|
|
22
|
Balkwill F: Tumor necrosis factor or tumor
promoting factor? Cytokine Growth Factor Rev. 13:135–141. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Naylor MS, Stamp GW, Foulkes WD, Eccles D
and Balkwill FR: Tumor necrosis factor and its receptors in human
ovarian cancer. Potential role in disease progression. J Clin
Invest. 91:2194–2206. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Szlosarek PW, Grimshaw MJ, Kulbe H, et al:
Expression and regulation of tumor necrosis factor alpha in normal
and malignant ovarian epithelium. Mol Cancer Ther. 5:382–390. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Homma S, Hasumura S, Nagamori S and Kameda
H: Establishment and characterization of a human gall bladder
carcinoma cell line NOZ. Hum Cell. 1:95–97. 1988.(In Japanese).
|
|
26
|
Carswell EA, Old LJ, Kassel RL, Green S,
Fiore N and Williamson B: An endotoxin-induced serum factor that
causes necrosis of tumors. Proc Natl Acad Sci USA. 72:3666–3670.
1975. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Waters JP, Pober JS and Bradley JR: Tumour
necrosis factor in infectious disease. J Pathol. 230:132–147. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Blonska M, Shambharkar PB, Kobayashi M, et
al: TAK1 is recruited to the tumor necrosis factor-alpha
(TNF-alpha) receptor 1 complex in a receptor-interacting protein
(RIP)-dependent manner and cooperates with MEKK3 leading to
NF-kappaB activation. J Biol Chem. 280:43056–43063. 2005.
View Article : Google Scholar
|
|
29
|
Devin A, Lin Y, Yamaoka S, Li Z, Karin M
and Liu Z: The alpha and beta subunits of IkappaB kinase (IKK)
mediate TRAF2-dependent IKK recruitment to tumor necrosis factor
(TNF) receptor 1 in response to TNF. Mol Cell Biol. 21:3986–3994.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen Y, Jiang L, She F, et al: Vascular
endothelial growth factor-C promotes the growth and invasion of
gallbladder cancer via an autocrine mechanism. Mol Cell Biochem.
345:77–89. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lin W, Jiang L, Chen Y, et al: Vascular
endothelial growth factor-D promotes growth, lymphangiogenesis and
lymphatic metastasis in gallbladder cancer. Cancer Lett.
314:127–136. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yao X, Zhou L, Han S and Chen Y: High
expression of CXCR4 and CXCR7 predicts poor survival in gallbladder
cancer. J Int Med Res. 39:1253–1264. 2011. View Article : Google Scholar : PubMed/NCBI
|