Introduction
Gastric cancer is among the most common malignancies
worldwide, with a 5-year survival rate of only 20% (1). Due to the fact that this type of
cancer has a poor prognosis, attention has been drawn to the
chemopreventive effect of celecoxib, a cyclooxygenase-2 (COX-2)
inhibitor. COX, also known as prostaglandin synthase peroxidase, is
the rate-limiting enzyme catalyzing arachidonic acid into
prostaglandins, with COX-2 being involved in inflammatory diseases
and certain types of tumor (2,3).
In vitro and in vivo studies have shown that the
long-term and widespread use of non-steroidal anti-inflammatory
drugs (NSAIDs) may contribute to the maintenance of gastric health,
with epidemiological studies showing possible risk reduction
(4,5).
Apoptosis, programmed cell death (PCD), occurs in
multicellular organisms, with autophagy also triggering PCD through
different apoptotic mechanisms (6,7).
Celecoxib and related compounds have been shown to induce cell
cycle arrest, inhibit tumor growth and suppress tumor angiogenesis,
with celecoxib potently inducing apoptosis in tumor cells (8–11).
An increased expression of COX-2 and Akt in gastric carcinomas
relative to normal gastric mucosa, with celecoxib treatment
inducing tumor apoptosis has also been shown (12). Further experimentation showed that
COX-2 inhibitors may induce apoptosis by affecting Akt
phosphorylation, thus activating the Akt signaling pathway
(13). In this study, we used the
selective COX-2 inhibitor celecoxib to treat the SGC-7901 gastric
cancer cells and to induce cell apoptosis in vitro. The
effects of celecoxib on apoptotic and autophagic cell death through
the monitoring of mRNA and protein levels of Akt, caspase-8 and -9
were also examined. This approach aids in further characterization
of the apoptotic effect of celecoxib via the PI3K/Akt signaling
pathway in order to gain a better understanding of its antitumor
effects.
Materials and methods
Cell culture
SGC-7901 human gastric cancer cells (Type Culture
Collection Committee, Chinese Academy of Sciences, Shanghai, China)
were cultured in RPMI-1640 medium (Gibco, Long Island, NY, USA)
with 2 mmol/l glutamine, supplemented with 10% fetal bovine serum
(FBS; HyClone, Logan, UT, USA), 50 U/ml penicillin, and 50 mg/ml
streptomycin. The cells were plated at a density of
1×105 cells/ml in 6-well tissue culture plates and grown
to confluency at 37°C with 5% CO2. When 50% confluency
was reached, serum-supplemented medium was replaced with the
recommended serum-free RPMI-1640 medium for overnight culturing
before celecoxib intervention. Celecoxib, provided by the Faculty
of Medicine, the Chinese University of Hong Kong, stock solution
was added to the serum-supplemented medium at different
concentrations and cultured until the detection time.
MTT assay
The inhibition of cell proliferation in SGC-7901
cells following celecoxib treatment was evaluated using an MTT
assay (Sigma-Aldrich, Shanghai, China) as per the manufacturer’s
instructions. Briefly, 5×103 cells/well were seeded in
96-well plates, incubated in culture medium for 24 h, and treated
with varying concentrations of celecoxib (0, 50, 75, 100 and 125
μmol/l) for 24, 48 and 72 h, with parallel samples treated with
DMSO only serving as controls. Following treatment, the formation
of formazan crystals was measured after 4 h of MTT incubation (10%
v/v) at an optical density (OD) of 490 nm, with each experiment
repeated in triplicate. The relative cell proliferation inhibition
rate was calculated as:
(1−OD490Test/OD490Control) ×100% to show a
percentage value.
TUNEL assay
DNA breaks occur late in the apoptotic pathway and
can be determined and analyzed by performing the TUNEL assay
(Roche, Basel, Switzerland). Firstly, cells were seeded on
coverslips and treated with 100 μmol/l celecoxib for 72 h.
Following treatment, the cells were washed, fixed and stained as
per the manufacturer’s instructions and apoptotic numbers evaluated
using a confocal laser scanning microscope (Leica, Wetzlar,
Germany) at 515–565 nm.
Flow cytometric (FCM) analysis of
apoptosis
Apoptosis was assessed by flow cytometric analysis
using the Annexin V-FITC/PI apoptosis detection kit (Invitrogen,
Life Technologies Ltd., Carlsbad, CA, USA). SGC-7901 cells were
seeded in 6-well plates at ~5×104 cells/well. Following
treatment with celecoxib, the cells were trypsinized, centrifuged
to remove the supernatant, washed with phosphate-buffered saline
(PBS), suspended in 100 μl of 1X binding buffer (10 mM HEPES, 140
mM NaCl, and 2.5 mM CaCl2), and stained with Annexin V
and PI as per the manufacturer’s instructions. FITC-positive and
PI-negative cells were considered apoptotic cells, PI-positive
cells were considered necrotic, and unstained cells were considered
normal viable cells. The apoptotic rates of the various cell groups
were calculated and comparisons of apoptotic rates were conducted
among the various groups.
Transmission electron microscope (TEM)
analysis of cell ultrastructure
Cells were seeded on coverslips, treated with 125
μmol/l celecoxib for 72 h (with a parallel untreated control),
cultured in RPMI-1640 for 72 h, collected and fixed with 3%
glutaraldehyde. The cells were washed with PBS, fixed in 1% osmium
tetroxide, dehydrated by graded ethanol and acetone, and routinely
embedded and polymerized. The slices were contrasted with an
aqueous solution of uranyl acetate and lead citrate and examined by
JEM-1230 transmission electron microscope (Jeol Ltd., Tokyo,
Japan).
Quantitative
reverse-transcription-polymerase chain reaction (qRT-PCR)
analysis
SGC-7901 cells were cultured at a density of
1×105 cells/ml in 6-well tissue culture plates. One
group was treated with various concentrations of celecoxib (0, 75,
100 and 125 μmol/l) and cultured for 72 h, while an additional test
group was treated with 125 μmol/l celecoxib for 0, 24, 48 and 72 h.
Total RNA was extracted by a column RNA extraction kit (Sangon,
Shanghai, China) and reverse-transcribed into cDNA at 37°C for 15
min, and 85°C for 5 sec. Diluted cDNA was subjected to qRT-PCR
using a SYBR® Premix Ex Taq™ II kit (Takara Bio, Inc.,
Shiga, Japan) in 25 μl of reaction solution containing 2 μl of cDNA
template, 1 μM of each primer, 10 μl of 2X SYBR-Green master mix,
and brought to the final volume with RNase-free water. Reactions
were performed in triplicate via a PCR thermal cycler (Roter-Gene
3000; Corbett, Sydney, Australia) under the following conditions:
pre-denaturation at 95°C for 30 sec, 40 cycles of denaturation at
95°C for 5 sec, and annealing at 62°C for 30 sec. The relative
expression was calculated by the 2−ΔΔCT formula. The
primer pairs for qRT-PCR are listed in Table I.
 | Table IPrimer pairs for qRT-PCR. |
Table I
Primer pairs for qRT-PCR.
| Gene name | Accession | Sequence (5′-3′) | Product size
(bp) |
|---|
| Caspase-9 | NM_001229 | F:
CCCATATGATCGAGGACATCCA
R: ACAACTTTGCTGCTTGCCTGTTAG | 186 |
| Caspase-8 | NM_001228 | F:
GGTACATCCAGTCACTTTGCCAGA
R: GTTCACTTCAGTCAGGATGGTGAGA | 83 |
| Akt | NM_005163 | F:
GTGGCAGCACGTGTACGAGAA
R: GTGATCATCTGGGCCGTGAA | 108 |
| β-actin | NM_001101 | F:
TGGCACCCAGCACAATGAA
R: CTAAGTCATAGTCCGCCTAGAAGCA | 126 |
Western blot analysis
Total protein was extracted and protein
concentrations established via bicinchoninic acid (BCA) assay.
Protein (25 μg) was denatured, separated by SDS-PAGE
electrophoresis and transferred to a PVDF membrane. After blocking
overnight at 4°C using 5% BSA, the membranes were incubated with
primary antibodies (anti-procaspase-8 1:2,500 and procaspase-9
1:2,000; both from Abcam, Cambridge, MA, USA), p-Akt 1:800
(Bioworld, St. Louis Park, MN, USA) for 2 h at room temperature,
washed by TBST and incubated with the corresponding horseradish
peroxidase (HRP)-conjugated secondary antibody at 1:2,000 dilution
for 2 h. Bands were visualized using enhanced chemiluminescence
(ECL; Applygen, Beijing, China) detection reagents and scanned
images were quantified using ImageJ software. Experiments were
performed in triplicate with β-actin used as a housekeeping control
for normalization. The ratio of target gene to β-actin was used for
semi-quantification and comparison between different groups.
Statistical analysis
Triplicate data are presented as mean values and
shown as the means ± standard deviation (SD). Samples were analyzed
by one-way ANOVA, with P<0.05 considered to indicate statistical
significance.
Results
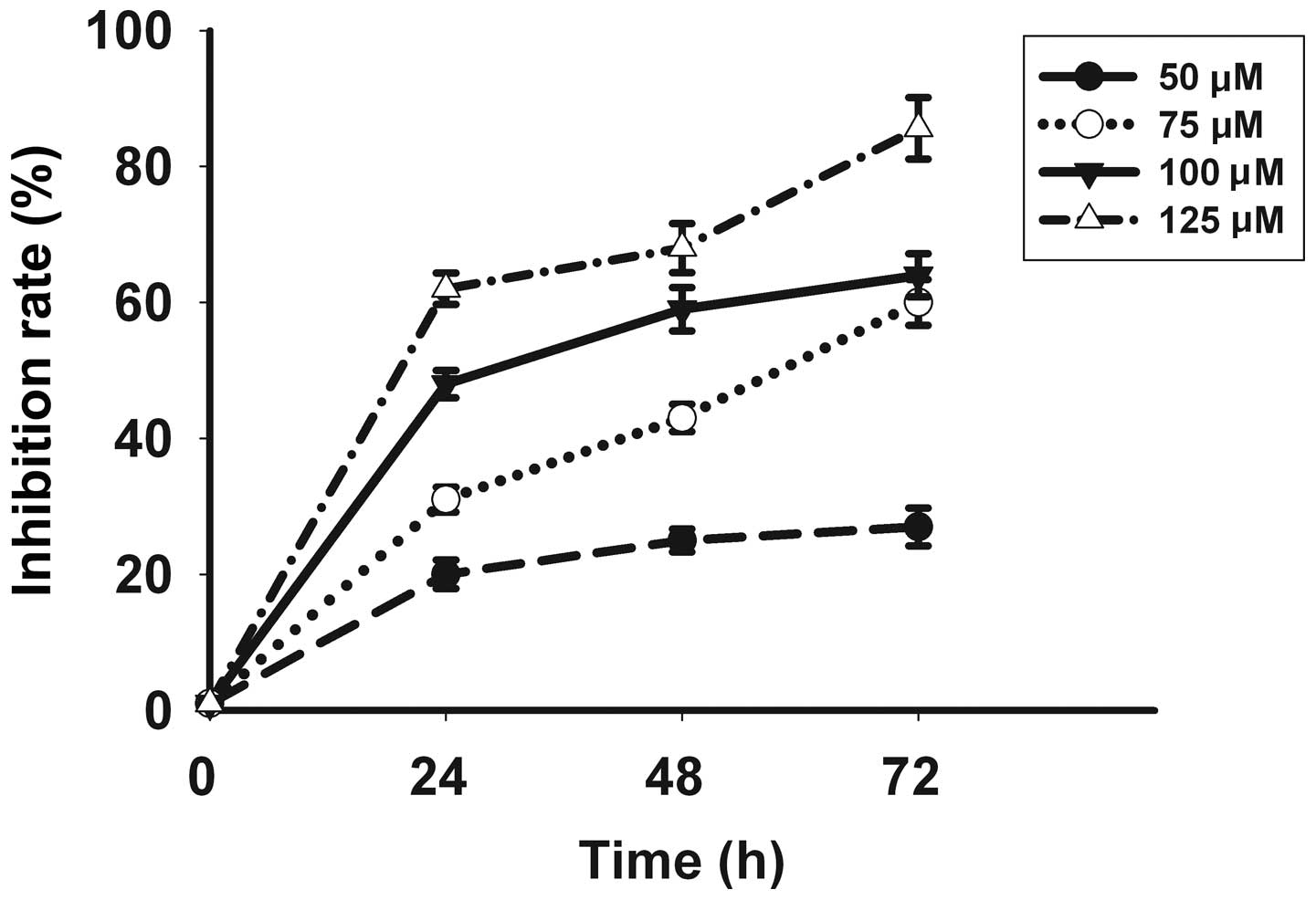
Celecoxib inhibits proliferation of
SGC-7901 cells
Following in vitro treatment with celecoxib,
the SGC-7901 gastric cancer cell line showed a significant
inhibition of cell proliferation in a time- and dose-dependent
manner, with the most pronounced effect evident at a concentration
of 125 μmol/l for 72 h as identified by a proliferation inhibition
rate of 85.6±4.51% (Fig. 1).
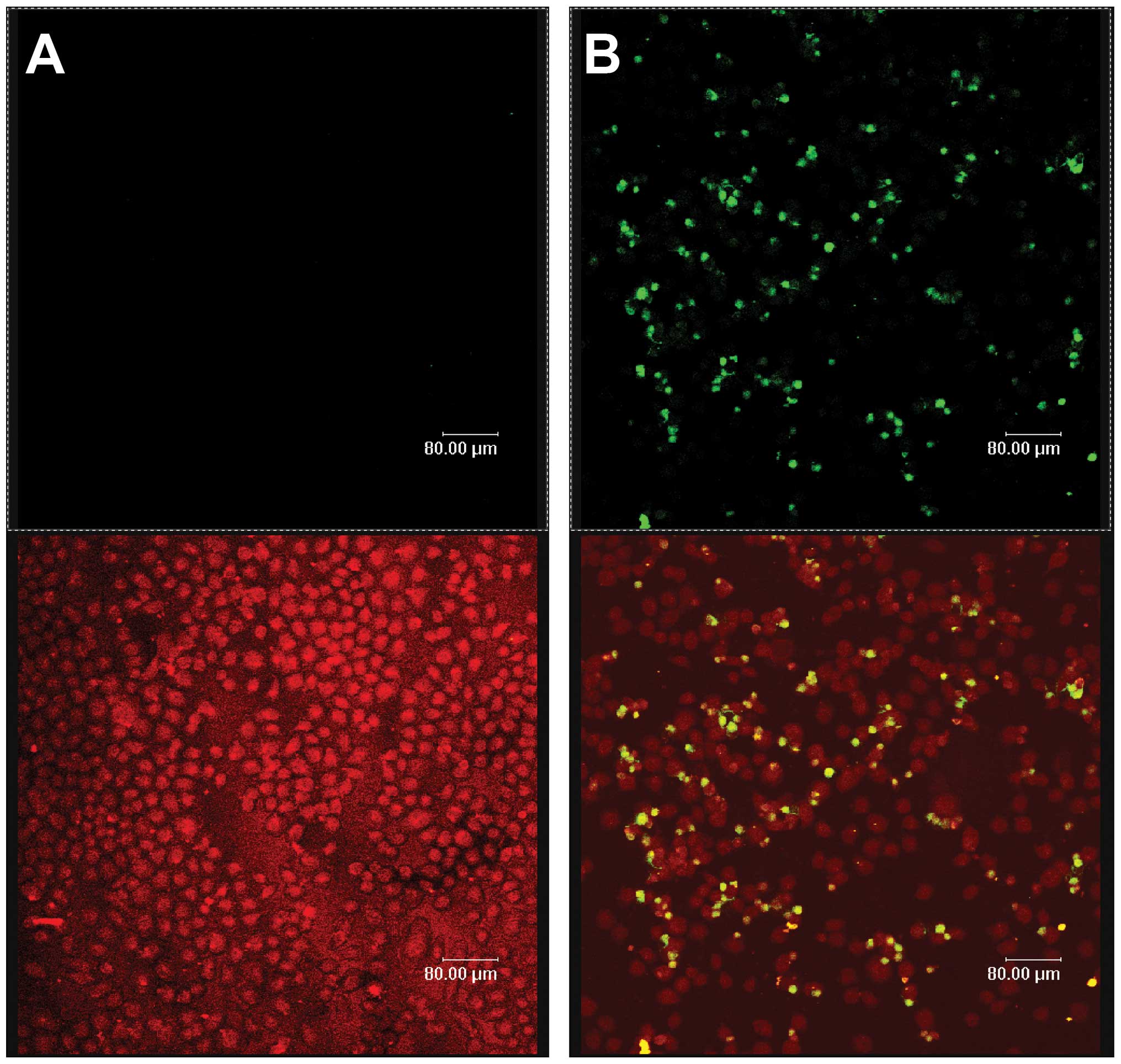
Celecoxib induces apoptosis of SGC-7901
cells
Fluorescein- labeled dUTP was connected to DNA 3′-OH
ends of apoptotic cells by the deoxynucleotidyl transferase enzyme.
Apoptotic cells with green fluorescence were detected by laser
scanning confocal microscopy at an excitation of 515–565 nm, while
all cells were exhibited as red under bright field microscopy. The
two images were superimposed to show the specificity of apoptotic
cells (yellow) and their position. Celecoxib-treated cells
(Fig. 2B) showed significant
levels of apoptosis relative to the control (Fig. 2A), with a statistical significance
of P<0.05.
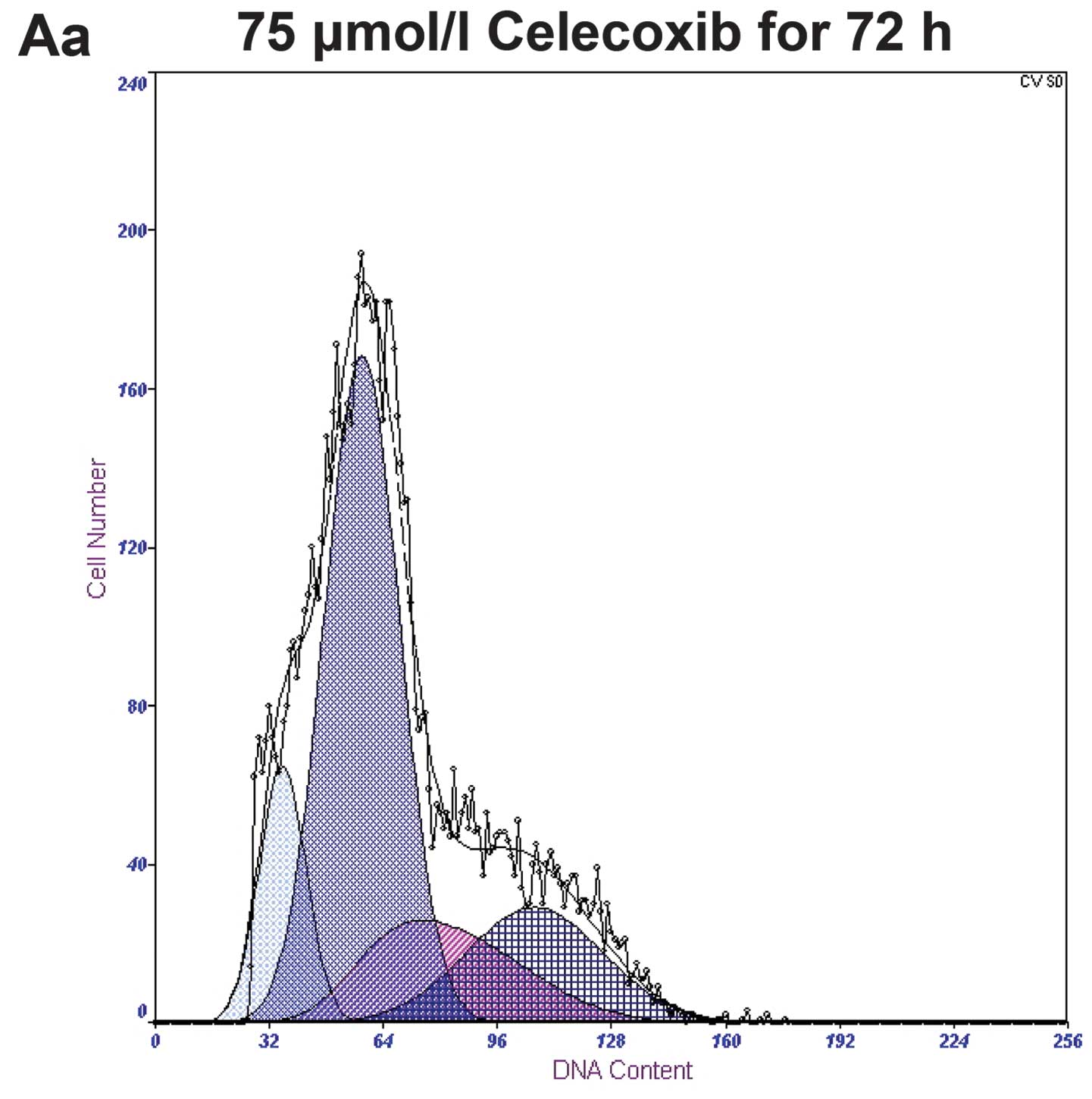
Treatment with 0, 75, 100 and 125 μmol/l of
celecoxib for 72 h yielded apoptotic rates of 4.0±0.91, 12.9±1.32,
24.6±3.63 and 35.7±2.73%, respectively, with a statistical
significance of P<0.05 when compared to the control group.
Treatment with 125 μmol/l of celecoxib for 0, 24, 48 and 72 h,
yielded apoptotic rates of 2.2±0.32, 8.5±1.57, 20.3±2.84 and
35.7±2.73%, respectively. Both study sets demonstrated a gradual
increase in apoptotic rates in a time-and dose-dependent manner
(Fig. 3).
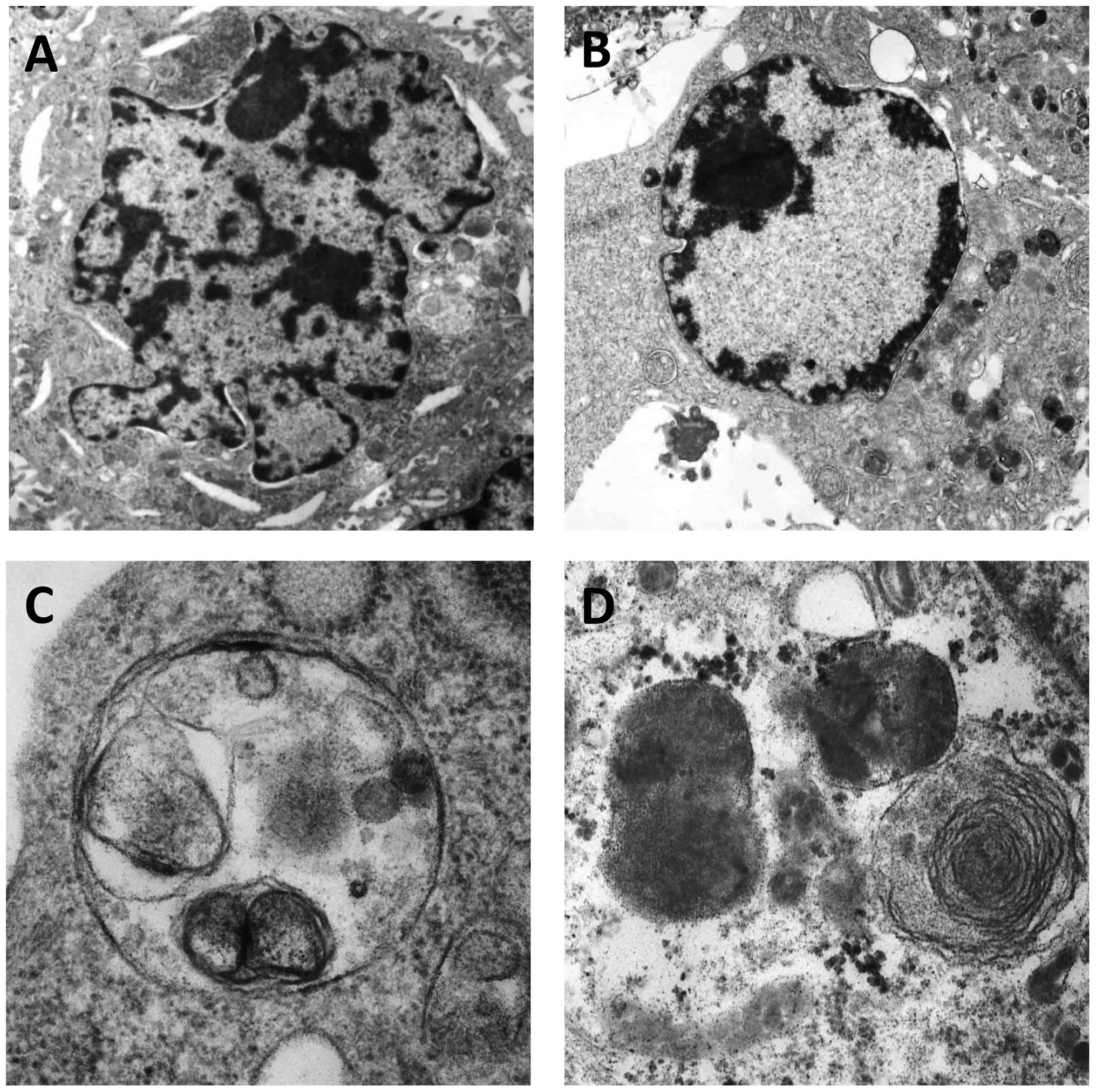
Celecoxib alters the ultrastructure of
SGC-7901 cells
Following treatment with 125 μmol/l celecoxib for 72
h, typical early apoptotic changes were found to include nuclear
membrane shrinkage and retraction (Fig. 4A), nuclear chromatin condensation,
marginalization and crescents, with late apoptotic changes observed
by nuclei cleavage into fragments and apoptotic body production
(Fig. 4B). Additionally, typical
autophagic structures were found to include several cytoplasmic
autophagic vacuoles and autophagosomes, which swallowed organelles
(Fig. 4C and D).
Effect of celecoxib on Akt, caspase-8 and
-9 expression
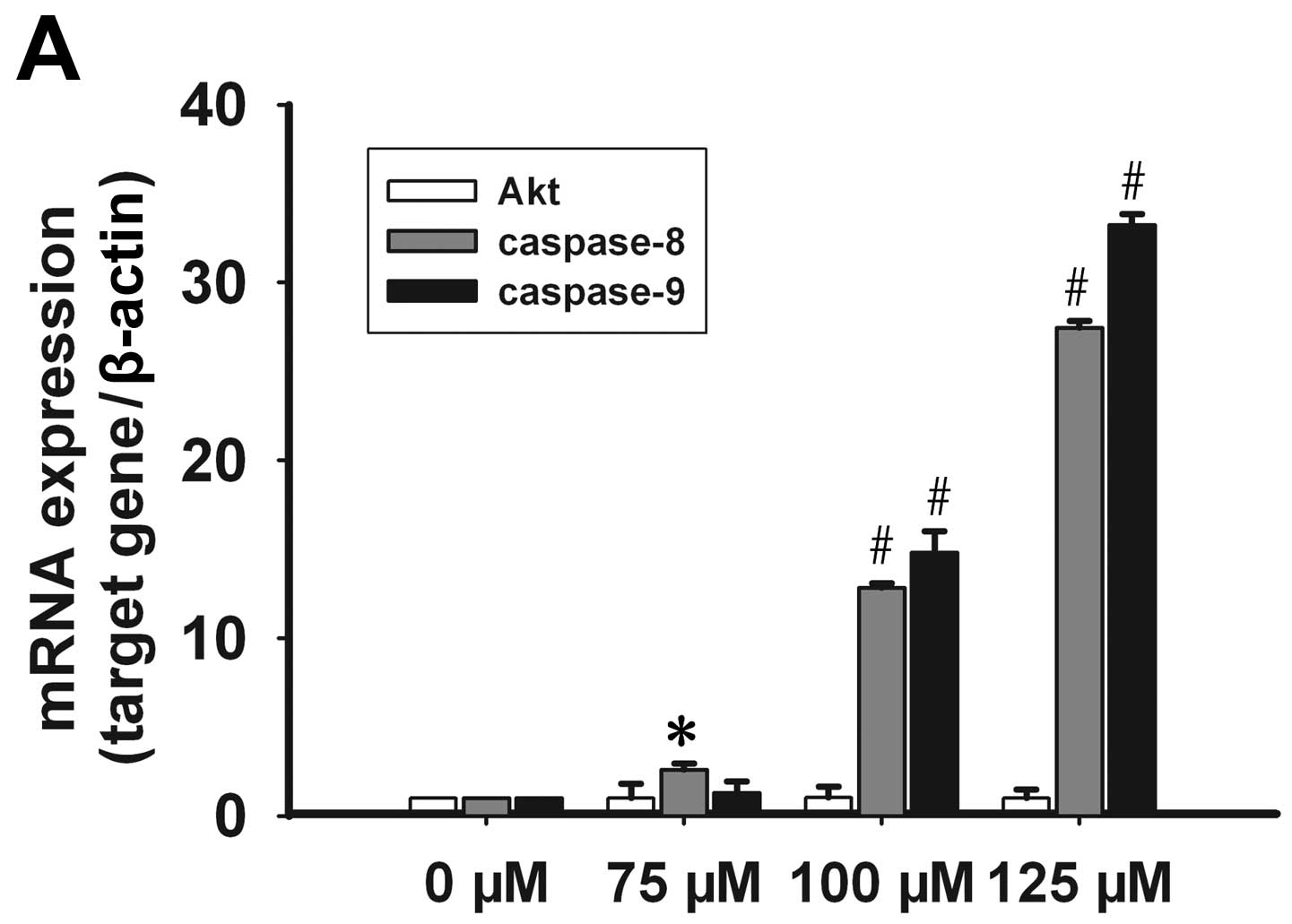
No significant change in the mRNA levels of Akt was
observed subsequent to treatment with celecoxib; however, the
presence of p-Akt decreased in a time- and dose-dependent manner.
Caspase-8 mRNA expression increased in a dose-dependent manner at
concentrations of 75, 100 and 125 μmol/l of celecoxib. Caspase-9
mRNA expression levels increased significantly at a concentration
of 100 and 125 μmol/l of celecoxib (Fig. 5A). Following treatment with 125
μmol/l of celecoxib for 24, 48 and 72 h, caspase-8 and -9 mRNA
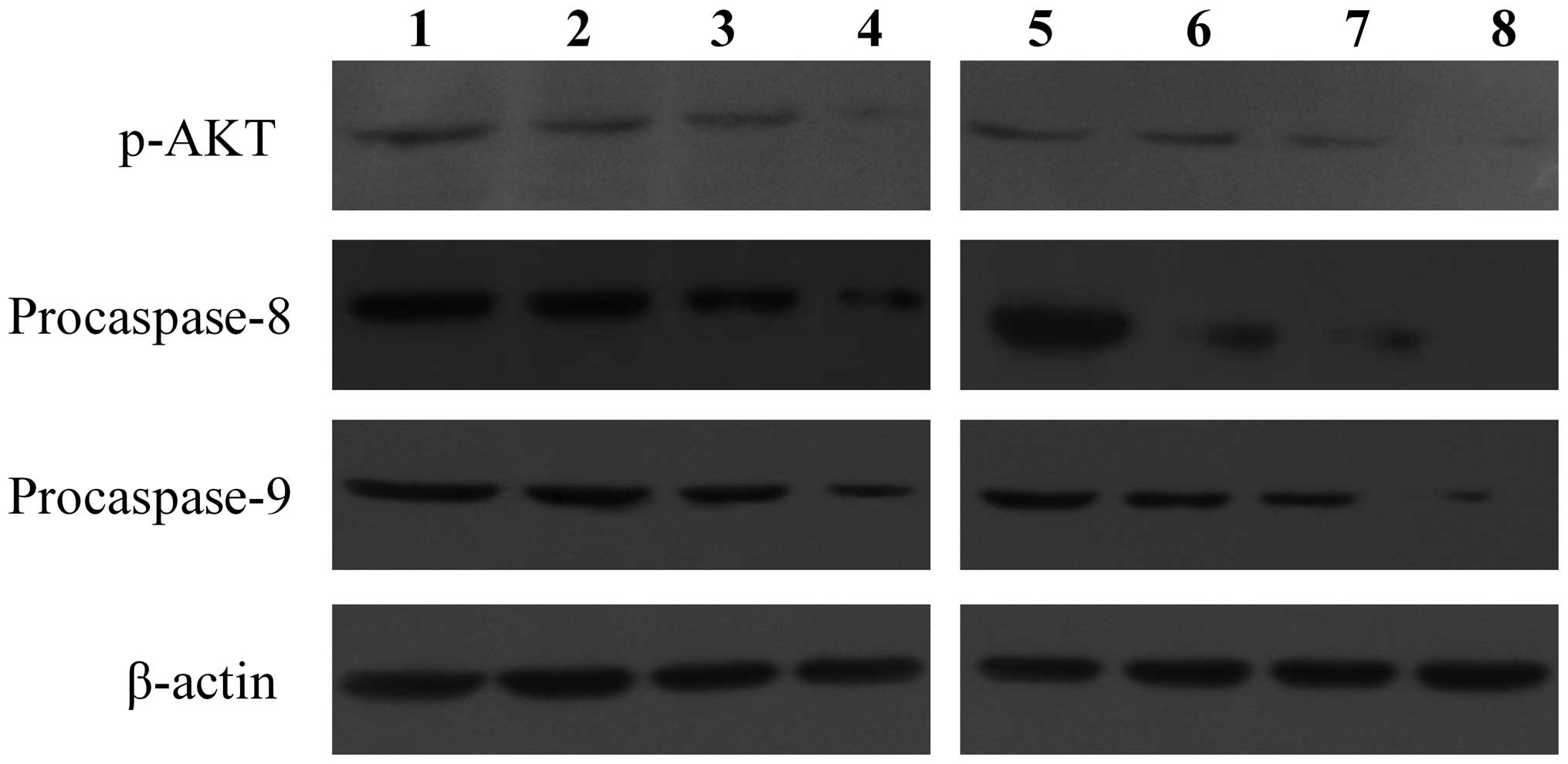
expression increased significantly (Fig. 5B). By contrast, procaspase-8 and
-9 protein expression was significantly lower than the control
group in a time- and dose-dependent manner (Fig. 6). These results showed that
celecoxib may inhibit Akt phosphorylation and promote caspase-8, -9
transcription and procaspase-8, -9 protein activation.
Discussion
Gastric carcinoma is among the most common
malignancies worldwide, with an elevated 5-year postoperative
mortality rate (14) thus
creating a need for an alternative treatment method. Currently, the
role of celecoxib, a non-cytotoxic COX-2 inhibitor, in cancer
therapy has been under scrutiny (14). COX, also known as prostaglandin
synthetase, has three known isoenzymes in mammals, COX-1, COX-2 and
COX-3 (15). COX-2 is present at
low levels of expression in most normal tissues, but tumor factors,
inflammatory cytokines and growth factors could promote its
expression (16). COX-2 is
related to the development of tumors by promoting tumor cell
proliferation, enabling tumor evasion of the host immune
surveillance and promoting tumor invasion/metastasis (5,17).
Multicellular organisms maintain their homeostasis
through cell proliferation and PCD, with an imbalance possibly
leading to the development of cancer. In this experiment, we found
that the selective COX-2 inhibitor celecoxib induced apoptosis of
SGC-7901 cells via reduced expression levels of COX-2, as obsreved
by inhibited cell proliferation using MTT analysis and an increased
number of apoptotic cells as detected by TUNEL and flow cytometry.
Moreover, typical apoptotic changes were shown to include nuclear
membrane shrinkage, nuclear chromatin condensation and apoptotic
bodies using TEM to support the apoptotic effects of celecoxib.
Caspases are a type of protease associated with
apoptosis and cytokine maturation, and are divided into initiator
caspases, effector caspases and inflammatory mediators. Caspases
are synthesized as relatively inactive zymogens and must undergo a
process of activation during apoptosis. Caspase-8 is the initiator
of the Fas-Fas ligand (FasL) pathway, and usually exists in the
form of procaspase-8. When FasL binds to the corresponding Fas
receptor, the intracellular death effector domain (DED) of the Fas
receptor attracts Fas associated with death domain protein (FADD)
and recruits procaspase-8 to form a death-inducing signaling
complex (DISC). Procaspase-8 is then hydrolyzed to generate
activated caspase-8, followed by the activation of procaspase-3 and
other effector caspases that eventually induce apoptosis (18). Caspase-9 is the initiator of the
mitochondrial pathway, also known as procaspase-9, an inactive
zymogen. The initiator caspase-9 is activated by the assembly of a
multimeric complex (dubbed apoptosome) involving Apaf-1 and
cytochrome c. Cleaved caspase-9 and -3 are activated and
these effector caspases degrade a large number of cell proteins,
ultimately inducing cell apoptosis (19,20). In this study, we found that
celecoxib significantly increased caspase-8 and -9 mRNA expression
in a time- and dose-dependent manner in SGC-7901 cells, suggesting
that celecoxib may activate caspase-8 and -9 to initiated apoptosis
through the death receptor and mitochondrial pathways,
respectively.
Autophagy is a crucial component of the cellular
stress adaptation response that maintains mammalian homeostasis
(21). There are three different
forms of autophagy that are commonly described: macroautophagy,
microautophagy and chaperone-mediated autophagy. Macroautophagy is
the predominant pathway occurring mainly to eradicate damaged
organelles or unused proteins. Macroautophagy is mediated by a
unique organelle, the autophagosome, which encloses long-lived
proteins and portions of organelles for delivery to the lysosome
(22,23). Autophagy may play different roles
in cancer occurrence and progression, while also potentially
promoting or inhibiting cell proliferation at different stages of
tumor growth (24). For example,
autophagy plays a protective role in tumor cells via degradation of
organelles under nutritional deficiency. Conversely, autophagy can
also inhibit tumor growth via beclin 1, UVRAG, Bif and Atg.
Findings of a recent study showed that berberine extracts promoted
autophagy by activating beclin 1 expression and activated caspase-9
to induce apoptosis in hepatoma cells (25). Plant lectin from Polygonatum
cyrtonema induced apoptosis and autophagy by inhibiting the
Ras/Raf and PI3K/Akt signaling pathways in murine fibrosarcoma
cells (26). In this study, the
selective COX-2 inhibitor celecoxib, not only generated
morphological changes indicative of apoptosis, but also typical
changes of autophagy to include cytoplasmic autophagic vacuoles and
autophagosomes.
The molecular mechanism by which celecoxib induces
apoptosis is not yet fully understood. The PI3K/Akt pathway widely
presents in normal cells, but is abnormally activated in many
malignant tumors (27–29). Akt, also known as protein kinase B
(PKB), is a central component of the PI3K/Akt pathway, with Akt
phosphoregulation impacting a variety of biological activities. In
healthy and tumorigenic cells, Akt can be activated in an
intracellular manner by hormones, growth factors, and extracellular
matrix components (30). Akt
regulates cell growth, survival and apoptosis through substrate
phosphorylation, with Akt phosphoregulation observed at the Thr308
and Ser473 site, which are both required for activation. Akt is
activated as follows: the activated PI3K produces a secondary
messenger PIP3 at the plasma membrane, PIP3 then binds an inactive
Akt inducing its shift from the cytoplasm to the plasma membrane
where Ser124 and Thr450 are phosphorylated, making Akt undergo a
conformational change exposing its Thr308 and Ser473 sites.
Immediately, phosphoinositide-dependent kinase 1 (PDK1) and
phosphoinositide-dependent kinase 2 (PDK2), which are in close
proximity to Akt, respectively catalyze the phosphorylation of the
exposed Thr308 and Ser473 sites, resulting in the complete
activation of Akt. This may trigger a phosphorylation cascade of
downstream targets, ultimately impacting the regulation of cell
growth and survival, proliferation and apoptosis, angiogenesis,
cell migration and numerous biological processes (30–33).
In the present study, following celecoxib treatment
in SGC-7901 cells, p-Akt, or activated Akt, was distinctly
downregulated, leading to the upregulation of caspase-8 and -9 mRNA
expression and increased procaspase-8 and -9 activation. Thus, we
hypothesized that celecoxib inhibited the PI3K/Akt pathway by
reducing the level of phosphorylation of Akt, which in turn
activated the expression and activation of caspase-8 and -9,
resulting in apoptosis through the death receptor and mitochondrial
pathways in SGC-7901 cells. Notably, we found changes in the cell
ultrastructure to include apoptosis and autophagy, suggesting that
celecoxib simultaneously induced apoptosis and autophagy, which is
consistent with results of previous studies (5,13)). Autophagy is an evolutionarily
conserved process that occurs during the growth and development
process in many animals, but its specific mechanism of PCD is
unclear. Autophagy and apoptosis could coadjust through p53
(35), PI3K/Akt (36) and Bcl-2-beclin 1 (37). Thus, celecoxib may impact both
apoptosis and autophagy via the PI3K/Akt signaling pathway in the
SGC-7901 gastric cancer cells. The results of this study provide a
new theoretical foundation for the antitumor mechanisms of
celecoxib and offers new targets for cancer therapy, although these
findings should be verified in future investigations.
Acknowledgements
This study was supported by a grant from the
National Science Foundation of China (no. 81172366). We would like
to thank LetPub for its linguistic assistance during the
preparation of this manuscript.
References
|
1
|
Saukkonen K, Rintahaka J, Sivula A, et al:
Cyclooxygenase-2 and gastric carcinogenesis. APMIS. 111:915–925.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jones DA, Carlton DP, McIntyre TM,
Zimmerman GA and Prescott SM: Molecular cloning of human
prostaglandin endoperoxide synthase type II and demonstration of
expression in response to cytokines. J Biol Chem. 268:9049–9054.
1993.PubMed/NCBI
|
|
3
|
Sarkar FH, Adsule S, Li Y and Padhye S:
Back to the future: COX-2 inhibitors for chemoprevention and cancer
therapy. Mini Rev Med Chem. 7:599–608. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Duan L, Wu AH, Sullivan-Halley J and
Bernstein L: Nonsteroidal anti-inflammatory drugs and risk of
esophageal and gastric adenocarcinomas in Los Angeles County.
Cancer Epidemiol Biomarkers Prev. 17:126–134. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fu SL, Wu YL, Zhang YP, Qiao MM and Chen
Y: Anti-cancer effects of COX-2 inhibitors and their correlation
with angiogenesis and invasion in gastric cancer. World J
Gastroenterol. 10:1971–1974. 2004.PubMed/NCBI
|
|
6
|
Lo AC, Woo TT, Wong RL and Wong D:
Apoptosis and other cell death mechanisms after retinal detachment:
implications for photoreceptor rescue. Ophthalmologica. 226(Suppl
1): 10–17. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Su M, Mei Y and Sinha S: Role of the
crosstalk between autophagy and apoptosis in cancer. J Oncol.
2013:1027352013.
|
|
8
|
Qadri SS, Wang JH, Coffey JC, et al:
Surgically induced accelerated local and distant tumor growth is
significantly attenuated by selective COX-2 inhibition. Ann Thorac
Surg. 79:990–995. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Grösch S, Maier TJ, Schiffmann S and
Geisslinger G: Cyclooxygenase-2 (COX-2)-independent
anticarcinogenic effects of selective COX-2 inhibitors. J Natl
Cancer Inst. 98:736–747. 2006.PubMed/NCBI
|
|
10
|
Baek JY, Hur W, Wang JS, Bae SH and Yoon
SK: Selective COX-2 inhibitor, NS-398, suppresses cellular
proliferation in human hepatocellular carcinoma cell lines via cell
cycle arrest. World J Gastroenterol. 13:1175–1181. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jendrossek V: Targeting apoptosis pathways
by celecoxib in cancer. Cancer Lett. 332:313–324. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fan XM, Jiang XH, Gu Q, et al: Inhibition
of Akt/PKB by a COX-2 inhibitor induces apoptosis in gastric cancer
cells. Digestion. 73:75–83. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim N, Kim CH, Ahn DW, et al: Anti-gastric
cancer effects of Celecoxib, a selective COX-2 inhibitor, through
inhibition of Akt signaling. J Gastroenterol Hepatol. 24:480–487.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Patru CL, Surlin V, Georgescu I and Patru
E: Current issues in gastric cancer epidemiology. Rev Med Chir Soc
Med Nat Iasi. 117:199–204. 2013.PubMed/NCBI
|
|
15
|
Futagami S, Suzuki K, Hiratsuka T, et al:
Chemopreventive effect of Celecoxib in gastric cancer.
Inflammopharmacology. 15:1–4. 2007. View Article : Google Scholar
|
|
16
|
Willoughby DA, Moore AR and Colville-Nash
PR: COX-1, COX-2, and COX-3 and the future treatment of chronic
inflammatory disease. Lancet. 355:646–648. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Greenhough A, Smartt HJ, Moore AE, et al:
The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and
adaptation to the tumour microenvironment. Carcinogenesis.
30:377–386. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu Y and Liu BA: Enhanced proliferation,
invasion, and epithelial-mesenchymal transition of
nicotine-promoted gastric cancer by periostin. World J
Gastroenterol. 17:2674–2680. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fulda S: Caspase-8 in cancer biology and
therapy. Cancer Lett. 281:128–133. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Würstle ML, Laussmann MA and Rehm M: The
central role of initiator caspase-9 in apoptosis signal
transduction and the regulation of its activation and activity on
the apoptosome. Exp Cell Res. 318:1213–1220. 2012.PubMed/NCBI
|
|
21
|
Allan LA and Clarke PR: Apoptosis and
autophagy: Regulation of caspase-9 by phosphorylation. FEBS J.
276:6063–6073. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
White E, Karp C, Strohecker AM, Guo Y and
Mathew R: Role of autophagy in suppression of inflammation and
cancer. Curr Opin Cell Biol. 22:212–217. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lorin S, Hamaï A, Mehrpour M and Codogno
P: Autophagy regulation and its role in cancer. Semin Cancer Biol.
23:361–379. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
White E: Deconvoluting the
context-dependent role for autophagy in cancer. Nat Rev Cancer.
12:401–410. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Helgason GV, Holyoake TL and Ryan KM: Role
of autophagy in cancer prevention, development and therapy. Essays
Biochem. 55:133–151. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xie BS, Zhao HC, Yao SK, et al: Autophagy
inhibition enhances etoposide-induced cell death in human hepatoma
G2 cells. Int J Mol Med. 27:599–606. 2011.PubMed/NCBI
|
|
27
|
Liu B, Wu JM, Li J, et al: Polygonatum
cyrtonema lectin induces murine fibrosarcoma L929 cell
apoptosis and autophagy via blocking Ras-Raf and PI3K-Akt signaling
pathways. Biochimie. 92:1934–1938. 2010. View Article : Google Scholar
|
|
28
|
Ye B, Jiang LL, Xu HT, Zhou DW and Li ZS:
Expression of PI3K/AKT pathway in gastric cancer and its blockade
suppresses tumor growth and metastasis. Int J Immunopathol
Pharmacol. 25:627–636. 2012.PubMed/NCBI
|
|
29
|
Lin X, Zhang X, Wang Q, et al: Perifosine
downregulates MDR1 gene expression and reverses multidrug-resistant
phenotype by inhibiting PI3K/Akt/NF-kappaB signaling pathway in a
human breast cancer cell line. Neoplasma. 59:248–256. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kang XH, Xu ZY, Gong YB, et al: Bufalin
reverses HGF-induced resistance to EGFR-TKIs in EGFR mutant lung
cancer cells via blockage of Met/PI3k/Akt pathway and induction of
apoptosis. Evid Based Complement Alternat Med.
2013:2438592013.PubMed/NCBI
|
|
31
|
Chang F, Lee JT, Navolanic PM, et al:
Involvement of PI3K/Akt pathway in cell cycle progression,
apoptosis, and neoplastic transformation: a target for cancer
chemotherapy. Leukemia. 17:590–603. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Song G, Ouyang G and Bao S: The activation
of Akt/PKB signaling pathway and cell survival. J Cell Mol Med.
9:59–71. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Falasca M: PI3K/Akt signalling pathway
specific inhibitors: a novel strategy to sensitize cancer cells to
anti-cancer drugs. Curr Pharm Des. 16:1410–1416. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Garcia-Echeverria C and Sellers WR: Drug
discovery approaches targeting the PI3K/Akt pathway in cancer.
Oncogene. 27:5511–5526. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu J, Lin Y, Yang H, Deng Q, Chen and He
J: The expression of p33(ING1), p53, and autophagy-related gene
Beclin1 in patients with non-small cell lung cancer. Tumor Biol.
32:1113–1121. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cheng Y, Ren X, Zhang Y, et al: eEF-2
kinase dictates cross-talk between autophagy and apoptosis induced
by Akt inhibition, thereby modulating cytotoxicity of novel Akt
inhibitor MK-2206. Cancer Res. 71:2654–2663. 2011. View Article : Google Scholar
|
|
37
|
Djavaheri-Mergny M, Maiuri MC and Kroemer
G: Cross talk between apoptosis and autophagy by caspase-mediated
cleavage of Beclin 1. Oncogene. 29:1717–1719. 2010. View Article : Google Scholar : PubMed/NCBI
|