1. Introduction
In most genetic systems, including humans,
translation termination occurs when one of three stop (nonsense)
codons (UAA, UAG and UGA) enters the ribosomal A-site (1–3).
In contrast to the recognition of sense codons which is carried out
by transfer RNA (tRNA), the recognition of stop codons is mediated
by extra-ribosomal proteins known as class 1 release factors (RFs).
Subsequent to the recognition of the nonsense codon, release
factors trigger the hydrolysis of the ester bond between the
nascent polypeptide chain and the tRNA in the ribosomal P-site,
resulting in translation termination (3) (Fig.
1).
Translation termination is not a perfect process and
its efficiency depends on competition between the recognition of
the stop codon by a release factor and decoding of the stop codon
by a near-cognate tRNA (paired using two of the three bases). The
fact that translation termination is not 100% efficient results in
a low level of natural nonsense suppression or translational
read-through of the termination codon. Translational read-through
results in an amino acid being incorporated in place of the stop
codon and the synthesis of a C-terminally extended protein that
terminates at the next stop codon present in the same reading
frame; all termination codons, whether natural or premature exhibit
low levels of translational read-through. Multiple factors
contribute to the efficiency of translational read-through,
including the identity of the nucleotides 5′ and 3′ of the
termination codon (4).
Additionally, the termination codons themselves mediate translation
termination with varying efficiencies (UAA ≥ UAG ≥ UGA), which is
inversely correlated with the extent of natural read-through that
occurs at each nonsense codon (UGA ≥ UAG ≥ UAA) (4,5).
While understanding the mechanism of protein
synthesis has always attracted considerable interest, the
realization that premature termination codons (PTCs) contribute to
human pathology has significantly increased the attention given to
the mechanism of translation termination and the mechanisms capable
of promoting translational read-through (or nonsense suppression).
PTCs have been implicated in several human diseases with ~2,400
distinct genetic disorders having at least one causative PTC allele
(6). Additionally, a recent
meta-analysis of the Human Gene Mutation Database (HGMD) estimated
that ~11% of HGMD lesions responsible for inherited disorders are
nonsense generating mutations (7). In addition to the C-terminally
truncated protein that is synthesized as a result of the PTC, the
presence of a PTC within a messenger RNA (mRNA) is often, though
not always, accompanied with an increased rate of mRNA decay via
nonsense mediated mRNA decay (NMD) (8). Thus, as a result of the reduction in
gene expression, coupled with the synthesis of a C-terminally
truncated protein product which may be harmful to cells, nonsense
mutations that trigger NMD are more likely to lead to a disease
phenotype (7).
Several small molecules have been identified that
are capable of modulating the efficiency of translation termination
(9, and references therein). More
recently, novel strategies based on targeting the nucleotides of
the PTC for post-transcriptional modification have been developed.
While these methods are still in their infancy, they represent an
exciting new avenue towards the development of therapeutics capable
of specifically suppressing PTCs. In this review, we summarize our
current understanding of the mechanism of translation termination
and discuss multiple strategies currently being investigated to
promote translational read-through of PTCs.
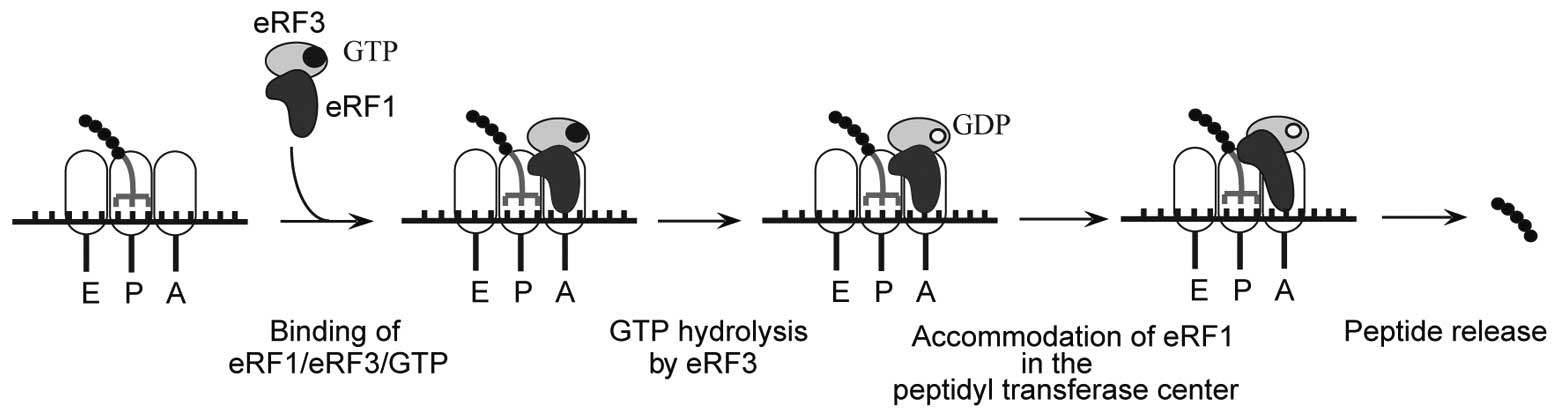
2. Mechanism of translation termination
Translation termination occurs when a stop codon
enters the ribosomal A-site. For simplicity, termination can be
thought of as two distinct steps, stop codon recognition and
peptide release. In eukaryotes, translation termination is mediated
by eukaryotic release factor 1 (eRF1), which is responsible for
stop codon recognition and triggering peptide release, and eRF3, a
GTPase that stimulates eRF1-mediated peptide release (10,11). eRF1, in turn, stabilizes binding
of GTP to eRF3 so that they form a stable ternary complex (12,13) (Fig.
1).
X-ray crystallographic and nuclear magnetic
resonance (NMR) data of the RFs have provided significant insight
regarding their mechanism of action (14–16). Of note, eRF1, responsible for stop
codon recognition, adopts a fold resembling that of a tRNA and is
comprised of three distinct domains, namely N-terminal (N), middle
(M) and C-terminal (C) (16).
Although the mechanism by which eRF1 engages each of the three stop
codons is unknown, mutational and genetic analyses have identified
an essential role in this process for the GTS31–33,
TASNIKS58–64 and YxCxxxF125–131 motifs
located within the N-domain (16–26). Upon stop codon recognition the
highly conserved Gly-Gly-Gln (GGQ) motif of eRF1, located within
the M domain, is positioned within the peptidyl transferase center
resulting in a rearrangement of rRNA, allowing for the entry of a
water molecule and subsequent triggering of peptidyl-tRNA
hydrolysis (27–30). The rearrangement and correct
positioning of the GGQ motif is partially driven by GTP hydrolysis
by eRF3. In line with this, in the presence of eRF3, eRF1 adopts a
structure more ‘tRNA-like’ than that observed for free eRF1
(23). It is important to note
that although the GGQ motif of eRF1 is inserted into the peptidyl
transferase center and is critical for peptide hydrolysis, a water
molecule actually serves as the nucleophile in the hydrolysis of
the peptidyl-tRNA ester bond. The inclusion of a water molecule in
the peptidyl transferase center during termination makes this
reaction particularly distinct from polypeptide elongation where
ribosomes employ mechanisms to keep water out during amide
formation (31).
eRF3 consists of two domains, the N- and C-terminal.
The C-terminal domain of eRF3 is responsible for its interaction
with eRF1. Additionally, the C-terminal domain contains a
GTP-binding domain and β-barrel domains 2 and 3, which are
homologous to translation elongation factors EF-Tu and eEF1A
(14). The N-terminal domain of
eRF3 appears not to be required for translation termination
(23,32,33). eRF3 can bind GTP independently of
eRF1; however, stimulation of eRF3 GTPase activity requires both
eRF1 and the ribosome. Of note, however, a stop codon is not
required for GTPase stimulation. eRF1 and eRF3 bind to ribosomal
pre-termination complexes as an eRF1•eRF3•GTP ternary complex with
peptide release being dependent on eRF3-mediated GTP hydrolysis
(34). GTP hydrolysis releases
eRF1’s M domain from eRF3 enabling the correct positioning of the
GGQ motif within the peptidyl transferase center, thereby promoting
peptide hydrolysis (16).
3. PTC suppressive therapeutics
According to the National Organization for Rare
Disorders (NORD), there are over 7,000 rare genetic diseases.
Additionally, as mentioned above, ~2,400 distinct genetic disorders
have at least one causative PTC allele. Thus, there is an immense
need for pharmacological agents that are capable of promoting
nonsense suppression. Ultimately, the goal of suppression therapy
is to enhance the ability of near-cognate aminoacyl tRNAs to out
compete release factors for the binding to PTCs, resulting in the
incorporation of an amino acid at the PTC. Through increasing the
frequency that PTCs are read-through, it is possible that enough
functional full-length protein can be produced to reduce disease
severity. Below, we review various strategies that have been shown
to promote the suppression of PTCs.
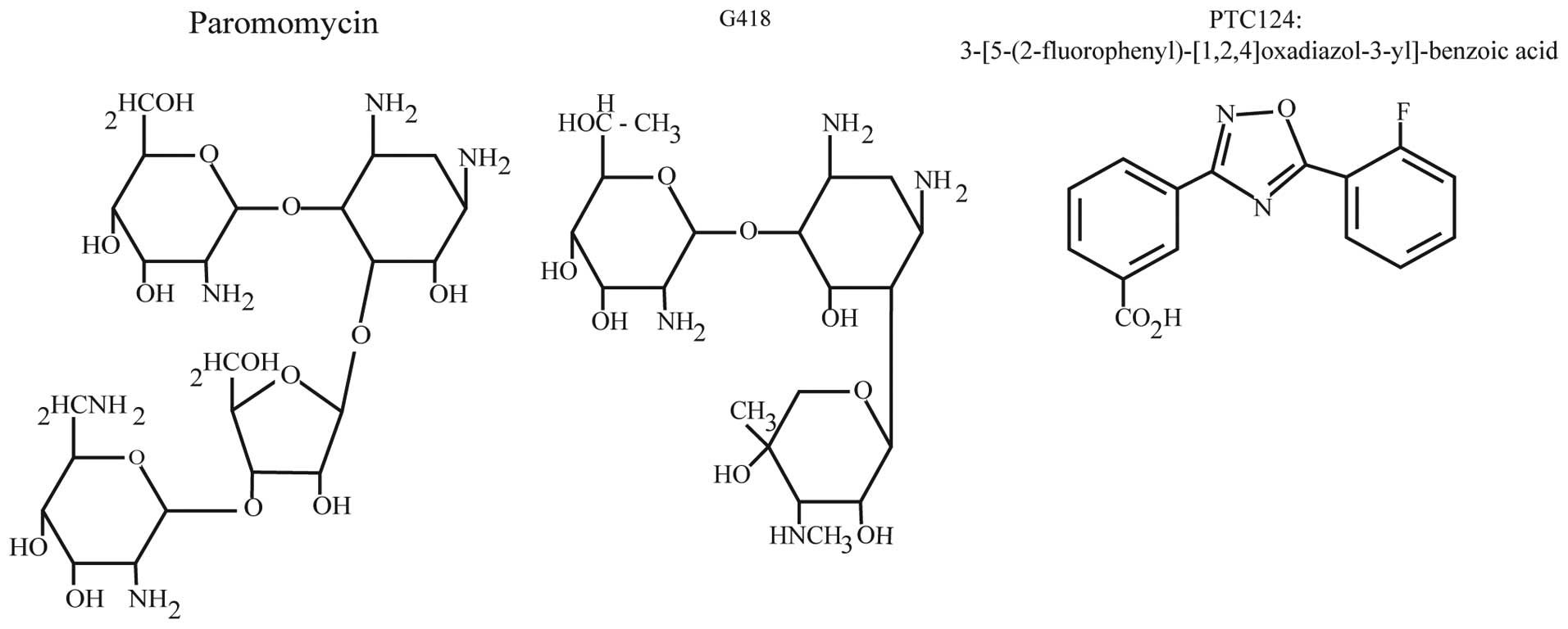
Aminoglycosides
In 1944 antibiotics of the aminoglycoside family
were first isolated from soil bacteria (35,36). Structurally, there are two major
classes of aminoglycosides, characterized by either 4,5- or
4,6-disubstituted 2-deoxystreptamine linked to an amino sugar
backbone (37) (Fig. 2). The antibacterial activity of
aminoglycosides is well established exhibiting broad activity
against many Gram-negative bacteria, select Gram-positive bacteria,
and non-tuberculous mycobacteria (38). The antibacterial activity of
aminoglycosides is a result of their binding to the decoding site
of the bacterial 16S rRNA (39).
The decoding center possesses a proofreading function where it
monitors base pairing between the mRNA codon and incoming aminoacyl
tRNAs (40,41). Through interacting with the
decoding center, aminoglycosides reduce the fidelity of the
proofreading process resulting in increased misincorporation of
near-cognate aminoacyl tRNAs into the ribosomal A-site, leading to
an accumulation of non-functional or truncated bacterial proteins
and culminating in bacterial cell death.
Although the decoding center of the ribosome is
fairly well conserved between prokaryotic and eukaryotic organisms,
the nucleotides responsible for the high affinity binding of
aminoglycosides to the prokaryotic 16S rRNA (A1408 and G1491) are
absent in the mammalian decoding center (18S rRNA; G1408 and A1491)
(42). This reduced affinity
forms part of the basis for their selectivity for the prokaryotic
ribosome. Nonetheless, a subset of aminoglycosides has been shown
to weakly bind eukaryotic ribosomes in a manner sufficient to
disrupt the normal proofreading function of the ribosome leading to
an increase in the insertion of a near-cognate aminoacyl-tRNA in
the ribosomal A-site (43). For
our discussion here, it is important to note that the reduction in
translation fidelity occurs at both sense and nonsense codons.
The general reduction in translation fidelity has
proved to be particularly useful in terms of promoting nonsense
suppression at PTCs. Of note, before the biochemical mechanism of
aminoglycoside antibacterial activity was known, aminoglycosides
were shown to possess the capacity to suppress PTCs and lead to the
production of full-length protein. This property was first
demonstrated for the aminoglycoside streptomycin when in 1964
Gorini and Kataja (44) showed
the phenotypic correction of defective genotypes induced by PTCs in
Escherichia coli. Extending upon these initial observations,
the nonsense suppressive activity of aminoglycosides, including
gentamicin, amikacin, paromomycin, geneticin (G418), lividomycin
and tobramycin, has now been demonstrated in numerous cell lines
and cell free extracts, including those derived from patients with
various genetic disorders (6,45–50). The success of aminoglycosides in
cell culture and mouse models of human disease quickly prompted
their testing in patients with disease caused by nonsense
mutations. Remarkably, the administration of gentamicin has been
shown to promote partial restoration of full-length functional
protein in clinical trials for a variety of diseases, including
cystic fibrosis (CF) (51),
Duchenne muscular dystrophy (DMD) (52), hemophilia A and B (53), and Hailey-Hailey disease (54). For instance, the intranasal
administration of gentamicin to the nasal mucosa for 14 days in
nonsense mutation CF patients has been shown to result in local CF
transmembrane conductance regulator (CFTR) protein production and
an improvement in chloride channel activity (51). Additionally, intravenous
gentamicin administration in patients with nonsense mutation DMD
resulted in an increase in full-length dystrophin in muscle
biopsies (52). While these
studies are promising it should be noted that not all patients
enrolled in these studies responded positively to treatment.
Although aminoglycosides clearly exhibit nonsense
suppressive properties, there are several obstacles that must be
overcome before they can be used for long-term suppressive therapy
in patients. For one, the efficiency at which aminoglycosides can
suppress PTCs is significantly affected by the identity of the
termination codon. For example, the efficiency of gentamicin for
suppressing various PTCs has been shown to vary greatly (5,50).
Additionally the context of the PTC, both 5′ and 3′ nucleotides,
also influences the efficiency of suppression, with the presence of
a cytosine in the +1 position (nucleotide immediately 3′ of the
termination codon) promoting the highest levels of basal and
drug-induced suppression (5,55).
This suggests that only a subset of PTC-carrying patients would be
likely to benefit from aminoglycoside treatment regimens.
Perhaps more troublesome is the fact that
aminoglycosides do exhibit significant toxicity (56,57). One way that aminoglycosides enter
cells is through the receptor, megalin (58). Megalin is a multi-ligand endocytic
receptor that is highly expressed in the proximal tubules of the
kidney and the cochlear hair cells of the inner ear, resulting in
the accumulation of aminoglycosides in these two locations. In line
with this, two of the more common complications of aminoglycoside
treatment are nephrotoxicity and ototoxicity (59–61). While the cellular toxicity is
likely due to many contributing factors, three distinct mechanisms
have been proposed. First, following endocytosis, aminoglycosides
become positively charged, allowing them to interact with
phospholipids and interfere with phospholipase signaling occurring
on the lysosomal membrane (62).
Secondly, due to their charged nature, aminoglycosides have been
shown to lead to the generation of reactive oxygen species (ROS)
(63). Finally, in individuals
with specific polymorphisms in their mitochondrial 12S rRNA,
aminoglycosides can interact with the mitochondrial ribosome
resulting in mitochondrial dysfunction (64). In conclusion, the toxicity
associated with aminoglycosides in conjunction with their
specificity for specific nonsense codons (and flanking nucleotides)
prevents their long-term use in all patients with nonsense
mutation-mediated disease. Additionally, as aminoglycosides work by
generally reducing the fidelity of proofreading, there is
significant potential to disrupt normal decoding at both sense and
nonsense codons. These issues highlight the importance of
identifying additional small molecules that are capable of
modulating the efficiency of translation termination.
PTC124
In an effort to identify novel small molecules
capable of selectively suppressing PTCs, Welch et al
(65) screened ~800,000 low
molecular weight compounds for their ability to specifically
suppress a PTC within an integrated luciferase gene in HEK293
cells, while not affecting termination at the normal stop codon.
Through these screens PTC124
{3-[5-(2-fluorophenyl)-[1,2,4]oxadiazol-3-yl]-benzoic acid;
C15H9FN2O3} was
identified (Fig. 2). Of note,
PTC124 lacks any structural similarity to aminoglycosides or other
clinically developed drugs.
The initial description of PTC124 demonstrated
dose-dependent read-through of all three stop codons. Remarkably,
PTC124 was more efficient than aminoglycosides at promoting
read-through. Specifically, low concentrations (0.01–10 μM) of
PTC124 promoted significant PTC suppression in tissue culture,
whereas 100 μM gentamicin failed to exhibit any read-through.
Furthermore, while aminoglycosides are known to globally reduce
translation fidelity and will thus affect translation termination
at normal termination codons, PTC124 demonstrated specificity for
the PTC within the luciferase open reading frame (ORF).
Additionally, global protein and mRNA profiles appear unaffected by
PTC124 (65).
In addition to demonstrating the nonsense
suppressive activity of PTC124 in tissue culture, Welch et
al (65) also demonstrated
the utility of PTC124 in the mdx mouse model of nonsense
mutation DMD. Using a treatment regimen that targeted a plasma
concentration of 5–10 μg/ml, treatment with PTC124 was able to
improve multiple phenotypes, including a functional strength
deficit, protection against contraction-induced injury, and a
reduction in serum creatine kinase levels. Accordingly, western
blot analyses demonstrated a 20–25% increase in dystrophin levels
in animals treated with PTC124. To date, PTC124 has been tested
preclinically in diverse models of nonsense-mediated disease,
including CF (66), Miyoshi
myopathy (67), Hurler syndrome
(6), Carnitine
palmitoyltransferase 1A deficiency (68), Usher syndrome (69) and Batten disease (70).
PTC124 has gone through phase I clinical trials
where it has been deemed safe for therapeutic uses (71). Consistent with the preclinical
animal data, phase II clinical trials for CF and DMD both reported
positive findings. For instance, CF patients treated with PTC124
for three months exhibited increased chloride channel activity, as
well as an improvement in pulmonary function (72,73). Additionally, in a separate
nonsense mutation DMD phase II study, the oral administration of
PTC124 increased dystrophin protein expression in 61% of the
patients (74). It is currently
in phase III clinical trials for both CF and DMD (75; http://clinicaltrials.gov/ct2/results?term=ataluren).
Currently, the mechanism by which PTC124 promotes
PTC selective nonsense suppression is unknown. For normal
translation termination to be unaffected by PTC124 it would suggest
that termination at a PTC is mechanistically different from
termination at a normal stop codon. Of note, consistent with this
notion, ribosomal toe-prints are much more pronounced at PTCs than
they are at normal stop codons (76). This suggests that translation
termination at a PTC is kinetically less efficient and that
ribosomes pause for a greater amount of time at PTCs than for
normal termination codons. Exactly how and whether this matters
with regard to the mechanism of PTC124-mediated nonsense
suppression is unclear. Undoubtedly, it will be interesting to
determine the mechanism of its nonsense suppressive activity.
Although the results of PTC124 have been nothing
short of remarkable, it has not gone without some controversy.
Initial concerns were primarily derived from the setup of the high
throughput screen that identified PTC124. As mentioned, the screen
utilized a luciferase construct containing a PTC that prevented the
synthesis of full-length firefly luciferase (FLuc) protein
(65). The logic behind this
assay is that suppression of the PTC would generate full-length
luciferase which can then be detected by an increase in
luminescence. However, shortly following the initial description of
PTC124, it was demonstrated that PTC124 interacts with ATP
generating the stable acyl-AMP mixed-anhydride adduct PTC124-AMP
(77,78). When bound to FLuc it results in
its stabilization and an increase in steady-state luciferase
activity, which in Welch’s screen (65) would score as a molecule with
nonsense suppressive activity. Interestingly, this interaction is
specific for FLuc as no stabilization was seen for Renilla
reniformis luciferase (RLuc). In line with the hypothesis that
PTC124 simply stabilized FLuc thereby promoting an increase in
luminescence, PTC124 failed to promote nonsense suppression of an
RLuc reporter (77). It is
important to note that rebuttals against the conclusion of this
study have been published (79).
More recently, McElroy et al (80) also failed to detect the nonsense
suppressive activity for PTC124 using a variety of reporter
systems. Regardless of the role of off-target effects of PTC124 on
FLuc activity during its initial identification, the fact is PTC124
has demonstrated positive results in multiple preclinical models of
disease, as well as in clinical trials involving patients with
nonsense mutation disease. The ability to promote functional
full-length protein expression in patients with nonsense mutation
disease is exactly what PTC124 was developed to do. Thus, PTC124
represents a potential pharmacological PTC suppression therapy that
could reduce disease severity for many patients and certainly
warrants further investigation in to its mechanism of action as
well as the breadth of patients that may benefit from its use as a
therapy.
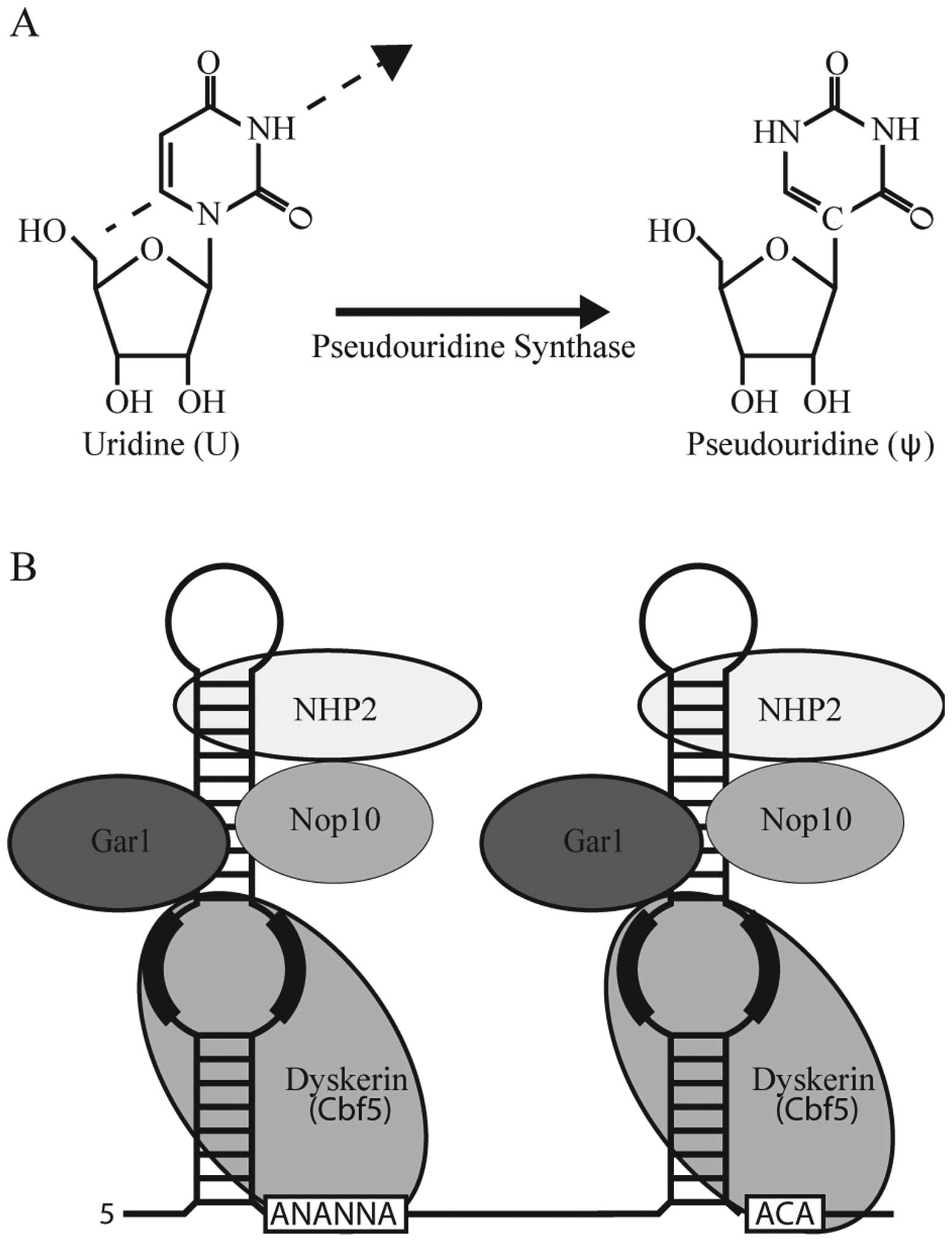
Pseudouridine (ψ)-mediated
suppression
In addition to the development of small
molecule-based approaches to promoting nonsense suppression,
recently, we have been successful in developing a novel strategy
that specifically targets the PTC for recoding through nucleotide
modification (81).
Post-transcriptional nucleotide modifications are naturally
abundant in various cellular RNAs. Pseudouridine is the C-5
glycoside isomer of uridine and is particularly abundant in rRNA
and the spliceosomal U small nuclear RNA (snRNA) (Fig. 3A). Generally speaking, ψ optimizes
the structure and function of rRNAs and U snRNAs in translation and
pre-mRNA splicing, respectively (82,83). It is well established that ψ has
distinct biochemical properties from uridine. In each case these
biochemical properties depend on the structural context and can
extend beyond the site of modification. For instance, multiple
studies have demonstrated that RNA fragments containing ψ are
significantly more stable than if the same RNA contained uridine
(84).
The fact that ψ is biochemical distinct from
uridine, coupled with the fact that a uridine is present at the
first position of all termination codons prompted us to test
whether the incorporation of a ψ into a termination codon could
affect translation termination in vitro (81). Remarkably, replacing the uridine
residue with a ψ drastically reduced the efficiency of translation
termination in both rabbit reticulocyte lysate and Escherichia
coli lysate (81,85). With the success of our in
vitro translation system, we set forth towards developing means
to target PTC containing transcripts in cells.
One mechanism by which pseudouridine can be
generated is through the action of Box H/ACA ribonucleoprotein
(RNP) complexes (86) (Fig. 3B). Box H/ACA RNPs consist of 4
core protein components, one of which is a pseudouridine synthase,
and a non-coding RNA referred to as a Box H/ACA RNA (Fig. 3B). While the protein components of
the RNP are responsible for stabilization of the RNP as well as
enzymatic activity, the non-coding RNA is responsible for substrate
recognition through complementary base pairing interactions. Thus,
in theory, through the construction and expression of artificial
Box H/ACA RNAs any uridine residue should be amenable to targeted
modification (87).
Taking advantage of the fact that one can ‘design’
novel Box H/ACA RNAs to target any RNA, we developed a system to
specifically detect pseudouridine-mediated nonsense suppression
in vivo (in S. cerevisiae), namely the
CUP1-PTC reporter system (81). The CUP1-PTC reporter system
utilizes the CUP1 gene which provides cells with resistance to
copper; hence the introduction of a PTC into the CUP1 gene renders
cells sensitive to copper. Remarkably, while cells expressing a
control box H/ACA RNA were unable to grow on media containing
copper, expression of a box H/ACA RNA targeting the PTC of the
CUP1-PTC transcript restored cellular growth (albeit
partially). Additionally, targeted pseudouridylation of PTCs
located within the yeast TRM4 gene similarly resulted in nonsense
suppression as determined by western blot analysis of full-length
protein. Interestingly, immunoprecipitation coupled with mass
spectrometry determined that the pseudouridylated termination
codons of TRM4 resulted in the incorporation of specific amino
acids. Specifically, ψAG and ψAA both resulted in the corporation
of serine and threonine, while ψGA directed the incorporation of
tyrosine and phenylalanine (81).
As the biochemical properties of ψ are largely affected by its
context, whether the local nucleotide context of the
pseudouridylated termination codon influences which amino acid is
incorporated is an interesting idea that deserves more
attention.
Recently. high resolution X-ray crystallographic
data has shed some light on the mechanism by which pseudouridine is
able to promote nonsense suppression (85). For example, during the normal
translation decoding process, the tRNA-mRNA base pair interaction
is monitored by A1493 of the ribosomal decoding center. This
nucleotide normally adopts the anti conformation; however,
the decoding of ψAG by tRNASer results in A1493 adopting
the syn conformation. Further non-canonical interactions
between the ψAG and the anticodon of tRNASer were
observed, including normally forbidden purine-purine base pairs at
the second and third positions. Interestingly, these interactions
were mediated by an unusual Watson-Crick/Hoogsteen geometry. Thus,
the ribosome appears to be capable of accommodating non-canonical
base pairs. Ultimately, determining the effect of ψ on the rate
constants of various steps in decoding, and on the efficiency of
termination will help to clarify the mechanism of
pseudouridine-mediated nonsense suppression.
While site-specific targeted pseudouridylation
represents a novel means of promoting nonsense suppression and
offers both unprecedented specificity and versatility, it too faces
great obstacles in terms of being clinically relevant. The greatest
challenge is how one would introduce the guide RNAs into cells in a
manner that would allow them to still function, as well as protect
them from recognition by the cellular innate immune system RNA
sensors (88). Additionally, it
is unknown whether all mRNA molecules will transit through cellular
locations that make them susceptible to efficient box H/ACA
RNA-mediated pseudouridylation.
4. Conclusion
PTCs can arise from a variety of mutations in either
germ or somatic cells. Overall, an estimated one third of all
genetic disorders are caused by PTCs (89). As the genes and specific mutations
that cause various diseases continue to be identified, there is no
doubt that more disease phenotypes will be attributed to the
presence of PTCs. Thus the need to develop drugs that are capable
of promoting nonsense suppression will only grow.
To date, therapeutic PTC suppression has been
demonstrated in both preclinical animal models and in clinical
trials for diseases such as CF and DMD. The suppression of PTCs
with small molecules represents an approach to treat nonsense
mutation disease that is growing in favor. In addition to small
molecule approaches strategies that aim to promote translational
read-through via targeted nucleotide modification of the PTC, such
as the recent discovery of ψ-mediated nonsense suppression, are
also being explored. It is unclear whether a single therapeutic
strategy will suffice to promote nonsense suppression at any PTC or
whether therapeutic PTC suppression will require a case by case
analysis. Nonetheless, with the continuing accumulation of
mechanistic insight into translation termination the field is
certainly poised to develop novel therapeutics.
Acknowledgements
This study was supported by grants GM104077 and
AG039559 from NIH (to Y.-T.Y.). J.K. is a Damon Runyon Postdoctoral
Fellow and is supported by the Damon Runyon Cancer Research
Foundation (DRG2121-12).
References
|
1
|
Brenner S, Barnett L, Katz ER and Crick
FH: UGA: a third nonsense triplet in the genetic code. Nature.
213:449–450. 1967. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brenner S, Stretton AO and Kaplan S:
Genetic code: the ‘nonsense’ triplets for chain termination and
their suppression. Nature. 206:994–998. 1965.
|
|
3
|
Dever TE and Green R: The elongation,
termination, and recycling phases of translation in eukaryotes.
Cold Spring Harb Perspect Biol. 4:a0137062012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bonetti B, Fu L, Moon J and Bedwell DM:
The efficiency of translation termination is determined by a
synergistic interplay between upstream and downstream sequences in
Saccharomyces cerevisiae. J Mol Biol. 251:334–345. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Manuvakhova M, Keeling K and Bedwell DM:
Aminoglycoside antibiotics mediate context-dependent suppression of
termination codons in a mammalian translation system. RNA.
6:1044–1055. 2000. View Article : Google Scholar
|
|
6
|
Peltz SW, Morsy M, Welch EM and Jacobson
A: Ataluren as an agent for therapeutic nonsense suppression. Annu
Rev Med. 64:407–425. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mort M, Ivanov D, Cooper DN and Chuzhanova
NA: A meta-analysis of nonsense mutations causing human genetic
disease. Hum Mutat. 29:1037–1047. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nagy E and Maquat LE: A rule for
termination-codon position within intron-containing genes: when
nonsense affects RNA abundance. Trends Biochem Sci. 23:198–199.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Keeling KM, Xue X, Gunn G and Bedwell DM:
Therapeutics based on stop codon readthrough. Annu Rev Genomics Hum
Genet. 15:8.1–8.24. 2014. View Article : Google Scholar
|
|
10
|
Salas-Marco J and Bedwell DM: GTP
hydrolysis by eRF3 facilitates stop codon decoding during
eukaryotic translation termination. Mol Cell Biol. 24:7769–7778.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Alkalaeva EZ, Pisarev AV, Frolova LY,
Kisselev LL and Pestova TV: In vitro reconstitution of eukaryotic
translation reveals cooperativity between release factors eRF1 and
eRF3. Cell. 125:1125–1136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pisareva VP, Pisarev AV, Hellen CU,
Rodnina MV and Pestova TV: Kinetic analysis of interaction of
eukaryotic release factor 3 with guanine nucleotides. J Biol Chem.
281:40224–40235. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mitkevich VA, Kononenko AV, Petrushanko
IY, Yanvarev DV, Makarov AA and Kisselev LL: Termination of
translation in eukaryotes is mediated by the quaternary
eRF1*eRF3*GTP*Mg2+complex.
The biological roles of eRF3 and prokaryotic RF3 are profoundly
distinct. Nucleic Acids Res. 34:3947–3954. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kong C, Ito K, Walsh MA, Wada M, Liu Y,
Kumar S, Barford D, Nakamura Y and Song H: Crystal structure and
functional analysis of the eukaryotic class II release factor eRF3
from S. pombe. Mol Cell. 14:233–245. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mantsyzov AB, Ivanova EV, Birdsall B,
Alkalaeva EZ, Kryuchkova PN, Kelly G, Frolova LY and Polshakov VI:
NMR solution structure and function of the C-terminal domain of
eukaryotic class 1 polypeptide chain release factor. FEBS J.
277:2611–2627. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Song H, Mugnier P, Das AK, Webb HM, Evans
DR, Tuite MF, Hemmings BA and Barford D: The crystal structure of
human eukaryotic release factor eRF1 - mechanism of stop codon
recognition and peptidyl-tRNA hydrolysis. Cell. 100:311–321. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bertram G, Bell HA, Ritchie DW, Fullerton
G and Stansfield I: Terminating eukaryote translation: domain 1 of
release factor eRF1 functions in stop codon recognition. RNA.
6:1236–1247. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chavatte L, Seit-Nebi A, Dubovaya V and
Favre A: The invariant uridine of stop codons contacts the
conserved NIKSR loop of human eRF1 in the ribosome. EMBO J.
21:5302–5311. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Frolova L, Seit-Nebi A and Kisselev L:
Highly conserved NIKS tetrapeptide is functionally essential in
eukaryotic translation termination factor eRF1. RNA. 8:129–136.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Seit-Nebi A, Frolova L and Kisselev L:
Conversion of omnipotent translation termination factor eRF1 into
ciliate-like UGA-only unipotent eRF1. EMBO Rep. 3:881–886. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ito K, Frolova L, Seit-Nebi A, Karamyshev
A, Kisselev L and Nakamura Y: Omnipotent decoding potential resides
in eukaryotic translation termination factor eRF1 of variant-code
organisms and is modulated by the interactions of amino acid
sequences within domain 1. Proc Natl Acad Sci USA. 99:8494–8499.
2002. View Article : Google Scholar
|
|
22
|
Fan-Minogue H, Du M, Pisarev AV, Kallmeyer
AK, Salas-Marco J, Keeling KM, Thompson SR, Pestova TV and Bedwell
DM: Distinct eRF3 requirements suggest alternate eRF1 conformations
mediate peptide release during eukaryotic translation termination.
Mol Cell. 30:599–609. 2008. View Article : Google Scholar
|
|
23
|
Cheng Z, Saito K, Pisarev AV, Wada M,
Pisareva VP, Pestova TV, Gajda M, Round A, Kong C, Lim M, Nakamura
Y, Svergun DI, Ito K and Song H: Structural insights into eRF3 and
stop codon recognition by eRF1. Genes Dev. 23:1106–1118. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Conard SE, Buckley J, Dang M, Bedwell GJ,
Carter RL, Khass M and Bedwell DM: Identification of eRF1 residues
that play critical and complementary roles in stop codon
recognition. RNA. 18:1210–1221. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kryuchkova P, Grishin A, Eliseev B,
Karyagina A, Frolova L and Alkalaeva E: Two-step model of stop
codon recognition by eukaryotic release factor eRF1. Nucleic Acids
Res. 41:4573–4586. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Merritt GH, Naemi WR, Mugnier P, Webb HM,
Tuite MF and von der Haar T: Decoding accuracy in eRF1 mutants and
its correlation with pleiotropic quantitative traits in yeast.
Nucleic Acids Res. 38:5479–5492. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Frolova LY, Tsivkovskii RY, Sivolobova GF,
Oparina NY, Serpinsky OI, Blinov VM, Tatkov SI and Kisselev LL:
Mutations in the highly conserved GGQ motif of class 1 polypeptide
release factors abolish ability of human eRF1 to trigger
peptidyl-tRNA hydrolysis. RNA. 5:1014–1020. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Laurberg M, Asahara H, Korostelev A, Zhu
J, Trakhanov S and Noller HF: Structural basis for translation
termination on the 70S ribosome. Nature. 454:852–857. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Weixlbaumer A, Jin H, Neubauer C, Voorhees
RM, Petry S, Kelley AC and Ramakrishnan V: Insights into
translational termination from the structure of RF2 bound to the
ribosome. Science. 322:953–956. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Santos N, Zhu J, Donohue JP, Korostelev AA
and Noller HF: Crystal structure of the 70S ribosome bound with the
Q253P mutant form of release factor RF2. Structure. 21:1258–1263.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kapp LD and Lorsch JR: The molecular
mechanics of eukaryotic translation. Ann Rev Biochem. 73:657–704.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ter-Avanesyan MD, Kushnirov VV,
Dagkesamanskaya AR, Didichenko SA, Chernoff YO, Inge-Vechtomov SG
and Smirnov VN: Deletion analysis of the SUP35 gene of the yeast
Saccharomyces cerevisiae reveals two non-overlapping
functional regions in the encoded protein. Mol Microbiol.
7:683–692. 1993.PubMed/NCBI
|
|
33
|
Kononenko AV, Mitkevich VA, Dubovaya VI,
Kolosov PM, Makarov AA and Kisselev LL: Role of the individual
domains of translation termination factor eRF1 in GTP binding to
eRF3. Proteins. 70:388–393. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Frolova L, Le Goff X, Zhouravleva G,
Davydova E, Philippe M and Kisselev L: Eukaryotic polypeptide chain
release factor eRF3 is an eRF1- and ribosome-dependent guanosine
triphosphatase. RNA. 2:334–341. 1996.PubMed/NCBI
|
|
35
|
Jones D, Metzger HJ, Schatz A and Waksman
SA: Control of gram-negative bacteria in experimental animals by
streptomycin. Science. 100:103–105. 1944. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schatz A, Bugie E and Waksman SA:
Streptomycin, a substance exhibiting antibiotic activity against
gram-positive and gram-negative bacteria. 1944. Clin Orthop Relat
Res. 437:3–6. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hermann T: Drugs targeting the ribosome.
Curr Opin Struct Biol. 15:355–366. 2005. View Article : Google Scholar
|
|
38
|
Hermann T: Aminoglycoside antibiotics: old
drugs and new therapeutic approaches. Cell Mol Life Sci.
64:1841–1852. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Moazed D and Noller HF: Interaction of
antibiotics with functional sites in 16S ribosomal RNA. Nature.
327:389–394. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Moazed D and Noller HF: Binding of tRNA to
the ribosomal A and P sites protects two distinct sets of
nucleotides in 16 S rRNA. J Mol Biol. 211:135–145. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yoshizawa S, Fourmy D and Puglisi JD:
Recognition of the codon-anticodon helix by ribosomal RNA. Science.
285:1722–1725. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
François B, Russell RJ, Murray JB,
Aboul-ela F, Masquida B, Vicens Q and Westhof E: Crystal structures
of complexes between aminoglycosides and decoding A site
oligonucleotides: role of the number of rings and positive charges
in the specific binding leading to miscoding. Nucleic Acids Res.
33:5677–5690. 2005.
|
|
43
|
Fan-Minogue H and Bedwell DM: Eukaryotic
ribosomal RNA determinants of aminoglycoside resistance and their
role in translational fidelity. RNA. 14:148–157. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gorini L and Kataja E: Phenotypic repair
by streptomycin of defective genotypes in E. coli. Proc Natl
Acad Sci USA. 51:487–493. 1964. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lai CH, Chun HH, Nahas SA, Mitui M, Gamo
KM, Du L and Gatti RA: Correction of ATM gene function by
aminoglycoside-induced read-through of premature termination
codons. Proc Natl Acad Sci USA. 101:15676–15681. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Keeling KM and Bedwell DM: Clinically
relevant aminoglycosides can suppress disease-associated premature
stop mutations in the IDUA and P53 cDNAs in a mammalian translation
system. J Mol Med (Berl). 80:367–376. 2002. View Article : Google Scholar
|
|
47
|
Sleat DE, Sohar I, Gin RM and Lobel P:
Aminoglycoside-mediated suppression of nonsense mutations in late
infantile neuronal ceroid lipofuscinosis. Eur J Paediatr Neurol.
5(Suppl A): 57–62. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Howard M, Frizzell RA and Bedwell DM:
Aminoglycoside antibiotics restore CFTR function by overcoming
premature stop mutations. Nat Med. 2:467–469. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bedwell DM, Kaenjak A, Benos DJ, Bebok Z,
Bubien JK, Hong J, Tousson A, Clancy JP and Sorscher EJ:
Suppression of a CFTR premature stop mutation in a bronchial
epithelial cell line. Nat Med. 3:1280–1284. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bidou L, Hatin I, Perez N, Allamand V,
Panthier JJ and Rousset JP: Premature stop codons involved in
muscular dystrophies show a broad spectrum of readthrough
efficiencies in response to gentamicin treatment. Gene Ther.
11:619–627. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wilschanski M, Yahav Y, Yaacov Y, Blau H,
Bentur L, Rivlin J, Aviram M, Bdolah-Abram T, Bebok Z, Shushi L,
Kerem B and Kerem E: Gentamicin-induced correction of CFTR function
in patients with cystic fibrosis and CFTR stop mutations. N Engl J
Med. 349:1433–1441. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Politano L, Nigro G, Nigro V, Piluso G,
Papparella S, Paciello O and Comi LI: Gentamicin administration in
Duchenne patients with premature stop codon. Preliminary results.
Acta Myol. 22:15–21. 2003.PubMed/NCBI
|
|
53
|
James PD, Raut S, Rivard GE, Poon MC,
Warner M, McKenna S, Leggo J and Lillicrap D: Aminoglycoside
suppression of nonsense mutations in severe hemophilia. Blood.
106:3043–3048. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kellermayer R, Szigeti R, Keeling KM,
Bedekovics T and Bedwell DM: Aminoglycosides as potential
pharmacogenetic agents in the treatment of Hailey-Hailey disease. J
Invest Dermatol. 126:229–231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Floquet C, Hatin I, Rousset JP and Bidou
L: Statistical analysis of readthrough levels for nonsense
mutations in mammalian cells reveals a major determinant of
response to gentamicin. PLoS Genet. 8:e10026082012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Turnidge J: Pharmacodynamics and dosing of
aminoglycosides. Infect Dis Clin North Am. 17:503–528. 2003.
View Article : Google Scholar
|
|
57
|
Fischel-Ghodsian N: Genetic factors in
aminoglycoside toxicity. Pharmacogenomics. 6:27–36. 2005.
View Article : Google Scholar
|
|
58
|
Moestrup SK, Cui S, Vorum H, Bregengard C,
Bjørn SE, Norris K, Gliemann J and Christensen EI: Evidence that
epithelial glycoprotein 330/megalin mediates uptake of polybasic
drugs. J Clin Invest. 96:1404–1413. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Guthrie OW: Aminoglycoside induced
ototoxicity. Toxicology. 249:91–96. 2008. View Article : Google Scholar
|
|
60
|
Mingeot-Leclercq MP and Tulkens PM:
Aminoglycosides: nephrotoxicity. Antimicrob Agents Chemother.
43:1003–1012. 1999.PubMed/NCBI
|
|
61
|
Avent ML, Rogers BA, Cheng AC and Paterson
DL: Current use of aminoglycosides: indications, pharmacokinetics
and monitoring for toxicity. Intern Med J. 41:441–449. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Laurent G, Carlier MB, Rollman B, Van Hoof
F and Tulkens P: Mechanism of aminoglycoside-induced lysosomal
phospholipidosis: in vitro and in vivo studies with gentamicin and
amikacin. Biochem Pharmacol. 31:3861–3870. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sha SH and Schacht J: Stimulation of free
radical formation by aminoglycoside antibiotics. Hear Res.
128:112–118. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Hobbie SN, Akshay S, Kalapala SK, Bruell
CM, Shcherbakov D and Böttger EC: Genetic analysis of interactions
with eukaryotic rRNA identify the mitoribosome as target in
aminoglycoside ototoxicity. Proc Natl Acad Sci USA.
105:20888–20893. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Welch EM, Barton ER, Zhuo J, Tomizawa Y,
Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S,
Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC,
Corson D, Turpoff AA, Campbell JA, Conn MM, Khan A, Almstead NG,
Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA,
Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL,
Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW and
Sweeney HL: PTC124 targets genetic disorders caused by nonsense
mutations. Nature. 447:87–91. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Du M, Liu X, Welch EM, Hirawat S, Peltz SW
and Bedwell DM: PTC124 is an orally bioavailable compound that
promotes suppression of the human CFTR-G542X nonsense allele in a
CF mouse model. Proc Natl Acad Sci USA. 105:2064–2069. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wang B, Yang Z, Brisson BK, Feng H, Zhang
Z, Welch EM, Peltz SW, Barton ER, Brown RH Jr and Sweeney HL:
Membrane blebbing as an assessment of functional rescue of
dysferlin-deficient human myotubes via nonsense suppression. J Appl
Physiol. 1985. 109:901–905. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Tan L, Narayan SB, Chen J, Meyers GD and
Bennett MJ: PTC124 improves readthrough and increases enzymatic
activity of the CPT1A R160X nonsense mutation. J Inherit Metab Dis.
34:443–447. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Goldmann T, Overlack N, Wolfrum U and
Nagel-Wolfrum K: PTC124-mediated translational readthrough of a
nonsense mutation causing Usher syndrome type 1C. Hum Gene Ther.
22:537–547. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Sarkar C, Zhang Z and Mukherjee AB: Stop
codon read-through with PTC124 induces palmitoyl-protein
thioesterase-1 activity, reduces thioester load and suppresses
apoptosis in cultured cells from INCL patients. Mol Genet Metab.
104:338–345. 2011. View Article : Google Scholar
|
|
71
|
Hirawat S, Welch EM, Elfring GL, Northcutt
VJ, Paushkin S, Hwang S, Leonard EM, Almstead NG, Ju W, Peltz SW
and Miller LL: Safety, tolerability, and pharmacokinetics of
PTC124, a nonaminoglycoside nonsense mutation suppressor, following
single- and multiple-dose administration to healthy male and female
adult volunteers. J Clin Pharmacol. 47:430–444. 2007. View Article : Google Scholar
|
|
72
|
Sermet-Gaudelus I, Boeck KD, Casimir GJ,
Vermeulen F, Leal T, Mogenet A, Roussel D, Fritsch J, Hanssens L,
Hirawat S, Miller NL, Constantine S, Reha A, Ajayi T, Elfring GL
and Miller LL: Ataluren (PTC124) induces cystic fibrosis
transmembrane conductance regulator protein expression and activity
in children with nonsense mutation cystic fibrosis. Am J Respir
Crit Care Med. 182:1262–1272. 2010. View Article : Google Scholar
|
|
73
|
Wilschanski M, Miller LL, Shoseyov D, Blau
H, Rivlin J, Aviram M, Cohen M, Armoni S, Yaakov Y, Pugatsch T,
Cohen-Cymberknoh M, Miller NL, Reha A, Northcutt VJ, Hirawat S,
Donnelly K, Elfring GL, Ajayi T and Kerem E: Chronic ataluren
(PTC124) treatment of nonsense mutation cystic fibrosis. Eur Respir
J. 38:59–69. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Finkel RS, Flanigan KM, Wong B, Bönnemann
C, Sampson J, Sweeney HL, Reha A, Northcutt VJ, Elfring G, Barth J
and Peltz SW: Phase 2a study of ataluren-mediated dystrophin
production in patients with nonsense mutation Duchenne muscular
dystrophy. PLoS One. 8:e813022013. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Kerem E, Konstan MW, De Boeck K, Accurso
FJ, Sermet-Gaudelus I, Wilschanski M, Elborn JS, Melotti P,
Bronsveld I, Fajac I, Malfroot A, Rosenbluth DB, Walker PA,
McColley SA, Knoop C, Quattrucci S, Rietschel E, Zeitlin PL, Barth
J, Elfring GL, Welch EM, Branstrom A, Spiegel RJ, Peltz SW, Ajayi T
and Rowe SM; for the Cystic Fibrosis Ataluren Study Group. Ataluren
for the treatment of nonsense-mutation cystic fibrosis: a
randomised, double-blind, placebo-controlled phase 3 trial. Lancet
Respir Med. pii: S2213-2600(14)70100-6. View Article : Google Scholar : 2014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Amrani N, Ganesan R, Kervestin S, Mangus
DA, Ghosh S and Jacobson A: A faux 3′-UTR promotes aberrant
termination and triggers nonsense-mediated mRNA decay. Nature.
432:112–118. 2004.
|
|
77
|
Auld DS, Thorne N, Maguire WF and Inglese
J: Mechanism of PTC124 activity in cell-based luciferase assays of
nonsense codon suppression. Proc Natl Acad Sci USA. 106:3585–3590.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Auld DS, Lovell S, Thorne N, Lea WA,
Maloney DJ, Shen M, Rai G, Battaile KP, Thomas CJ, Simeonov A,
Hanzlik RP and Inglese J: Molecular basis for the high-affinity
binding and stabilization of firefly luciferase by PTC124. Proc
Natl Acad Sci USA. 107:4878–4883. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Peltz SW, Welch EM, Jacobson A, Trotta CR,
Naryshkin N, Sweeney HL and Bedwell DM: Nonsense suppression
activity of PTC124 (ataluren). Proc Natl Acad Sci USA.
106:E64author reply E65. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
McElroy SP, Nomura T, Torrie LS, Warbrick
E, Gartner U, Wood G and McLean WH: A lack of premature termination
codon read-through efficacy of PTC124 (Ataluren) in a diverse array
of reporter assays. PLoS Biol. 11:e10015932013. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Karijolich J and Yu YT: Converting
nonsense codons into sense codons by targeted pseudouridylation.
Nature. 474:395–398. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Karijolich J, Kantartzis A and Yu YT: RNA
modifications: a mechanism that modulates gene expression. Methods
Mol Biol. 629:1–19. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Karijolich J and Yu YT: Spliceosomal snRNA
modifications and their function. RNA Biol. 7:192–204. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Kierzek E, Malgowska M, Lisowiec J, Turner
DH, Gdaniec Z and Kierzek R: The contribution of pseudouridine to
stabilities and structure of RNAs. Nucleic Acids Res. 42:3492–3501.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Fernández IS, Ng CL, Kelley AC, Wu G, Yu
YT and Ramakrishnan V: Unusual base pairing during the decoding of
a stop codon by the ribosome. Nature. 500:107–110. 2013.PubMed/NCBI
|
|
86
|
Ganot P, Bortolin ML and Kiss T:
Site-specific pseudouridine formation in preribosomal RNA is guided
by small nucleolar RNAs. Cell. 89:799–809. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Huang C, Karijolich J and Yu YT:
Post-transcriptional modification of RNAs by artificial Box H/ACA
and Box C/D RNPs. Methods Mol Biol. 718:227–244. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Barbalat R, Ewald SE, Mouchess ML and
Barton GM: Nucleic acid recognition by the innate immune system.
Ann Rev Immunol. 29:185–214. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Frischmeyer PA and Dietz HC:
Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet.
8:1893–1900. 1999. View Article : Google Scholar : PubMed/NCBI
|