1. Introduction
Cigarette smoking is the most preventable risk
factor for human health. According to a WHO report, 1.3 billion
individuals are active smokers worldwide and smoking kills six
million individuals each year; eventually, half of these smokers
die due to smoking-related diseases (1). To this end, cigarette smoking is
known to be associated with cardiovascular diseases (2), cancers (3,4),
lung diseases (5), chronic renal
disorders (6) and other diseases
remained to be defined. It has been regarded as a main killer and
induces serious problems in humans with major concerns in public
health.
Over 5,000 ingredients are found in cigarette smoke
(7,8). Among these, at least 150 compounds
found in cigarette smoke are known to induce free radicals and
possess toxic and carcinogenic activities. Based on their
structures, these toxic and carcinogenic ingredients are divided
into several chemical classes. These include alkaloids, phenolic
compounds, volatile aldehydes, polycyclic aromatic hydrocarbons
(PAHs), tobacco-specific nitrosamines (TNSAs), as well as heavy
metals (8). These chemical induce
high levels of oxidative stress in smokers (7), and trigger and augment lipid
peroxidation, which causes low-density lipoprotein (LDL) oxidation
and atherosclerosis (9). These
active ingredients also cause a high incidence of lung cancer
accounting for approximately 90% of small cell lung cancer (SCLC)
cases and 70% of non-small cell lung cancer (NSCLC) cases worldwide
(10).
A number of studies have provided evidence that
cigarette smoking is a major cause of gastrointestinal (GI)
disorders, which include chronic inflammation, such as peptic
ulcers and inflammatory bowel disease (IBD), and cancers of the GI
tract (1,3–8).
In this review, we mainly discuss the relationship between smoking
and GI disorders, and the underlying mechanisms through which
cigarette smoke and its active ingredients affect the pathogenic
processes of some of these diseases of the GI tract.
2. Cigarette smoking increases the risk of
ulcers and inflammatory diseases of the GI tract
Cigarette smoking increases the risk of
peptic ulcer disease
Peptic ulcers are histologically identified as
necrosis of the mucosa, which produces lesions. This disease is
mainly caused by Helicobacter pylori (H. pylori)
infection, as well as the excessive use of non-steroidal
anti-inflammatory drugs (NSAIDs), such as aspirin and ibuprofen
(11). Cigarette smoking is also
considered to be one of the major contributors to ulcer diseases. A
large US population-based study (1997–2003) revealed that the
prevalence of ulcer disease in current and former smokers (11.43
and 11.52%) is almost doubled that of never smokers (6.00%)
(12). It is also clear that the
risk of peptic ulcers is associated with the quantity of tobacco
use (13).
According to clinical observations, cigarette
smokers are more likely to develop ulcers which are more difficult
to heal (14). The risk of peptic
ulcers also increases in smokers who have a large daily intake of
tobacco compared with never smokers (15). However, cigarette smoking is not
an independent ulcerogenic. It adversely affects the gastroduodenal
mucosal protective mechanisms and increases the risk of H.
pylori infection (14).
Cigarette smoking allows the reflux of harmful duodenal contents
back into the stomach. Furthermore, smokers appear to be at higher
risk of becoming infected with H. pylori. This increased
risk may be due to the adverse effects of smoking on the reduction
of antioxidants or the defensive immune system locally present in
the gastroduodenal mucosa. All these actions can interfere with the
natural defensive mechanisms against H. pylori infection in
the stomach and duodenum.
Effects of smoking on IBD
IBD is known as chronic inflammation of the GI
tract, particularly the colon and small intestine. IBD includes
Crohn’s disease and ulcerative colitis (16). Cigarette smoking exerts a
dichotomous effect in the progression of IBD. Smokers seem to be
more likely to develop Crohn’s disease, with a higher recurrence
after surgery and poor response to medications (17,18). However, people who smoke have a
lower risk of developing ulcerative colitis (19). Cigarette smoking is known as an
independent risk factor for Crohn’s disease. As early as 1984, a
case-control study involving 82 patients with Crohn’s disease and
matched controls was conducted in the UK to determine the
relationship between Crohn’s disease and cigarette smoking
(20). Patients with this disease
were more likely to have a smoking habit. The relative risk for
smokers to develop Crohn’s disease was significantly higher than in
non-smokers (20). In a separate
study involving 2,795 patients with Crohn’s disease, patients were
classified into non-smokers, light smokers (1–10 cigarettes/day)
and heavy smokers (>10 cigarettes/day). Researchers found that
the percentage of years with active Crohn’s disease for
non-smokers, light smokers and heavy smokers were 37, 46 and 48%,
respectively. Besides, the number of years of immumosuppressant
mediation was higher in the heavy smokers than the light smokers
and non-smokers (36 vs. 34 vs. 32%, respectively) (21). These results suggest that smoking
may worsen the severity of the disease and prolong the disease
course and drug treatment with immunosuppressants.
Conversely, smoking has been demonstrated to reduce
the risk of ulcerative colitis. Mounting evidence has indicated
that patients with ulcerative colitis tend to be non-smokers
(22). In a population-based
incident cohort study, smoking was more common in male patients
(P=0.002), and positively correlated with an increased risk of
Crohn’s disease [odds ratio, 1.96; 95% confidence interval (CI),
1.63–2.37; P<0.001]. By contrast, current smoking was protective
against ulcerative colitis (odds ratio, 0.33; 95% CI, 0.27–0.41).
In addition, in ulcerative colitis, cigarette smoking was
associated with less extensive disease (P=0.01) and a decreased
need for colectomy (P=0.06) (23). The underlying mechanisms of action
of smoking and its bidirectional effects on the progression of
Crohn’s disease and ulcerative colitis have not yet been fully
elucidated and further studies are required using human and animal
models.
Possible mechanisms of action of smoking
in inflammatory diseases of the GI tract
As discussed, cigarette smoking is a major risk
factor for the development of inflammation-related diseases, such
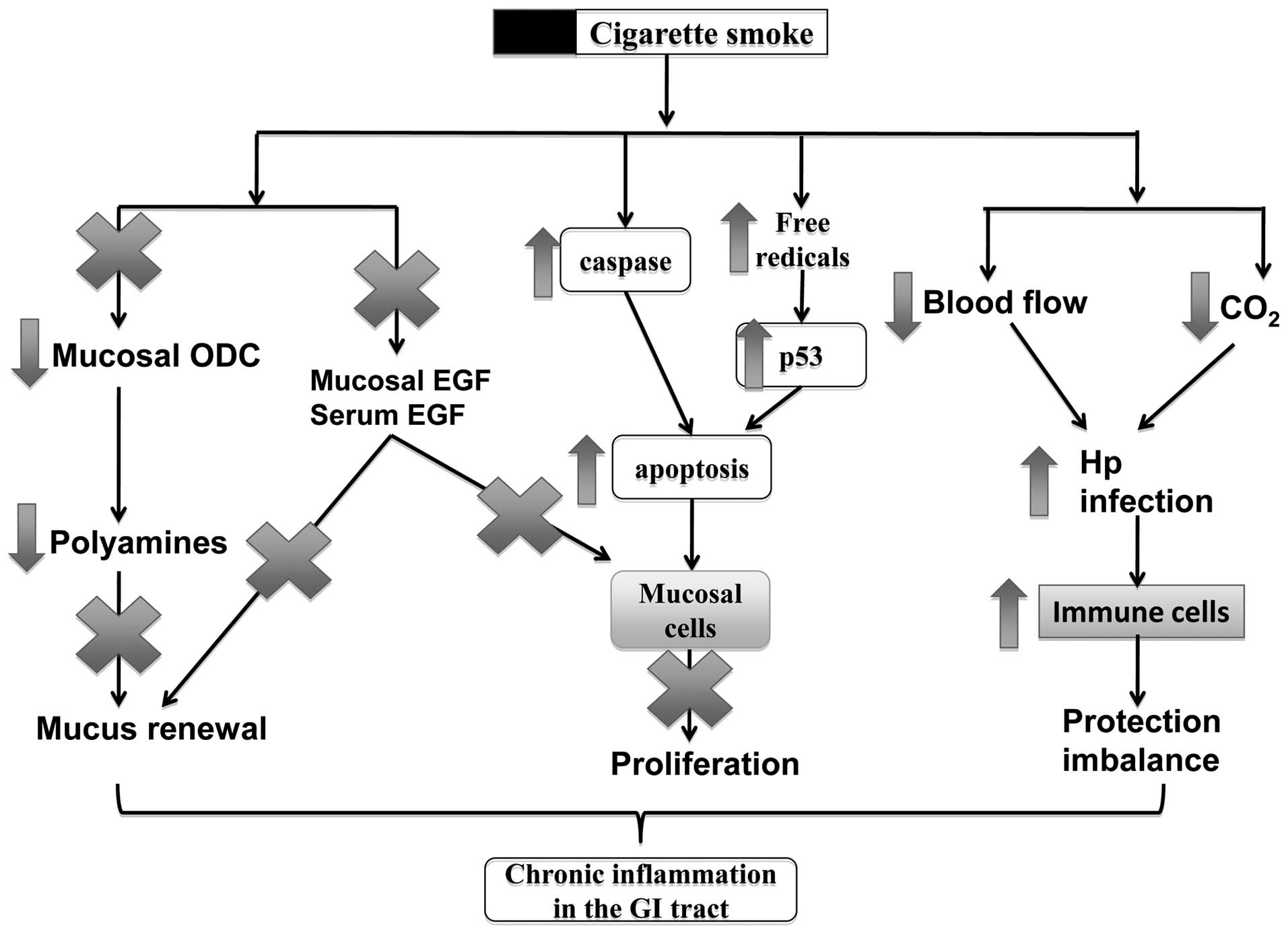
as ulcers and Crohn’s disease. In Fig. 1, the mechanisms of action of
smoking in these disorders include the alteration of mucosal cell
proliferation, change of blood flow in the inflammatory sites, the
increase of viral or bacterial infections and the dysfunction of
the immune system in the GI mucosa.
Smoking induces cell death in the
mucosa
The mucosa is the inner layer of the GI tract, which
surrounds the lumen. The innermost layer is known as the
epithelium, which forms a continuous layer of protection against
noxious agents from the lumen. The induction of cell apoptosis is
an adverse effect of cigarette smoking, which results in tissue
injury and dysfunction in the GI tract. A number of studies have
shown that cigarette smoke can induce cell apoptosis in the
esophagus and gastric mucosa (24), as well as in the inner layers of
the small intestine and colon (25). Exposure to cigarette smoke induces
a time- and concentration-dependent increase in apoptosis in the
gastric mucosa (26).
Pre-treatment with allopurinol (a xanthine oxidase inhibitor) or
dimethyl sulfoxide (DMSO) (a hydroxyl free radical scavenger) can
block the apoptotic activity induced by smoking, and does not
affect the p53 level of the mucosa (26), suggesting that the apoptosis
induced by cigarette smoking is mediated through reactive oxygen
species (ROS) and occurs independently of the p53 pathway. Chronic
exposure to cigarette smoke can also induce apoptosis in the
follicle-associated epithelium, possibly through the CCL20-CCR6
cascade (25). Benzo(e)pyrene, a
toxic compound found in cigarette smoke, also causes cell death in
human retinal pigment epithelial cells (ARPE-19), and induces
apoptosis through the involvement of multiple caspase pathways
(27).
Smoking inhibits epithelial cell renewal
in the GI tract
Epithelial cell renewal in the GI tract is an
effective progress for protecting the surface epithelial cells from
various aggressive factors coming from the lumen. The effects of
cigarette smoking on cell renewal in the GI tract have been
reviewed by a number of studies showing that cigarette smoke and
its active ingredients not only inhibit mucosal cell proliferation,
but also induce cell apoptosis during ulcer healing (8,24).
Cell renewal is a protective process for the GI tract, the
dysfunction of which plays a vital role in ulceration and ulcer
healing (28). We have previously
reviewed that cigarette smoke and its active ingredients can
suppress mucosal cell proliferation and induce apoptosis during
ulceration and the healing processes (8,24).
Epidermal growth factor (EGF)
In our previous studies we demonstrated that
cigarette smoke or its extracts significantly inhibit mucosal cell
proliferation in human and animal mucosal cells, associated with
the reduction of EGF and polyamine release (29,30). EGF plays an important role in
mucosal cell proliferation and modulates mucosal integrity. During
ulceration, the synthesis of EGF, as well as its expression are
markedly upregulated in epithelial cells adjacent to the ulcer
crater (31). Data from our
previous study also demonstrated that cigarette smoke significantly
inhibited EGF synthesis and its mRNA expression in salivary glands
and gastric mucosa in rats with acetic acid ulcers (29). In addition, gastric ulcer healing
was also delayed along with a reduced mucosal cell proliferation,
suggesting that the delay of ulcer healing induced by cigarette
smoke is possibly caused by the reduction of EGF release at the
ulcer site.
Polyamines
Polyamines are found to be associated with mucosal
cell proliferation during the ulcer healing process (32,33). Polyamines are involved in
EGF-mediated cell proliferation and acid secretion in the stomach
(34). Ornithine decarboxylase
(ODC) is the primary enzyme for the biosynthesis of polyamines,
including putrescine, spermine and spermidine. To further elucidate
the association between smoking and peptic ulcer disease, in
previous studies, we also examined ODC activity, which is crucial
for promoting mucosal growth and has gastroprotective effects
during gastric ulcer healing. Following the intragastric
administration with cigarette smoke extracts once daily for three
days, ulcer sizes were markedly enlarged and the myeloperoxidase
activity was also increased. Cigarette smoke also significantly
inhibited cell migration and cell proliferation with a reduction in
ODC activity in an in vitro wound model. Moreover, the
inhibitory effect on cell proliferation and ODC activity induced by
cigarette smoke may be reversed by exogenous spermidine, indicating
that the delayed wound healing in the stomach induced by cigarette
smoke was at least in part due to a reduction in polyamine
synthesis (30,32).
Smoking interferes with GI mucosal
protective mechanisms
Stomach acid secretion
Under normal conditions, large amounts of
hydrochloric acid exist in the stomach, which help to break down
food into smaller particles for further digestion in the digestive
tract. Gastric acid is neutralized in the duodenum by sodium
bicarbonate produced by the pancreas. The increased secretion of
stomach acid and/or a reduction in sodium bicarbonate production in
the pancreas can interfere with the protective mechanisms of the
gastric mucosa and the inner layer of the duodenum, where ulcers
are normally formed. Ample evidence suggests that smoking can
increase the production of gastric acid, accompanied by a reduction
in bicarbonate production. The role of cigarette smoke and its
active compounds, such as nicotine on acid production and sodium
bicarbonate production has been reviewed (35). Researchers have found that the
intravenous injection of nicotine hydrogen tartrate (0.012–0.020
mg/kg body weight) increases the concentration of hydrogen and
chloride ions in the gastric juice (36). In an early study, Ligny et
al (37) demonstrated that
the magnitude of acid secretion was associated with the number of
cigarettes smoked. They also found that tobacco smoking over a long
period of time stimulated vagus nerves and induced functional
parietal cells to increase pentagastrin-induced acid output in
smokers.
Biliary reflux
Bile is a digestive fluid produced by the liver, and
normally flows into the duodenum, where it digests fats and removes
toxins. Bile salts also function as detergents and damage the
mucosal barrier. The pylorus is a one-way valve between the stomach
and the duodenum that prevents bile and other contents of the small
intestine going back into the stomach (38). In a clinical study, it was
demonstrated that cigarette smoking induces pyloric incompetence
and increases the duodenogastric reflux (39), which may be due to the reduction
in basal pyloric pressure induced by smoking (40), leading to mucosal injury in the
stomach.
Pancreatic bicarbonate secretion
Pancreatic bicarbonate plays an important role in
neutralizing extra acid coming from the stomach. The increased
secretion of gastric acid, as well as a reduction in sodium
bicarbonate production would interfere with the protective
mechanisms in the stomach and the duodenum, possibly leading to the
development of ulcers in these organs. Several clinical studies
have shown that the secretion of bicarbonate is diminished after
cigarette smoking (41,42). Furthermore, the degree of
inhibition on basal pancreatic secretion has been shwon to have a
good correlation with the blood nicotine concentrations in humans
(43).
Smoking increases susceptibility to H.
pylori infection
H. pylori is known as one of the most common
infectious bacteria found in humans (44). Growing evidence points to a
potential association between H. pylori infection and GI
disorders, including gastroduodenal ulcers and cancer (45). Although some researchers have
found that cigarette smoking is negatively associated with H.
pylori infection, particularly in younger subjects (46), other epidemiological and
experimental studies have indicated that smoking is also a risk
factor for H. pylori infection at least under certain
clinical conditions (47,48).
Free radicals
Free radicals have been related to a wide spectrum
of GI disorders, including ulcers, IBDs and GI cancers.
Oxygen-derived free radicals play an important role in the
pathogenesis of peptic ulcers and IBD induced by smoking, alcohol,
as well as NSAIDs (49–51). Cigarette smoke contains large
amounts of free radicals (52).
The quinone/hydroquinone complex, for example, is an active redox
system which is capable of decreasing molecular oxygen to produce
superoxide, eventually transforming to hydrogen peroxide and
hydroxyl radicals (52). The
blood concentrations of free radicals in smokers are higher than
those of non-smokers, indicating that smoking-induced free radicals
promote gastric mucosal injury (53).
Smoking regulates immune cells in the GI
tract
The GI tract is also protected by the local mucosal
immune system operating in the GI mucosa against various internal
and external pathogens (8).
Chronic exposure to cigarette smoke and its active ingredients has
also been demonstrated to lead to alterations in the immune system
(54). Macrophages, neutrophils,
lymphocytes and dendritic cells may be involved in the pathogenesis
of inflammatory disorders in the GI tract. A research group found
that chronic smoke exposure was positively associated with immune
cell accumulation in Peyer’s patches. The total number of dendritic
cells, CD4+ T cells (including regulatory T cells) and
CD8+ T cells was significantly increased following
exposure to cigarette smoke for 24 weeks (25). Furthermore, the expression of
chemokines, including CCL9 and CCL20 was also upregulated, which
may play an important role in the pathogenesis of Crohn’s disease.
Smoke exposure also increases xanthine oxidase activity and
histamine release in the gastric mucosa. This may further lead to
neutrophil aggregation and vascular damage, thus promoting gastric
ulcers in rats (55).
3. Smoking increases the risk of cancer of
the GI tract
As stated in the previous section, tobacco smoking
induces various chronic inflammatory diseases of the GI tract,
including ulcers. It is clearly understood that chronic
inflammation can cause tumor initiation through the induction of
genomic instability, leading to mutagenesis (56). In addition, cigarette smoke
contains a broad spectrum of toxic and carcinogenic components,
such as aromatic amines, phenolic compounds, alkaloids, PAHs,
TNSAs, as well as heavy metals (7,8).
Among these, aromatic amines are thought to be the inducers of
bladder cancer, and TNSAs are thought to contribute to lung cancer
in smokers (57). Nicotine, taken
as an example, is known as the most active ingredient in cigarette
smoke, which is as high as 0.3–5% of the dry weight in tobacco
leaves (58). It has been found
that nicotine plays an important role in gastroduodenal ulceration
(35) and Crohn’s disease.
Furthermore, it also promotes cancer development in the esophagus
(59), stomach (4), colon (60) and liver (61).
Epidemiological studies
Cigarette smoking causes esophageal
cancer
Cigarette smoking is one of the risk factors for
esophageal cancer (62–65). Recently, a cohort study with a
20-year follow-up period conducted by Japanese researchers found
that individuals who began smoking at a younger age and consumed
larger amounts of alcohol more had a higher risk of developing
esophageal cancer compared with the normal population. The
esophageal cancer mortality risk was as high as 9.33 (95% CI,
2.55–34.2) for smokers who began smoking between the ages of 10 and
19 years and consuming three units of alcohol per day (64).
Cigarette smoking and cancer of the
oral cavity
Supporting data have demonstrated that cigarette
smoking is a major risk factor for cancer of the oral cavity
(66–68). A recent study demonstrated that
the odds ratios for current smokers and former smokers were 11.8
(95% CI, 8.6–16.3) and 2.2 (95% CI, 1.6–3.1), respectively when
compared to non-smokers. The risk of developing cancer of the oral
cavity increased with the quantity and duration of cigarette
smoking (66). In addition, the
risk of developing cancer of the oral cavity in former smokers
decreased with time. The buccal mucosa and the floor of the mouth
were the most sensitive sites with lesions induced by smoking
(67).
Cigarette smoking and gastric
cancer
The relationship between the occurrence of stomach
cancer and cigarette smoking has been studied since the 1950s.
Cigarette smoking has been considered as one of the key risk
factors for gastric cancer, which increases the incidence of the
disease by approximately 1.5- to 2.5-fold among current smokers
(69). Nicotine, the active
compound in cigarette smoke, has been demonstrated to be capable of
promoting gastric tumor growth and neovascularization (3). In a 20-year follow-up study
involving 18,244 middle-aged and older men conducted in Shanghai,
China, researchers found that the risk of gastric cancer was
statistically significantly higher in ever smokers [hazard ratio
(HR), 1.59; 95% CI, 1.27–1.99] than in non-smokers (70). Furthermore, among the
non-drinkers, the ever smokers experienced an 80% higher risk of
gastric cancer (HR, 1.81; 95% CI, 1.36–2.41). All these
observations indicate that cigarette smoking may exert independent
effects on the development of gastric cancer.
Cigarette smoking causes pancreatic
cancer
Tobacco consumption is considered an established
risk factor for pancreatic cancer (71,72). In a 10-year cohort study,
researchers found that cigarette smoking was related to an
increased risk of the disease [relative risk (RR), 1.7; 95% CI,
1.6–1.9] and mortality (RR, 1.6; 95% CI, 1.4–1.7) in patients with
pancreatic cancer (72).
Cigarette smoking causes colorectal
cancer
Phipps et al (74) carried out a study in 1,968
patients with stage III colon cancer in order to examine the
relationship between smoking and cancer outcome. They found that
smoking history was significantly associated with a shorter
disease-free survival (DFS), and time to recurrence (73) in patients with colon cancer
(74). Compared with
never-smokers, ever smokers experienced a significantly shorter DFS
with a three-year DFS proportion of 70 vs. 74% (HR, 1.21; 95% CI,
1.02–1.42). Compared with never-smokers, participants who were
former or current smokers were older and were more likely to be
male, and to have colon tumors that were dMMR and/or BRAF mutated
(74).
Possible effects of smoking on
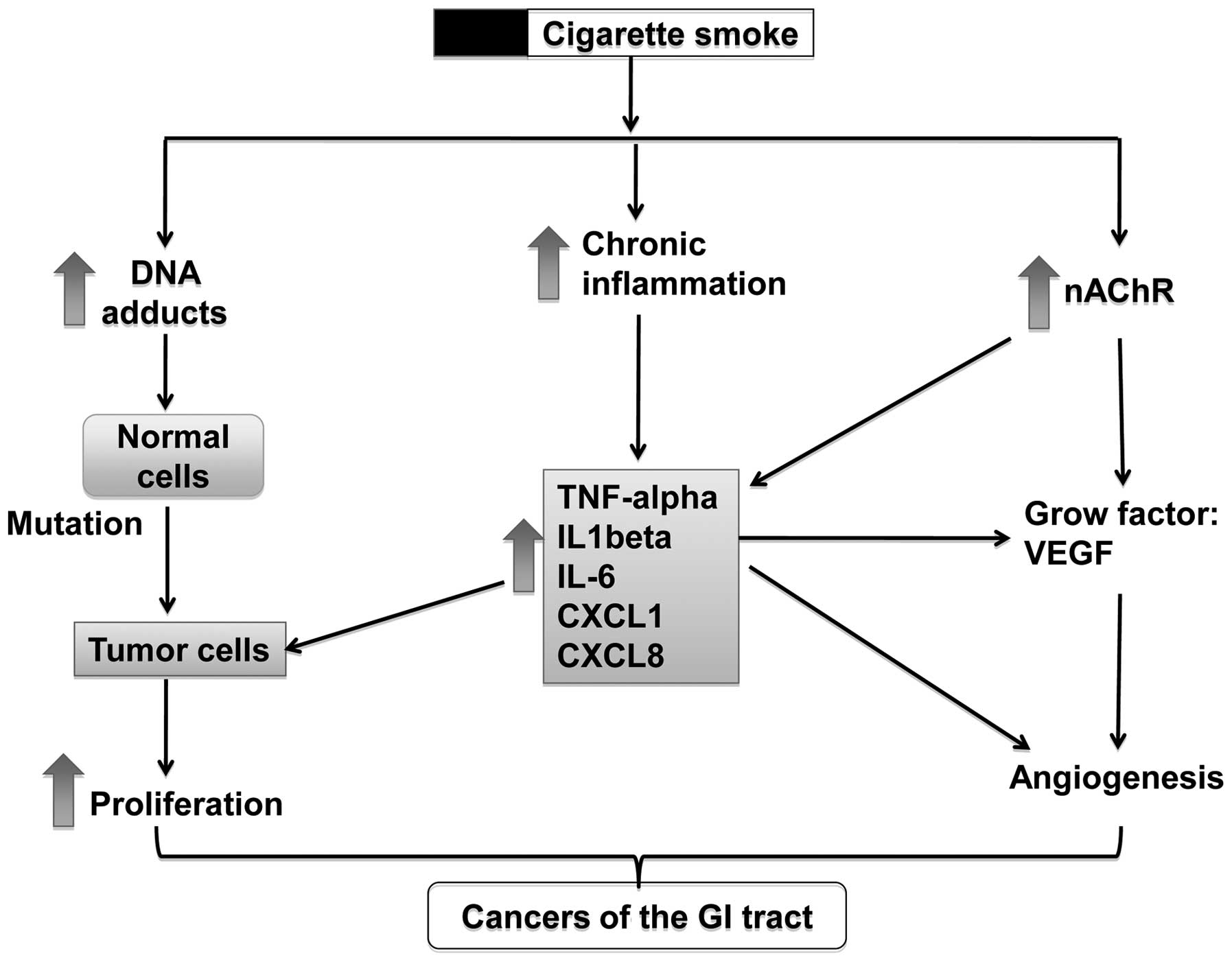
tumorigenesis in the GI tract
Cigarette smoke contains a broad spectrum of toxic
and carcinogenic components, such as aromatic amines, phenolic
compounds, alkaloids, PAHs, TNSAs, as well as heavy metals
(7,8). These toxic and carcinogenic
ingredients induce tumorigenesis in the GI tract through several
possible mechanisms, including the activation of nicotinic
acetylcholine receptors (nAChRs), the formation of DNA adducts,
stimulation of tumor angiogenesis, the involvement of immune
response and others (Fig. 2).
Normally, these mechanisms co-exist and have synergistic effects on
the promotion of tumorigenesis. For example, nicotine can activate
the nAChRs on cancer cells and induce the release of growth
factors, such as vascular endothelial growth factor (VEGF) and
IL-1β into the tumor microenvironment, which can increase tumor
angiogenesis and therefore promote tumor growth.
nAChRs
nAChRs are a family of ligand gate ion channels that
function as the key regulators of nicotinic and cholinergic
signaling in cells (75). nAChRs
are known to participate in cellular adhesion and migration through
the interactions with rapsyn and herparan sulphate proteoglycan
(76,77). Increasing evidence suggests that
nicotine and its derivatives, such as N-nitrosonornicotine and
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanonee can directly
activate nAChRs to promote cell growth and angiogenesis and inhibit
the drug-induced apoptosis of cancer cells (75).
A recent study demonstrated that nicotine activates
Yes-associated protein 1 (YAP1) through nAChR-mediated signaling in
esophageal squamous cell cancer (ESCC) (76). Zhao et al (76) reported that nicotine
administration increased cell proliferation and migration, and
promoted resistance to apoptosis in ESCC. In addition, nicotine
administration was also found to induce the nuclear translocation
and activation of YAP1 in ESCC. Nicotine signaling can also inhibit
the interaction of YAP1 with p63 that contributes to the inhibitory
effects of nicotine on cell apoptosis. The association between
cigarette smoking and YAP1 activation was also observed in clinical
esophageal cancer samples (76).
These results suggest that nicotine may be the active compound in
cigarette smoke responsible for carcinogenesis in the
esophagus.
In our previous studies, we found that nicotine in
cigarette smoke may be the most active ingredient responsible for
the tumorigenesis of colon cancer cells (78,79). Nicotine was demonstrated to
stimulate the proliferation of human colon adenocarcinoma HT-29
cells through the activation of α7-nAChR followed by the
catecholamine-synthesis pathway and adrenaline release and finally,
β-adrenergic activation (78).
Furthermore, in an animal study, nicotine was shown to promote
tumor growth, mainly by activating the β-adrenoceptors and the
subsequent expression of cyclooxygenase-2, prostaglandin E2, and
VEGF in tumor tissues (79).
These results demonstrate for the first time the contributory role
of α7-bAChR and β-adrenoceptors in the tumorigenesis of colon
cancer with significant involvement of some stress hormones.
Formation of DNA adducts
Many toxic compounds in cigarette smoke can interact
with DNA to form DNA adducts, which are believed to be another
important mechanism for carcinogenesis induced by cigarette smoke
(80). Among various ingredients
in cigarette smoke, TNSAs are known as the responsible compounds
for the formation of DNA adducts (81). In an early clinical study, Dyke
et al (81) found that in
males only, DNA adducts in gastric tumor tissues from smokers were
significantly higher than in those from non-smokers. Nitrosamines
and other nitroso compounds in cigarette smoke are capable of
covalently interacting with DNA, which alters the normal biological
function of DNA and eventually induces carcinogenesis in the GI
tract and in urinary bladder (81,82). To date, the formation of DNA
adducts has been found in cancer tissues from the oral cavity
(83), esophagus (84), stomach (81), pancreas (85) and colon (86).
The tobacco-specific nitrosamine
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) has been
demonstrated to be one of contributors for smoke-induced pancreatic
cancers. It is clear that NNK can react with DNA to form DNA methyl
and pyridyloxobutyl adducts (87). These DNA adducts can induce an
activating point mutation of the Ki-ras gene in codon 12, which is
common in human pancreatic adenocarcinomas (88,89). Askari et al (89) further demonstrated that NNK
induced the transactivation of the EGF receptor, increased the
accumulation of intracellular cyclic AMP, and activated the
phosphorylation of mitogen-activated protein kinase (MAPK) and
ERK1/2. These results indicate that the NNK-mediated β-adrenergic
cAMP-dependent signaling pathway may contribute to the development
of pancreatic carcinogenesis in smokers.
Chronic inflammation
The relationship between cancer and inflammation was
perceived as early as the 19th century. It is clear that chronic
inflammation predisposes to cancer at the proximity of the site of
inflammation (90). Chronic
inflammation in the GI tract can be caused by H. pylori
infection, autoimmune diseases, such as IBD and inflammatory
conditions, such as peptic ulcers. Various types of immune cells
are involved in the formation of the tumor inflammatory
microenvironment, such as macrophages, neutrophils, mast cells and
lymphocytes. During the inflammatory process, various inflammatory
components acting as messengers of inflammation are released by the
immune cells and tumor cells in the microenvironment of the
neoplastic tissues. These include cytokines, such as tumor necrosis
factor-α (TNF-α), interleukin (IL)-1 and IL-6, and chemokines, such
as CXCL8. Cigarette smoking is a risk factor for the development of
chronic inflammation in the GI tract as reviewed in the previous
section, which also promotes inflammation-associated
adenoma/adenocarcinoma formation (91).
Cytokines: a) TNF-α
TNF-α, as a pro-inflammatory cytokines, can not only
induce hemorrhagic necrosis of tumors, but also has protumoral
functions. It has been found that a high dose of TNF can destroy
the tumor vasculature and cut-off the supplement of O2
and nutrition for tumor growth to exert necrotic effects in tumors
(92). However, TNF-α can also
induce DNA damage (93), suppress
DNA repair (93) and promote the
growth of tumor cells (94).
Increasing evidence also suggests that TNF-α enhances tumor growth
and invasion, angiogenesis, leukocyte recruitment and facilitates
epithelial to mesenchymal transition (90). The bidirectional role of TNF-α in
tumor progression and cell death is due to the fact that TNF can
bind to different membrane-bound homotrimeric receptors, TNFRI and
TNFRII, to trigger opposite pathways (95). As regards tumor promotion, TNF-α
can inhibit the expression of glycogen synthase kinase-3β, and
consequently activate the Wnt/β-catenin signaling pathway to induce
tumor development (96).
Furthermore, TNF family members can also suppress the immune
response in the tumor environment, which may be due to the
inhibition of the major histocompatibility complex class II in
tumor-associated macrophages through the decoy receptor-3 (97).
b) IL-6
IL-6 plays an important role in tumor development,
such as colorectal cancer, in the GI tract (98). Clinical data have shown that IL-6
serum levels from patients with colorectal cancers are
significantly increased and positively correlate with tumor load,
including tumor size and liver metastases (98). It was demonstrated that two major
signaling pathways, the signal transducers and activators of
transcription 1 and 3 (STAT1/3) and the Src-homology tyrosine
phosphatase 2 (SHP2)-Ras-ERK, are involved in the IL-6-mediated
proliferation of intestinal epithelial cells (99). In addition, IL-6 can also promote
tumor growth by increasing the colony formation of human colon
carcinoma cells (100). These
biological actions of IL-6 in colorectal cancer progression were
further elucidated by mediating through the soluble IL-6 receptors
derived from tumor cells rather than from the membrane-bound
receptors (101).
c) IL-1β
IL-1β has been found to be capable of promoting
tumor cells to metastasize, by activating the cancer-related
inflammation cascade (102,103). In models of
3-methylcholanthrene-induced carcinogenesis, it was IL-1β, rather
than IL-1α in the tumor microenvironment that was capable of
determining the invasive potential of malignant cells, including
increased tumor adhesion and invasion, angiogenesis and immune
suppression (104).
Microenvironmental IL-1β is a required factor for tumor
invasiveness and angiogenesis, which may contribute to the
production of TNF-α and vascular endothelial cell growth factor by
IL-1β (105). Recently, Carmi
et al (106) found that
myeloid cells released IL-1β and induced endothelial cells to
produce proangiogenic factors, such as VEGF, and subsequently
provided the inflammatory microenvironment for tumor progression
and angiogenesis. Furthermore, they also observed that IL-1β
inhibition significantly reduced tumor growth by suppressing
inflammation and inducing the maturation of immature myeloid cells
into M1 macrophages (106).
Chemokines
Chemokines in the tumor microenvironment are another
important factors for modifying tumor growth, and promoting
angiogenesis (107) and tumor
metastatic spread (108). CXCL1
(growth-regulated oncogene α) for example, produced by human
colorectal cancer cells is capable of inducing microvascular
endothelial cell migration and tube formation in vitro.
PGE2-induced CXCL1 in the tumor microenvironment has also been
found to increase microvessel density and stimulate LS-174T cell
proliferation in an in vivo model (109). Together with CXCL-1, the
angiogenic chemokine CXCL8 (IL-8) was also significantly
unregulated in tumor tissues from patients with colorectal cancer
(110). CXCL8 signals are mainly
activated through the interaction with CXCR1 and CXCR2 present in
cancer cells and other cells. To date, CXCR1 and CXCR2 receptors
are widely expressed in cancer cells, tumor-associated macrophages,
neutrophils and endothelial cells (111). Therefore, the increased CXCL8
levels caused by cigarette smoking could nurture the tumor
microenvironment to promote cancer growth (112). Studies have shown that CXCL8
induces cell proliferation by the activation of classical MAPK and
downstream phosphorylation of ERK1/2 in neutrophils and cancer
cells (113,114). CXCL8 also regulates angiogenesis
by the induction of matrix metalloproteinase 9 (MMP-9) through the
activation of VEGFR-2 in endothelial cells, and subsequently
promotes cancer growth and metastasis (115).
4. Conclusions
Mounting evidence demonstrates that cigarette
smoking can induce pathogenic and carcinogenic processes in the GI
tract. These may lead to severe chronic inflammation and
subsequently, the development of cancer at the inflammation sites.
Clinical and experimental data have also shown that cigarette
smoking is a main risk factor for the induction of inflammatory
diseases, such as ulcers and Crohn’s disease. Cigarette smoke and
its active compounds impair the fundamental structure of the GI
tract through the induction of cellular apoptosis and the
inhibition of mucosal cell renewal. Cigarette smoke also interferes
with the protective mechanisms of the GI tract by decreasing the
blood flow in the mucosa and modulating the mucosal immune system.
Furthermore, cigarette smoke also inhibits the synthesis and
release of EGF and polyamines and thereby, mucus secretion, which
plays an important role in protecting mucosal integrity. Chronic
inflammation induced by cigarette smoke exposure releases various
inflammatory components, including the cytokines, TNF-α, IL-1 and
IL-6, and the chemokines, CXCL1 and CXCL8. These inflammatory
components are capable of promoting tumor growth, tumor adhesion
and invasion. Moreover, these mediators also induce angiogenesis
and immune suppression in the tumor microenvironment. Along with
the induction of chronic inflammation, cigarette smoke and its
active ingredients can directly activate nAChRs, and form DNA
adducts to initiate tumorigenesis in the GI tract. In conclusion,
cigarette smoke is a detrimental factor affecting the pathogenesis
and tumorigenesis of certain disorders in the GI tract. Detailed
mechanistic studies may aid in the development of more effective
therapies for various disorders of the GI tract.
References
|
1
|
WHO urges more countries to require large,
graphic health warnings on tobacco packaging: the WHO report on the
global tobacco epidemic, 2011 examines anti-tobacco mass-media
campaigns. Cent Eur J Public Health. 19:1331512011.
|
|
2
|
Peters SA, Huxley RR and Woodward M:
Smoking as a risk factor for stroke in women compared with men: a
systematic review and meta-analysis of 81 cohorts, including
3,980,359 individuals and 42,401 strokes. Stroke. 44:2821–2828.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shin VY and Cho CH: Nicotine and gastric
cancer. Alcohol. 35:259–264. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chu KM, Cho CH and Shin VY: Nicotine and
gastrointestinal disorders: its role in ulceration and cancer
development. Curr Pharm Des. 19:5–10. 2013.PubMed/NCBI
|
|
5
|
Peluso ME, Munnia A, Srivatanakul P,
Jedpiyawongse A, Sangrajrang S, Ceppi M, Godschalk RW, van Schooten
FJ and Boffetta P: DNA adducts and combinations of multiple lung
cancer at-risk alleles in environmentally exposed and smoking
subjects. Environ Mol Mutagen. 54:375–383. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jain G and Jaimes EA: Nicotine signaling
and progression of chronic kidney disease in smokers. Biochem
Pharmacol. 86:1215–1223. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li W, Zhou J, Chen L, Luo Z and Zhao Y:
Lysyl oxidase, a critical intra- and extra-cellular target in the
lung for cigarette smoke pathogenesis. Int J Environ Res Public
Health. 8:161–184. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang L, Ren JW, Wong CC, Wu WK, Ren SX,
Shen J, Chan RL and Cho CH: Effects of cigarette smoke and its
active components on ulcer formation and healing in the
gastrointestinal mucosa. Curr Med Chem. 19:63–69. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ross R: The pathogenesis of
atherosclerosis: a perspective for the 1990s. Nature. 362:801–809.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hecht SS: Tobacco carcinogens, their
biomarkers and tobacco-induced cancer. Nat Rev Cancer. 3:733–744.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ootani H, Iwakiri R, Shimoda R, Nakahara
S, Amemori S, Fujise T, Kikkawa A, Tsunada S, Sakata H and Fujimoto
K: Role of Helicobacter pylori infection and nonsteroidal
anti-inflammatory drug use in bleeding peptic ulcers in Japan. J
Gastroenterol. 41:41–46. 2006.
|
|
12
|
Garrow D and Delegge MH: Risk factors for
gastrointestinal ulcer disease in the us population. Dig Dis Sci.
55:66–72. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen MH, Wu MS, Lee WC, Wang HP and Lin
JT: A multiple logistic regression analysis of risk factors in
different subtypes of gastric ulcer. Hepatogastroenterology.
49:589–592. 2002.PubMed/NCBI
|
|
14
|
Parasher G and Eastwood GL: Smoking and
peptic ulcer in the Helicobacter pylori era. Eur J
Gastroenterol Hepatol. 12:843–853. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Andersen IB, Jorgensen T, Bonnevie O,
Gronbaek M and Sorensen TI: Smoking and alcohol intake as risk
factors for bleeding and perforated peptic ulcers: a
population-based cohort study. Epidemiology. 11:434–439. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ananthakrishnan AN: Environmental risk
factors for inflammatory bowel disease. Gastroenterol Hepatol (NY).
9:367–374. 2013.PubMed/NCBI
|
|
17
|
Zaharie R, Zaharie F, Mocan L, Andreica V,
Tantau M, Zdrehus C, Iancu C and Tomus C: Surgical outcome of
inflammatory bowel disease - experience of a tertiary center.
Chirurgia (Bucur). 108:812–815. 2013.PubMed/NCBI
|
|
18
|
Lawrance IC, Murray K, Batman B, Gearry
RB, Grafton R, Krishnaprasad K, Andrews JM, Prosser R, Bampton PA,
Cooke SE, Mahy G, et al: Crohn’s disease and smoking: Is it ever
too late to quit? J Crohns Colitis. 7:e665–e671. 2013.
|
|
19
|
Lunney PC and Leong RW: Review article:
Ulcerative colitis, smoking and nicotine therapy. Aliment Pharmacol
Ther. 36:997–1008. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Somerville KW, Logan RF, Edmond M and
Langman MJ: Smoking and crohn’s disease. Br Med J (Clin Res Ed).
289:954–956. 1984.
|
|
21
|
Seksik P, Nion-Larmurier I, Sokol H,
Beaugerie L and Cosnes J: Effects of light smoking consumption on
the clinical course of Crohn’s disease. Inflamm Bowel Dis.
15:734–741. 2009.PubMed/NCBI
|
|
22
|
Harries AD, Baird A and Rhodes J:
Non-smoking: A feature of ulcerative colitis. Br Med J (Clin Res
Ed). 284:7061982. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lakatos PL, Vegh Z, Lovasz BD, David G,
Pandur T, Erdelyi Z, Szita I, Mester G, Balogh M, Szipocs I, Molnar
C, et al: Is current smoking still an important environmental
factor in inflammatory bowel diseases? Results from a
population-based incident cohort. Inflamm Bowel Dis. 19:1010–1017.
2013. View Article : Google Scholar
|
|
24
|
Wu WK and Cho CH: The pharmacological
actions of nicotine on the gastrointestinal tract. J Pharmacol Sci.
94:348–358. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Verschuere S, Bracke KR, Demoor T,
Plantinga M, Verbrugghe P, Ferdinande L, Lambrecht BN, Brusselle GG
and Cuvelier CA: Cigarette smoking alters epithelial apoptosis and
immune composition in murine GALT. Lab Invest. 91:1056–1067. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang H, Ma L, Li Y and Cho CH: Exposure to
cigarette smoke increases apoptosis in the rat gastric mucosa
through a reactive oxygen species-mediated and p53-independent
pathway. Free Radic Biol Med. 28:1125–1131. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sharma A, Neekhra A, Gramajo AL, Patil J,
Chwa M, Kuppermann BD and Kenney MC: Effects of Benzo(e)Pyrene, a
toxic component of cigarette smoke, on human retinal pigment
epithelial cells in vitro. Invest Ophthalmol Vis Sci. 49:5111–5117.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
FitzGerald AJ, Mandir N and Goodlad RA:
Leptin, cell proliferation and crypt fission in the
gastrointestinal tract of intravenously fed rats. Cell Prolif.
38:25–33. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ma L, Wang WP, Chow JY, Yuen ST and Cho
CH: Reduction of EGF is associated with the delay of ulcer healing
by cigarette smoking. Am J Physiol Gastrointest Liver Physiol.
278:G10–G17. 2000.PubMed/NCBI
|
|
30
|
Shin VY, Liu ES, Koo MW, Wang JY, Matsui H
and Cho CH: Cigarette smoke extracts delay wound healing in the
stomach: Involvement of polyamine synthesis. Exp Biol Med
(Maywood). 227:114–124. 2002.PubMed/NCBI
|
|
31
|
Konturek JW, Bielanski W, Konturek SJ,
Bogdal J and Oleksy J: Distribution and release of epidermal growth
factor in man. Gut. 30:1194–1200. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ma L, Wang WP, Chow JY, Lam SK and Cho CH:
The role of polyamines in gastric mucus synthesis inhibited by
cigarette smoke or its extract. Gut. 47:170–177. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brzozowski T, Konturek SJ, Majka J,
Dembinski A and Drozdowicz D: Epidermal growth factor, polyamines,
and prostaglandins in healing of stress-induced gastric lesions in
rats. Dig Dis Sci. 38:276–283. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Konturek JW, Brzozowski T and Konturek SJ:
Epidermal growth factor in protection, repair, and healing of
gastroduodenal mucosa. J Clin Gastroenterol. 13(Suppl 1): S88–S97.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Maity P, Biswas K, Roy S, Banerjee RK and
Bandyopadhyay U: Smoking and the pathogenesis of gastroduodenal
ulcer - recent mechanistic update. Mol Cell Biochem. 253:329–338.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mertz DP and Thongbhoubesra T: Effect of
nicotine on the production of gastric acid (author’s transl). Med
Klin. 71:147–155. 1976.(In German).
|
|
37
|
Ligny G, Van Ccauter J and Henry JP: The
effect of cigarette smoking on the cicatrization of duodenal ulcers
in patients treated with cimetidine. The role of acid
hypersecretion. Rev Med Brux. 10:233–238. 1989.(In French).
|
|
38
|
Fiddian-Green R, Russell RC and Hobsley M:
Pyloric reflux in duodenal ulceration and its relationship to
smoking. Br J Surg. 60:3211973.PubMed/NCBI
|
|
39
|
Read NW and Grech P: Effect of cigarette
smoking on competence of the pylorus: preliminary study. Br Med J.
3:313–316. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Valenzuela JE, Defilippi C and Csendes A:
Manometric studies on the human pyloric sphincter. Effect of
cigarette smoking, metoclopramide, and atropine. Gastroenterology.
70:481–483. 1976.PubMed/NCBI
|
|
41
|
Bynum TE, Solomon TE, Johnson LR and
Jacobson ED: Inhibition of pancreatic secretion in man by cigarette
smoking. Gut. 13:361–365. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bochenek WJ and Koronczewski R: Effects of
cigarette smoking on bicarbonate and volume of duodenal contents.
Am J Dig Dis. 18:729–733. 1973.PubMed/NCBI
|
|
43
|
Murthy SN, Dinoso VP Jr, Clearfield HR and
Chey WY: Simultaneous measurement of basal pancreatic, gastric acid
secretion, plasma gastrin, and secretin during smoking.
Gastroenterology. 73:758–761. 1977.PubMed/NCBI
|
|
44
|
Deng B, Li Y, Zhang Y, Bai L and Yang P:
Helicobacter pylori infection and lung cancer: a review of
an emerging hypothesis. Carcinogenesis. 34:1189–1195. 2013.
View Article : Google Scholar
|
|
45
|
Bures J, Kopacova M, Skodova Fendrichova M
and Rejchrt S: Epidemiology of Helicobacter pylori
infection. Vnitr Lek. 57:993–999. 2011.(In Czech).
|
|
46
|
Ogihara A, Kikuchi S, Hasegawa A, Kurosawa
M, Miki K, Kaneko E and Mizukoshi H: Relationship between
Helicobacter pylori infection and smoking and drinking
habits. J Gastroenterol Hepatol. 15:271–276. 2000.
|
|
47
|
Arkkila PE, Kokkola A, Seppälä K and
Sipponen P: Size of the peptic ulcer in Helicobacter
pylori-positive patients: association with the clinical and
histological characteristics. Scand J Gastroenterol. 42:695–701.
2007.PubMed/NCBI
|
|
48
|
Endoh K and Leung FW: Effects of smoking
and nicotine on the gastric mucosa: a review of clinical and
experimental evidence. Gastroenterology. 107:864–878. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Smith SM and Kvietys PR: Gastric ulcers:
Role of oxygen radicals. Crit Care Med. 16:892–898. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hirota M, Inoue M, Ando Y and Morino Y:
Inhibition of stress-induced gastric mucosal injury by a long
acting superoxide dismutase that circulates bound to albumin. Arch
Biochem Biophys. 280:269–273. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Calvino Fernández M and Parra Cid T: H.
pylori and mitochondrial changes in epithelial cells. The role
of oxidative stress. Rev Esp Enferm Dig. 102:41–50. 2010.
|
|
52
|
Church DF and Pryor WA: Free-radical
chemistry of cigarette smoke and its toxicological implications.
Environ Health Perspect. 64:111–126. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kalra J, Chaudhary AK and Prasad K:
Increased production of oxygen free radicals in cigarette smokers.
Int J Exp Pathol. 72:1–7. 1991.PubMed/NCBI
|
|
54
|
Sopori M: Effects of cigarette smoke on
the immune system. Nat Rev Immunol. 2:372–377. 2002. View Article : Google Scholar
|
|
55
|
Chow JY, Ma L and Cho CH: Involvement of
free radicals and histamine in the potentiating action of cigarette
smoke exposure on ethanol-induced gastric mucosal damage in rats.
Free Radic Biol Med. 24:1285–1293. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Grivennikov SI, Greten FR and Karin M:
Immunity, inflammation, and cancer. Cell. 140:883–899. 2010.
View Article : Google Scholar
|
|
57
|
Tang Y, Kassie F, Qian X, Ansha B and
Turesky RJ: DNA adduct formation of 2-amino-9H-pyrido[2,3-b)indole
and 2-amino-3,4-dimethylimidazo[4,5-f)quinoline in mouse liver and
extrahepatic tissues during a subchronic feeding study. Toxicol
Sci. 133:248–258. 2013.
|
|
58
|
Martin JW, Mousa SS, Shaker O and Mousa
SA: The multiple faces of nicotine and its implications in tissue
and wound repair. Exp Dermatol. 18:497–505. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zong Y, Zhang ST and Zhu ST: Nicotine
enhances migration and invasion of human esophageal squamous
carcinoma cells which is inhibited by nimesulide. World J
Gastroenterol. 15:2500–2505. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Cucina A, Dinicola S, Coluccia P, Proietti
S, D’Anselmi F, Pasqualato A and Bizzarri M: Nicotine stimulates
proliferation and inhibits apoptosis in colon cancer cell lines
through activation of survival pathways. J Surg Res. 178:233–241.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Seitz HK and Cho CH: Contribution of
alcohol and tobacco use in gastrointestinal cancer development.
Methods Mol Biol. 472:217–241. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Castellsagué X, Muñoz N, De Stefani E,
Victora CG, Castelletto R, Rolón PA and Quintana MJ: Independent
and joint effects of tobacco smoking and alcohol drinking on the
risk of esophageal cancer in men and women. Int J Cancer.
82:657–664. 1999.PubMed/NCBI
|
|
63
|
Freedman ND, Abnet CC, Leitzmann MF, Mouw
T, Subar AF, Hollenbeck AR and Schatzkin A: A prospective study of
tobacco, alcohol, and the risk of esophageal and gastric cancer
subtypes. Am J Epidemiol. 165:1424–1433. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yaegashi Y, Onoda T, Morioka S, Hashimoto
T, Takeshita T, Sakata K and Tamakoshi A: Joint effects of smoking
and alcohol drinking on esophageal cancer mortality in Japanese
men: Findings from the Japan collaborative cohort study. Asian Pac
J Cancer Prev. 15:1023–1029. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Brown LM, Hoover R, Silverman D, Baris D,
Hayes R, Swanson GM, Schoenberg J, Greenberg R, Liff J, Schwartz A,
Dosemeci M, et al: Excess incidence of squamous cell esophageal
cancer among US Black men: role of social class and other risk
factors. Am J Epidemiol. 153:114–122. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Radoï L, Paget-Bailly S, Cyr D,
Papadopoulos A, Guida F, Schmaus A, Cénée S, Menvielle G, Carton M,
Lapôtre-Ledoux B, Delafosse P, et al: Tobacco smoking, alcohol
drinking and risk of oral cavity cancer by subsite: results of a
French population-based case-control study, the ICARE study. Eur J
Cancer Prev. 22:268–276. 2013.PubMed/NCBI
|
|
67
|
Pentenero M, Giaretti W, Navone R, Rostan
I, Gassino L, Broccoletti R, Arduino PG, Malacarne D and Gandolfo
S: Evidence for a possible anatomical subsite-mediated effect of
tobacco in oral potentially malignant disorders and carcinoma. J
Oral Pathol Med. 40:214–217. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Muwonge R, Ramadas K, Sankila R, Thara S,
Thomas G, Vinoda J and Sankaranarayanan R: Role of tobacco smoking,
chewing and alcohol drinking in the risk of oral cancer in
Trivandrum, India: a nested case-control design using incident
cancer cases. Oral Oncol. 44:446–454. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Sasazuki S, Sasaki S and Tsugane S:
Cigarette smoking, alcohol consumption and subsequent gastric
cancer risk by subsite and histologic type. Int J Cancer.
101:560–566. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Moy KA, Fan Y, Wang R, Gao YT, Yu MC and
Yuan JM: Alcohol and tobacco use in relation to gastric cancer: a
prospective study of men in Shanghai, China. Cancer Epidemiol
Biomarkers Prev. 19:2287–2297. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Eguchi H and Nakachi K: Smoking as a risk
factor for pancreatic cancer. Nihon Rinsho. 64(Suppl 1): S10–S13.
2006.(In Japanese).
|
|
72
|
Yun JE, Jo I, Park J, Kim MT, Ryu HG,
Odongua N, Kim E and Jee SH: Cigarette smoking, elevated fasting
serum glucose, and risk of pancreatic cancer in korean men. Int J
Cancer. 119:208–212. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Farris SM, Pettrey C and Daly KC: A
subpopulation of mushroom body intrinsic neurons is generated by
protocerebral neuroblasts in the tobacco hornworm moth, Manduca
sexta (Sphingidae, Lepidoptera). Arthropod Struct Dev.
40:395–408. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Phipps AI, Shi Q, Newcomb PA, Nelson GD,
Sargent DJ, Alberts SR and Limburg PJ: Associations between
cigarette smoking status and colon cancer prognosis among
participants in North Central Cancer Treatment Group Phase III
Trial N0147. J Clin Oncol. 31:2016–2023. 2013. View Article : Google Scholar
|
|
75
|
Schuller HM: Is cancer triggered by
altered signalling of nicotinic acetylcholine receptors? Nat Rev
Cancer. 9:195–205. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhao Y, Zhou W, Xue L, Zhang W and Zhan Q:
Nicotine activates YAP1 through nAChRs mediated signaling in
esophageal squamous cell cancer (ESCC). PLoS One. 9:e908362014.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Chernyavsky AI, Arredondo J, Vetter DE and
Grando SA: Central role of alpha9 acetylcholine receptor in
coordinating keratinocyte adhesion and motility at the initiation
of epithelialization. Exp Cell Res. 313:3542–3555. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wong HP, Yu L, Lam EK, Tai EK, Wu WK and
Cho CH: Nicotine promotes cell proliferation via alpha7-nicotinic
acetylcholine receptor and catecholamine-synthesizing
enzymes-mediated pathway in human colon adenocarcinoma ht-29 cells.
Toxicol Appl Pharmacol. 221:261–267. 2007. View Article : Google Scholar
|
|
79
|
Wong HP, Yu L, Lam EK, Tai EK, Wu WK and
Cho CH: Nicotine promotes colon tumor growth and angiogenesis
through beta-adrenergic activation. Toxicol Sci. 97:279–287. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Jarabek AM, Pottenger LH, Andrews LS,
Casciano D, Embry MR, Kim JH, Preston RJ, Reddy MV, Schoeny R,
Shuker D, Skare J, et al: Creating context for the use of DNA
adduct data in cancer risk assessment: I. Data organization. Crit
Rev Toxicol. 39:659–678. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Dyke GW, Craven JL, Hall R and Garner RC:
Smoking-related DNA adducts in human gastric cancers. Int J Cancer.
52:847–850. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Bartsch H, Ohshima H, Pignatelli B and
Calmels S: Human exposure to endogenous N-nitroso compounds:
quantitative estimates in subjects at high risk for cancer of the
oral cavity, oesophagus, stomach and urinary bladder. Cancer Surv.
8:335–362. 1989.PubMed/NCBI
|
|
83
|
Pabiszczak M, Szmeja Z, Szyfter K and
Szyfter W: Analysis of aromatic DNA adducts in oral cavity and
pharyngeal cancer. Otolaryngol Pol. 54:151–156. 2000.(In
Polish).
|
|
84
|
Lee JM, Liu TY, Wu DC, Tang HC, Leh J, Wu
MT, Hsu HH, Huang PM, Chen JS, Lee CJ and Lee YC: Safrole-DNA
adducts in tissues from esophageal cancer patients: Clues to
areca-related esophageal carcinogenesis. Mutat Res. 565:121–128.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Wang M, Abbruzzese JL, Friess H, Hittelman
WN, Evans DB, Abbruzzese MC, Chiao P and Li D: DNA adducts in human
pancreatic tissues and their potential role in carcinogenesis.
Cancer Res. 58:38–41. 1998.PubMed/NCBI
|
|
86
|
Al-Saleh I, Arif J, El-Doush I, Al-Sanea
N, Jabbar AA, Billedo G, Shinwari N, Mashhour A and Mohamed G:
Carcinogen DNA adducts and the risk of colon cancer: case-control
study. Biomarkers. 13:201–216. 2008. View Article : Google Scholar
|
|
87
|
Hecht SS: Recent studies on mechanisms of
bioactivation and detoxification of
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), a
tobacco-specific lung carcinogen. Crit Rev Toxicol. 26:163–181.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Belinsky SA, Devereux TR, Maronpot RR,
Stoner GD and Anderson MW: Relationship between the formation of
promutagenic adducts and the activation of the K-ras protooncogene
in lung tumors from A/J mice treated with nitrosamines. Cancer Res.
49:5305–5311. 1989.PubMed/NCBI
|
|
89
|
Askari MD, Tsao MS and Schuller HM: The
tobacco-specific carcinogen,
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone stimulates
proliferation of immortalized human pancreatic duct epithelia
through beta-adrenergic transactivation of EGF receptors. J Cancer
Res Clin Oncol. 131:639–648. 2005. View Article : Google Scholar
|
|
90
|
Colotta F, Allavena P, Sica A, Garlanda C
and Mantovani A: Cancer-related inflammation, the seventh hallmark
of cancer: links to genetic instability. Carcinogenesis.
30:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Liu ES, Ye YN, Shin VY, Yuen ST, Leung SY,
Wong BC and Cho CH: Cigarette smoke exposure increases ulcerative
colitis-associated colonic adenoma formation in mice.
Carcinogenesis. 24:1407–1413. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
van Horssen R, Ten Hagen TL and Eggermont
AM: TNF-alpha in cancer treatment: molecular insights, antitumor
effects, and clinical utility. Oncologist. 11:397–408.
2006.PubMed/NCBI
|
|
93
|
Wheelhouse NM, Chan YS, Gillies SE,
Caldwell H, Ross JA, Harrison DJ and Prost S: TNF-α induced DNA
damage in primary murine hepatocytes. Int J Mol Med. 12:889–894.
2003.
|
|
94
|
Charles KA, Kulbe H, Soper R,
Escorcio-Correia M, Lawrence T, Schultheis A, Chakravarty P,
Thompson RG, Kollias G, Smyth JF, Balkwill FR and Hagemann T: The
tumor-promoting actions of TNF-alpha involve TNFR1 and IL-17 in
ovarian cancer in mice and humans. J Clin Invest. 119:3011–3023.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Idriss HT and Naismith JH: TNF alpha and
the TNF receptor superfamily: structure-function relationship(s).
Microsc Res Tech. 50:184–195. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Oguma K, Oshima H, Aoki M, Uchio R, Naka
K, Nakamura S, Hirao A, Saya H, Taketo MM and Oshima M: Activated
macrophages promote Wnt signalling through tumour necrosis
factor-alpha in gastric tumour cells. EMBO J. 27:1671–1681. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Chang YC, Chen TC, Lee CT, Yang CY, Wang
HW, Wang CC and Hsieh SL: Epigenetic control of MHC class II
expression in tumor-associated macrophages by decoy receptor 3.
Blood. 111:5054–5063. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Chung YC and Chang YF: Serum interleukin-6
levels reflect the disease status of colorectal cancer. J Surg
Oncol. 83:222–226. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Tebbutt NC, Giraud AS, Inglese M, Jenkins
B, Waring P, Clay FJ, Malki S, Alderman BM, Grail D, Hollande F,
Heath JK and Ernst M: Reciprocal regulation of gastrointestinal
homeostasis by SHP2 and STAT-mediated trefoil gene activation in
gp130 mutant mice. Nat Med. 8:1089–1097. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Schneider MR, Hoeflich A, Fischer JR, Wolf
E, Sordat B and Lahm H: Interleukin-6 stimulates clonogenic growth
of primary and metastatic human colon carcinoma cells. Cancer Lett.
151:31–38. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Becker C, Fantini MC, Schramm C, Lehr HA,
Wirtz S, Nikolaev A, Burg J, Strand S, Kiesslich R, Huber S, Ito H,
et al: TGF-beta suppresses tumor progression in colon cancer by
inhibition of IL-6 trans-signaling. Immunity. 21:491–501. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Candido J and Hagemann T: Cancer-related
inflammation. J Clin Immunol. 33(Suppl 1): S79–S84. 2013.
View Article : Google Scholar
|
|
103
|
Luo JL, Tan W, Ricono JM, Korchynskyi O,
Zhang M, Gonias SL, Cheresh DA and Karin M: Nuclear
cytokine-activated IKKalpha controls prostate cancer metastasis by
repressing Maspin. Nature. 446:690–694. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Krelin Y, Voronov E, Dotan S, Elkabets M,
Reich E, Fogel M, Huszar M, Iwakura Y, Segal S, Dinarello CA and
Apte RN: Interleukin-1beta-driven inflammation promotes the
development and invasiveness of chemical carcinogen-induced tumors.
Cancer Res. 67:1062–1071. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Voronov E, Shouval DS, Krelin Y, Cagnano
E, Benharroch D, Iwakura Y, Dinarello CA and Apte RN: IL-1 is
required for tumor invasiveness and angiogenesis. Proc Natl Acad
Sci USA. 100:2645–2650. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Carmi Y, Dotan S, Rider P, Kaplanov I,
White MR, Baron R, Abutbul S, Huszar M, Dinarello CA, Apte RN and
Voronov E: The role of IL-1β in the early tumor cell-induced
angiogenic response. J Immunol. 190:3500–3509. 2013.
|
|
107
|
Dimberg A: Chemokines in angiogenesis.
Curr Top Microbiol Immunol. 341:59–80. 2010.
|
|
108
|
Wilson J and Balkwill F: The role of
cytokines in the epithelial cancer microenvironment. Semin Cancer
Biol. 12:113–120. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Wang D, Wang H, Brown J, Daikoku T, Ning
W, Shi Q, Richmond A, Strieter R, Dey SK and DuBois RN: CXCL1
induced by prostaglandin E2 promotes angiogenesis in colorectal
cancer. J Exp Med. 203:941–951. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Erreni M, Bianchi P, Laghi L, Mirolo M,
Fabbri M, Locati M, Mantovani A and Allavena P: Expression of
chemokines and chemokine receptors in human colon cancer. Methods
Enzymol. 460:105–121. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Gales D, Clark C, Manne U and Samuel T:
The chemokine CXCL8 in carcinogenesis and drug response. ISRN
Oncol. 2013:8591542013.PubMed/NCBI
|
|
112
|
Vandercappellen J, Van Damme J and Struyf
S: The role of CXC chemokines and their receptors in cancer. Cancer
Lett. 267:226–244. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Luppi F, Longo AM, de Boer WI, Rabe KF and
Hiemstra PS: Interleukin-8 stimulates cell proliferation in
non-small cell lung cancer through epidermal growth factor receptor
transactivation. Lung Cancer. 56:25–33. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Richardson RM, Ali H, Pridgen BC, Haribabu
B and Snyderman R: Multiple signaling pathways of human
interleukin-8 receptor A. Independent regulation by
phosphorylation. J Biol Chem. 273:10690–10695. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Inoue K, Slaton JW, Eve BY, Kim SJ,
Perrotte P, Balbay MD, Yano S, Bar-Eli M, Radinsky R, Pettaway CA
and Dinney CP: Interleukin 8 expression regulates tumorigenicity
and metastases in androgen-independent prostate cancer. Clin Cancer
Res. 6:2104–2119. 2000.PubMed/NCBI
|