Introduction
Endothelial-mesenchymal cell transdifferentiation
(EndoMT) is the process in which endothelial cells lose their cell-
type-specific characteristics and gain a mesenchymal or
myofibroblastic phenotype (1).
EndoMT may be initiated by cytokines or growth factors secreted by
peri-vascular cells (2). The
basement membrane under the endothelial cells is likely to be
degraded by matrix metalloproteinases (MMPs), then the
transitioning endothelial cells become motile and invade the
surrounding tissues (3). During
EndoMT, endothelial cells lose the expression of their markers,
such as vascular endothelial (VE)-cadherin and von Willebrand
factor (vWF), and gain the expression of mesenchymal cell markers
including vimentin, α-smooth muscle actin (SMA) and type I collagen
(1,2,4).
EndoMT was first observed in studies on cardiac development
(5,6). EndoMT has emerged as a possible
mechanism in the pathogenesis of various diseases, including
diabetic nephropathy, cardiac fibrosis, intestinal fibrosis,
pulmonary hypertension and systemic sclerosis (7–10).
Despite the notable importance of EndoMT for
embryonic development and pathologic conditions, the underlying
molecular mechanisms involved in EndoMT have yet to be fully
elucidated. Substantial evidence has indicated the crucial role of
TGF-β signaling in the initiation of EndoMT (11). A number of signaling transduction
pathways, including VEGF, NFAT, BMP, Wnt/β-catenin, ErbB, and
NF1/Ras, play a role in EndoMT during cardiac development (12). In addition, EndoMT can be
modulated in response to manipulations of the Notch pathways in
many different endothelial cell types (13). It is also suggested that a number
of signaling pathways interact with TGF-β and Notch to mediate
EndoMT during heart valve development (14,15).
The Notch signaling pathway is evolutionarily
conserved and plays a fundamental role in a number of mechanisms
(16,17). Activation of Notch signaling is
initiated through ligand-receptor interactions which lead to
proteolytic cleavage of the receptor (18). Mammals have four receptors
(Notch1, 2, 3, 4) and five ligands (Jagged 1 and 2, and δ-like 1, 3
and 4). Following activation, the intracellular domain of Notch
(IC-Notch) translocates into the nucleus and binds the DNA-binding
protein CSL (CBF1/suppressor of hairless/Lag-1) through its RAM23
domain (19). The CSL protein
(also known as CBF-1/RBP-Jκ) binds to the DNA sequence GTGGGAA in
the promoter region of Notch-regulated genes (20). The components of the Notch
signaling pathway are crucial for cell fate decisions during
morphogenesis and embryonic development (17,21). Notch signaling was thought to be
involved in the regulation of vascular smooth muscle
differentiation during heart valve and cardiac cushion development
(22). Subsequent studies have
confirmed the involvement of Notch signaling in the EndoMT process
(23–25).
Notch proteins are expressed in most cell types and
are involved in a broad spectrum of disorders (26). Studies of the Notch pathway in
mice using gain- and loss-of-function approaches have been
restricted due to the development of embryonic lethal phenotypes
(27–29). To investigate the function of
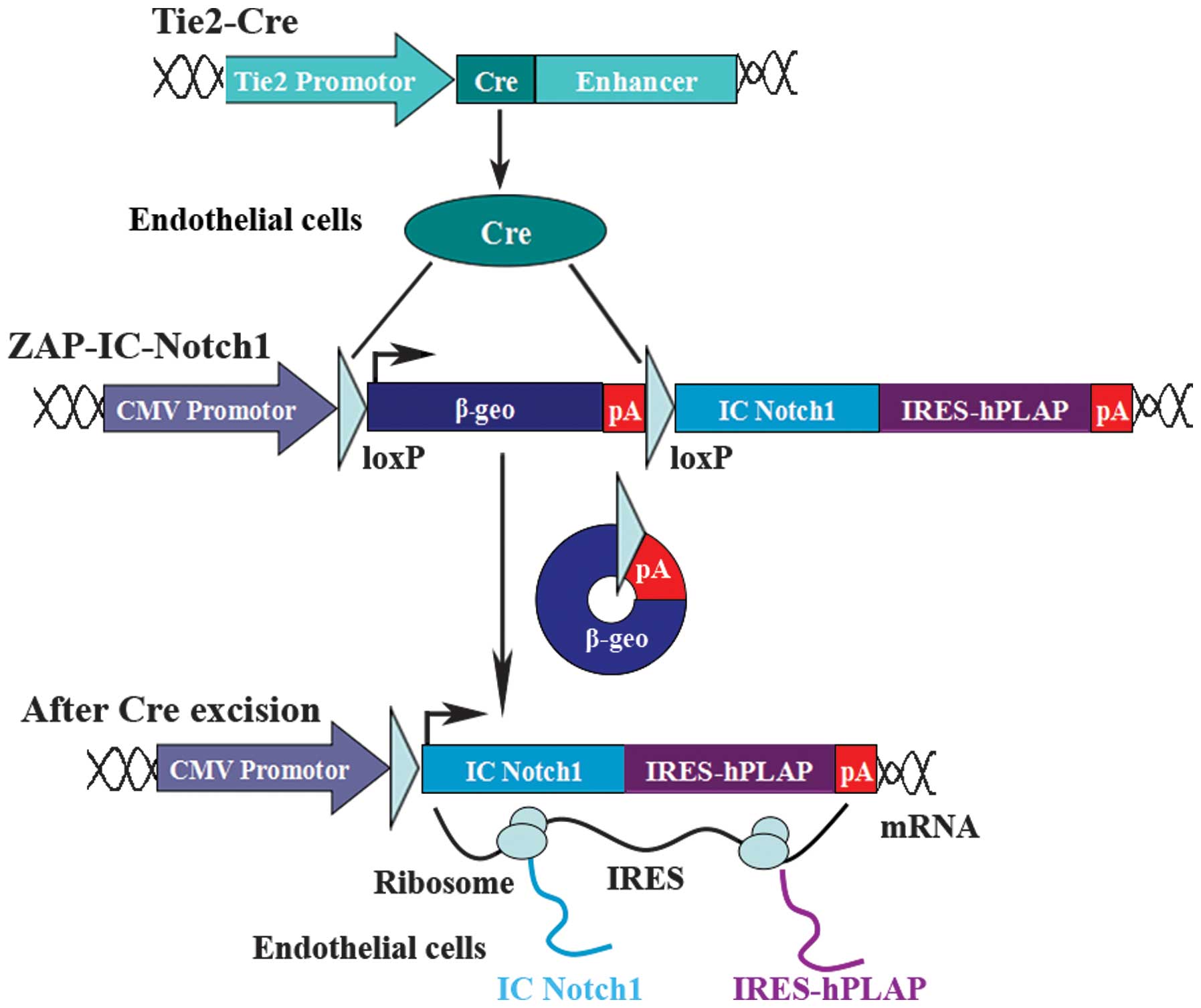
Notch signaling in specific tissues, we established ZAP-IC-Notch1
transgenic mice to take advantage of the Cre recombinase-expressing
system to tailor IC-Notch1 expression to particular cell types
(30). The ZAP-IC-Notch1
construct utilizes a CMV enhancer-chicken β-actin promoter followed
by a loxP-flanked β-geo fusion gene and three polyadenylation (pA)
sequences. Downstream of the pA sequence is the coding sequence for
the IC-Notch1 protein with an internal ribosomal entry site
(IRES)-linked human placental alkaline phosphatase (hPLAP).
Therefore, IC-Notch1 is silent in the transgenic mice but can be
activated by the introduction of Cre recombinase and excision of
the stop signal. IC-Notch1 expression can be monitored by the
co-expression with hPLAP following Cre excision.
In this study, ZAP-IC-Notch1 mice were crossed with
Tie2-Cre mice, which drive Cre recombination in the entire vascular
endothelium (31). The expression
of IC-Notch1 was activated in endothelial cells during early
development. Constitutively active Notch1 signaling induced
disruption of the vasculature, enlargement of myocardium and
embryonic lethality at E9.5–10.5. VE-cadherin expression was
decreased while EphrinB2 expression was increased. Mesenchymal cell
marker α-SMA was expressed in IC-Notch1-expressing cells. In
addition, Snail, the key effector in mediating EndoMT, was
upregulated in the ZAP-IC-Notch1/Tie2-Cre double transgenic mouse
embryo heart. Results of this study therefore support the role of
Notch signaling in the promotion of EndoMT.
Materials and methods
Mice
ZAP-IC-Notch1 transgenic mice were previously
generated in our laboratory (30). Tie2-Cre transgenic mice were
generously provided by Dr Yanagisawa (University of Texas
Southwestern Medical Center, Dallas, TX, USA) (31). The ZAP IC-Notch1 transgene was
genotyped by staining ear clips for lacZ expression and by pCCALL
PCR using genomic DNA isolated from mouse ear biopsies (32). The Tie2-Cre mice were genotyped by
Cre PCR as previously described (31).
Alkaline phosphatase (AP) staining
The embryos were rinsed in PBS prior to fixing in
lacZ fix solution (0.2% glutaraldehyde, 50 mM EGTA, pH 7.3,
100 mM MgCl2 in 100 mM sodium phosphate, 0.02% NP-40 and
0.01% sodium deoxycholate, pH 7.4) for 5 min. The endogenous APs
were inactivated by incubation in PBS at 70–75°C for 30 min.
Following washing in AP buffer (100 mM Tris-HCl, pH 9.5, 100 mM
NaCl, 10 mM MgCl2) for 10 min, the samples were stained
with AP staining solution (100 mM Tris-HCl, pH 9.5, 100 mM NaCl, 50
mM MgCl2, 0.01% sodium deoxycholate, 0.02% NP-40, 337
mg/ml nitro blue tetrazolium salt (NBT), and 175 mg/ml
5-bromo-4-chloro-3-indolyl phosphate, toluidinium salt (BCIP) (both
from Roche Diagnostics, Basel, Switzerland). The staining reaction
was allowed to proceed for 10–30 min at room temperature. The
samples were then washed extensively in PBS and stored at 4°C. The
AP staining on frozen sections was performed based on the same
procedure as above except that the sections were counterstained
with Nuclear Fast Red (Sigma-Aldrich, St. Louis, MO, USA).
Immunohistochemistry
Tissue preparation and immunohistochemistry were
performed as previously described (33). The primary antibodies used were:
anti-platelet endothelial cell adhesion molecule-1 (PECAM-1)
monoclonal antibody (1:100; BD Pharmingen, San Diego, CA, USA),
anti-SMA antibody (1:200; Sigma-Aldrich), anti-EphrinB2 polyclonal
antibody (1:1,000), and anti-Snail polyclonal antibody
(1:2,000) (both from Santa Cruz Biotechnology, Inc., Santa Cruz,
CA, USA). Secondary antibodies were biotinylated rabbit anti-rat
(1:500) and goat anti-rabbit (1:200) antibodies from Vector
Laboratories, Inc., Burlingame, CA, USA. The peroxidase activities
were visualized using streptavidin-horseradish peroxidase (HP) and
the diaminobenzidine (DAB) detection system (Vector Laboratories,
Inc.). The slides were then counterstained with hematoxylin
(Surgipath; Leica Microsystems, Wetzlar, Germany).
Western blot analysis
The mouse embryo hearts were lysed in ice-cold RIPA
buffer (20 mM Tris pH 7.5, 150 mM NaCl, 50 mM NaF, 1% NP-40, 0.1%
DOC, 0.1% SDS, 1 mM EDTA and supplemented with 1 mM PMSF and 1
μg/ml leupeptin). The protein concentration was determined using
the BCA assay (Bio-Rad, Hercules, CA, USA). Equal amounts of
protein were separated by a 10% SDS-PAGE and transferred onto a
PVDF membrane. The membranes were blocked with 2.5% BSA, and
incubated with the primary antibodies at 4°C overnight in PBS-T.
Primary antibodies used were: rabbit anti-VE-cadherin antibody
(Abcam, Cambridge, MA, USA), rabbit anti-EphrinB2 antibody, rabbit
anti-Snail antibody (both from Santa Cruz Biotechnology, Inc.), and
mouse anti-β-actin antibody (Sigma-Aldrich). Immunoreactivity was
visualized with HRP-linked secondary antibodies and
chemiluminescence.
Semi-quantitative PCR analysis
Total RNA isolation from mouse embryo hearts was
performed using TRIzol reagent (Invitrogen, Carlsbad, CA, USA)
according to the manufacturer’s instructions. An aliquot of 2 μg
total RNA from each sample was used for the synthesis of cDNA using
a High-Capacity cDNA Reverse Transcription kits (Applied Biosystems
Inc., Foster City, CA, USA). The cDNA was amplified in a final
volume of 20 μl with 1 unit of Taq DNA polymerase (Invitrogen) and
10 pmol of each primer. Oligonucleotide primer sequences are shown
in Table I. The PCR products were
visualized by ethidium bromide staining following a 1.2% agarose
gel electrophoresis.
 | Table IPCR primer sequences. |
Table I
PCR primer sequences.
| Gene | Sequences | Size (bp) | Tm (°C) |
|---|
| mVE-cadherin | Forward:
TCCTCTGCATCCTCACTATCACA | 122 | 60.63 |
| Reverse:
GTAAGTGACCAACTGCTCGTGA | | 60.55 |
| mSnail |
| Forward:
GCCGGAAGCCCAACTATAGCGA | 469 | 64.68 |
| Reverse:
TTCAGAGCGCCCAGGCTGAGGTACT | | 69.30 |
| mEphrin-B2 |
| Forward:
CAAGTTCTGCTGGATCAGCCA | 124 | 60.61 |
| Reverse:
TCGGTGCTAGAACCTGGATTT | | 59.09 |
| β-actin |
| Forward:
GGCACCACACCTTCTACAATG | 352 | 59.19 |
| Reverse:
GTGGTGGTGAAGCTGTAGCC | | 60.96 |
Results
Embryos with endothelial cell-specific
expression of IC-Notch1 exhibit disorganized vasculature
ZAP-IC-Notch mice were crossed with Tie2-Cre mice to
activate IC-Notch1 expression in endothelial cells (Fig. 1). The embryos were taken at
various stages, dissected and photographed. The genotypes of the
embryos were determined by PCR of yolk sac samples. Double
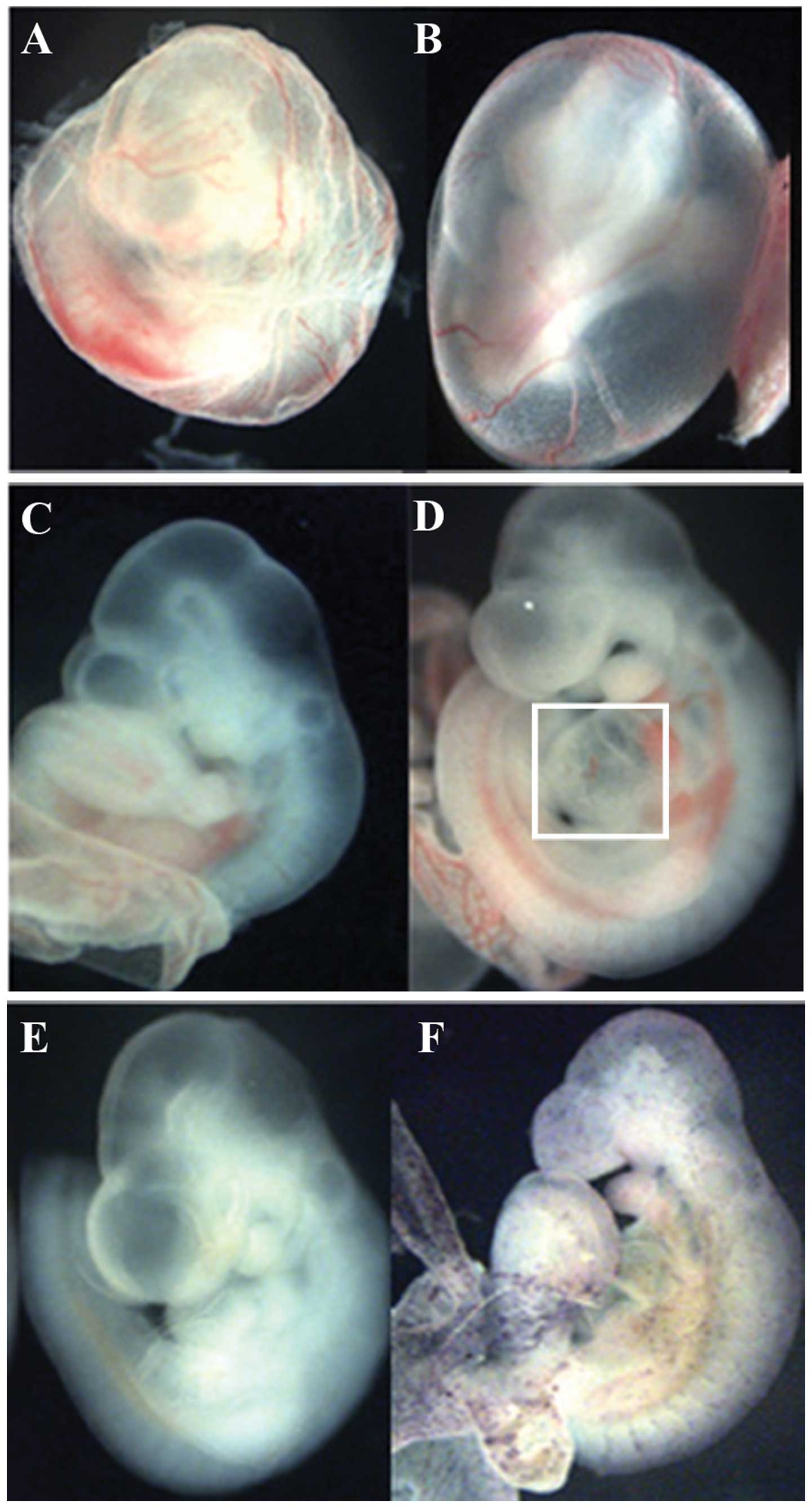
transgenic ZAP-IC-Notch1/Tie2-Cre embryos died at E9.5–10.5 showing
pale yolk sacs with fewer blood vessels than the littermates
(Fig. 2A and B). These embryos
also exhibited an enlarged heart and hemorrhaging around the
vessels (Fig. 2D). After
whole-mount AP staining, ZAP-IC-Notch1/Tie2-Cre embryos exhibited
purple/blue color on the blood vessels in the embryos and yolk sacs
(Fig. 2F), indicating that Cre
excision of the STOP signal successfully activated constitutive
Notch1 signaling together with hPLAP expression.
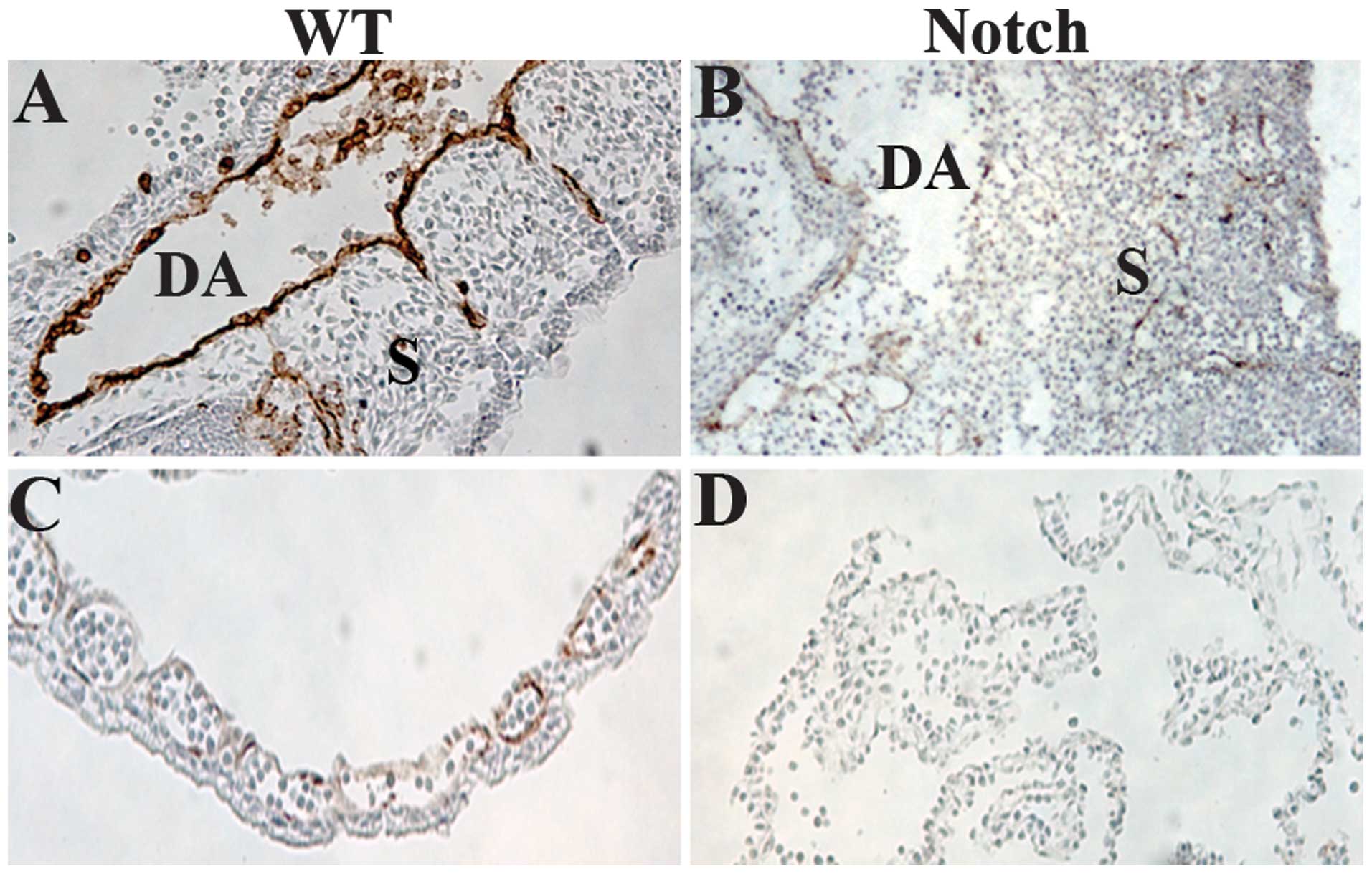
The embryos were sectioned and stained with a
monoclonal antibody to PECAM-1, a marker for VE cells (34) (Fig.
3). We observed that blood vessels were collapsed in
ZAP-IC-Notch1/Tie2-Cre double transgenic embryos, leading to
bleeding and the death of the embryos. In the trunk of wild-type
embryos at E9.5, intersomitic blood vessels were apparent along the
boundaries between adjacent somites (Fig. 3A), but in ZAP-IC-Notch1/Tie2-Cre
embryos intersomitic vessels were severely disorganized and
irregularly positioned (Figure
3B). Yolk sacs demonstrated the presence of a well-organized
capillary bed in wild-type embryos (Fig. 3C), whereas ZAP-IC-Notch1/Tie2-Cre
yolk sacs exhibited a disorganized vascular plexus lacking intact
blood vessels (Fig. 3D).
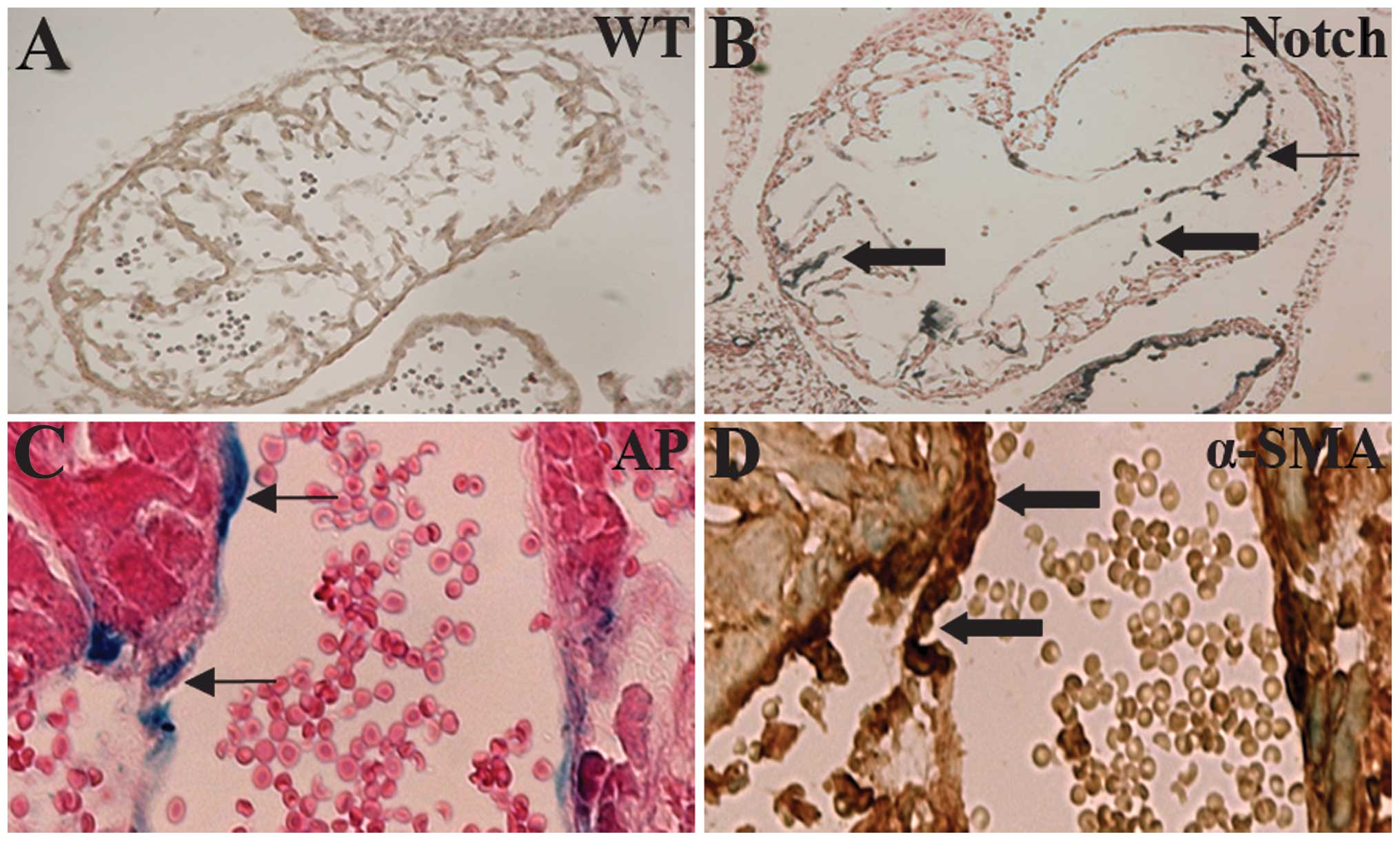
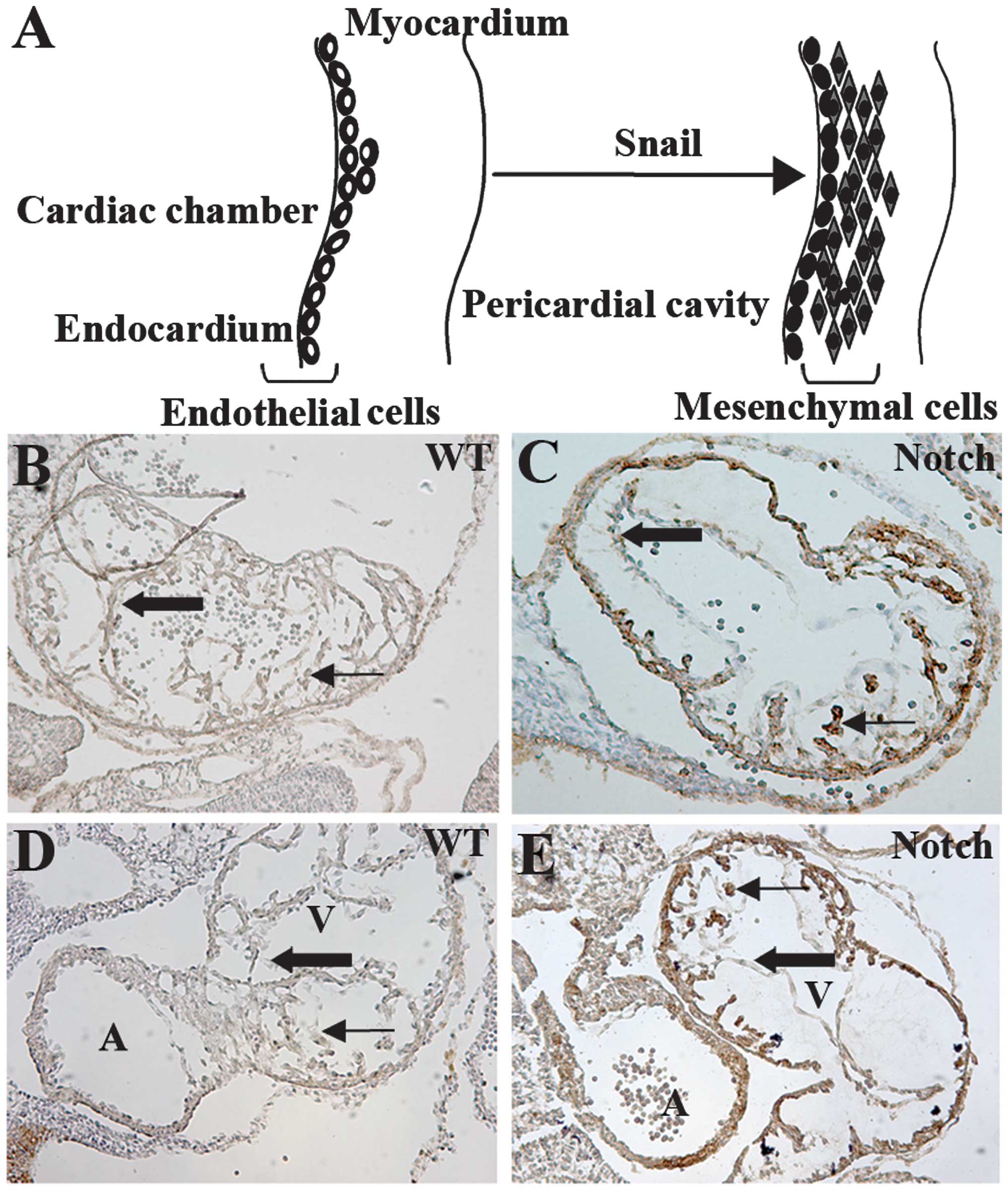
IC-Notch1 promotes cardiac cushion
formation and EndoMT in embryo heart
During cardiac cushion formation from the heart
tube, endothelial cells of the endocardium lead to interstitial
mesenchymal cells through EndoMT (35). In the ZAP-IC-Notch1/Tie2-Cre mouse
embryos, the endocardium of the embryonic heart was intact;
however, the cardiac cushion showed hypercellularity and advanced
development of heart valves (Fig. 4A
and B). On the sections, cells with positive AP staining were
observed in the endocardium and myocardium, suggesting that
IC-Notch1-expressing endothelial cells migrated into the myocardium
(Fig. 4B). α-SMA is expressed in
mesenchymal cells such as myofibroblasts, and is not normally
expressed in endothelial cells. However, endothelial cells of the
ZAP-IC-Notch1/Tie2-Cre mouse embryos, which express IC-Notch1 as
shown by AP staining, were stained positive for antibody against
α-SMA (Fig. 4C and D). Therefore,
these endothelial cells underwent transdifferentiation and gained
the characteristics of mesenchymal cells
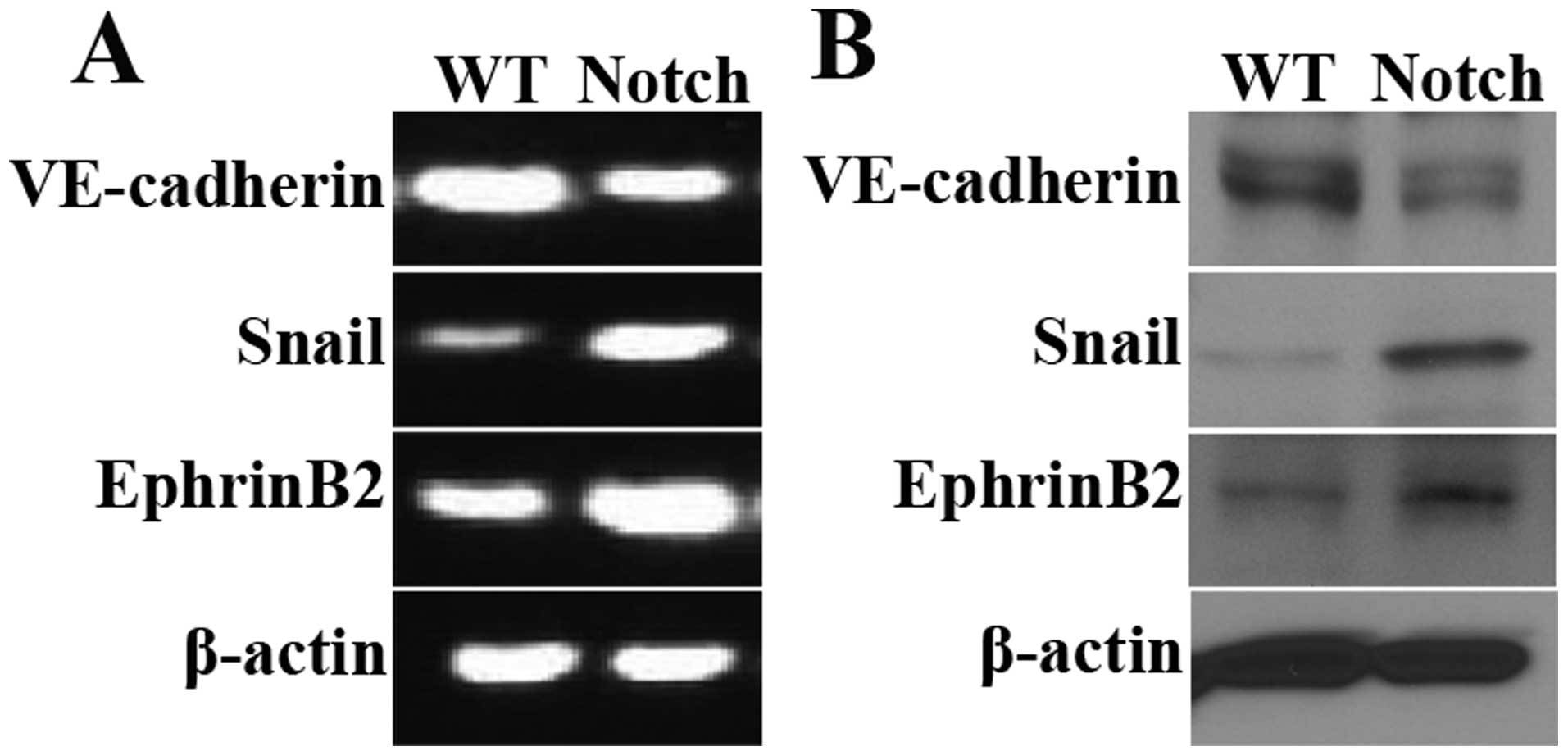
IC-Notch1 promotes Snail expression in
embryo heart
Snail, a Zinc-finger-containing transcriptional
repressor, has been identified as a key promoter of EMT (36,37) (Fig.
6A). Results of semi-quantitative PCR and western blot
analysis, revealed that the mRNA and protein levels of Snail
were elevated in the ZAP-IC-Notch1/Tie2-Cre mouse embryo hearts
(Fig. 5A and B). The expression
of endothelial cell markers was also examined. VE cadherin is an
endothelial cell-specific junction molecule and its expression is
reduced during EMT. EphrinB2, together with its receptor EphB4, are
known to play a crucial role in arteriovenous differentiation
(38,39). In the ZAP-IC-Notch1/Tie2-Cre mouse
embryo hearts, VE-cadherin expression was decreased while EphrinB2
expression was increased, suggesting that IC-Notch1 differentially
regulated endothelial cell markers during EndoMT. Moreover, a
strong increase in the Snail protein expression was observed in
mesenchymal cells in the cardiac cushion by immunohistochemistry
(Fig. 6B and C), and the
upregulation of Snail was accompanied by an increase in EphrinB2
expression (Fig. 6D and E).
Discussion
The Notch signaling pathway is essential for
cardiovascular development and is involved in the pathogenesis of
many cardiovascular diseases (40). Recently, Notch signaling has been
shown to regulate cardiac cushion formation and EndoMT. However,
its roles have not been well studied through gain-of-function mouse
models. Using the Cre/loxP system, we were able to establish
transgenic mouse lines carrying a silent transgene for
constitutively active IC-Notch1 (ZAP-IC-Notch1). Expression of the
IC-Notch1 transgene can be activated in a tissue-specific manner
and monitored by an hPLAP reporter. In this study, ZAP-IC-Notch1
mice were crossed with Tie2-Cre mice and constitutively active
Notch signaling was triggered specifically in endothelial cells.
This unique conditional expression model permitted us to examine
the role of Notch signaling in angiogenesis and EndoMT in
vivo.
During embryonic development, endothelial precursors
assemble in a primitive network through vasculogenesis, and the
network expands through angiogenesis (41). Angiogenesis is a process of
sprouting new capillaries from pre-existing vessels, and is tightly
controlled by various cell signaling cascades (42). The Notch signaling pathway plays
crucial roles in angiogenesis during embryonic development
(21). In mice, the targeted
deletion of Notch family genes including Notch receptors or their
ligands lead to vascular defects and embryonic lethality at
E9.5–10.5 (27–29,43). The gain-of-function studies of
Notch signaling are relatively limited. The IC-Notch14 expressed
under the flk-1 locus caused embryonic lethality at E9.5
with a disorganized vascular network (44). In the current study, the Notch
signaling pathway was activated specifically in endothelial cells
by crossing ZAP-IC-Notch1 mice with Tie2-Cre mice. The double
transgenic embryos died before E10.5 with disruption of vasculature
in both embryos and yolk sacs. The same outcome was observed for
another line of conditional IC-Notch mice ZEG-IC-Notch1 crossed
with Tie2-Cre mice (data not shown). The disruption in vasculature
produced by constitutively active Notch signaling was similar to
the defects in angiogenesis produced by deficient Notch signaling
in mouse embryos, suggesting a balance of Notch signaling is
required for correct vessel formation during development.
EndoMT is required for embryonic heart development
and frequently observed in adult cardiac fibrosis (45). Previous studies have demonstrated
an essential role for Notch signaling in the control of endocardial
cushion EndoMT (46). In human,
mutations of the Notch1 gene are associated with mitral
valve anomalies, bicuspid aortic valve disease and tetralogy of
fallot (47). Patients with
mutations of the Jagged1 gene develop Alagille syndrome with
cardiac cushion defects (48,49). In mice, the targeted deletion of
Notch1 or its key nuclear partner CSL results in cardiac cushion
EndoMT defects (50,51). Additionally, the targeted deletion
of the downstream Notch effector Hairy/enhancer-of-split associated
with the YRPW motif 2 (Hey2) or double-deficiency of Hey1 and Hey2
results in various congenital heart anomalies including cardiac
cushion defects (43,52). Notch inhibition in zebrafish
embryos similarly prevents cardiac valve development, whereas the
transient ectopic expression of activated Notch1 leads to
hypercellular valves (51).
Similar to zebrafish embryos, results of the present study show
that constitutively active Notch1 in endothelial cells increased
EndoMT in the mouse embryo heart. In addition, IC-Notch1-expressing
cells exhibited the expression of myoblast marker α-SMA and
downregulation of the endothelial cell marker VE cadherin. These
findings provide further evidence of the involvement of Notch
signaling in embryonic development by regulating EndoMT.
The Snail family proteins are zinc finger-containing
transcriptional repressors that trigger EndoMT during embryonic
development by regulating the expression of junctional proteins
such as cadherins (36,53). In mice, Snail is expressed
in the cardiac cushions after E9.5 (51). Mouse embryos with targeted
deletion of Notch1 or CSL lack cardiac Snail expression, and
show abortive endocardial EndoMT with abnormal maintenance of
intercellular adhesion complexes (51). In the embryo heart, Notch
functions via lateral induction to promote Snail-mediated
EndoMT which leads to the cellularization of the developing cardiac
valvular primordium (54). In
this study, we found that ZAP-IC-Notch1/Tie2-Cre mouse embryo
hearts showed a higher expression of Snail in the mRNA and
protein levels. In addition, an increase in Snail protein
expression was observed in mesenchymal cells of cardiac cushion.
These findings confirm that Notch signaling promotes Snail
expression in embryo heart through the gain-of-function mouse
model.
VE cadherin is a strictly endothelial-specific
adhesion molecule located at junctions between endothelial cells
(55). During EndoMT, VE-cadherin
expression is reduced in endothelial cells undergoing
transdifferentiation (56). The
expression of cadherin 5 gene, which encodes VE-cadherin
protein, can be suppressed by Snail transcription factor and Notch
signaling components (57). Our
results indicate that the mRNA and protein levels of VE cadherin is
decreased in ZAP-IC-Notch1/Tie2-Cre mouse embryo heart, which
confirms that constitutively active Notch signaling downregulates
VE-cadherin expression. EphrinB2 and its receptor EphB4 are
involved in determining the boundaries between arteries and veins
(39). Although its expression is
known to be arterial endothelial cell-specific, EphrinB2 is also
expressed in perivascular mesenchymal cells (58). Dll4/Notch1 is upstream of
EphB4/ephrinB2 signaling during cardiovascular development
(59,60). In this study, EphrinB2 expression
was upregulated in ZAP-IC-Notch1/Tie2-Cre mouse embryo heart and
immunostaining showed an increased EphrinB2 expression in cardiac
mesenchymal cells, suggesting that Notch signaling differentially
regulates endothelial cell markers during EndoMT.
In summary, we employed a Cre/loxP conditional mouse
model and specifically activated IC-Notch1 expression in
endothelial cells. The results demonstrate that constitutively
active Notch signaling inhibits angiogenesis and promotes EndoMT in
mouse embryos through the gain-of-function mouse model. In
addition, IC-Notch1 promotes Snail expression and differentially
regulates endothelial/mesenchymal cell markers. The present study
provides insights into the role of Notch signaling in EndoMT and
cardiovascular development.
Acknowledgements
This study was supported by a grant from the Heart
and Stroke Foundation of Canada. We are grateful for the support
from Shandong Taishan Scholarship (to Ju Liu).
References
|
1
|
Kovacic JC, Mercader N, Torres M, Boehm M
and Fuster V: Epithelial-to-mesenchymal and
endothelial-to-mesenchymal transition: from cardiovascular
development to disease. Circulation. 125:1795–1808. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zeisberg EM, Tarnavski O, Zeisberg M, et
al: Endothelial-to- mesenchymal transition contributes to cardiac
fibrosis. Nat Med. 13:952–961. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Garcia J, Sandi MJ, Cordelier P, et al:
Tie1 deficiency induces endothelial-mesenchymal transition. EMBO
Rep. 13:431–439. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Arciniegas E, Neves CY, Carrillo LM,
Zambrano EA and Ramírez R: Endothelial-mesenchymal transition
occurs during embryonic pulmonary artery development. Endothelium.
12:193–200. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Markwald RR, Fitzharris TP and Smith WN:
Structural analysis of endocardial cytodifferentiation. Dev Biol.
42:160–180. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Markwald RR, Fitzharris TP and Manasek FJ:
Structural development of endocardial cushions. Am J Anat.
148:85–119. 1977. View Article : Google Scholar
|
|
7
|
Arciniegas E, Frid MG, Douglas IS and
Stenmark KR: Perspectives on endothelial-to-mesenchymal transition:
potential contribution to vascular remodeling in chronic pulmonary
hypertension. Am J Physiol. 293:L1–L8. 2007.PubMed/NCBI
|
|
8
|
Li J and Bertram JF: Review:
Endothelial-myofibroblast transition, a new player in diabetic
renal fibrosis. Nephrology (Carlton). 15:507–512. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu H, Zaidi M, Struve J, et al: Abnormal
fibrillin-1 expression and chronic oxidative stress mediate
endothelial mesenchymal transition in a murine model of systemic
sclerosis. Am J Physiol Cell Physiol. 300:C550–C556. 2011.
View Article : Google Scholar
|
|
10
|
Kizu A, Medici D and Kalluri R:
Endothelial-mesenchymal transition as a novel mechanism for
generating myofibroblasts during diabetic nephropathy. Am J Pathol.
175:1371–1373. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yoshimatsu Y and Watabe T: Roles of TGF-β
signals in endothelial-mesenchymal transition during cardiac
fibrosis. Int J Inflam. 2011:7240802011.
|
|
12
|
Armstrong EJ and Bischoff J: Heart valve
development: endothelial cell signaling and differentiation. Circ
Res. 95:459–470. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
High FA, Jain R, Stoller JZ, et al: Murine
Jagged1/Notch signaling in the second heart field orchestrates Fgf8
expression and tissue-tissue interactions during outflow tract
development. J Clin Invest. 119:1986–1996. 2009.
|
|
14
|
Garside VC, Chang AC, Karsan A and
Hoodless PA: Co-ordinating Notch, BMP, and TGF-β signaling during
heart valve development. Cell Mol Life Sci. 70:2899–2917.
2013.PubMed/NCBI
|
|
15
|
Fu Y, Chang A, Chang L, et al:
Differential regulation of transforming growth factor beta
signaling pathways by Notch in human endothelial cells. J Biol
Chem. 284:19452–19462. 2009. View Article : Google Scholar
|
|
16
|
Lewis J: Notch signalling. A short cut to
the nucleus. Nature. 393:304–305. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gridley T: Notch signaling in vascular
development and physiology. Development. 134:2709–2718. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gridley T: Notch signaling during vascular
development. Proc Natl Acad Sci USA. 98:5377–5378. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fortini ME and Artavanis-Tsakonas S: The
suppressor of hairless protein participates in notch receptor
signaling. Cell. 79:273–282. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tun T, Hamaguchi Y, Matsunami N, Furukawa
T, Honjo T and Kawaichi M: Recognition sequence of a highly
conserved DNA binding protein RBP-J kappa. Nucleic Acids Res.
22:965–971. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Krebs LT, Xue Y, Norton CR, et al: Notch
signaling is essential for vascular morphogenesis in mice. Genes
Dev. 14:1343–1352. 2000.PubMed/NCBI
|
|
22
|
Noseda M, McLean G, Niessen K, et al:
Notch activation results in phenotypic and functional changes
consistent with endothelial-to-mesenchymal transformation. Circ
Res. 94:910–917. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Grieskamp T, Rudat C, Lüdtke TH, Norden J
and Kispert A: Notch signaling regulates smooth muscle
differentiation of epicardium-derived cells. Circ Res. 108:813–823.
2011. View Article : Google Scholar
|
|
24
|
Chang AC, Fu Y, Garside VC, et al: Notch
initiates the endothelial-to-mesenchymal transition in the
atrioventricular canal through autocrine activation of soluble
guanylyl cyclase. Dev Cell. 21:288–300. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Noseda M, Fu Y, Niessen K, et al: Smooth
muscle alpha-actin is a direct target of Notch/CSL. Circ Res.
98:1468–1470. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Penton AL, Leonard LD and Spinner NB:
Notch signaling in human development and disease. Semin Cell Dev
Biol. 23:450–457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Duarte A, Hirashima M, Benedito R, et al:
Dosage-sensitive requirement for mouse Dll4 in artery development.
Genes Dev. 18:2474–2478. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Krebs LT, Shutter JR, Tanigaki K, Honjo T,
Stark KL and Gridley T: Haploinsufficient lethality and formation
of arteriovenous malformations in Notch pathway mutants. Genes Dev.
18:2469–2473. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xue Y, Gao X, Lindsell CE, et al:
Embryonic lethality and vascular defects in mice lacking the Notch
ligand Jagged1. Hum Mol Genet. 8:723–730. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu J and Lobe CG: Cre-conditional
expression of constitutively active Notch1 in transgenic mice.
Genesis. 45:259–265. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kisanuki YY, Hammer RE, Miyazaki J,
Williams SC, Richardson JA and Yanagisawa M: Tie2-Cre transgenic
mice: a new model for endothelial cell-lineage analysis in vivo.
Dev Biol. 230:230–242. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lobe CG, Koop KE, Kreppner W, Lomeli H,
Gertsenstein M and Nagy A: Z/AP, a double reporter for cre-mediated
recombination. Dev Biol. 208:281–292. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu J, Kanki Y, Okada Y, et al: A +220
GATA motif mediates basal but not endotoxin-repressible expression
of the von Willebrand factor promoter in Hprt-targeted transgenic
mice. J Thromb Haemost. 7:1384–1392. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Baldwin HS, Shen HM, Yan HC, et al:
Platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31):
alternatively spliced, functionally distinct isoforms expressed
during mammalian cardiovascular development. Development.
120:2539–2553. 1994.
|
|
35
|
Eisenberg LM and Markwald RR: Molecular
regulation of atrioventricular valvuloseptal morphogenesis. Circ
Res. 77:1–6. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nieto MA, Sargent MG, Wilkinson DG and
Cooke J: Control of cell behavior during vertebrate development by
Slug, a zinc finger gene. Science. 264:835–839. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bolós V, Peinado H, Pérez-Moreno MA, Fraga
MF, Esteller M and Cano A: The transcription factor Slug represses
E-cadherin expression and induces epithelial to mesenchymal
transitions: a comparison with Snail and E47 repressors. J Cell
Sci. 116:499–511. 2003.PubMed/NCBI
|
|
38
|
Adams RH, Wilkinson GA, Weiss C, et al:
Roles of ephrinB ligands and EphB receptors in cardiovascular
development: demarcation of arterial/venous domains, vascular
morphogenesis, and sprouting angiogenesis. Genes Dev. 13:295–306.
1999. View Article : Google Scholar
|
|
39
|
Gerety SS and Anderson DJ: Cardiovascular
ephrinB2 function is essential for embryonic angiogenesis.
Development. 129:1397–1410. 2002.PubMed/NCBI
|
|
40
|
Niessen K and Karsan A: Notch signaling in
the developing cardiovascular system. Am J Physiol. 293:C1–C11.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Carmeliet P: Mechanisms of angiogenesis
and arteriogenesis. Nat Med. 6:389–395. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Risau W: Mechanisms of angiogenesis.
Nature. 386:671–674. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fischer A, Schumacher N, Maier M, Sendtner
M and Gessler M: The Notch target genes Hey1 and Hey2 are required
for embryonic vascular development. Genes Dev. 18:901–911. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Uyttendaele H, Ho J, Rossant J and
Kitajewski J: Vascular patterning defects associated with
expression of activated Notch4 in embryonic endothelium. Proc Natl
Acad Sci USA. 98:5643–5648. 2001. View Article : Google Scholar
|
|
45
|
Kang Y and Massague J:
Epithelial-mesenchymal transitions: twist in development and
metastasis. Cell. 118:277–279. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Iso T, Hamamori Y and Kedes L: Notch
signaling in vascular development. Arterioscler Thromb Vasc Biol.
23:543–553. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Garg V, Muth AN, Ransom JF, et al:
Mutations in NOTCH1 cause aortic valve disease. Nature.
437:270–274. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Eldadah ZA, Hamosh A, Biery NJ, et al:
Familial tetralogy of fallot caused by mutation in the jagged1
gene. Hum Mol Genet. 10:163–169. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Li L, Krantz ID, Deng Y, et al: Alagille
syndrome is caused by mutations in human Jagged1, which encodes a
ligand for Notch1. Nat Genet. 16:243–251. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Oka C, Nakano T, Wakeham A, et al:
Disruption of the mouse RBP-J kappa gene results in early embryonic
death. Development. 121:3291–3301. 1995.PubMed/NCBI
|
|
51
|
Timmerman LA, Grego-Bessa J, Raya A, et
al: Notch promotes epithelial-mesenchymal transition during cardiac
development and oncogenic transformation. Genes Dev. 18:99–115.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Donovan J, Kordylewska A, Jan YN and Utset
MF: Tetralogy of fallot and other congenital heart defects in Hey2
mutant mice. Curr Biol. 12:1605–1610. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Nieto MA: The snail superfamily of
zinc-finger transcription factors. Nat Rev Mol Cell Biol.
3:155–166. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
54
|
Murray SA and Gridley T: Snail family
genes are required for left-right asymmetry determination, but not
neural crest formation, in mice. Proc Natl Acad Sci USA.
103:10300–10304. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Vestweber D: VE-cadherin: the major
endothelial adhesion molecule controlling cellular junctions and
blood vessel formation. Arterioscler Thromb Vasc Biol. 28:223–232.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar
|
|
57
|
Cheng JC, Chang HM and Leung PC:
Transforming growth factor-β1 inhibits trophoblast cell invasion by
inducing Snail-mediated down-regulation of vascular
endothelial-cadherin protein. J Biol Chem. 288:33181–33192.
2013.
|
|
58
|
Pérez-Pomares JM and de la Pompa JL:
Signaling during epicardium and coronary vessel development. Circ
Res. 109:1429–1442. 2011.PubMed/NCBI
|
|
59
|
del Monte G, Casanova JC, Guadix JA, et
al: Differential Notch signaling in the epicardium is required for
cardiac inflow development and coronary vessel morphogenesis. Circ
Res. 108:824–836. 2011.PubMed/NCBI
|
|
60
|
Iso T, Maeno T, Oike Y, et al:
Dll4-selective Notch signaling induces ephrinB2 gene expression in
endothelial cells. Biochem Biophys Res Commun. 341:708–714. 2006.
View Article : Google Scholar : PubMed/NCBI
|