Introduction
Left ventricular remodeling (LVR), including cardiac
hypertrophy, myocardial fibrosis and changes in left ventricular
shape, is an adaptation to preserve cardiac output in a number of
pathological conditions, such as hypertension and myocardial
infarction (MI). However, deteriorated LVR may eventually lead to
congestive heart failure (CHF) (1,2).
There are several factors involved in the progression of LVR to
CHF; however, endothelial function has received increasing
attention. The vascular endothelium is widely distributed, and it
plays a very important role in the pathophysiological process of
cardiovascular diseases. The important roles of the coronary
endothelium include not only the control of blood flow, but also
the regulation of cardiomyocyte structure and function through
autocrine or paracrine signaling via many vasoactive substances
(3). Additionally, previous
studies have demonstrated that deteriorative left ventricular
dysfunction is associated with coronary endothelial dysfunction
(4–6). Moreover, in the study by Shiojima
et al (7), it was
suggested that the imbalance between endothelial function and the
adaptation of cardiac hypertrophy causes the transition from LVR to
failure. Therefore, correcting coronary endothelial dysfunction in
the residual myocardium is a very important therapeutic strategy
for the clinical treatment of patients with post-infarction
CHF.
The ATP-sensitive potassium channel
(KATP) is widely distributed in the cardiovascular
system and plays important roles. KATP consists of
inward rectifier potassium channel subunits (Kir6s) and regulative
sulfonylurea receptors (SURs) (8); the SUR2B/Kir6.1 subtype is mainly
found in the vascular endothelium (9). It has previously been reported that
the protein expression of Kir6.1 is increased in the ischemic
myocardium and that the dysfunction of Kir6.1/SUR2 is involved in
cardiac hypertrophy and heart failure (1). Our laboratory has previously
reported that the activation of the SUR2B/Kir6.1 subtype prevents
cardiac hypertrophy and the progression from hypertrophy to failure
induced by pressure overload (11,12). Therefore, it can by hypothesized
that correcting endothelial dysfunction by activating Kir6.1/SUR2B
channels in the endothelium may prevent post-infarction CHF.
Natakalim, a novel KATP channel opener, can reverse
vascular endothelial cell dysfunction caused by homocysteine and
hypoxia and prevent the progression of cardiac hypertrophy to
failure induced by pressure overload by selectively opening the
SUR2B/Kir6.1 channel subtype (12–14). Based on this evidence, we
hypothesized that natakalim may improve post-infarction LVR and CHF
by protecting against coronary endothelial dysfunction in the
residual myocardium.
The progression from occlusion of the left anterior
descending coronary artery (LAD) to post-infarction CHF in rats is
similar to what occurs when a patient survives a large MI but
subsequently develops heart failure (15,16). However, few experimental studies
have attempted to clarify the effects of activating the
SUR2B/Kir6.1 channel subtype on LVR and CHF using this model.
Therefore, to the bets of our knowledge, for the first time, we
evaluated the pharmacological characteristics and experimental
therapeutic effects of natakalim on LVR and CHF in a rat model of
ligation of the LAD. We also explored whether natakalim improves
post-infarction CHF by restoring the coordinated balance between
endothelial function and cardiac hypertrophy in the residual
myocardium.
Materials and methods
Reagents
Natakalim was synthesized at the Thadweik Academy of
Medicine (Beijing, China). All other chemicals and materials were
obtained from local commercial sources.
Modle of MI and study protocol
All procedures were performed in accordance with the
protocol outlined in the Guide for the Care and Use of Laboratory
Animals published by the US National Institutes of Health (NIH
publication no. 85-23, revised in 1996) and approved by the local
animal care and use committee. Experiments were performed using
healthy adult male Wistar rats (weight range, 200±30 g), and the
animals were divided into 6 groups (n=9–11) as follows: the
sham-operated group, which included animals that underwent a
similar procedure without left coronary artery ligation and were
treated with distilled water; the model group, which included rats
with MI induced by LAD ligation that were treated with distilled
water; rats with MI that were treated with 1, 3 or 9 mg/kg/day of
natakalim; and rats with MI treated with a positive control drug
(lisinopril) at 15 mg/kg/day. No significant difference was found
among all the experimental groups as regards age or body weight
(BW) prior to surgery.
The rat model of MI was implemented as previously
described (15), with slight
modifications. First, following anesthesia with pentobarbital (40
mg/kg, intraperitoneal injection), the trachea of the rat was
incised and cannulated to allow artificial ventilation by a
volume-constant rodent ventilator under sterile conditions. Second,
a left thoracotomy was performed at the fourth intercostal space,
and the ribs were opened with a hemostatic clamp to expose the
heart. After opening the pericardium, the left coronary artery was
ligated using a 7.0 silk suture close to the artery origin (2 mm
below). Subsequently, the rib cage, muscle layer and skin were
closed separately. Successful occlusion was immediately confirmed
by ECG (ST-elevation) and the observation of an arising pale
ischemic zone with weak activity. The drugs were dissolved in
normal saline and administered orally via a gastric tube once a day
for 8 weeks from the third post-operative day. At the end of the 8
weeks, the rats were sacrificed for histological evaluation.
Hemodynamics and cardiac remodeling
index
On day 56, following anesthesia with a mixture of
xylazine (5 mg/kg) and pentobarbital (15 mg/kg, intraperitoneal
injection), the right carotid was cannulated with a polyethylene
catheter (outer diameter of 0.96 mm) to measure the carotid
arterial pressure. Subsequently, a catheter was inserted along the
right carotid artery into the left ventricle, and the signals were
recorded on an 8-channel direct-writing oscillograph (RM-6000;
Nihon Kohden Corporation, Tokyo, Japan) and digitally sampled (1
kHz) on a personal computer equipped with an analog-to-digital
interface (SMUP-PC bioanalysis system; Jialong Instrument,
Shanghai, China). The heart rate (HR), systolic blood pressure
(SBP), diastolic blood pressure (DBP), left ventricular systolic
pressure (LVSP), left ventricular end diastolic pressure (LVEDP),
maximal rate of left ventricular systolic and diastolic pressure
(±dp/dtmax), physiological velocity of
contractile element shorting (Vpm) and maximal velocity of
contractile element shorting (Vmax) were recorded.
Thereafter, blood samples were collected and the
animals were sacrificed by exsanguination. The still-beating heart
was exposed after thoracic cavity opening. The hearts and lungs
were rapidly removed, rinsed in ice-cold 0.9% NaCl solution,
blotted and weighed. The heart weight (HW)/BW ratio (HW/BW) was
calculated by dividing the HW by the BW; the left ventricles
(including interventricular septum) and right ventricular free
walls were then collected separately and weighed. The left
ventricular weight (LVW)/BW (LVW/BW) ratio, the right ventricular
weight (RVW)/BW ratio (RVW/BW) and the lung weight (LW)/BW ratio
(LW/BW) were calculated.
Histological analysis and transmission
electron microscopy
The hearts were immersion-fixed in neutral 10%
buffered formalin and paraffin sections (5 μm) were cut. The
myocyte cross-sectional area and myocardial fibrosis were
quantitatively analyzed using NIH Image 1.61 software (National
Institutes of Health Service Branch) in digitized microscopic
images. For measurement of the cross-sectional area, 100 cells (per
animal, hematoxylin and eosin-stained, ×400 magnification) from the
left ventricular mid-lateral free wall (including epicardial and
endocardial portions) were randomly selected and analyzed.
Interstitial myocardial fibrosis in the tissue sections was
quantitatively analyzed by morphometry (in Masson’s
trichrome-stained sections, ×100 magnification). The collagen in
the myocardial interstitial spaces was visualized and the whole
areas of the sections were scanned. The total interstitial fibrosis
index was defined as the sum of the total area of collagen in the
entire visual field divided by the sum of total connective tissue
area plus the myocardial area in the entire visual field. All the
images were digitized and transformed into binary images, and the
areas occupied by collagen were calculated by an automatic
area-quantification program using NIH Image software.
Myocardial samples were routinely fixed in 2.5%
glutaraldehyde in 0.1 M phosphate buffer (pH 7.3) and then fixed in
buffered 1% osmium tetroxide for 1 h. The samples were then
dehydrated in a series of ethanol (50% ethanol, 5 min; 75% ethanol,
5 min; 90% ethanol, 5 min; and 100% ethanol, 5 min) and embedded in
Epon 812. Thin sections were stained with uranyl acetate and lead
citrate and examined under a Philips CM-120 electron microscope
(Royal Dutch Philips Electronics Ltd, Amsterdam, The Netherlands).
Muscle fiber and mitochondrial abnormalities were evaluated.
Measurement of nitric oxide (NO),
endothelin (ET)-1, prostacyclin [prostaglandin I2
(PGI2), thromboxane A2 (TXA2) and
hydroxyproline levels
Blood was obtained from the right carotid artery
with a polyethylene catheter at the time of sacrifice. The level of
NO in serum was measured using the NO Detection kit (Nanjing
Jiancheng Bioengineering Institute, Nanjing, China) according to
the manufacturer’s instructions. ET-1, PGI2 and
TXA2 levels were measured using commercial
radioimmunoassay kits (Eastern Asia Radioimmunity Research
Institute, Beijing, China) according to the manufacturer’s
instructions. The hydroxyproline content in the hearts was measured
using a commercial kit (Nanjing Jiancheng Bioengineering Institute)
according to the manufacturer’s instructions.
Immunohistochemistry
Immunohistochemical staining was performed using the
UltraSensitive™ S-P kit (Wuhan Boster Bio-engineering Co, Ltd,
Wuhan, China) according to the manufacturer’s instructions.
Anti-endothelin receptor A and B (Sigma-Aldrich) were used as
primary antibodies at a 1:100 dilution. Images from the entire
sections were acquired using a digital camera system (Leica DFC295;
Leica, Solms, Germany). Confocal images were then transferred to a
personal computer using the NIH Image software package for image
analysis, as previously reported (11) and with minor modifications.
Western blot analysis
Protein was isolated from frozen non-infarcted left
ventricular tissue using cell lysis buffer for western blotting
(Beytime Institute of Biotechnology, Haimen, China) in accordance
with the manufacturer’s instructions. Equal amounts of protein
extracts were separated by 8% SDS-PAGE gel and then transferred
onto PVDF membranes using a Bio-Rad transfer blotting system
(Bio-Rad Laboratories, Hercules, CA, USA). The membranes were
incubated with antibodies against endothelial NO synthase (eNOS)
and inducible NO synthase (iNOS) (Abcam, Cambridge, MA, USA).
Proteins were visualized using an enhanced chemiluminescence
detection system (ECL; Cell Signaling Technology Inc). Anti-GAPDH
antibody (BioEasy) was used to control for equal protein
loading.
Quantitative reverse transcription PCR
(RT-qPCR)
Total RNA was extracted from the cardiac tissue of
rats using TRIzol reagent according to the manufacturer’s
instructions (Invitrogen, Carlsbad, CA, USA) and converted to cDNA
using the Quantscript RT kit (Tiangen Biotech Co., Ltd., Beijing,
China). Quantitative PCR was performed using the Real-Time PCR
iCycler iQ5 System (Bio-Rad Laboratories) and SYBR-Green PCR Master
Mix (Toyobo, Osaka, Japan) in accordance to the manufacturer’s
instructions. The expression levels of GAPDH were used for sample
normalization. Primers for rat atrial natriuretic peptide (ANP),
brain natriuretic peptide (BNP) and GAPDH were as follows: ANP
sense, 5′-ATT TCA AGA ACC TGC TAG ACC-3′ and antisense, 5′-CAA TCC
TGT CAA TCC TAC CC-3′, 300 bp; BNP sense, 5′-GGA CCA AGG CCC TAC
AAA AGA ACT-3′ and antisense, 5′-ACA ACC TCA GCC CGTCAC AGC-3′, 176
bp; GAPDH sense, 5′-AAA CCT CCA AGT ATG ATG AC-3′ and antisense,
5′-TTG TCA TAC CAG GAA ATG AGC-3′, 197 bp. The reactions were
incubated at 95°C for 1 min, followed by 40 cycles at 95°C for 15
sec, 66.5°C for 15 sec and 72°C for 45 sec.
Statistical analysis
In this stuyd, the results are expressed as the
means ± SD. Statistical analysis of the data was performed using
one-way ANOVA with the SPSS 13 software program. Probability values
(P-values) ≤0.05 were considered to indicate statistically
significant differences.
Results
Hemodynamic effects of natakalim
Carotid arterial pressure and cardiac function were
measured 8 weeks following LAD occlusion. As shown in Table I, compared with the sham-operated
rats, the mean arterial blood pressure (MABP) of the right carotid
aorta was significantly reduced in the rats with MI. This was
prevented by treatment with natakalim at daily oral doses of 1, 3
or 9 mg/kg for 8 weeks. The in vivo left ventricular
function measurements for all the groups are shown in Table I. Systolic cardiac parameters,
including LVSP, +dp/dtmax, Vpm, Vmax and
the diastolic cardiac parameter −dp/dtmax,
were all significantly decreased in the rats with MI. By contrast,
the diastolic cardiac parameter, LVEDP, was significantly
increased. These changes were prevented by treatment with natakalim
in a dose-dependent manner. Long-term treatment with natakalim did
not affect the HR of the rats in the MI group.
 | Table IEffects of natakalim on HR, MABP and
hemodynamic parameters in the rats with myocardial infarction
(MI). |
Table I
Effects of natakalim on HR, MABP and
hemodynamic parameters in the rats with myocardial infarction
(MI).
| Group | n | HR/min | MABP (kPa) | LVSP (kPa) |
+dp/dtmax
(kPa/s) | Vpm(/sec) | Vmax(/sec) | LVEDP (kPa) |
−dp/dtmax
(kPa/sec) |
|---|
| Sham-operated | 11 | 405±21 | 14.69±1.07 | 20.10±0.81 | 440.0±48.0 | 0.919±0.100 | 0.542±0.176 | 6.04 ±0.48 | 409.4±57.9 |
| MI | 12 | 388±34 |
13.75±1.20a |
19.03±0.70a |
382.6±51.9a |
0.753±0.120b |
0.319±0.186b |
7.23±0.81b |
346.0±61.6a |
| Nat 1
mg/kg/day | 11 | 399±29 |
14.98±1.41d |
19.82±0.72d |
438.2±49.2d |
0.890±0.116d | 0.444±0.192 |
6.41±0.54d |
407.2±51.2c |
| Nat 3
mg/kg/day | 12 | 393±33 |
15.08±0.89d |
19.88±0.33c |
431.5±28.0d |
0.899±0.084d |
0.530±0.168d |
6.26±0.54d |
403.3±34.4c |
| Nat 9
mg/kg/day | 12 | 406±30 |
15.17±0.70d |
19.78±0.78c |
455.0±34.0d |
0.977±0.076d |
0.639±0.156d |
5.82±0.34d |
433.6±44.2d |
| Lis 15
mg/kg/day | 11 | 413±31 |
12.16±1.15b,d |
18.64±0.55b |
366.7±25.9b |
0.724±0.058b |
0.214±0.108b |
7.40±0.30b |
345.4±26.8b |
Natakalim improves LVR induced by MI
The results at 8 weeks for all groups are shown in
Figs. 1 and 2. LVR in the remote zone was
characterized by augmentation of the HW/BW and LVW/BW ratios
(Fig. 1B and C), the myocyte
cross-sectional area (Fig. 2E)
and interstitial myocardial fibrosis (Fig. 2D, E and G). Histological
examination (H&E and Mason’s staining) revealed that myocardial
tissue damage, compensatory cardiac hypertrophy and myocardial
fibrosis of the heart occurred in the left ventricular myocardium
(Fig. 2A, B and D). Histological
analysis of the hearts of the rats from the MI group indicated that
the myocyte cross-sectional area and the extent of myocardial
fibrosis were significantly increased compared with the hearts of
the rats in the control group (Fig.
2E and F). The hydroxyproline content reflects the amount of
collagen in cardiac tissue, and it was increased by 36.4% in the MI
group compared with the sham-operated group (Fig. 2G). Ultrastructural examination
revealed myofibril disarray, fewer mitochondria and the destruction
of mitochondrial membranes in the rats in the MI group (Fig. 2C). Natakalim administered at all
doses for 8 weeks reversed these changes.
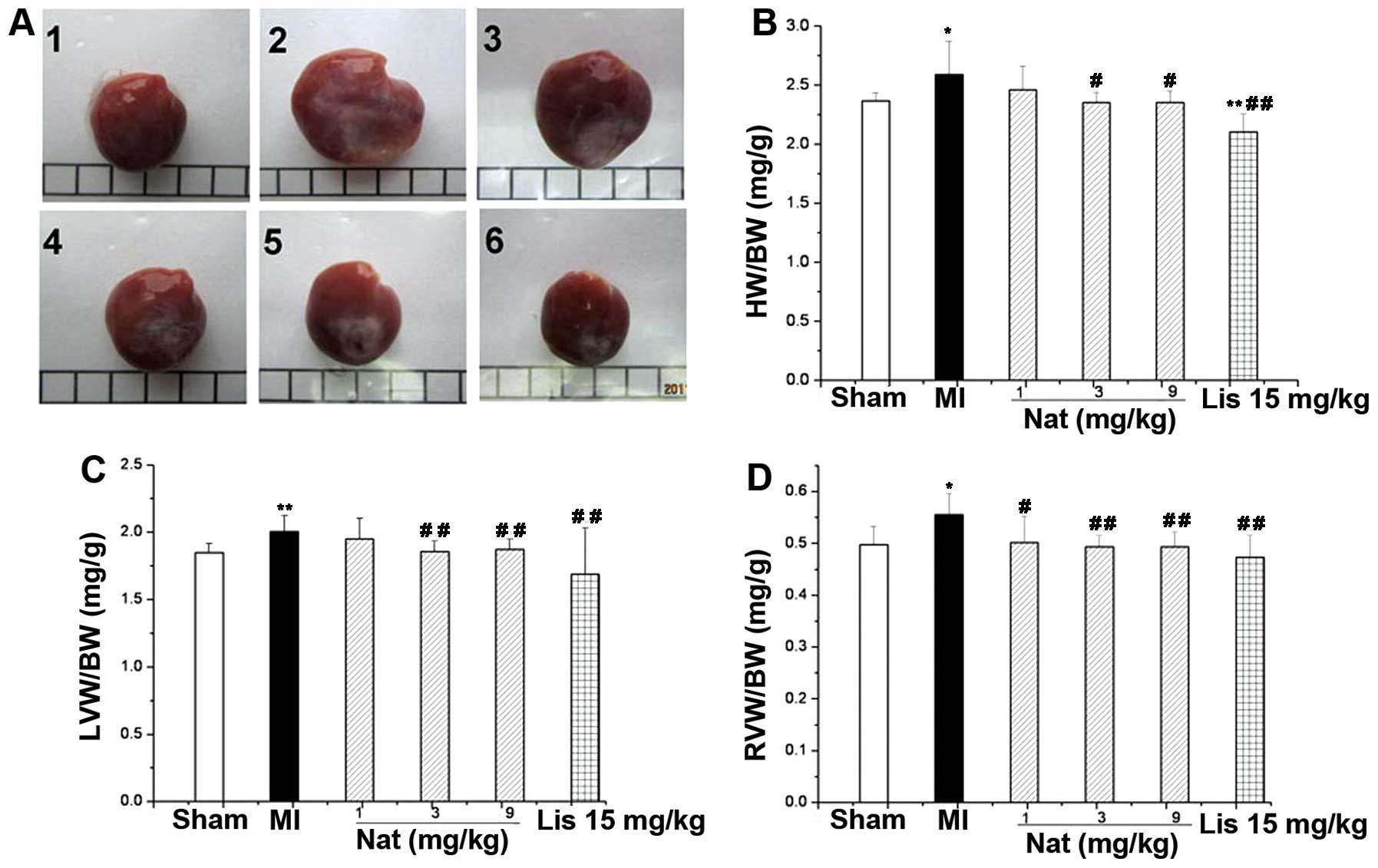 | Figure 1Effects of natakalim on (A) heart
weight (HW), (B) HW/body weight (BW) ratio, (C) left ventricular
weight (LVW)/BW ratio and (D) right ventricular weight (RVW)/BW
ratio in rats with myocardial infarction (MI). (A) Representative
macroscopic image of hearts; Panel 1, sham-operated group; panel 2,
MI group; panel 3, natakalim 1 mg/kg/day group; panel 4, natakalim
3 mg/kg/day group; panel 5, natakalim 9 mg/kg/day group; panel 6,
positive control (lisinopril 15 mg/kg/day). (B-D) Quantification
results. Left ventricular hypertrophy was characterized by an
increase in the HW/BW, LVW/BW and RVW/BW ratios. These effects were
reversed by treatment with natakalim at all doses for 8 weeks. Data
are expressed as the means ± SD, n=10–12; *P<0.05 and
**P<0.01 vs. sham-operated rats (sham);
#P<0.05 and ##P<0.01 vs. rats with MI.
Nat, natakalim; Lis, lisinopril. |
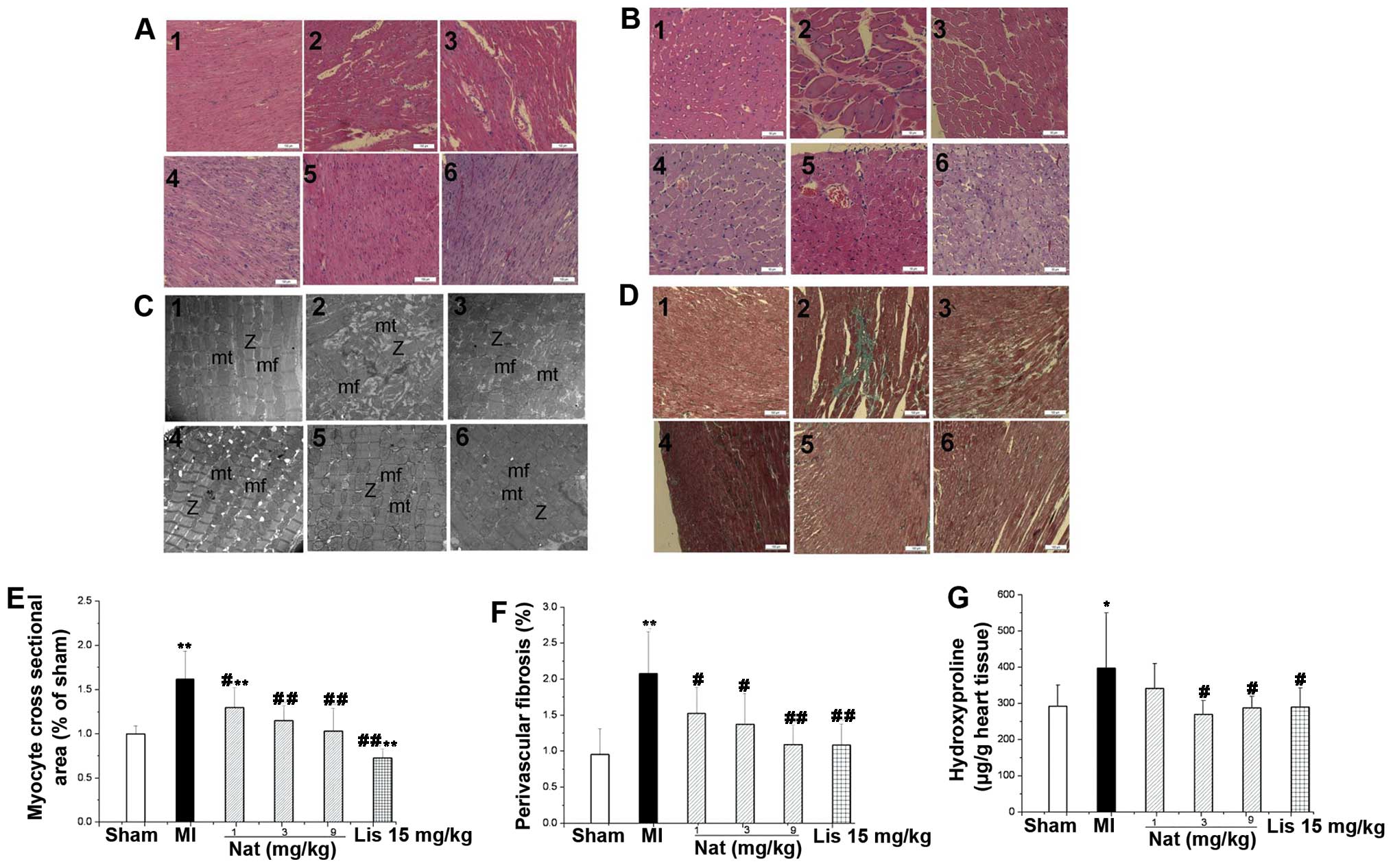 | Figure 2Effects of natakalim on myocyte
cross-sectional area, myocardial fibrosis, ultrastructural
pathological changes and hydroxyproline content of cardiac tissue
in rats with myocardial infarction (MI). (A) Representative myocyte
long axis images (H&E staining, ×100 magnification); (B)
representative myocyte cross-sectional images (H&E staining,
×200 magnification); (C) representative ultrastructural
pathological changes in cardiac tissues (transmission electron
microscope micrographs, ×15,000 magnification); (D) representative
myocardial fibrosis (Masson’s staining, ×100 magnification); panel
1, sham-operated group; panel 2, MI group; panel 3, natakalim 1
mg/kg/day group; panel 4, natakalim 3 mg/kg/day group; panel 5,
natakalim 9 mg/kg/day group; panel 6, positive control (lisinopril
15 mg/kg/day). (E) Quantification of myocyte cross-sectional area;
(F) quantification of myocardial fibrosis; (G) quantification of
hydroxyproline content in cardiac tissue. Myocyte cross-sectional
area, myocardial fibrosis and cardiac hydroxyproline content both
increased significantly when compared with the rats with heart
failure. Treatment with natakalim at all doses for 8 weeks reversed
these pathological changes in left ventricular hypertrophyic
parameters. Ultrastructural examination of hearts revealed
well-organized myofibrils with mitochondria grouped along the
periphery of longitudinally oriented fibers in natakalim group
rats. Myocyte cross-sectional area and myocardial fibrosis data are
expressed as the means ± SD, n=6; hydroxyproline content data are
expressed as the means ± SD, n=11–12; *P<0.05,
**P<0.01 vs. sham-operated rats (sham);
#P<0.05, ##P<0.01 vs. rats with MI. mf,
myofibrils; mi, mitochondria; Z, Z-line. Nat, natakalim; Lis,
lisinopril. |
Natakalim prevents the progression of
hypertrophy to cardiac failure
The transition from LVR to failure in experimental
models is characterized by pulmonary, chronic, right ventricular
hypertrophy and the overexpression of ANP and BNP mRNAs, 2
molecular markers of heart failure (11,12). As shown in Figs. 1D and 3, the LW/BW and RVW/BW ratios were
increased, and ANP and BNP mRNA expression was markedly increased
in the MI group compared with the sham-operated group. These
changes were completely reversed by treatment with natakalim at all
doses for 8 weeks.
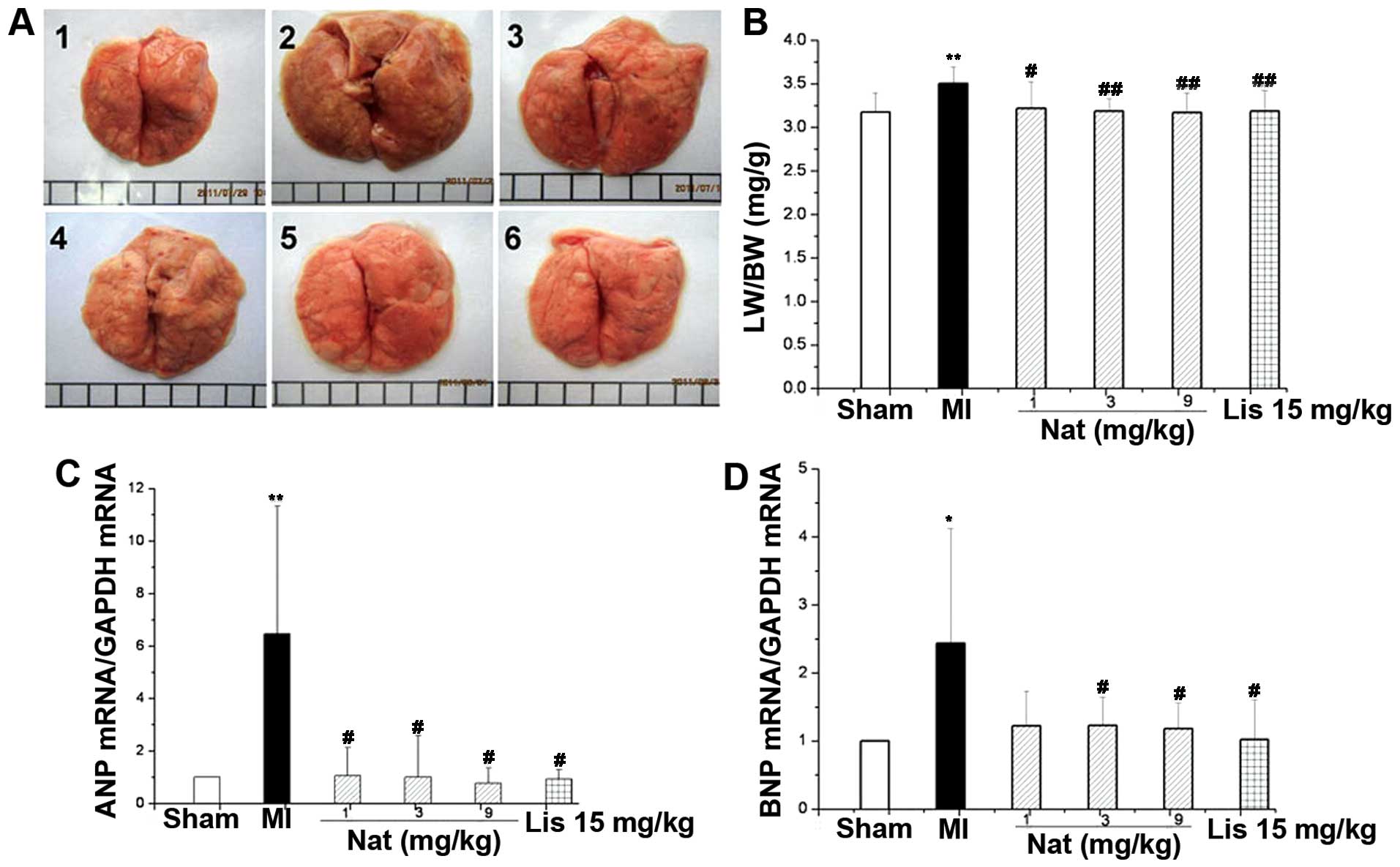 | Figure 3Effects of natakalim on lung index
[lung weight (LW)/body weight (BW)] ratio and the expression of
atrial natriuretic peptide (ANP) and brain natriuretic peptide
(BNP) mRNA in rats with MI. (A) Representative lung macroscopic
images; panel 1, sham-operated group; panel 2, MI group; panel 3,
natakalim 1 mg/kg/day group; panel 4, natakalim 3 mg/kg/day group;
panel 5, natakalim 9 mg/kg/day group; panel 6, positive control
(lisinopril 15 mg/kg/day). (B) Quantification of LW/BW ratio; (C
and D) quantification of relative expression levels of ANP and BNP
mRNA. The progression of left ventricular hypertrophy to heart
failure was characterized by an increased LW/BW ratio, the
overexpression of ANP and BNP mRNA and increased right ventricular
weight (RVW)/BW ratio. The LW/BW and RVW/BW ratios increased and
the ANP and BNP mRNA expression was markedly increased in the rats
with MI compared with the sham-operated rats. These effects were
completely reversed by treatment with natakalim at all doses for 8
weeks. Data are expressed as the means ± SD, n=10–12;
*P<0.05 and **P<0.01 vs. sham-operated
rats (sham); #P<0.05, ##P<0.01 vs. rats
with MI. Nat, natakalim; Lis, lisinopril. |
Effects of natakalim on nitric oxide and
NO synthases
eNOS protein expression was significantly reduced in
the MI group; however, the serum NO concentration and iNOS protein
expression were significantly increased (P<0.01; Fig. 4). Treatment with natakalim for 8
weeks restored the concentration of serum NO to normal levels; the
protein expression of eNOS increased to almost normal levels and
the protein expression of iNOS in cardiac tissue decreased to
almost normal levels.
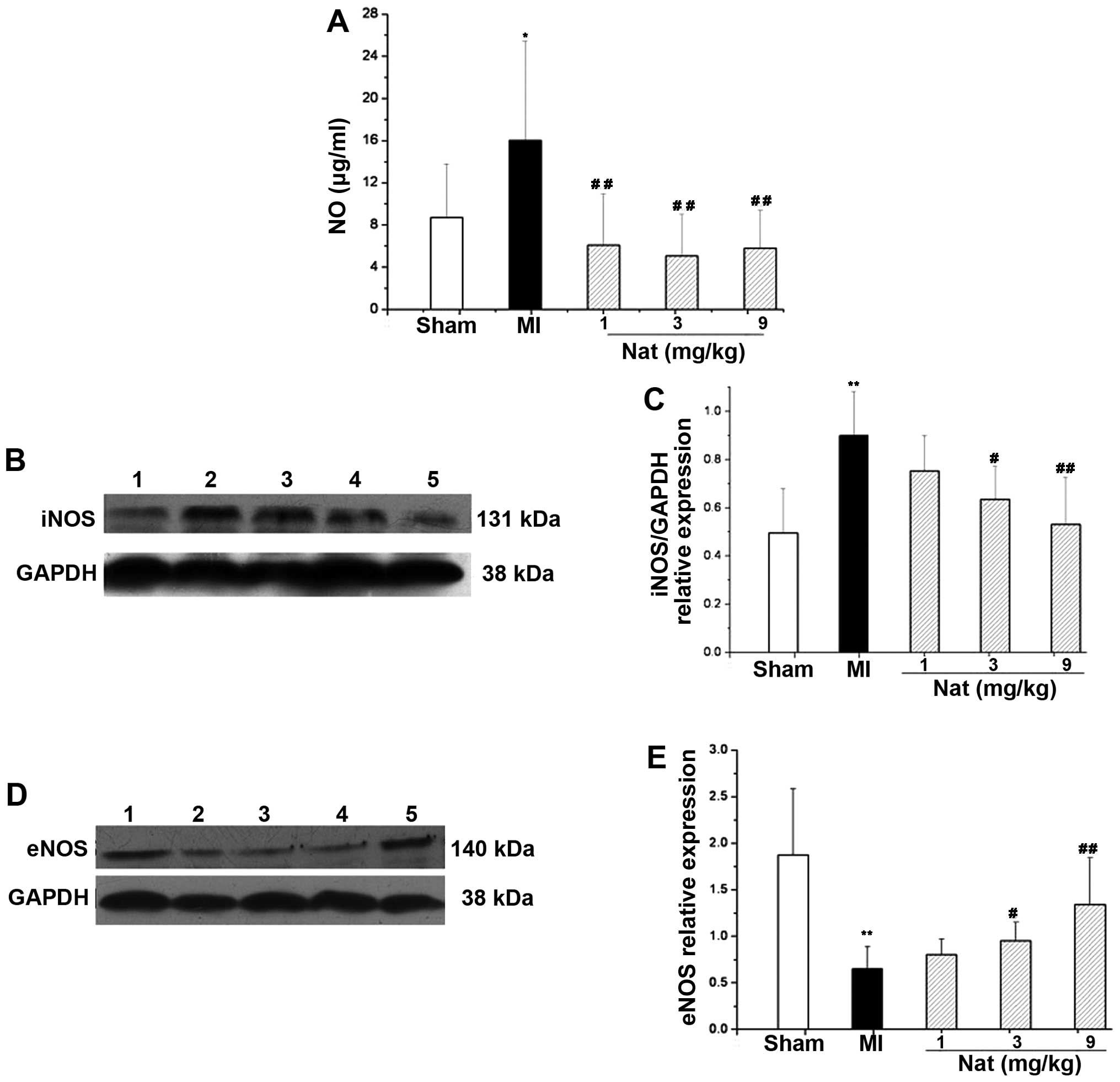 | Figure 4Effects of natakalim on serum
concentration of nictic oxide (NO), inducible nitric oxide synthase
(iNOS) and endothelial nitric oxide synthase (eNOS) protein
expression in rats with myocardial infarction (MI). (A)
Quantification of serum content of nitric oxide; (B and D)
representative blots of iNOS and eNOS protein expression (western
blot analysis); lane 1, sham-operated group; lane 2, MI group; lane
3, natakalim 1 mg/kg/day group; lane 4, natakalim 3 mg/kg/day
group; lane 5, natakalim 9 mg/kg/day group; (C and E)
quantification of relative values of iNOS and eNOS protein
expression. The relative values of eNOS and eNOS protein expression
normalized to GAPDH as an internal protein control. Serum NO and
iNOS protein expression was increased, whereas eNOS protein
expression was decreased in the rats with MI. These effects were
reversed by treatment with natakalim at all doses for 8 weeks.
Serum NO data are expressed as the means ± SD, n=10–12; eNOS and
iNOS data are expressed as the means ± SD, n=6;
**P<0.01 vs. sham-operated rats (sham);
#P<0.05 and ##P<0.01 vs. rats with MI.
Nat, natakalim. |
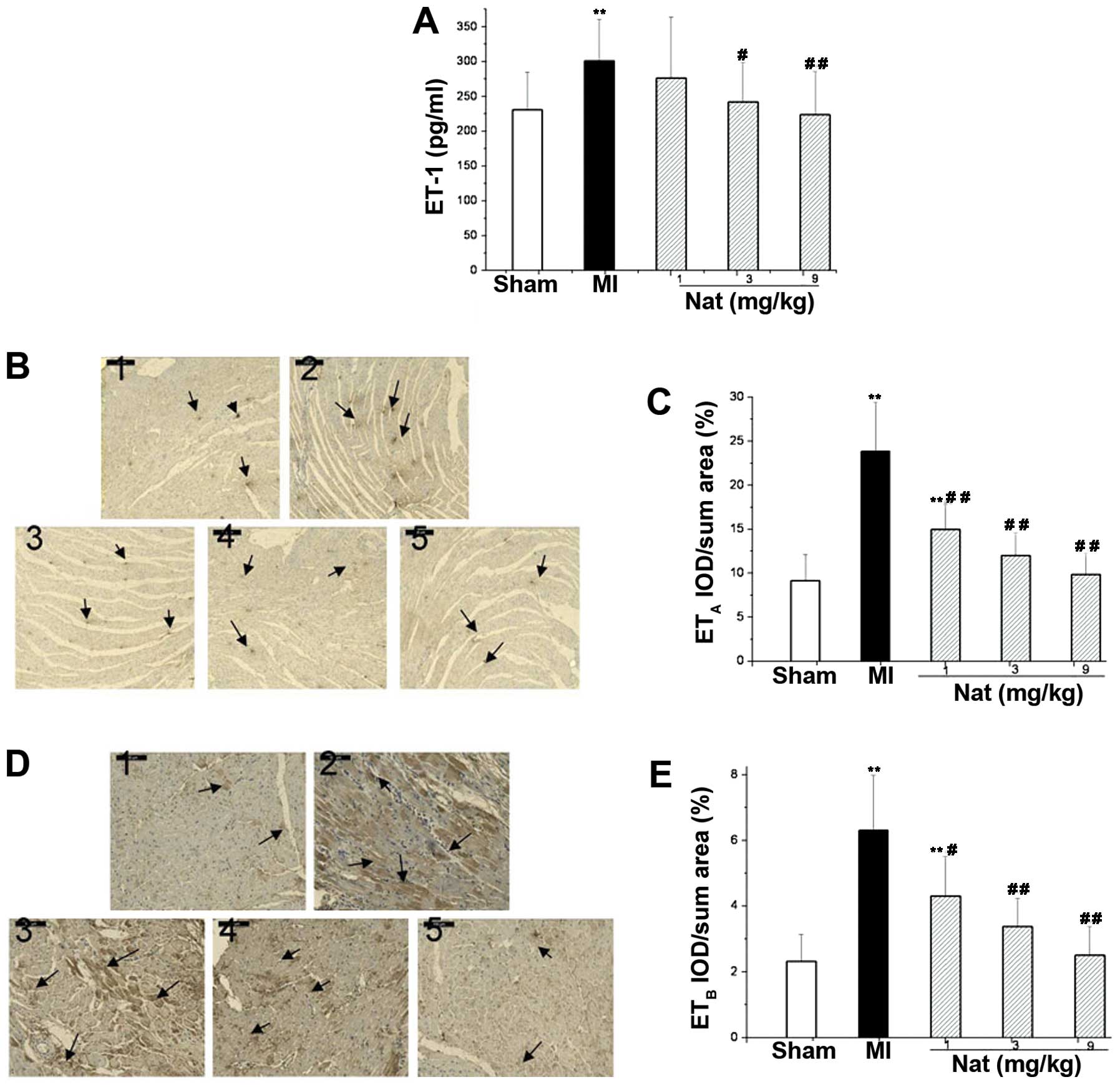
Effects of natakalim on the endothelial
system
The serum concentration of ET-1 and the cardiac
tissue protein levels of ETA and ETB were
significantly increased in the rats following MI (P<0.01;
Fig. 5). These findings indicate
that the MI-induced CHF led to an increase in the synthesis and
release of ET-1 and to the increased expression of ET receptors.
Treatment with natakalim for 8 weeks markedly attenuated the
elevated serum levels of ET-1 and the production of ETA
and ETB proteins in the cardiac tissue; the levels
returned to almost normal levels.
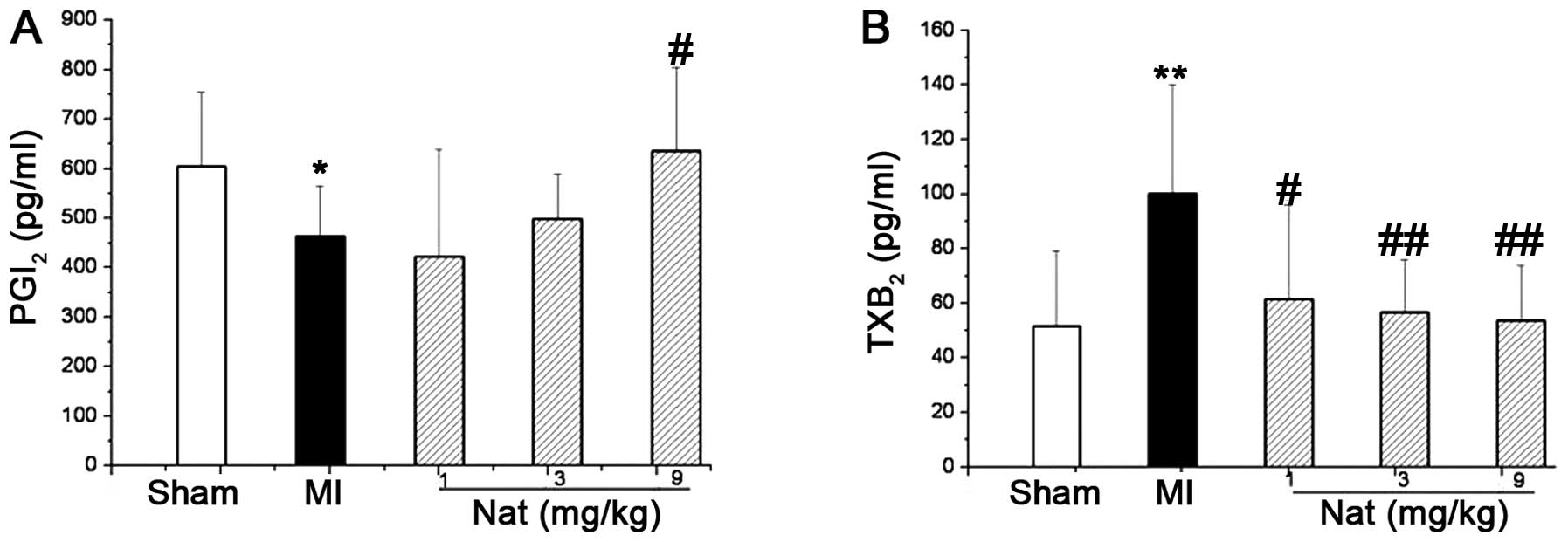
Effects of natakalim on the concentration
of plasma PGI2 and TXA2
The plasma levels of prostaglandin F1a
(6-keto-PGF1a) and thromboxane B2
(TXB2), metabolites of PGI2 and
TXA2, respectively, were used to estimate the plasma
levels of PGI2 and TXA2, respectively, due to
their short half-lives. In the rats with CHF induced by MI, the
concentration of plasma 6-keto-PGF1a was significantly
decreased (Fig. 6A), while the
concentration of plasma TXB2 was significantly increased
(Fig. 6B). Treatment with
natakalim for 8 weeks restored the concentrations of plasma
6-keto-PGF1a and TXB2 to almost normal
levels.
Discussion
To the best of our knowledge, the present study is
the first to evaluate the effects of natakalim, a novel
SUR2B/Kir6.1 channel opener, on post-infarction LVR and CHF in
rats. The major findings of the present study include the following
observations: i) natakalim reverses pathological LVR in a rat model
of LAD ligation; ii) natakalim prevents the progression from
hypertrophy to CHF; iii) the underlying mechanisms of action of
natakalim may be attributed to the restoration of a coordinated
balance between endothelial function and cardiac hypertrophy by the
correction of endothelial dysfunction, the decrease in NO and the
increase in PGI2 secretion, the inhibition of ET-1 and
TXA2 biosynthesis and secretion, the increase in cardiac
tissue eNOS protein expression, and the decrease in cardiac tissue
iNOS, ETA and ETB receptor protein
expression.
CHF is caused by multiple factros that result from
the heart being unable to pump sufficient blood to meet the
metabolic requirements of the body. The heart undergoes a
compensatory remodeling process in response to initial myocardial
insults. The remodeling initially helps the heart to preserve pump
function; however, the heart ultimately becomes maladaptive and
fails to function. Ischemic heart disease, the most important
etiology of CHF, accounts for up to two-thirds of all CHF cases.
Hypertension is another important factor; thus, the rat model of
pressure overload that leads to the development of concentric
hypertrophy and systolic dysfunction is widely used to simulate the
progression of hypertensive heart disease (16). We have previously demonstrated
that natakalim prevents LVR and the progression from hypertrophy to
failure induced by pressure overload (12). Therefore, in this study, we
explored the effects of natakalim on LVR and heart failure in a rat
model of MI.
The LAD ligation model involves the MI-induced
progression from hypertrophy to cardiac failure and has been
characterized extensively (15–18). In the present study, we found that
following LAD ligation, the infarcted wall of the heart was
gradually replaced by scar tissue, and the non-infarcted myocardium
exhibited LVR, including cardiac hypertrophy, interstitial
myocardial fibrosis and changes in left ventricular shape; these
findings are consistent with those of previous studies (15–18). The hypertrophy of the
non-infarcted myocardium, which may not appear to be damaged at the
time of the infarction, cannot compensate sufficiently to prevent
the progression of heart failure, and this inability to compensate
is identical to our findings, such as pulmonary congestion, right
ventricular hypertrophy, overexpression of the heart failure
indicators, ANP and BNP mRNA, and the depression of cardiac
systolic and diastolic function (11,12). To the best of our knowledge, the
present study is the first to evaluate the effects of natakalim on
LVR and CHF induced by MI. Following treatment with natakalim for 8
weeks, the LVW/BW ratio, cardiomyocyte cross-sectional area, the
extent of interstitial myocardial fibrosis, hydroxyproline content
and pathological damage in cardiac tissue were all reversed, and
the progression from LVR to failure was prevented. These results
suggest that natakalim can prevent LVR and the progression from
hypertrophy to failure induced by MI. Taken together, these
findings suggest that natakalim may be a suitable therapy for
patients with post-infarction CHF.
However, the mechanisms responsible for the
progression of LVR to heart failure remain poorly understood. An
increasing body of evidence has drawn attention to the role of the
coronary endothelium (6,19,20). Coronary endothelial dysfunction is
related to the development of myocardial ischemia, impairment in
contractility and the progression to CHF (6). Furthermore, it has been reported
that the imbalance between endothelial function and the adaptation
of cardiac hypertrophy causes the transition of LVR to heart
failure (7). Therefore, a novel
therapeutic strategy for post-MI heart failure may be to correct
coronary endothelial dysfunction in the non-infarcted myocardium
and restore the coordinated balance between endothelial function
and cardiac hypertrophy.
The vascular endothelium produces and releases a
wide range of vasoactive factors to regulate vascular homeostasis,
such as NO, ET-1, angiotensin II (AngII), PGI2 and
TXA2 (3,4,19,20). Endothelial dysfunction is an
important cardiovascular risk factor, and it may result from a
variety of factors, including augmented ET-1 synthesis and reduced
endothelium-derived NO synthesis. Endothelial dysfunction may also
result from the imbalance between the formation and release of
PGI2 and TXA2 into the circulation. A growing
body of evidence suggests that NO, ET-1 and PGI2 are
associated with cardiac hypertrophy and heart failure following MI
(20–22).
Previous studies have suggested that LVR and the
development of failure are regulated by eNOS, iNOS and NO. At a low
concentration, NO synthesized by eNOS in the endotheliocyte
contributes to the maintenance of vascular tone and function,
protects against myocyte hypertrophy and apoptosis and preserves
cardiomyocyte contractility (5).
Compared with wild-type mice, eNOS knockout mice display
significantly aggravated LVR and impaired myocardial function
following MI (23), whereas eNOS
overexpression improves survival and cardiac function following MI
(24). In the normal adult heart,
iNOS has not been found to be constitutively present, but iNOS can
be induced by ischemia, heart failure, stroke and infection
(25). iNOS may lead to the
production of large amounts of NO for a sustained period of time,
but the excessive amounts of NO are sufficiently cytotoxic,
inducing the apoptosis of cardiomyocytes and impairing cardiac
contractile function, which has been demonstrated in patients with
CHF and animal models (26,27). The protective effects exerted by
natakalim against post-MI CHF are related to the reduction in
iNOS-derived NO and the increase in eNOS-derived NO. Further
evidence indicates that inflammation is involved in post-MI CHF and
that iNOS and eNOS participate in the inflammation of CHF (28). Iptakalim can suppress the
inflammatory reaction by counteracting endothelial dysfunction
(29); however, determining
whether the protective effects exerted by its derivative,
natakalim, involve the suppression of inflammation in post-MI CHF
requires further investigation.
The endothelin system, particularly ET-1 and the
ETA and ETB receptors, has been implicated in
pathological states, such as ventricular remodeling and CHF
following MI (30). ET-1 is the
most potent endothelium-derived vasoconstrictor peptide, and it can
stimulate myocardial hypertrophy, myocardial fibrosis and
cardiomyocyte injury. The actions of ET-1 are mediated through the
activation of ETA and ETB receptors, which
are found in a variety of cells of the cardiovascular system
(31). Elevated circulating ET-1
levels are observed in the failing hearts of patients and animals
with CHF, whereas suppressing circulating ET-1 levels during the
acute phase of MI prevents post-MI LVR in patients (30). Moreover, ETA and
ETB receptor antagonists prevent progressive LVR and
improve cardiac function in rats with a large MI (30,32,33). Taken together, this evidence
suggests that the downregulation of the endothelin system
alleviates cardiomyocyte hypertrophy and prevents LVR and CHF
following acute MI.
PGI2 and TXA2 are active
metabolites of arachidonic acid and PGI2 is thought to
be a physiological antagonist of TXA2. PGI2
is generated by vascular endothelial cells, which possess strong
vasodilator, anti-aggregatory and antihypertrophic effects.
TXA2 is generated by activated platelets and
neutrophilic granulocytes when vascular endothelial cells are
injured, and TXA2 exhibits pro-aggregatory,
vasoconstrictor properties and stimulates vascular remodeling
(34). PGI2 receptor
IP knockout mice develop significant LVR and severe myocardial
fibrosis following MI, aortic banding or salt-sensitive
hypertension (35–37). Thus, an imbalance between
PGI2 and TXA2 is deleterious for the
circulatory system and can lead to vascular diseases, such as
atherosclerosis, MI, post-MI LVR and hypertension.
KATP channels are membrane sensors of
energy metabolism, and play an important role in a number of
diseases, such as MI and CHF. It has been reported that the
SUR2B/Kir6.1 subtype is mainly found in the endothelium, and it
affects the production and release of endothelial autacoids, such
as NO, PGI2 and ET-1 (38). A series of studies carried out in
our laboratory have suggested that natakalim has a high selectivity
for the SUR2B/Kir6.1 subtype of KATP channels in
endotheliocytes and promotes the secretion of NO and
PGI2 and inhibits the production of ET-1 in
endotheliocytes (12–14,39). In our rat model of post-MI heart
failure, natakalim increased eNOS protein expression and the serum
concentration of endothelium-derived NO, reduced iNOS protein
expression, and attenuated the pathologically abnormal increase in
NO concentration, all of which counteracted the deregulation of the
endothelium-derived NO system. In addition, natakalim reduced
plasma ET-1 levels and protein expression of ETA and
ETB receptors, suppressing the endothelin system.
Additionally, natakalim increased the plasma PGI2
concentration and decreased the plasma TXA2
concentration, which corrected the imbalance between
PGI2 and TXA2. Therefore, as previously
noted, natakalim can correct coronary endothelial dysfunction in
the non-infarcted myocardium following post-MI heart failure.
In conclusion, to the best of our knowledge, the
present study demonstrates for the first time that natakalim
counteracts cardiac hypertrophy and prevents the progression to
heart failure induced by MI. The underlying molecular mechanisms
involved the restoration of the coordinated balance between
endothelial function and cardiac hypertrophy by correcting
endothelial dysfunction, including the amelioration of the
endothelium-derived NO system, the inhibition of the endothelin
system and the rectification of the imbalance between
PGI2 and TXA2. Thus, natakalim may provide a
novel therapeutic strategy for correcting endothelial dysfunction.
Although natakalim appears to be suitable for the clinical
treatment of coronary heart disease and post-infarction heart
failure, a series of well-designed trials is required in order to
confirm our results.
Acknowledgements
The present study was supported by grants from the
State Key Research Project of China (AWS11J003), the National Basic
Research ‘973’ Program (2012CB518200) and the National New Drug
Research and Development Key Project (2010ZX09401-307).
References
|
1
|
McMurray JJ, Adamopoulos S, Anker SD,
Auricchio A, Böhm M, Dickstein K, Falk V, Filippatos G, Fonseca C,
Gomez-Sanchez MA, Jaarsma T, Køber L, Lip GY, Maggioni AP,
Parkhomenko A, Pieske BM, Popescu BA, Rønnevik PK, Rutten FH,
Schwitter J, Seferovic P, Stepinska J, Trindade PT, Voors AA,
Zannad F and Zeiher A; Task Force for the Diagnosis and Treatment
of Acute and Chronic Heart Failure 2012 of the European Society of
Cardiology. Bax JJ, Baumgartner H, Ceconi C, Dean V, Deaton C,
Fagard R, Funck-Brentano C, Hasdai D, Hoes A, Kirchhof P, Knuuti J,
Kolh P, McDonagh T, Moulin C, Popescu BA, Reiner Z, Sechtem U,
Sirnes PA, Tendera M, Torbicki A, Vahanian A, Windecker S, McDonagh
T, Sechtem U, Bonet LA, Avraamides P, Ben Lamin HA, Brignole M,
Coca A, Cowburn P, Dargie H, Elliott P, Flachskampf FA, Guida GF,
Hardman S, Iung B, Merkely B, Mueller C, Nanas JN, Nielsen OW, Orn
S, Parissis JT and Ponikowski P; ESC Committee for Practice
Guidelines. ESC guidelines for the diagnosis and treatment of acute
and chronic heart failure 2012: The Task Force for the Diagnosis
and Treatment of Acute and Chronic Heart Failure 2012 of the
European Society of Cardiology. Developed in collaboration with the
Heart Failure Association (HFA) of the ESC. Eur Heart J.
33:1787–1847. 2012.
|
|
2
|
Konstam MA, Kramer DG, Patel AR, Maron MS
and Udelson JE: Left ventricular remodeling in heart failure:
current concepts in clinical significance and assessment. JACC
Cardiovasc Imaging. 4:98–108. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lerman A and Zeiher AM: Endothelial
function: cardiac events. Circulation. 111:363–368. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Adamopoulos S, Parissis JT and Kremastinos
DT: Endothelial dysfunction in chronic heart failure: clinical and
therapeutic implications. Eur J Intern Med. 13:233–239. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Scherrer-Crosbie M, Ullrich R, Bloch KD,
Nakajima H, Nasseri B, Aretz HT, Lindsey ML, Vançon AC, Huang PL,
Lee RT, Zapol WM and Picard MH: Endothelial nitric oxide synthase
limits left ventricular remodeling after myocardial infarction in
mice. Circulation. 104:1286–1291. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Prasad A, Higano ST, Al Suwaidi J, Holmes
DR Jr, Mathew V, Pumper G, Lennon RJ and Lerman A: Abnormal
coronary microvascular endothelial function in humans with
asymptomatic left ventricular dysfunction. Am Heart J. 146:549–554.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shiojima I, Sato K, Izumiya Y, Schiekofer
S, Ito M, Liao R, Colucci WS and Walsh K: Disruption of coordinated
cardiac hypertrophy and angiogenesis contributes to the transition
to heart failure. J Clin Invest. 115:2108–2118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Seino S and Miki T: Physiological and
pathophysiological roles of ATP-sensitive K+ channels.
Prog Biophys Mol Biol. 81:133–176. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yoshida H, Feig JE, Morrissey A, Ghiu IA,
Artman M and Coetzee WA: KATP channels of primary human
coronary artery endothelial cells consist of a heteromultimeric
complex of kir6.1, kir6.2, and sur2b subunits. J Mol Cell Cardiol.
37:857–869. 2004.
|
|
10
|
Nichols CG, Singh GK and Grange DK:
KATP channels and cardiovascular disease: suddenly a
syndrome. Circ Res. 112:1059–1072. 2013.
|
|
11
|
Gao S, Long CL, Wang RH and Wang H:
KATP activation prevents progression of cardiac
hypertrophy to failure induced by pressure overload via protecting
endothelial function. Cardiovasc Res. 83:444–456. 2009.
|
|
12
|
Tang Y, Long CL, Wang RH, Cui W and Wang
H: Activation of SUR2B/Kir6.1 subtype of adenosine
triphosphate-sensitive potassium channel improves pressure
overload-induced cardiac remodeling via protecting endothelial
function. J Cardiovasc Pharmacol. 56:345–353. 2010. View Article : Google Scholar
|
|
13
|
Chen Y, Pan Z, Cui W and Wang H:
Selectivity of new iptakalim derivatives on the subtypes of
ATP-sensitive potassium channels. Chin Pharmacol Bull.
24:1427–1430. 2008.
|
|
14
|
Wei H, Cao YQ, Zhang JD, Peng Z, Yang DF,
Liu JY, Shi YP, Yang YL and Wang H: Protective effects of
SUR2B/Kir6.1 potassium channel opener natakalim against RAVECs
injuries induced by hypoxia. Chin J Appl Physiol. 28:241–244.
2012.PubMed/NCBI
|
|
15
|
Goldman S and Raya TE: Rat infarct model
of myocardial infarction and heart failure. J Card Fail. 1:169–177.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Velagaleti RS, Pencina MJ, Murabito JM,
Wang TJ, Parikh NI, D’Agostino RB, Levy D, Kannel WB and Vasan RS:
Long-term trends in the incidence of heart failure after myocardial
infarction. Circulation. 118:2057–2062. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ezekowitz JA, Kaul P, Bakal JA, Armstrong
PW, Welsh RC and McAlister FA: Declining in-hospital mortality and
increasing heart failure incidence in elderly patients with first
myocardial infarction. J Am Coll Cardiol. 53:13–20. 2009.
View Article : Google Scholar
|
|
18
|
Krzemiński TF, Nozyński JK, Grzyb J and
Porc M: Wide-spread myocardial remodeling after acute myocardial
infarction in rat. Features for heart failure progression. Vascul
Pharmacol. 48:100–108. 2008.PubMed/NCBI
|
|
19
|
Marti CN, Gheorghiade M, Kalogeropoulos
AP, Georgiopoulou VV, Quyyumi AA and Butler J: Endothelial
dysfunction, arterial stiffness, and heart failure. J Am Coll
Cardiol. 60:1455–1469. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Khazaei M, Moien-Afshari F and Laher I:
Vascular endothelial function in health and diseases.
Pathophysiology. 15:49–67. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yin WH, Chen YH, Wei J, Jen HL, Huang WP,
Young MS, Chen DC and Liu PL: Associations between endothelin-1 and
adiponectin in chronic heart failure. Cardiology. 118:207–216.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Francois H, Athirakul K, Howell D, Dash R,
Mao L, Kim HS, Rockman HA, Fitzgerald GA, Koller BH and Coffman TM:
Prostacyclin protects against elevated blood pressure and cardiac
fibrosis. Cell Metab. 2:201–207. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Feng Q, Song W, Lu X, Hamilton JA, Lei M,
Peng T and Yee SP: Development of heart failure and congenital
septal defects in mice lacking endothelial nitric oxide synthase.
Circulation. 106:873–879. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Janssens S, Pokreisz P, Schoonjans L,
Pellens M, Vermeersch P, Tjwa M, Jans P, Scherrer-Crosbie M, Picard
MH, Szelid Z, Gillijns H, Van de Werf F, Collen D and Bloch KD:
Cardiomyocyte-specific overexpression of nitric oxide synthase 3
improves left ventricular performance and reduces compensatory
hypertrophy after myocardial infarction. Circ Res. 94:1256–1262.
2004. View Article : Google Scholar
|
|
25
|
Takimoto Y, Aoyama T, Keyamura R, Shinoda
E, Hattori R, Yui Y and Sasayama S: Differential expression of
three types of nitric oxide synthase in both infarcted and
non-infarcted left ventricles after myocardial infarction in the
rat. Int J Cardiol. 76:135–145. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu YH, Carretero OA, Cingolani OH, Liao
TD, Sun Y, Xu J, Li LY, Pagano PJ, Yang JJ and Yang XP: Role of
inducible nitric oxide synthase in cardiac function and remodeling
in mice with heart failure due to myocardial infarction. Am J
Physiol Heart Circ Physiol. 289:H2616–H2623. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Starling RC: Inducible nitric oxide
synthase in severe human heart failure: impact of mechanical
unloading. J Am Coll Cardiol. 45:1425–1427. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tousoulis D, Charakida M and Stefanadis C:
Inflammation and endothelial dysfunction as therapeutic targets in
patients with heart failure. Int J Cardiol. 100:347–353. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhao RJ and Wang H: Chemerin/ChemR23
signaling axis is involved in the endothelial protection by
KATP channel opener iptakalim. Acta Pharmacol Sin.
32:573–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kolettis TM, Barton M, Langleben D and
Matsumura Y: Endothelin in coronary artery disease and myocardial
infarction. Cardiol Rev. 21:249–256. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Böhm F and Pernow J: The importance of
endothelin-1 for vascular dysfunction in cardiovascular disease.
Cardiovasc Res. 76:8–18. 2007.PubMed/NCBI
|
|
32
|
Spieker LE, Noll G, Ruschitzka FT and
Lüscher TF: Endothelin receptor antagonists in congestive heart
failure: a new therapeutic principle for the future? J Am Coll
Cardiol. 37:1493–1505. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nguyen QT, Cernacek P, Sirois MG,
Calderone A, Lapointe N, Stewart DJ and Rouleau JL: Long-term
effects of nonselective endothelin A and B receptor antagonism in
postinfarction rat: importance of timing. Circulation.
104:2075–2081. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Harding P and Murray DB: The contribution
of prostaglandins versus prostacyclin in ventricular remodeling
during heart failure. Life Sci. 89:671–676. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xiao CY, Hara A, Yuhki K, Fujino T, Ma H,
Okada Y, Takahata O, Yamada T, Murata T, Narumiya S and Ushikubi F:
Roles of prostaglandin I2 and thromboxane A2
in cardiac ischemia-reperfusion injury: a study using mice lacking
their respective receptors. Circulation. 104:2210–2015.
2001.PubMed/NCBI
|
|
36
|
Chan EC, Dusting GJ, Guo N, Peshavariya
HM, Taylor CJ, Dilley R, Narumiya S and Jiang F: Prostacyclin
receptor suppresses cardiac fibrosis: role of CREB phosphorylation.
J Mol Cell Cardiol. 49:176–185. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yuhki K, Kojima F, Kashiwagi H, Kawabe J,
Fujino T, Narumiya S and Ushikubi F: Roles of prostanoids in the
pathogenesis of cardiovascular diseases: novel insights from
knockout mouse studies. Pharmacol Ther. 129:195–205. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Minamino T and Hori M: Protecting
endothelial function: a novel therapeutic target of atp-sensitive
potassium channel openers. Cardiovasc Res. 73:448–449. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang H, Tang Y, Wang L, Long CL and Zhang
YL: ATP-sensitive potassium channel openers and
2,3-dimethyl-2-butylamine derivatives. Curr Med Chem. 14:133–155.
2007. View Article : Google Scholar : PubMed/NCBI
|