Introduction
S-nitrosylation is a protein modification in which a
nitrosyl group covalently attaches to the thiol group on protein
cysteine residues, forming an S-nitrosothiol. S-nitrosylation is a
redox mechanism for the dynamic and post-translational regulation
of most or all of the major classes of proteins and is independent
of enzyme catalysis, labile modification, and on/off switch-like
photophosphorylation, involved in cellular signal transduction
(1–3). The S-nitrosylation of proteins is
involved in the regulation of cardiovascular functions (2). Classically, nitric oxide (NO) is
generated through the activation of endothelial NO synthase (eNOS),
and NO diffuses from the endothelial cells to the vascular smooth
muscle cells, where it binds to its intracellular receptor, soluble
guanylyl cyclase, and activates protein kinase G to mediate
vasorelaxation (1–3). However, NO also regulates widespread
and diverse biologic functions in the endothelium, such as ion
channel activity (4),
antioxidative stress (5),
anti-apoptotic mechanisms (6);
and anti-inflammatory responses (3,7).
These functions have been attributed to the protein S-nitrosylation
(3–7). It has been shown that many factors
are involved in the regulation of protein S-nitrosylation in
endothelial cells. Tumor necrosis factor-α (8), oxidized low-density lipoprotein
(8); and high glucose (9) decrease protein S-nitrosylation,
whereas shear stress (10),
17b-estradiol (3); and acute
hypoxia (11) upregulate protein
S-nitrosylation in endothelial cells.
Homocysteine (Hcy) is a sulfur-containing amino acid
formed during the transmethylation of methionine. High plasma
levels of Hcy, termed hyperhomocysteinemia (HHcy), is an
independent risk factor for cardiovascular disease. Elevated plasma
levels of Hcy are found in 5–10% of the general population and in
up to 40% of patients with vascular disease (12). It has been reported that Hcy
reduced the levels of NO (13).
However, whether Hcy also impairs protein S-nitrosylation in
endothelial cells by mediating the levels of NO remains to be
determined. Thus, we hypothesized that Hcy may decrease the levels
of protein S-nitrosylation in endothelial cells. In the present
study, we examined the effects of Hcy on human umbilical vein
endothelial cells (HUVECs) and rat aortas. The results showed that
Hcy significantly reduced the cellular protein S-nitrosylation.
Molecular mechanisms that are involved in the process of
S-nitrosylation were also examined. These findings provide new
insights into understanding of the pathway for Hcy-mediated
vascular damage.
Materials and methods
Cell culture
HUVECs were isolated from human umbilical cords. The
investigation conformed to the principles outlined in the
Declaration of Helsinki for the use of human tissue or subjects.
The HUVECs were isolated using 0.1% (w/v) collagenase
(Gibco/Invitrogen, Carlsbad, CA, USA). The cells were collected and
cultured in M199 medium supplemented with 20% fetal bovine serum
(FBS) (both from Gibco/Invitrogen), and 1% (v/v) endothelial cell
growth supplement (ScienCell Research Laboratories, Carlsbad, CA,
USA) at 37°C in a humidified atmosphere of 5% CO2 and
95% air. The growth medium was changed every three days until the
cells reached confluence. The identity of the HUVECs was performed
by positive staining with an anti-von Willebrand factor (vWF)
antibody (Thermo Fisher Scientific/Pierce, Rockford, IL, USA)
(3). After the HUVECs became
confluent, they were incubated in M199 medium containing 2% FBS for
16 h prior to different treatments.
Animal model
Moderate HHcy was induced in male Sprague-Dawley
rats (180–200 g; supplied by the Laboratory Animal Center of Xi’an
Jiaotong University, Xi’an, China) (n=12) by oral administration of
L-methionine (1 g/kg body weight per day) (Fluka/Sigma-Aldrich, St.
Louis, MO, USA) and succinylsulfathiazole (0.5 g/kg body weight per
day) (Fluka/Sigma-Aldrich) for 4 weeks as previously described
(14). The animal experiments in
this study were approved by the Laboratory Animal Administration
Committee of Xi’an Jiaotong University and performed according to
the Guidelines for Animal Experimentation of Xi’an Jiaotong
University, the Guidelines on the Care and Use of Laboratory
Animals issued by the Chinese Council on Animal Research, and the
Guide for the Care and Use of Laboratory Animals published by the
US National Institutes of Health (NIH publication no. 85–23,
revised 1996).
Reactive oxygen species (ROS) assay
Intracellular ROS generation was assessed using
2′,7′-dichlorofluorescein diacetate (DCFH-DA). ROS in the cells
oxidize DCFH-DA, yielding the fluorescent compound
2′,7′-dichlorofluorescein (DCF). The levels of ROS were detected
using the ROS assay kit (Beyotime Institute of Biotechnology,
Nantong, China).
Nitrate/nitrite assay
The levels of NO of the conditioned medium were
determined by estimating the total concentration of nitrate/nitrite
using a nitrate/nitrite colorimetric assay kit (Jiancheng
Technology Co., Ltd., Nanjing, China).
Plasma Hcy assay
The rats were anesthetized with an intraperitoneal
injection of sodium pentobarbital (50 mg/kg) (14). The blood was collected in
centrifuge tubes (containing EDTA) from the aortas of fasted rats.
The blood was immediately cooled on ice and centrifuged at 3,000 ×
g for 20 min at 4°C. Plasma was then stored at −20°C until assayed.
Total plasma Hcy concentrations were measured using an ELISA kit
(R&D Systems, Inc., Minneapolis, MN, USA).
Immunofluorescence
The HUVECs were fixed with 4% paraformaldehyde for
15 min at room temperature. The cells were permeabilized with 0.1%
(v/v) Triton X-100 for 15 min, and blocked with 5% horse serum for
30 min, and incubated with anti-nitrosocysteine rabbit polyclonal
antibody (Abcam, Cambridge, MA, USA) or anti-p65 antibody (Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA) at 4°C. After
overnight incubation the cells were treated with a FITC-conjugated
goat anti-rabbit IgG (Santa Cruz Biotechnology, Inc.) for 30 min at
room temperature. Nuclei were stained with
4,6-diamidino-phenylindole (DAPI; 0.2 mg/ml in 10 mM Tris-HCl, pH
7.0, 10 mM EDTA, 100 mM NaCl). To validate the
anti-S-nitrosocysteine antibody, we treated HUVECs for 10 min with
1 mM DL-dithiothreitol (DTT), which caused severe loss of
S-nitrosocysteine staining. For the negative control, fixed and
permeabilized cells were preincubated with 0.8% HgCl2
for 1 h at 37°C (8).
To stain the aorta, 10-μm sections were placed on
glass slides and fixed for 10 min in acetone at room temperature.
After permeabilization, the sections were incubated with rabbit
polyclonal anti-nitrosocysteine antibody and a mouse monoclonal
anti-vWF antibody overnight at 4°C. The incubation with a
TRITC-conjugated goat anti-rabbit IgG (H+L) (Santa Cruz
Biotechnology) and a FITC-conjugated goat anti-mouse IgG (H+L) was
performed. Nuclei were stained with DAPI. The results were
visualized using fluorescence microscopy (Olympus, Tokyo, Japan).
The mean fluorescence intensity was analyzed by Image-Pro Plus 6.0
software (Media Cybernetics Inc., Bethesda, MD, USA) (3).
Detection of S-nitrosylated proteins by
the biotin switch assay
The HUVECs were lysed in HEN buffer (100 mM Hepes, 1
mM EDTA, and 0.1 mM neocuproine, pH 8.0) containing 1% (w/v) SDS
and 1 mM PMSF plus protease inhibitors (Hoffmann-La Roche, Basel,
Switzerland). The proteins extracted from the HUVECs were
quantified using a BCA assay kit (Thermo Fisher Scientific/Pierce).
Equal amounts of protein were incubated with HEN buffer containing
2.5% (w/v) SDS and 0.1% (v/v) S-methyl-methanethiosulfonate
(Fluka/Sigma-Aldrich) at 50°C in the dark for 20 min. The extracts
were precipitated with cold acetone. The proteins were re-suspended
in HEN buffer plus 1% SDS. S-nitrosothiols were decomposed by
adding ascorbate followed by incubation with 2.5 mg/ml biotin-HPDP
(Thermo Fisher Scientific/Pierce) for 1 h at room temperature. The
proteins were subsequently precipitated again using acetone and
resuspended in non-reducing Laemmli loading buffer. For
purification of the biotinylated proteins, the proteins
precipitated by acetone were diluted with neutralization buffer [20
mM HEPES (pH 7.7, 100 mM NaCl, 1 mM EDTA, and 0.5% (v/v) Triton
X-100] and 50% streptavidin agarose suspension
(Fluka/Sigma-Aldrich) and incubated for 1 h at room temperature.
The proteins were eluted with elution buffer.
Samples from the biotin switch assay were separated
on 12% SDS polyacrylamide gels and transferred to PVDF membranes
(EMD Millipore, Billerica, MA, USA). PVDF membranes were blocked
with 5% non-fat dried milk for 1 h at 37°C and incubated with
specific antibodies, including horseradish peroxidase-conjugated
anti-biotin (Cell Signaling Technology, Danvers, MA, USA),
anti-β-actin antibody (Abcam), and anti-p65 antibody, overnight at
4°C. Membranes were then incubated with a horseradish
peroxidase-conjugated goat against mouse IgG (H+L), followed by
enhanced chemiluminescence using the SuperSignal West Pico
Substrate kit (both from Thermo Fisher Scientific/Pierce). The
protein bands were detected and analyzed using the Chemi-Doc-it HR
410 imaging system (Ultra-Violet Products, Ltd., Upland, CA, USA)
(15).
Western blot analysis
The cells were lysed on ice for 1 h in RIPA buffer
[50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% Triton X-100 (v/v), 1%
deoxycholic acid (w/v), and 0.1% sodium dodecyl sulfate)]
containing 0.5 mM PMSF and protease inhibitors (Hoffmann-La Roche).
The protein concentration was measured. Each sample was
subsequently denatured by boiling for 10 min in Laemmli loading
buffer. Equal amounts of protein were separated by 12%
SDS-polyacrylamide gel electrophoresis and transferred to PVDF
membranes. After blocking with 5% non-fat dried milk for 1 h at
37°C, the membranes were incubated overnight at 4°C with the
following antibodies: anti-phospho-Akt (Ser-473) antibody,
anti-phospho-eNOS (Ser-1177) antibody (Cell Signaling Technology),
anti-Akt antibody, anti-eNOS antibody (Abcam), anti-vascular cell
adhesion molecule-1 (VCAM-1) antibody (Santa Cruz Biotechnology,
Inc.), and anti-β-actin antibody. After washing, the membranes were
incubated using horseradish peroxidase-conjugated goat anti-mouse
or anti-rabbit IgG (Thermo Fisher Scientific/Pierce) for 1 h at
37°C, followed by enhanced chemiluminescence using the SuperSignal
West Pico Substrate kit. The protein bands were analyzed using the
Chemi Doc-it HR 410 imaging system (16).
Electrophoretic mobility shift assay
(EMSA)
Nuclear and cytoplasmic protein extraction were
investigated. NE-PER Nuclear and Cytoplasmic Extraction Reagents
(Thermo Fisher Scientific/Pierce) were used to extract the cell
proteins according to the manufacturer’s instructions. The nuclear
proteins were quantified and stored at −80°C until they were used
for the EMSA.
Nuclear factor κB (NF-κB) DNA-binding
activity
The EMSA was performed with IRDye 700 infrared
dye-labeled double-stranded oligonucleotides containing an NF-κB
consensus site (5′-AGT TGA GGG GAC TTT CCC AGG C-3′,
3′-TCA ACT CCC CTG AAA
GGG TCC G-5′) (LI-COR Biosciences, Lincoln, NE, USA). The
nuclear extracts (8 μg per reaction) and 50 fmol of the
oligonucleotides were preincubated in 20 μl binding buffer [10 mM
Tris-HCl (pH 7.5), 50 mM NaCl, 3 mM DTT, 0.5 μg poly dI-dC, 5 mM
MgCl2, and 0.1% Tween-20)] for 30 min at room
temperature. The specificity of the bands was confirmed by
supershifting with specific antibodies of p65 and p50 (Santa Cruz
Biotechnology, Inc.) and by competition with 100 × specific cold
competitor (100 × SPC) and 100 × non-specific cold competitor (100
× NSC) (LI-COR Biosciences). The DNA protein complexes were
separated on 5% native polyacrylamide gels for 60 min at 100 V in
0.5X TBE buffer. The signals were scanned and quantified using the
Odyssey infrared imaging system (LI-COR Biosciences) (17).
Statistical analysis
Data are presented as the means ± SEM. One-way ANOVA
followed by an LSD test and Student’s t-tests were used for the
comparison between the groups. P<0.05 was considered to be
statistically significant. The analysis was performed using the
SPSS 13.0 software (SPSS Inc., Chicago, IL, USA).
Results
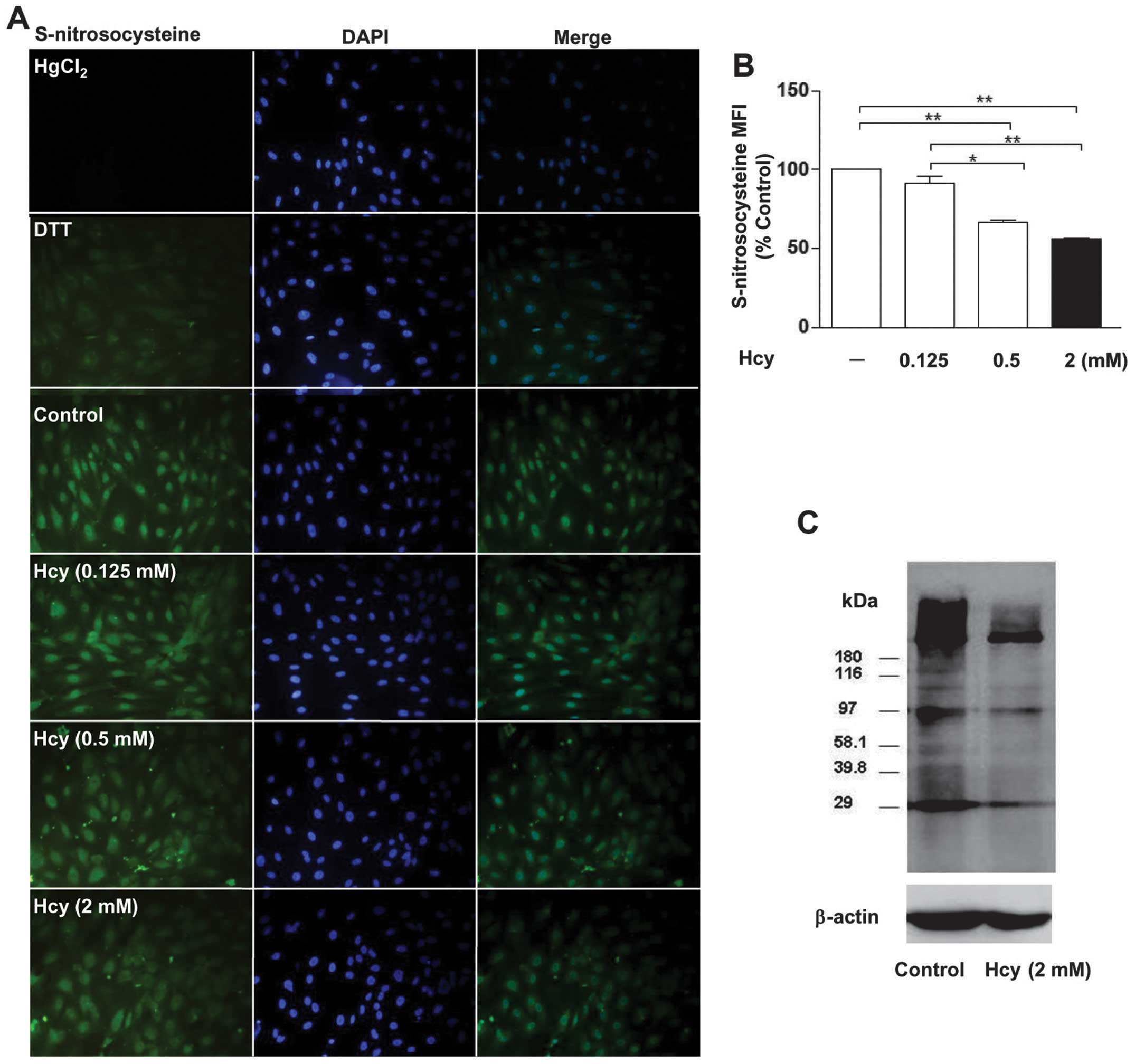
Hcy reduces protein S-nitrosylation in
HUVECs
HUVECs were treated with different doses of Hcy
(0.125, 0.5 and 2 mM) for 24 h. Cell morphology was normal
following treatment with Hcy (data not shown). The protein
S-nitrosylation was detected for S-nitrosocysteine using
immunofluorescence and was further confirmed by the biotin switch
method. We found that treatment with different doses of Hcy (2, 0.5
and 0.125 mM) resulted in the reductions (44>34>8%,
respectively) in the protein S-nitrosylation compared with the
control group (Fig. 1A and B).
However, there were no significant differences between 0.125 mM and
the control. Protein S-nitrosylation was significantly decreased in
HUVECs treated with Hcy (0.5 or 2 mM). In addition, we found that
S-nitrosocysteine was present in the nuclei and cytosol of the
HUVECs (Fig. 1A). The biotin
switch method on HUVECs further confirmed our finding (Fig. 1C).
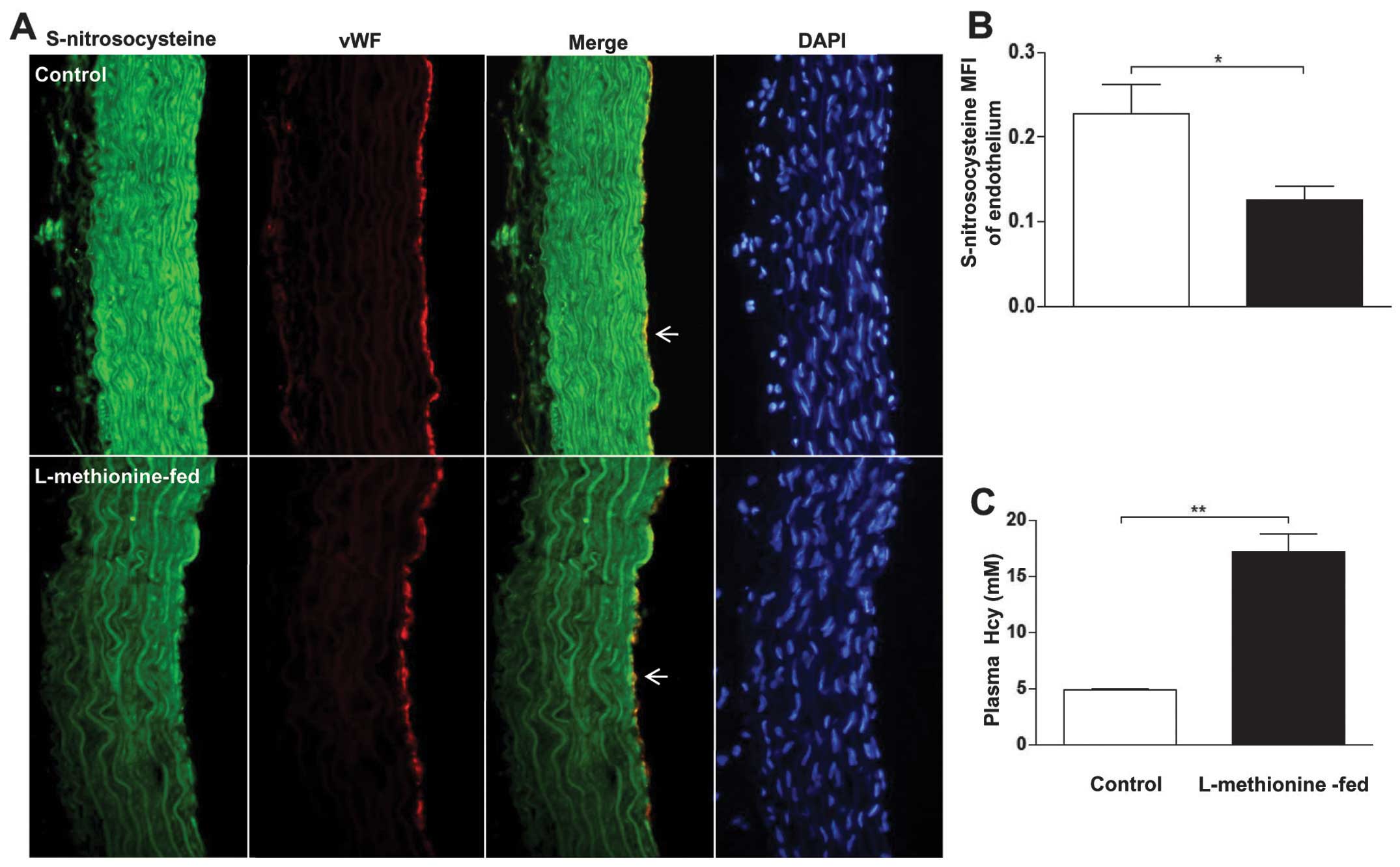
Hyperhomocysteinemia attenuates
endothelial S-nitrosylation of aortas in the rat
We assessed whether HHcy promoted a decrease in
S-nitrosylation levels in the endothelium. HHcy was induced in rats
by feeding a high-methionine diet to male Sprague-Dawley rats. As a
result, the high-methionine diet resulted in a significant 3.6-fold
increase in the plasma Hcy levels (17.2 vs. 4.8 mM in control rats)
(Fig. 2C). Furthermore, we
detected a significant decrease in nitrosocysteine staining in the
endothelium of thoracic aorta sections of L-methionine-fed rats
compared with the controls (Fig. 2A
and B).
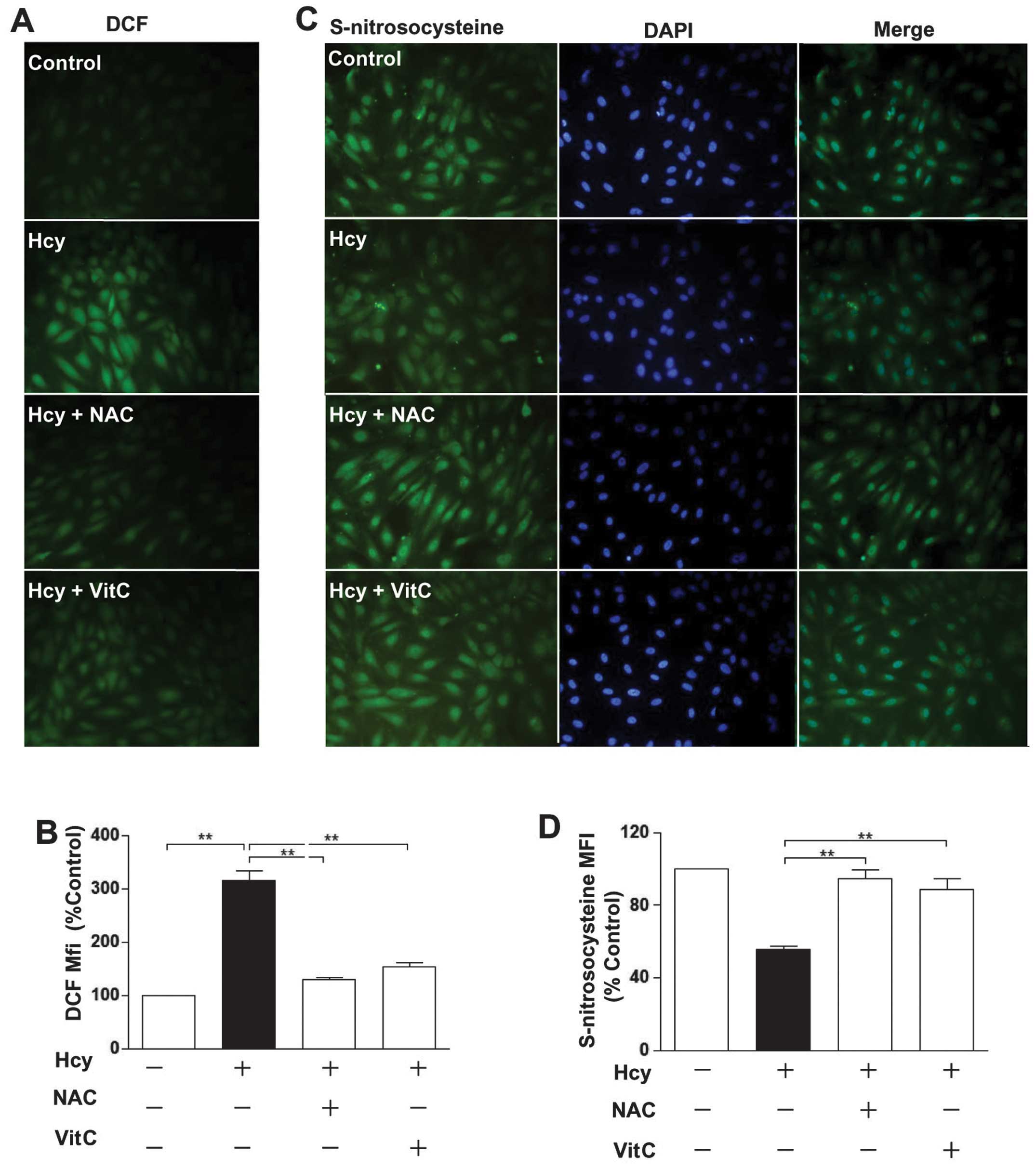
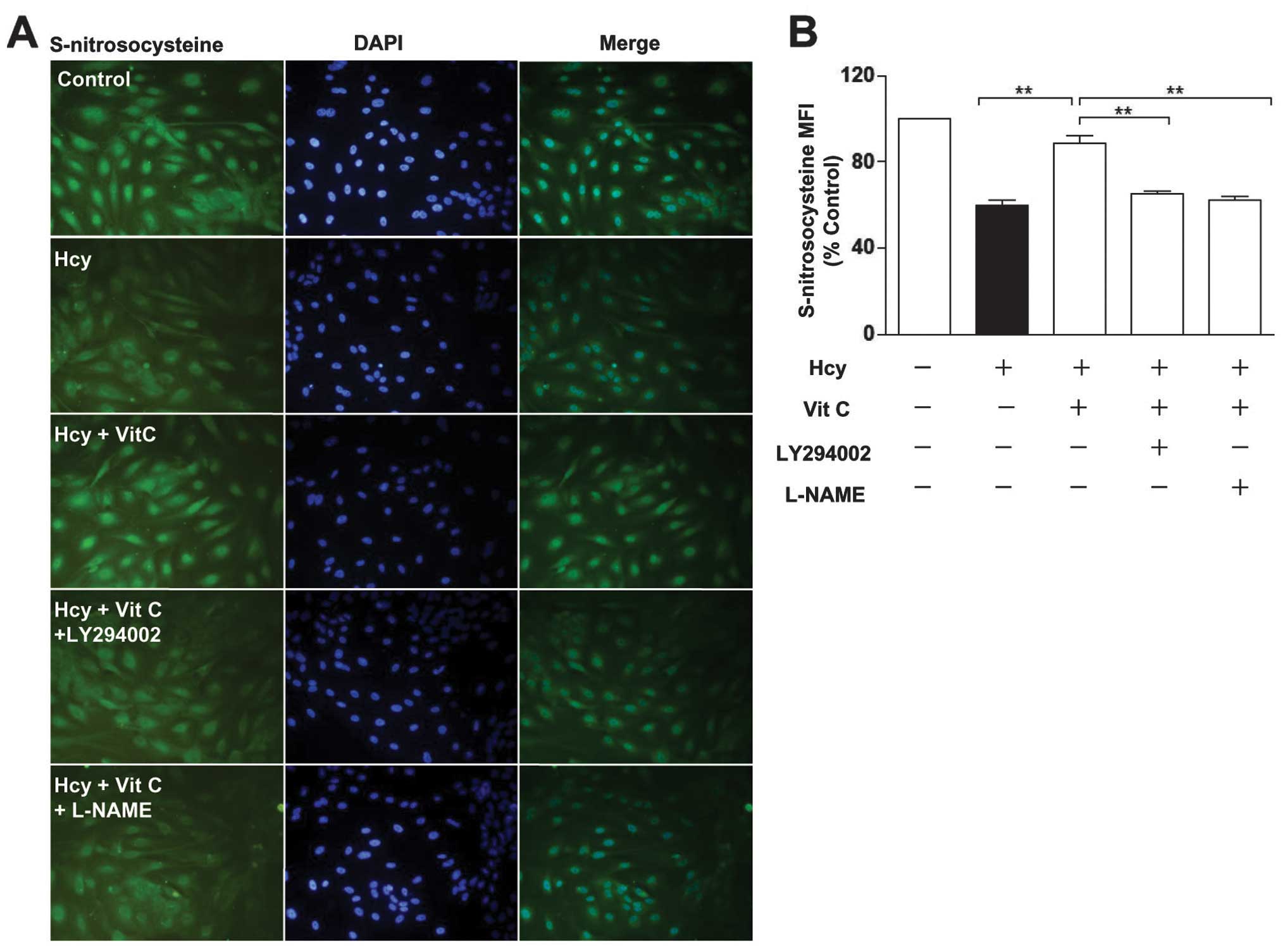
ROS scavengers reverse the effect of Hcy
on protein S-nitrosylation
ROS are involved in the deleterious effects of Hcy
on endothelial functions (13).
It is unclear whether Hcy reduces protein S-nitrosylation by ROS.
The results showed that Hcy significantly increased ROS levels.
This effect was blocked by vitamin C (VitC) and N-acetylcysteine
(NAC), which are both ROS scavengers (18) (Fig.
3A and B). Furthermore, VitC and NAC significantly increased
the decreased levels of protein S-nitrosylation reduction by Hcy
(Fig. 3C and D), suggesting that
ROS play a crucial role in the Hcy-induced reduction of protein
S-nitrosylation in endothelial cells.
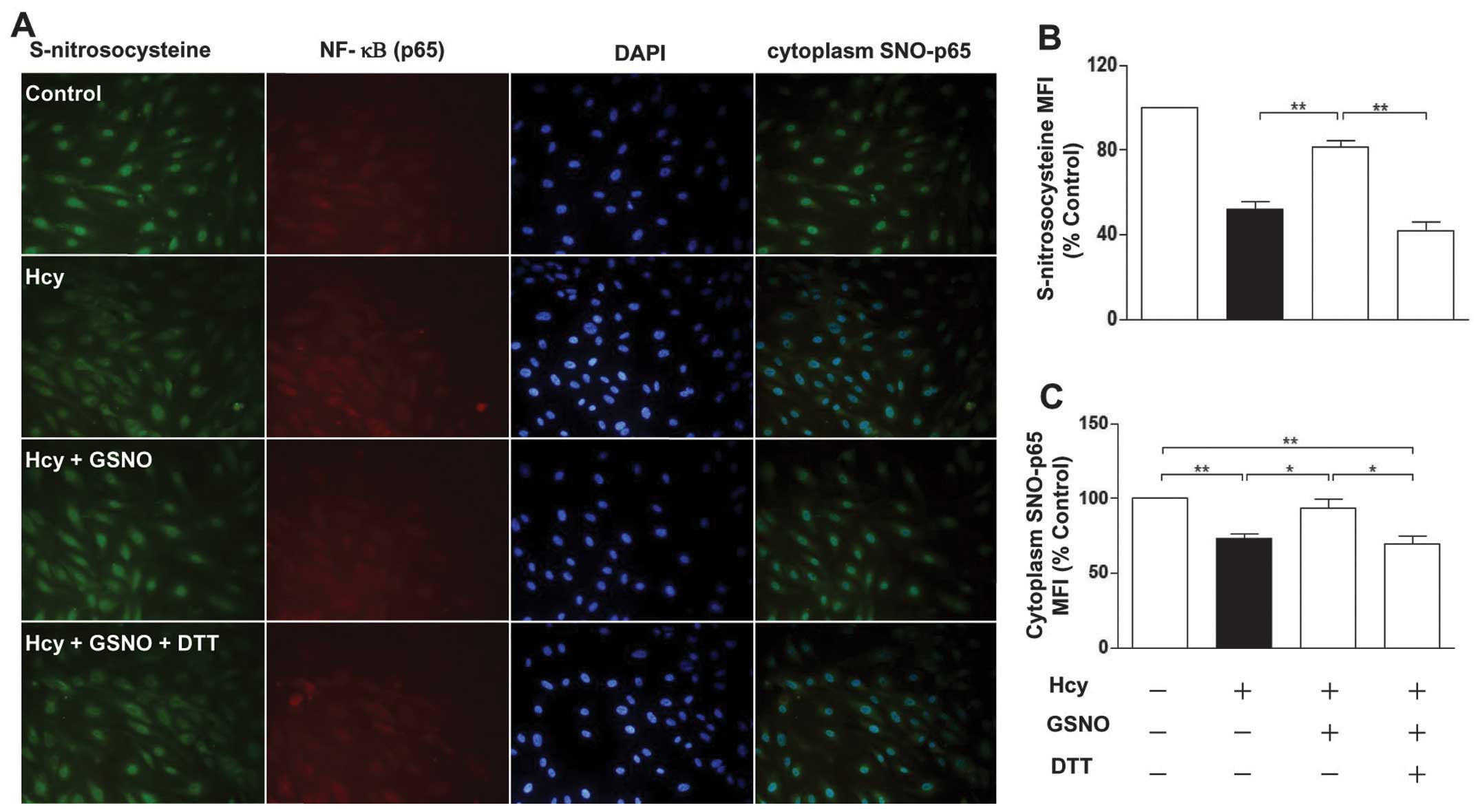
Furthermore, the results showed that protein
S-nitrosylation levels were partially restored in the Hcy-treated
endothelial cells in the presence of the NO donor
S-nitrosoglutathione (GSNO). However, these levels of
S-nitrosylation were abrogated by DTT (Fig. 4A and B). These data suggested that
Hcy reduces protein S-nitrosylation by decreasing the levels of
NO.
Involvement of oxidative stress and the
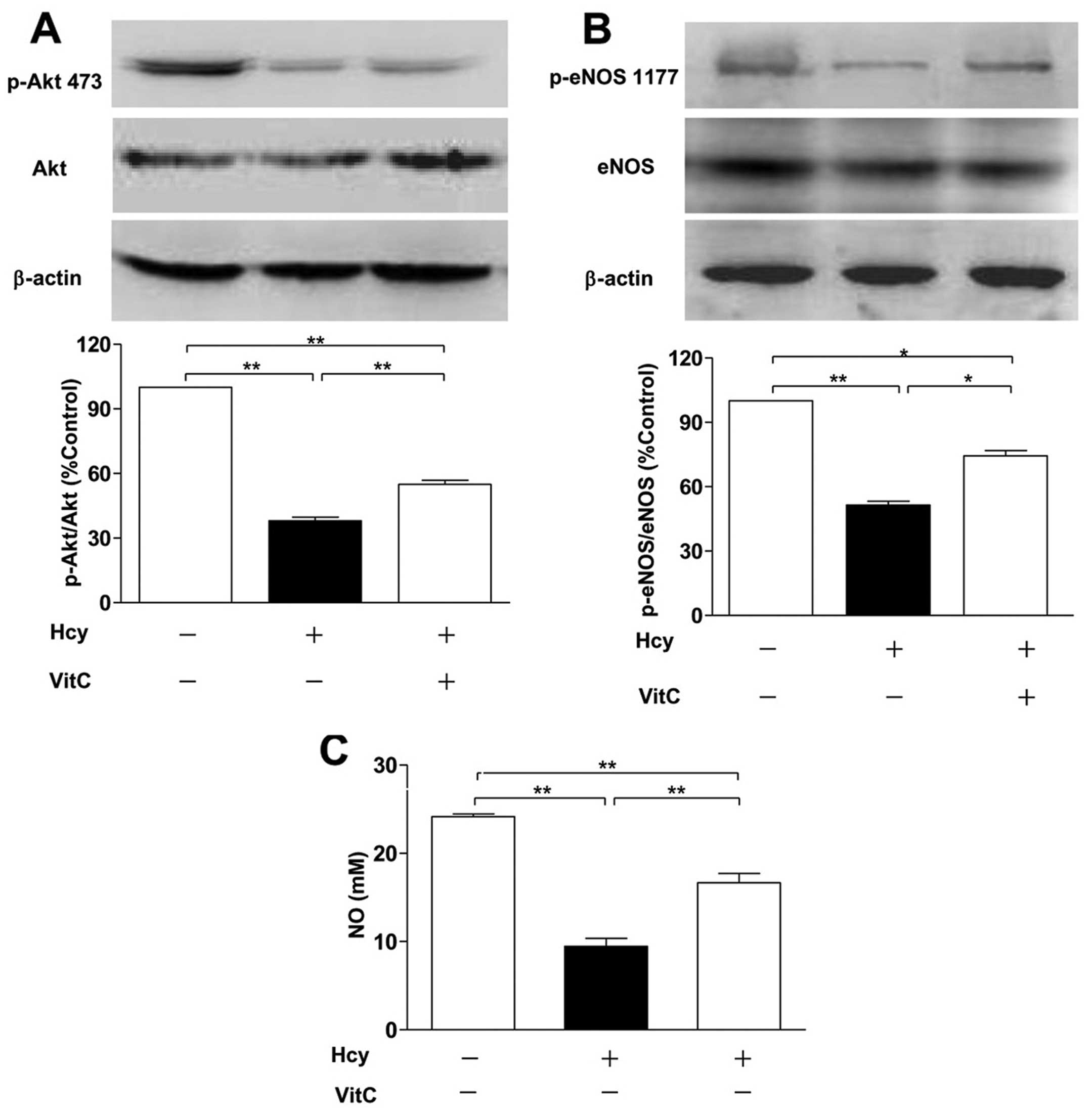
Akt/eNOS/NO signaling pathway
We investigated whether the Akt/eNOS/NO pathway is
involved in Hcy-induced protein S-nitrosylation in endothelial
cells. The protein levels of Akt and eNOS were not affected by Hcy.
(19). Thus, after treatment with
Hcy, we determined the phosphorylation of Akt at Ser-473 and eNOS
at Ser-1177 and the NO levels of cell-free supernatants in HUVECs.
The results showed that Hcy downregulated the phosphorylation of
Akt at Ser-473 and eNOS at Ser-1177 and decreased the levels of NO,
while the effects were partially reversed by the addition of VitC
(Fig. 5A–C). Furthermore, the
restoration of protein S-nitrosylation by VitC was abrogated by the
LY294002 (PI3K/Akt inhibitor) and L-NAME (NOS inhibitor; it
reversibly inhibits eNOS activity) (Fig. 6A–C). These results suggested that
Hcy increases the levels of intracellular ROS and subsequently
decreases NO production by suppressing the Akt/eNOS/NO signaling
pathway, which inhibits protein S-nitrosylation in HUVECs.
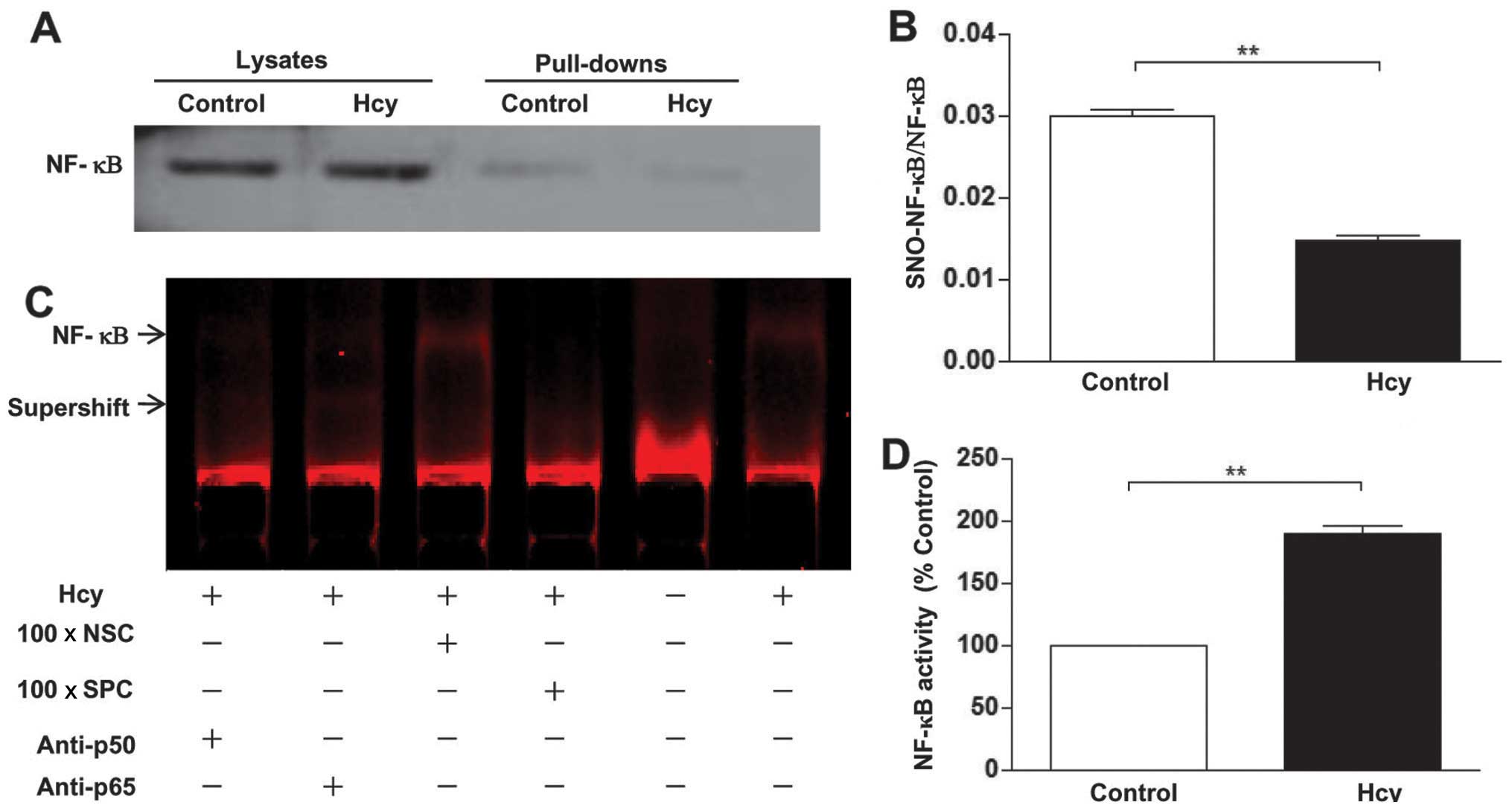
Hcy upregulates NF-κB activity
To investigate whether Hcy-induced protein
S-nitrosylation affects NF-κB activity, we detected the levels of
S-nitrosylation and the activity of NF-κB. The results showed that
Hcy abrogated NF-κB S-nitrosylation, which was accompanied by an
increase in the activity of NF-κB (Fig. 7A–D).
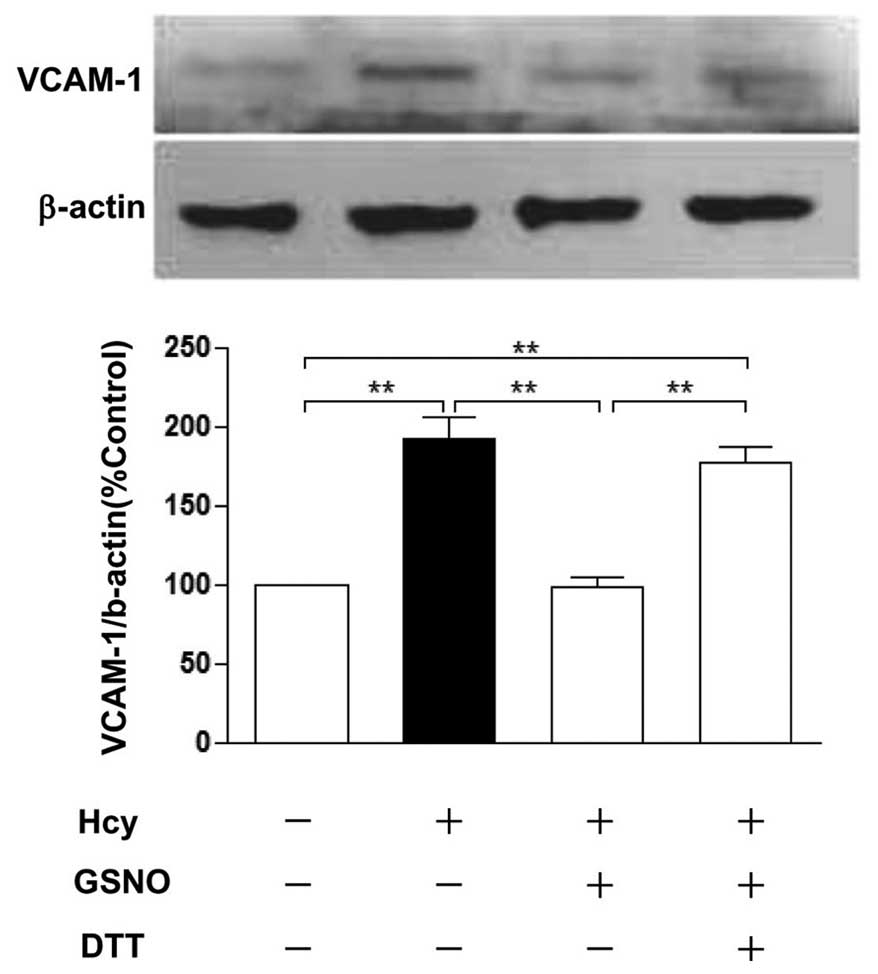
Hcy effect on levels of S-nitrosylation
of p65 and VCAM-1
To determine the relationship between S-nitrosylated
NF-κB (p65) and VCAM-1, our data showed that Hcy increased the
protein expression of VCAM-1, whereas GSNO blocked the increase of
VCAM-1 protein levels induced by Hcy, and DTT restored the
expression of VCAM-1 (Fig. 8).
However, the expression of VCAM-1 is upregulated along with the
levels of cytoplasm S-nitrosylated NF-κB (p65) decreasing (Fig. 4A and B).
Discussion
To the best of our knowledge, the present study has
demonstrated for the first time that, Hcy attenuated the protein
S-nitrosylation levels in HUVECs and an in vivo model.
Attenuation of the S-nitrosylation induced by Hcy was mediated by
the ROS/Akt/eNOS/NO signaling pathway. Hcy induces ROS production,
followed by the inhibition of Akt and eNOS phosphorylation. As a
result, the reduced intracellular NO leads to the inhibition of
protein S-nitrosylation. Moreover, we found that Hcy may upregulate
the expression of VCAM-1 via the reduction of cytoplasm
S-nitrosylation of NF-κB in endothelial cells.
S-nitrosylation is a reversible covalent bonding to
the SH group of a reactive cysteine on the target protein (2). Protein S-nitrosylation is also a
ubiquitous post-translational modification that is responsible for
a broad spectrum of biological functions. In endothelial cells,
S-nitrosylation is able to affect cellular metabolic processes and
regulate vascular functions such as inflammation (6,9),
apoptosis (8), permeability
(20), migration (21), cell growth (22), and vascular stiffness (23). Over 100 S-nitrosylation proteins
have been identified in endothelial cells, many of which are
important in regulating endothelial functions (24). For example, S-nitrosylation can
inhibit the activity of nicotinamide adenine dinucleotide, whereas
it promotes the activity of thioredoxin-1, thus resulting in the
prevention of oxidative stress in endothelial cells (5). S-nitrosylation of p65 or p50
inhibits NF-κB activation, and suppresses inflammation in
endothelial cells (9). Hcy
decreases the levels of NO and induces endothelial dysfunction
(13). In the present study, the
levels of protein S-nitrosylation were reduced by Hcy in HUVECs,
whereas the endogenous NO donor GSNO was able to rescue this
reduction of protein S-nitrosylation. Thus, Hcy reduces the levels
of protein S-nitrosylation by decreasing the levels of NO in
endothelial cells. It was emphasized earlier that low Hcy does not
evidently attenuate protein S-nitrosyaltion in a short period of
time in vitro. However, when the concentration of Hcy was
>0.5 mM, the degree of attenuation regarding S-nitrosylation
protein was only marginally deepened. HHcy also decreased
endothelium S-nitrosyaltion in a rat model. Although plasma Hcy
concentration is low, chronic plasma Hcy stimulation on the
endothelium over a long period of time may be a major cause.
Oxidative stress has been widely accepted as an
important mechanism of Hcy-induced endothelial dysfunction and
damage (25). Hcy leads to the
accumulation of ROS (26). We
found that Hcy significantly increased the levels of ROS, however,
this effect was blocked by VitC and NAC. In addition, VitC and NAC
reversed the reduction of protein S-nitrosylation induced by Hcy,
suggesting that Hcy reduces protein S-nitrosylation levels due to
the excessive generation of ROS. We also observed that Hcy
decreases the production of NO, which is consistent with previous
studies (26). Moreover, the
reduction of NO is blocked by VitC. We hypothesized that Hcy
reduces protein S-nitrosylation through the reduction of NO release
which is induced by the overproduction of ROS. Under certain
circumstances, ROS can also induce the dephosphorylation of Akt at
Ser-473 (26), followed by Akt
inactivation, leading to the dephosphorylation of eNOS at Ser-1177
and the reduction of NO generation in endothelial cells (27). Hcy did not affect the expression
levels of Akt and eNOS, but decreased the levels of phosphorylated
Akt at Ser-473 and eNOS at Ser-1177 in endothelial cells (19). We also found that Hcy may affect
the phosphorylation of the Akt and eNOS proteins. Athough
incubation with VitC increased the phosphorylation levels of Akt
and eNOS proteins, the reduced levels of phosphorylation protein in
Hcy-treated cells did not restore the control levels following
incubation with VitC. This indicates that decreased phosphorylation
levels of the Akt and eNOS proteins did not solely occur due to the
accumulation of ROS. Furthermore, we found that the protein
S-nitrosylation that was partially restored by VitC was abrogated
by LY294002 and L-NAME in the endothelial cells treated with Hcy.
Therefore, the protein S-nitrosylation inhibitory effect of Hcy may
be associated with increased ROS levels together with a blockage of
the Akt/eNOS/NO pathway.
NF-κB is important in regulating endothelial
pro-inflammatory genes, including VCAM-1 (28), while Hcy activates NF-κB via
oxidative stress (29).
Accumulating evidence suggests that NF-κB activity is inhibited by
S-nitrosylation. The p65 component of NF-κB is a target for
S-nitrosylation, which inhibits its translocation into the cell
nucleus (30). The p50 subunit of
NF-κB has also been reported to be S-nitrosylated, causing the
inhibition of its binding to DNA (7,9,31).
In addition, inhibitory κB kinase, the catalytic subunit required
for the activation of NF-κB, was S-nitrosylated, resulting in loss
of its activity (32). Thus, NO
attenuated NF-κB activity through a diverse S-nitrosylation
pathway. Hcy decreased the levels of cytoplasm S-nitrosylated p65,
which may inhibit the translocation of NF-κB into the nucleus. GSNO
rescued Hcy-induced decrease in levels of cytoplasm S-nitrosylated
p65. Furthermore, Hcy enhanced the activity of NF-κB, suggesting
that inhibition of S-nitrosylated p65 is one of numerous pathways
that activate NF-κB following treatment with Hcy. Consequently, the
protein expression of VCAM-1 was also enhanced.
In conclusion, findings of the present study
demonstrate that Hcy attenuates protein S-nitrosylation in
endothelial cells and rat aortas. This may be associated with
increased levels of ROS and a blockage of the Akt/eNOS/NO pathway
and promotion of inflammation. This finding provides new insight
into HHcy-induced vascular damage.
Acknowledgements
This study was partly supported by the National
Natural Science Foundation of China (81070250) and by a Public
Service Platform Grant of Shaanxi Province (2010FWPT-1).
Abbreviations:
|
DCF
|
2′,7′-dichlorofluorescein
|
|
EMSA
|
electrophoretic mobility shift
assay
|
|
eNOS
|
endothelial nitric oxide synthase
|
|
Hcy
|
homocysteine
|
|
HUVECs
|
human umbilical vein endothelial
cells
|
|
HHcy
|
hyperhomocysteinemia
|
|
NAC
|
N-acetylcysteine
|
|
NO
|
nitric oxide
|
|
NF-κB
|
nuclear factor κB
|
|
ROS
|
reactive oxygen species
|
|
GSNO
|
S-nitrosoglutathione
|
References
|
1
|
Martínez-Ruiz A and Lamas S: Signalling by
NO-induced protein S-nitrosylation and S-glutathionylation:
convergences and divergences. Cardiovasc Res. 75:220–228.
2007.PubMed/NCBI
|
|
2
|
Lima B, Forrester MT, Hess DT and Stamler
JS: S-nitrosylation in cardiovascular signaling. Circ Res.
106:633–646. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chakrabarti S, Lekontseva O, Peters A and
Davidge ST: 17beta-Estradiol induces protein S-nitrosylation in the
endothelium. Cardiovasc Res. 85:796–805. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sun J, Picht E, Ginsburg KS, Bers DM,
Steenbergen C and Murphy E: Hypercontractile female hearts exhibit
increased S-nitrosylation of the L-type Ca2t channel alpha1 subunit
and reduced ischemia/reperfusion injury. Circ Res. 98:403–411.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Selemidis S, Dusting GJ, Peshavariya H,
Kemp-Harper BK and Drummond GR: Nitric oxide suppresses NADPH
oxidase-dependent superoxide production by S-nitrosylation in human
endothelial cells. Cardiovasc Res. 75:349–358. 2007. View Article : Google Scholar
|
|
6
|
Kang-Decker N, Cao S, Chatterjee S, Yao J,
Egan LJ, Semela D, Mukhopadhyay D and Shah V: Nitric oxide promotes
endothelial cell survival signaling through S-nitrosylation and
activation of dynamin-2. J Cell Sci. 120:492–501. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Marshall HE and Stamler JS: Inhibition of
NF-kappa B by S-nitrosylation. Biochemistry. 40:1688–1693. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hoffmann J, Haendeler J, Zeiher AM and
Dimmeler S: TNFalpha and oxLDL reduce protein S-nitrosylation in
endothelial cells. J Biol Chem. 276:41383–41387. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wadham C, Parker A, Wang L and Xia P: High
glucose attenuates protein S-nitrosylation in endothelial cells:
role of oxidative stress. Diabetes. 58:2715–2721. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hoffmann J, Dimmeler S and Haendeler J:
Shear stress increases the amount of S-nitrosylated molecules in
endothelial cells: important role for signal transduction. FEBS
Lett. 551:153–158. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen SC, Huang B, Liu YC, Shyu KG, Lin PY
and Wang DL: Acute hypoxia enhances proteins’ S-nitrosylation in
endothelial cells. Biochem Biophys Res Commun. 377:1274–1278.
2008.
|
|
12
|
Hankey GJ and Eikelboom JW: Homocysteine
and vascular disease. Lancet. 354:407–413. 1999. View Article : Google Scholar
|
|
13
|
Suematsu N, Ojaimi C, Kinugawa S, et al:
Hyperhomocysteinemia alters cardiac substrate metabolism by
impairing nitric oxide bioavailability through oxidative stress.
Circulation. 115:255–262. 2007. View Article : Google Scholar
|
|
14
|
Ungvari Z, Sarkadi-Nagy E, Bagi Z, Szollár
L and Koller A: Simultaneously increased TxA(2) activity in
isolated arterioles and platelets of rats with
hyperhomocysteinemia. Arterioscler Thromb Vasc Biol. 20:1203–1208.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Forrester MT, Foster MW, Benhar M and
Stamler JS: Detection of protein S-nitrosylation with the
biotin-switch technique. Free Radic Biol Med. 46:119–126.
2009.PubMed/NCBI
|
|
16
|
Ota H, Eto M, Kano MR, Ogawa S, Iijima K,
Akishita M and Ouchi Y: Cilostazol inhibits oxidative
stress-induced premature senescence via upregulation of Sirt1 in
human endothelial cells. Arterioscler Thromb Vasc Biol.
28:1634–1639. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu Y, Zhang W, Liu W, Zhuo X, Zhao Z and
Yuan Z: The double-faced metabolic and inflammatory effects of
standard drug therapy in patients after percutaneous treatment with
drug-eluting stent. Atherosclerosis. 215:170–175. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Michaud SE, Dussault S, Groleau J, Haddad
P and Rivard A: Cigarette smoke exposure impairs VEGF-induced
endothelial cell migration: role of NO and reactive oxygen species.
J Mol Cell Cardiol. 41:275–284. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lan TH, Xu ZW, Wang Z, Wu YL, Wu WK and
Tan HM: Ginsenoside Rb1 prevents homocysteine-induced endothelial
dysfunction via PI3K/Akt activation and PKC inhibition. Biochem
Pharmacol. 82:148–155. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Thibeault S, Rautureau Y, Oubaha M,
Faubert D, Wilkes BC, Delisle C and Gratton JP: S-nitrosylation of
beta-catenin by eNOS-derived NO promotes VEGF-induced endothelial
cell permeability. Mol Cell. 39:468–476. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pi X, Wu Y, Ferguson JE III, Portbury AL
and Patterson C: SDF-1alpha stimulates JNK3 activity via
eNOS-dependent nitrosylation of MKP7 to enhance endothelial
migration. Proc Natl Acad Sci USA. 106:5675–5680. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Oliveira CJ, Curcio MF, Moraes MS, Tsujita
M, Travassos LR, Stern A and Monteiro HP: The low molecular weight
S-nitrosothiol, S-nitroso-N-acetylpenicillamine, promotes cell
cycle progression in rabbit aortic endothelial cells. Nitric Oxide.
18:241–255. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Santhanam L, Tuday EC, Webb AK, et al:
Decreased S-nitrosylation of tissue transglutaminase contributes to
age-related increases invascular stiffness. Circ Res. 107:117–125.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Martínez-Ruiz A and Lamas S: Detection and
proteomic identification of S-nitrosylated proteins in endothelial
cells. Arch Biochem Biophys. 423:192–199. 2004.
|
|
25
|
McCully KS: Chemical pathology of
homocysteine. IV Excitotoxicity, oxidative stress, endothelial
dysfunction, and inflammation. Ann Clin Lab Sci. 39:219–232.
2009.PubMed/NCBI
|
|
26
|
Chavakis E, Dernbach E, Hermann C, Mondorf
UF, Zeiher AM and Dimmeler S: Oxidized LDL inhibits vascular
endothelial growth factor-induced endothelial cell migration by an
inhibitory effect on the Akt/endothelial nitric oxide synthase
pathway. Circulation. 103:2102–2107. 2001. View Article : Google Scholar
|
|
27
|
Dimmeler S, Fleming I, Fisslthaler B,
Hermann C, Busse R and Zeiher AM: Activation of nitric oxide
synthase in endothelial cells by Akt-dependent phosphorylation.
Nature. 399:601–605. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Carluccio MA, Ancora MA, Massaro M, et al:
Homocysteine induces VCAM-1 gene expression through NF-kappaB and
NAD(P)H oxidase activation: protective role of Mediterranean diet
polyphenolic antioxidants. Am J Physiol Heart Circ Physiol.
293:H2344–H2354. 2007. View Article : Google Scholar
|
|
29
|
Au-Yeung KK, Woo CW, Sung FL, Yip JC, Siow
YL and OK: Hyperhomocysteinemia activates nuclear factor-kappaB in
endothelial cells via oxidative stress. Circ Res. 94:28–36. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kelleher ZT, Matsumoto A, Stamler JS and
Marshall HE: NOS2 regulation of NF-kappaB by S-nitrosylation of
p65. J Biol Chem. 282:30667–30672. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Marshall HE, Hess DT and Stamler JS:
S-nitrosylation: physiological regulation of NF-kappaB. Proc Natl
Acad Sci USA. 101:8841–8842. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Reynaert NL, Ckless K, Korn SH, et al:
Nitric oxide represses inhibitory kappaB kinase through
S-nitrosylation. Proc Natl Acad Sci USA. 101:8945–8950. 2004.
View Article : Google Scholar : PubMed/NCBI
|