Introduction
Parkinson’s disease (PD) is a common
neurodegenerative disorder characterized by the gradually
progressive and selective loss of dopaminergic neurons in the
substantia nigra (1). The
progressive loss of dopaminergic neurons is a complex process, and
multiple pathological events are involved in this process (2–6).
While the underlying mechanisms of nigrostriatal dopaminergic
neuron degeneration are not yet completely understood, accumulating
evidence indicates that mitochondrial dysfunction may be a central
event in neurodegenerative diseases (7–9).
The mitochondria are multifunctional organelles that are important
for living cells. Mitochondrial dysfuntion has a multitude of
consequences for cells, including apoptosis (10). The activation of the mitochondrial
permeability transition pore (mPTP) and the collapse of the
mitochondrial membrane potential may be major contributors to
mitochondrial-dependent cell death and at least partly reponsible
for the pathogenesis of PD and several other neurodegenerative
disorders (7–9).
PC12 cells treated with 1-methyl-4-phenylpyridinium
(MPP+) provide a reliable in vitro model for
investigating the pathogenesis of PD. MPP+ is an active
metobolite of the neurotoxin,
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which is known
to selectively kill dopaminergic neurons and cause irreversible
Parkinson-like symptoms in humans and primates (11–13). MPTP is a lipophilic molecule that
can rapidly cross the blood-brain barrier; it is subsequently
oxidized in the brain to its toxic metabolite, MPP+, by
type B monoamine oxidase (14).
The neurotoxic action of MPP+ is related to the
activation of the mPTP and the collapse of mitochondrial membrane
potential through oxidative damage, which together initiate the
downstream apoptotic pathway, including the release of cytochome c
and the activation of caspases, finally leading to neuronal cell
death (7–9). Damage to the mitochondria is
considered as an initial and irreversible step towards apoptosis;
thus, mitochondrial-targeted therapeutic strategies may be a
promising treatment for PD. Guanosine, a non-adenine-based purine,
is an intercellular signaling molecule affecting multiple cellular
processes, including cellular growth, differentiation and survival
(15–17). In multiple cell types, it exerts
protective effects against apoptosis induced by a number of agents,
such as staurosporine (18),
β-amyloid (19) and MPTP
(20). The neuroprotective
effects of guanosine in the central nervous system have also been
recognized (15). The present
study was designed to investigate the effects of guanosine on
MPP+-induced apoptosis in PC12 cells and the underlying
mechanisms for these actions. Our results demonstrated that
guanosine effectively prevented MPP+-induced PC12 cell
apoptosis by stabilizing the mitochondrial membrane potential and
attenuating the subsequent activation of caspases. In addition,
guanosine inhibited the production of reactive oxygen species (ROS)
and increased the expression levels of glutathione (GSH), further
supporting the protective role of guanosine in oxidative
conditions. Overall, these findings indicate the protective role of
guanosine in mitochondrial stress-induced dopaminergic neuronal
damage, thus providing potential effective strategies for the
treatment of PD.
Materials and methods
Drugs and chemicals
All reagents and chemicals were purchased from
Sigma-Aldrich (St. Louis, MO, USA) unless stated otherwise.
PC12 cell cultures
The PC12 cells were obtained from the Cell Bank of
the Chinese Academy of Sciences (Shanghai, China) and maintained in
high glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented
with 10% heat-inactivated fetal bovine serum, 4.00 mM L-glutamine,
100 U/ml of penicillin and 100 μg/ml of streptomycin (Gibco, Grand
Island, NY, USA). The cultures were maintained in a humidified 5%
CO2 atmosphere at 37°C. The culture medium was changed
every 3–4 days and the cells were seeded at a density of 30,000
cells/cm2.
Cell viability assay
Cell viability was measured using the modified
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
a mitochondrial dye which is converted into a blue formazan product
by mitochondrial dehydrogenases in metabolically active cells. The
PC12 cells were plated at a density of 30,000 cells/cm2
in 96-well plates and incubated for 24 h. To assess the
neuroprotective effects of guanosine on MPP+-induced
toxicity in PC12 cells, the cells were pre-treated with various
concentrations of guanosine (0.01–1,000 μM) for 3 h and were then
exposed to 500 μM MPP+ for 72 h, under optimal
conditions for the assessment of the neuroprotective effects as
previously described (21). MTT
(5 mg/ml) solution was added to the wells and the cells were
incubated for 4 h. Subsequently, the culture medium was removed,
and dimethyl sulfoxide was added to each well to solubilize the
formazan into a colored solution. The absorbance of colored
solution was measured at 570 nm using a microplate reader (Epoch;
BioTek, Winooski, VT, USA). The results were expressed as the
percentage of the absorbance of the control culture wells. Based on
these results, we used guanosine at a dose of 10 μM in all the
subsequent experiments.
Nuclear staining assay
The morphological signs of apoptosis induced by
MPP+ were detected using acridine orange (AO)/ethidium
bromide (EB) staining of the PC12 cells. The cells were plated in
6-well plates at a density of 30,000 cells/cm2 and were
incubated in DMEM medium at 37°C. After 3 h of pre-treatment with
guanosine (10 μM), MPP+ (500 μM) was added to the medium
for 72 h. The cells were washed and resuspended in
phosphate-buffered saline (PBS) and AO/EB was added at a final
concentration of 1 μg/ml. Subsequently, the number of apoptotic
cells was randomly counted under a fluorescence microscope (IX71;
Olympus, Tokyo, Japan). Viable cells with intact structures stained
with AO only showed bright green nuclear staining; the early
apoptotic cells were bright green and later apoptotic cells were
red-orange with condensed chromatin. The number of apoptotic cells
is expressed as a percentage of the total cells counted.
Measurement of apoptosis in cells
Apoptosis was assessed by measuring DNA
fragmentation with single-stranded DNA (ssDNA) apoptosis
enzyme-linked immunosorbent assay (ELISA) kits (Chemicon
International, Temecula, CA, USA) according to the manufacture’s
instructions. The cells plated at a concentration of 30,000
cells/cm2 were cultured for 24 h, followed by treatment
with 10 μM guanosine prior to the addition of 500 μM
MPP+ for 3 h. The cells were washed 3 times with PBS and
formamide was then added which selectively denaturates DNA in
apoptotic cells. Anti-ssDNA monoclonal antibody and
peroxidase-conjugated secondary antibody were then added to the
cells; the ssDNA was then measured at 450 nm using a microplate
reader (Epoch; BioTek).
Measurement of mitochondrial
transmembrane potential
Mitochondrial membrane potential is a key indicator
of mitochondrial function and cell death or injury, which can be
detected using the mitochondrial dye, 3,3-dihexyloxacarbocyanine
iodide [DiOC6(3)]. This dye is a lipophilic fluorescent
stain and becomes highly fluorescent when incorporated into
membranes. The cells at a concentration of 30,000
cells/cm2 were cultured in 24-well plates for 24 h,
followed by treatment with 10 μM guanosine prior to the addition of
500 μM MPP+ for 3 h. Following 72 h of incubation, 1 ml
of serum-free culture medium containing DiOC6(3) was
added to each well with the final concentration of 1 μM, and the
cells were cultured in a humidified incubator for 15 min. The cells
were collected and centrifuged at 1,000 × g for 5 min, and the cell
pellets were resuspended in PBS containing 0.5 mM EDTA. The
intensity of DiOC6(3) fluorescence was recorded using a
flow cytometer (Becton-Dickinson, San Diego, CA, USA).
Western blot analysis
Following treatment, the PC12 cells were collected
and lysed with cell lysis solution containing 4% sodium dodecyl
sulfate (SDS), 2 mM EDTA and 50 mM Tris-HCl, pH 6.8. Equal amounts
of protein were loaded onto a 12% SDS-polyacrylamide gel. Following
electrophoretic separation, the polyacrylamide gels were
transferred onto PVDF transfer membranes (Amersham Biosciences,
Uppsala, Sweden). The membranes were incubated in Tris-buffered
saline/Tween-20 (TBST) supplemented with 5% fat-free milk for 1 h
to block non-specific binding. The blots were incubated using
rabbit anti-Bax, anti-B-cell lymphoma 2 (Bcl-2) antibodies.
Horseradish peroxidase (HRP)-conjugated anti-rabbit antibodies were
used as the secondary antidodies.
Measurement of ROS production
Intracellular ROS produced during the inhibition of
mitochondrial complex I was detected using
2′–7′-dichlorofluorescein diacetate (DCFH-DA). This is a
non-fluorescent cell-permeating compound that can easily diffuse
into cells and be converted into dichlorofluorescin (DCFH) by
intracellular esterase. DCFH is then trapped within the cell and
oxidized into fluorescent dichlorofluorescein (DCF) by
intracellular ROS. Following treatment, the cells were incubated in
BSA-free DMEM with DCFH-DA at a final concentration of 20 μM for 30
min at 37°C. Thereafter, 10,000 cells of each group were analyzed
by flow cytometry using the FL1 flow cytometer detection channels.
The excitation wavelength was 485 nm and the reading was performed
at 530 nm.
Measurement of GSH levels
GSH levels were measured using GSH reductase, as
previously described (22).
Briefly, following centrifugation and washing with PBS, the cells
were dissovled with 2% 5-sulfosalicylic acid and incubated in 100
μl of the reaction mixture containing 20 mM sodium EDTA, 600 μM
nicotinamide adenine dinucleotide phosphate (NADPH), 12 mM
5,5′-dithiobis(2-nitrobenzoic acid) and 105 mM
NaH2PO4. GSH reductase was added to each
well, and the cells were cultured in a humidified incubator for 10
min. Absorbance was measured at 450 nM, and the calibration curve
was performed with standard GSH solutions. The results are
expressed as percentages of the control condition.
Evaluation of caspase-3 activity
Caspase-3 activity was measured using an ApoAlert
caspase-3 assay kit according to the manufacturer’s instructions.
Briefly, the cells were lysed and centrifuged at 1,000 × g for 10
min, then the supernatant was added to the reaction mixture
containing dithiothreitol and caspase-3 substrate
(N-acetyl-Asp-Glu-Val-Asp p-nitroanilide). The cells were incubated
for 1 h at 37°C, and the absorbance of the chromophore
p-nitroanilide produced was measured at 450 nm. The standard curves
were obtained from the absorbance of p-nitroanilide standard
reagent diluted with cell lysis buffer. One unit of the enzyme was
defined as the activity producing 1 nmol of p-nitroanilide.
Statistical analysis
Data are expressed as the means ± standard error of
the mean (SEM). Statistical analysis was performed by one-way
analysis of variance, followed by Dunnett’s multiple-comparisons
test. Differences between mean values were considered statistically
different at p<0.05.
Results
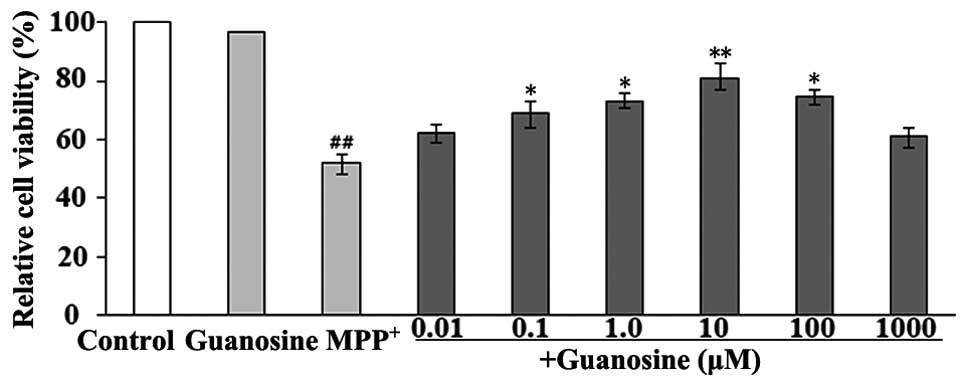
Guanosine reduces the
MPP+-induced loss of cell viability
The ability of guanosine to reverse the cytotoxicity
to PC12 cells induced by MPP+ was investigated using
MTT, which is a mitochondrial dye and can be converted into a blue
formazan product by mitochondrial dehydrogenases; therefore, it can
partially detect the levels of metabolically active cells. The
measurements revealed a significant decrease in the viability of
the PC12 cells following exposure to 500 μM MPP+ for 72
h; however, the cells treated with guanosine alone did not show a
decrease in cell viability. Pre-treatment with 10 μM guanosine
significantly decreased the MPP+-induced cytotoxicity
(Fig. 1).
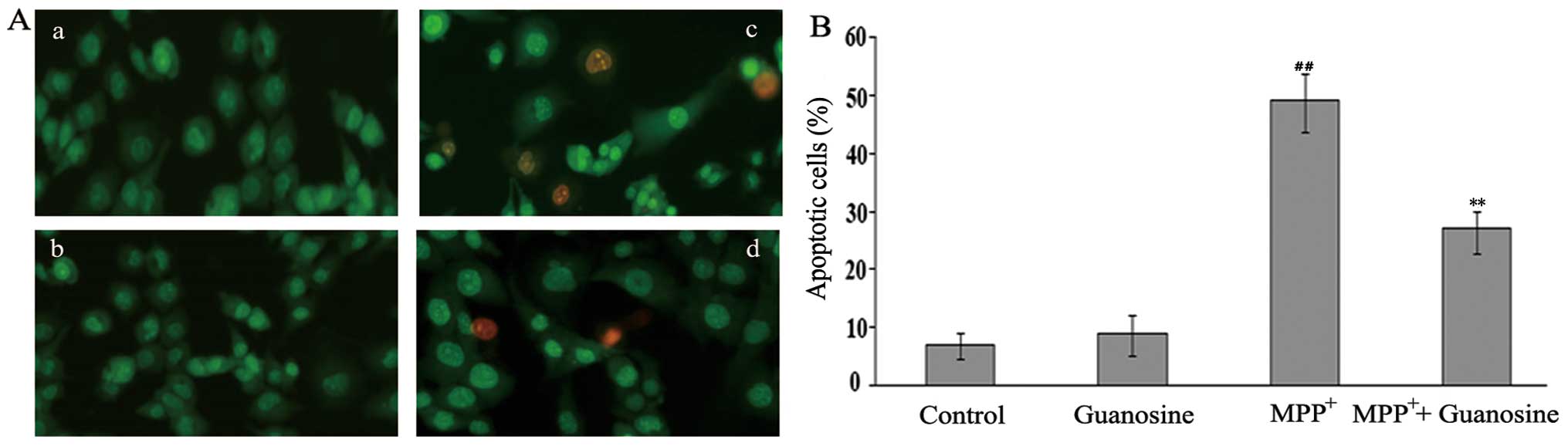
Guanosine attenuates
MPP+-induced apoptosis in PC12 cells
To determine whether guanosine prevents
MPP+-induced apoptosis in PC12 cells, AO/EB and DNA
fragmentation assays were performed. Apoptosis is a process of
programmed cell death characterized by a series of distinct nuclear
morphological changes. These changes can be detected by AO/EB
staining. This assay identified 3 types of cells under a
fluorescence microscope: live cells (green), early apoptotic cells
(bright green with condensed chromation) and later apoptotic cells
(red-orange with condensed chromation). The administration of
guanosine alone did not induce changes in the number of apoptotic
cells, while the administration of MPP+ significantly
increased the number of apoptotic cells compared to the control
group (p<0.01). Pre-treatment with 10 μM guanosine significantly
decreased the number of apoptotic cells induced by exposure to
MPP+ (p<0.01; Fig.
2), indicating that guanosine plays an anti-apoptotic role. To
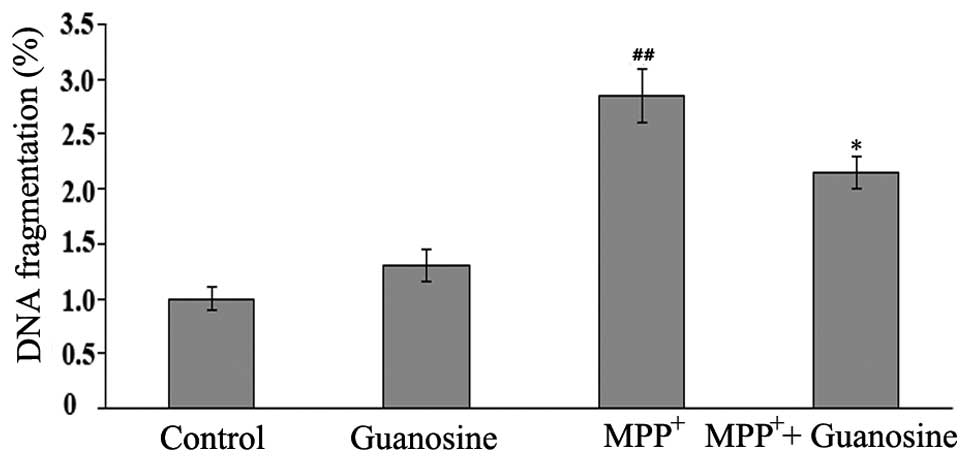
clarify the neuroprotective role of guanosine on
MPP+-induced toxicity in PC12 cells, DNA fragmentation,
a marker of late apoptosis, was further investigated by ssDNA
assay. The results revealed that the increase in DNA fragmentation
induced by exposure to MPP+ was markedly attenuated by
pre-treatment with guanosine (Fig.
3), supporting the protective role of guanosine in conditions
of oxidative stress.
Guanosine modulates Bax and Bcl-2 protein
expression
Bax and Bcl-2 are key members of the Bcl-2 family of
proteins that contribute to the opening of mPTP, leading to the
induction of apoptosis. To investigate the changes in Bax and Bcl-2
protein expression levels, western blot analysis was performed on
the untreated cells and the cells treated with 500 μM
MPP+ alone or 500 μM MPP+ in the presence of
10 μM of guanosine. The administration of MPP+
significantly increased the levels of Bax expression and decreased
Bcl-2 expression. These changes were be markedly reversed by
pre-treatment with guanosine. Treatment with guanosine alone did
not induce changes in the expression levels of these proteins
(Fig. 4), thus further
demonstrating the protective role of guanosine in
mitochondrial-stress induced cell damage.
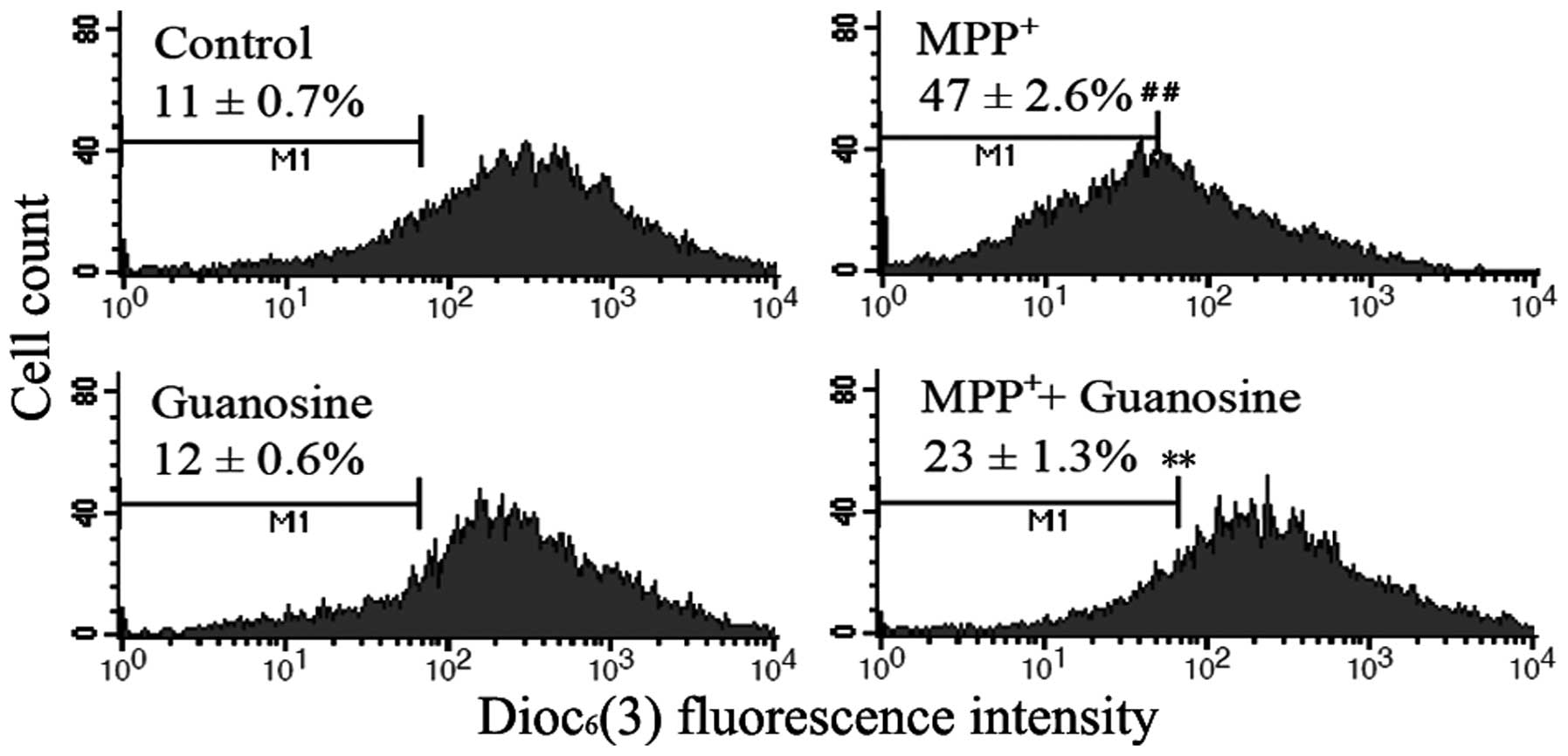
Guanosine prevents the
MPP+-induced collapse of mitochondrial transmembrane
potential
Mitochondrial membrane potential maintenance is
essential for living cells, and its collapse is a key event in the
activation of the mitochondrial-dependent pathway. The collapse of
mitochondrial transmembrane potential was assessed by measuring the
response to the mitochondrial dye, DiOC6(3), which is
converted into a highly green fluorescent dye following
incorporation into mitochondrial membranes, thereby allowing the
qualitative assessment of mitochondrial membrane potential. The
administration of MPP+ in comparison with the control
cells induced a significant decrease in fluorescence intensity,
indicating the increasing percentage of the cells with collapse of
mitochondrial membrane potential. The results also revealed a
marked reduction in the number of cells with the collapse of
mitochondrial membrane potential, when guanosine was administered
prior to exposure to MPP+; no significant change was
observed following treatment with guanosine alone (Fig. 5). These results suggest that the
mitochondrial dysfunction induced by MPP+ can be partly
restored by the administration of guanosine.
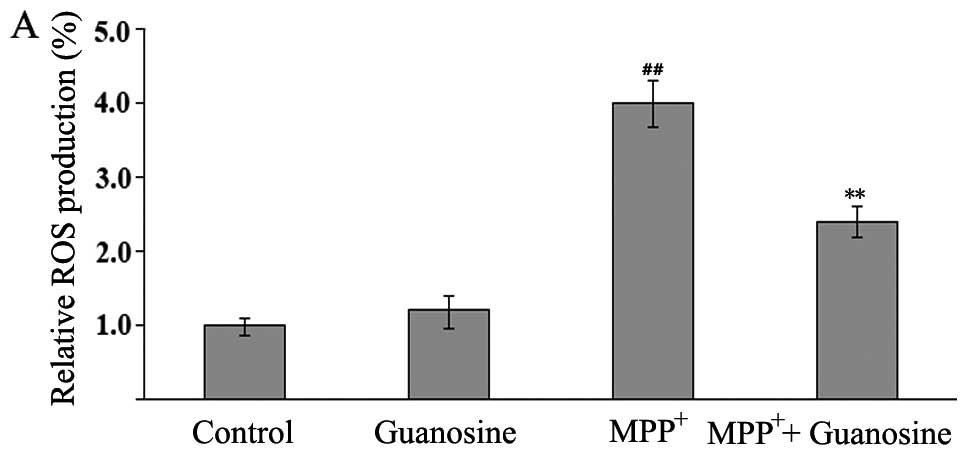
Guanosine inhitis the
MPP+-induced production of ROS
The levels of ROS production were evaluated by flow
cytometry with DCFH-DA. DCFH-DA is a stable compound that can
easily diffuse into cells, where it is converted into DCFH by
intracellular esterase. DCFH is then trapped within cells and
oxidized to highly fluorescent DCF by intracellular ROS; thus, the
intensity of fluorescence produced by DCF may reflect an
intracellular oxidative state.
The administration of guanosine alone, compared with
the control group, did not elicit changes in the levels of DCFH
oxidation. The administration of MPP+ induced a
significant increase in DCFH oxidation in the PC12 cells, which was
markedly reversed by pre-treatment with guanosine (Fig. 6A), thus indicating that guanosine
may play an antioxidant role.
Guanosine reverses the
MPP+-induced reduction in GSH levels
GSH protein is a major non-enzymatic antioxidant
that plays a crucial role in protecting neurons from oxidative
damage in the central nervous system (23). To assess the protective role of
guanosine in MPP+-induced oxidative damage, the levels
of GSH were measured in the PC12 cells. The administration of
MPP+ in comparison with the control cells induced a
significant decrease in GSH levels; this effect was markedly
reversed by pre-treatment with guanosine. The administration of
guanosine alone did not elicit any changes in the levels of GSH
(Fig. 6B).
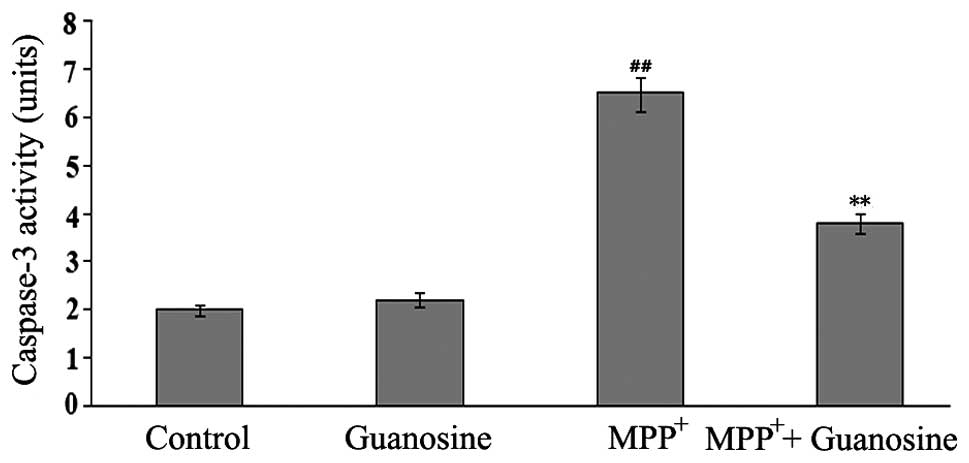
Guanosine reduces caspase-3 activity
Caspase-3 is an effector caspase that cleaves a wide
range of signal transduction proteins in the apoptotic process
(24). To determine whether
guanosine protects neuronal PC12 cells against
MPP+-induced cell death, the activity of caspase-3 was
measured by ELISA with an ApoAlert caspase-3 assay kit. The PC12
cells exposed to 500 μM MPP+ showed a significant
increase in caspase-3 activity; however, treatment with guanosine
alone did not induce any changes in caspase-3 activity.
Pre-treatment with guanosine markedly inhibited the
MPP+-induced the increment in caspase-3 activity
(Fig. 7), illustrating the
protective role of guanosine against MPP+-induced
toxicity in PC12 cells.
Discussion
The non-adenine-based purine, guanosine, is a
multifaceted intercellular signaling molecule affecting multiple
cellular processes, including cellular growth, differentiation and
survival (15). Its protective
roles have been reported in previous studies. It protects several
cell types against apoptosis induced by a number of agents, such as
staurosporine, β-amyloid and MPP+ through its
interactions with several steps of the biochemical and cellular
cascade (18,19). The protective role of guanosine
has also been reported in neurodegenerative diseases (20). The present study demonstrates that
guanosine exerts protective effects against apoptotic cell death
elicited by MPP+ by alleviating mitochondrial
dysfunction, inhibiting the activation of caspase-3 and,
subsequently, attenuating cytotoxic cell damage in a reliable
cellular model of PD.
PD is a common neurodegenerative disease clinically
characterized by rigidity, resting tremor, bradykinesia and
postural instability caused by the degeneration and death of
dopaminergic neurons in the pars compacta of the substantia nigra
(25). Althought the cellular and
molecular events underlying the loss of dopaminergic neurons remain
unclear, accumulating evidence indicates that mitochondrial
dysfunction may be a central event in the pathogenesis of PD
(2,8). The mitochondria are the most
important cytoplasmic organelles responsible for the life and death
of cells (26). The maintenance
of membrane potential and the low-conductance of the mPTP are the
major properties of mitochondria in living cells, and any changes
related to these properties are considered to be critical factors
associated with mitochondrial dysfunction and cell death in
neurodegenerative diseases (7,8).
Multiple proteins are involved in intrinsic apoptotic events
associated with mitochondrial dysfunction. The Bcl-2 family of
proteins are recognized as key messengers for delivering the
apoptotic signal to the mitochondria in response to various insults
(27). Pro-apoptotic Bax and
anti-apoptotic Bcl-2 are key members of the Bcl-2 family in
apoptosis mediated by mitochondrial stress. Under pathogenic
conditions, Bax is upregulated and translocates from the cytoplasm
to the mitochondria. Once located in the mitochondrial membrane,
this protein causes mitochondrial membrane disruption by
sequestering Bcl-2 and oligomerizing within the mitochondrial
membrane, leading to the opening of the mPTP, the collapse of
mitochondrial membrane potential and the subsequent release of
pro-apoptotic molecules into the cytoplasm (28,29). Compared to Bax, Bcl-2 is a key
protein that preserves mitochondrial integrity, thereby preventing
stress-induced mitochondrial damage in cells (30). Bcl-2 proteins are crucial
effectors in the opening of the mPTP and the collapse of
mitochondrial potential, thus determining the induction of
downstream events in the mitochondrial-dependent cell death
pathway, including the release of pro-apoptotic molecules and the
activation of caspases (28).
Our results revealed that treatment with
MPP+ induced the adverse expression levels of two Bcl-2
proteins and the disruption of the mitochondrial membrane
potential, supporting the involvement of mitochondrial dysfunction
in dopaminergic neuronal degeneration. These changes were reversed
by the administration of guanosine prior to exposure to
MPP+, demonstrating the protective role of guanosine in
mitochondrial-stress induced cell damage, which was partly mediated
through the regulation of the expression of proteins involved in
the mitochondrial stage of the apoptotic cascade. However, the
underlying mechanism responsible for this effect of guanosine is
unclear.
A number of studies have indicated that the
anti-apoptotic effects of guanosine are mediated by modulating the
phosphatidylinositol 3-kinase (PI3K)/Akt/protein kinase B (PKB) and
the mitogen-activated protein kinase (MAPK) cell survival pathways
(18,19,31,32). PI3K is an upstream signal of
glycogen synthase kinase 3 (GSK-3) that plays a central role in the
mitochondrial-dependent cell death pathway through the regulation
of anti-apoptotic and pro-apoptotic Bcl-2 family proteins,
including Bcl-2 and Bax (33–35). GSK-3β can directly phosphorylate
Bax on serine 163, which results in the activation of Bax, and,
subsequently, in its translocation from the cytoplasm to the
mitochondria. The inhibition of GSK-3β suppresses the levels of Bax
expression, but increases Bcl-2 expression, thereby promoting cell
survival by alleviating the mitochondrial dysruption under multiple
pathological conditions (34,36,37). Thus, the neuroprotective effects
of guanosine may be mediated through the activation of PI3K, which
inactivates the downstream signal protein, GSK-3β, leading to the
attenuation of the opening of the mPTP through the regulation of
Bcl-2 family proteins. This hypothesis is surported by our results
that guanosine reversed the collapse of mitochondrial membrane
potential, the downstream event of the opening of the mPTP in
mitochondrial-mediated cell death. The opening of the mPTP causes
the collapse of mitochondrial membrane potential and, subsequently,
the release of apoptotic proteins from the mitochondria into the
cytoplasm. Perhaps the most intriguing pro-apoptotic protein that
is released is cytochrome c, which triggers the activation
of the caspase cascade (28).
Caspase-3 is a key effector in the mitochondrial-stress-induced
apoptotic pathway, and its activation leads to the irreversible
process toward apoptosis (38,39). Our results also demonstrated that
guanosine, when administered to MPP+-treated neuronal
PC12 cells, effectively prevented the collapse of mitochondrial
potential and inhibited caspase-3 activity, further supporting its
protective role in mitichondrial stress-induced neuronal cell
damage.
Oxidative stress is another pathological event
associated with cell death mechanisms in PD. Studies using
postmortem samples of PD have demonstrated that oxidative markers,
including soluble protein carbonyl modifications, lipid
peroxidation and DNA oxidative damage are selectively observed in
the dopaminergic neurons in the pars compacta of the substantia
nigra, indicating the correlation of oxidative damage with striatal
dopaminergic neurodegeneration (4,5,40).
The inhibition of mitochondrial complex I with MPP+ and
rotenone, well-established inducers of Parkinson-like symptoms in
humans and primates, can lead to an increase in ROS production and
selective dopaminergic neuronal loss in the substantia nigra,
supporting the involvement of oxidative stress in the pathogenesis
of PD (41). ROS are mainly
produced as by-products of oxidative phosphorylation in the
mitochondria, and many mitochondrial proteins, which possess
iron-sulfur clusters for oxidation-reduction reactions and lack
protective histones, are particularly vulnerable to ROS attack
(42). Generally, cells develop
complex antioxidant systems to scavenge ROS. The GSH protein is
recognized as the major non-enzymatic antioxidant in the central
nervous system (23). A number of
studies have indicated that increased levels of GSH exert
protective effects in various neurodegenerative diseases, such as
Alzheimer’s disease and PD (22,43,44). The reduction of GSH levels
contributes to the dysfunction of the mitochondria and increases
the sensitivity of neurons to toxic insults (45). The neurotoxin, MPP+, is
an inhibitor of the mitochondrial respiratory chain and an inducer
of ROS in the mitochondria. Our results revealed that pre-treatment
with guanosine reduced the MPP+-induced increase in the
production of ROS, the crucial contributors to mitochondrial
dysfunction through oxidative damage and the opening of the mPTP.
Moreover, guanosine alleviated the decreased levels of GSH induced
by the administration of MPP+, reinforcing its
protective role in oxidative conditions and its role as a potential
neuroprotectant in mitochondrial-mediated neurodegenerative
diseases.
In conclusion, this study clearly demonstrates that
the neuroprotective effects of guanosine promote dopaminergic
neuronal survival by alleviating mitochontrial dysfunction in a
cellular model of PD. These neuroprotective effects are partly
mediated through the stabilization of mitochondrial membrane
potential via the modulation of the expression levels of intrinsic
apoptotic proteins involved in the mitochondrial apoptotic pathway.
Further studies are required to fully elucidate the mechanisms
responsible for the protective effects of guanosine in
neurodegenerative diseases, which may promote the development of
potentially effective treatments for neurodegenerative diseases by
targeting mitochondrias-mediated neuronal damage.
Abbreviations:
|
PD
|
Parkinson’s disease
|
|
MPTP
|
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
|
|
MPP+
|
1-methyl-4-phenylpyridinium
|
|
mPTP
|
mitochodrial permeability transition
pore
|
|
DCF
|
2′7′-dichlorodihydrofluorescein
|
|
Bcl-2
|
B-cell lymphoma 2
|
|
ROS
|
reactive oxygen species
|
References
|
1
|
Forno LS: Neuropathology of Parkinson’s
disease. J Neuropathol Exp Neurol. 55:259–272. 1996.
|
|
2
|
Abou-Sleiman PM, Muqit MM and Wood NW:
Expanding insights of mitochondrial dysfunction in Parkinson’s
disease. Nat Rev Neurosci. 7:207–219. 2006.
|
|
3
|
McGeer PL and McGeer EG: Glial reactions
in Parkinson’s disease. Mov Disord. 23:474–483. 2008.
|
|
4
|
Olanow CW: The pathogenesis of cell death
in Parkinson’s disease - 2007. Mov Disord. 22(Suppl 17): S335–S342.
2007.
|
|
5
|
Zhou C, Huang Y and Przedborski S:
Oxidative stress in Parkinson’s disease: a mechanism of pathogenic
and therapeutic significance. Ann NY Acad Sci. 1147:93–104.
2008.
|
|
6
|
Li DW, Liu ZQ, Chen W, Yao M and Li GR:
Association of glycogen synthase kinase-3β with Parkinson’s disease
(Review). Mol Med Rep. 9:2043–2050. 2014.
|
|
7
|
Vila M and Przedborski S: Targeting
programmed cell death in neurodegenerative diseases. Nat Rev
Neurosci. 4:365–375. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Perier C, Tieu K, Guégan C, et al: Complex
I deficiency primes Bax-dependent neuronal apoptosis through
mitochondrial oxidative damage. Proc Natl Acad Sci USA.
102:19126–19131. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Roucou X and Martinou JC: Conformational
change of Bax: a question of life or death. Cell Death Differ.
8:875–877. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shiba-Fukushima K, Imai Y, Yoshida S, et
al: PINK1-mediated phosphorylation of the Parkin ubiquitin-like
domain primes mitochondrial translocation of Parkin and regulates
mitophagy. Sci Rep. 2:10022012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kopin IJ and Markey SP: MPTP toxicity:
implications for research in Parkinson’s disease. Annu Rev
Neurosci. 11:81–96. 1988.
|
|
12
|
Heikkila RE, Sieber BA, Manzino L and
Sonsalla PK: Some features of the nigrostriatal dopaminergic
neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in
the mouse. Mol Chem Neuropathol. 10:171–183. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Calon F, Lavertu N, Lemieux AM, et al:
Effect of MPTP-induced denervation on basal ganglia GABA(B)
receptors: correlation with dopamine concentrations and dopamine
transporter. Synapse. 40:225–234. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chiba K, Trevor A and Castagnoli N Jr:
Metabolism of the neurotoxic tertiary amine, MPTP, by brain
monoamine oxidase. Biochem Biophys Res Commun. 120:574–578. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rathbone MP, Middlemiss PJ, Gysbers JW, et
al: Trophic effects of purines in neurons and glial cells. Prog
Neurobiol. 59:663–690. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dal-Cim T, Martins WC, Santos AR and Tasca
CI: Guanosine is neuroprotective against oxygen/glucose deprivation
in hippocampal slices via large conductance
Ca2+-activated K+ channels,
phosphatidilinositol-3 kinase/protein kinase B pathway activation
and glutamate uptake. Neuroscience. 183:212–220. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chang R, Algird A, Bau C, Rathbone MP and
Jiang S: Neuro-protective effects of guanosine on stroke models in
vitro and in vivo. Neurosci Lett. 431:101–105. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Di Iorio P, Ballerini P, Traversa U, et
al: The antiapoptotic effect of guanosine is mediated by the
activation of the PI 3-kinase/AKT/PKB pathway in cultured rat
astrocytes. Glia. 46:356–368. 2004.PubMed/NCBI
|
|
19
|
Pettifer KM, Kleywegt S, Bau CJ, et al:
Guanosine protects SH-SY5Y cells against beta-amyloid-induced
apoptosis. Neuroreport. 15:833–836. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pettifer KM, Jiang S, Bau C, et al:
MPP(+)-induced cytotoxicity in neuroblastoma cells: Antagonism and
reversal by guanosine. Purinergic Signal. 3:399–409. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li DW, Li GR, Lu Y, et al: α-lipoic acid
protects dopaminergic neurons against MPP+-induced
apoptosis by attenuating reactive oxygen species formation. Int J
Mol Med. 32:108–114. 2013.
|
|
22
|
Yim SB, Park SE and Lee CS: Protective
effect of glycyrrhizin on 1-methyl-4-phenylpyridinium-induced
mitochondrial damage and cell death in differentiated PC12 cells. J
Pharmacol Exp Ther. 321:816–822. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Banerjee R, Vitvitsky V and Garg SK: The
undertow of sulfur metabolism on glutamatergic neurotransmission.
Trends Biochem Sci. 33:413–419. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fischer U, Jänicke RU and Schulze-Osthoff
K: Many cuts to ruin: a comprehensive update of caspase substrates.
Cell Death Differ. 10:76–100. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
de Lau LM and Breteler MM: Epidemiology of
Parkinson’s disease. Lancet Neurol. 5:525–535. 2006.
|
|
26
|
Budd SL and Nicholls DG: Mitochondria in
the life and death of neurons. Essays Biochem. 33:43–52.
1998.PubMed/NCBI
|
|
27
|
Akhtar RS, Ness JM and Roth KA: Bcl-2
family regulation of neuronal development and neurodegeneration.
Biochim Biophys Acta. 1644:189–203. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Armstrong JS: Mitochondrial membrane
permeabilization: the sine qua non for cell death. Bioessays.
28:253–260. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Martinou JC and Green DR: Breaking the
mitochondrial barrier. Nat Rev Mol Cell Biol. 2:63–67. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gollapudi S, McCormick MJ and Gupta S:
Changes in mitochondrial membrane potential and mitochondrial mass
occur independent of the activation of caspase-8 and caspase-3
during CD95-mediated apoptosis in peripheral blood T cells. Int J
Oncol. 22:597–600. 2003.PubMed/NCBI
|
|
31
|
Tarozzi A, Merlicco A, Morroni F, et al:
Guanosine protects human neuroblastoma cells from oxidative stress
and toxicity induced by Amyloid-beta peptide oligomers. J Biol
Regul Homeost Agents. 24:297–306. 2010.PubMed/NCBI
|
|
32
|
Molz S, Dal-Cim T, Budni J, et al:
Neuroprotective effect of guanosine against glutamate-induced cell
death in rat hippocampal slices is mediated by the
phosphatidylinositol-3 kinase/Akt/glycogen synthase kinase 3β
pathway activation and inducible nitric oxide synthase inhibition.
J Neurosci Res. 89:1400–1408. 2011.PubMed/NCBI
|
|
33
|
Chuang DM, Chen RW, Chalecka-Franaszek E,
et al: Neuro-protective effects of lithium in cultured cells and
animal models of diseases. Bipolar Disord. 4:129–136. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen RW and Chuang DM: Long term lithium
treatment suppresses p53 and Bax expression but increases Bcl-2
expression. A prominent role in neuroprotection against
excitotoxicity. J Biol Chem. 274:6039–6042. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Manji HK and Chen G: PKC, MAP kinases and
the bcl-2 family of proteins as long-term targets for mood
stabilizers. Mol Psychiatry. 7(Suppl 1): S46–S56. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen G, Zeng WZ, Yuan PX, et al: The
mood-stabilizing agents lithium and valproate robustly increase the
levels of the neuroprotective protein bcl-2 in the CNS. J
Neurochem. 72:879–882. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kaga S, Zhan L, Altaf E and Maulik N:
Glycogen synthase kinase-3beta/beta-catenin promotes angiogenic and
anti-apoptotic signaling through the induction of VEGF, Bcl-2 and
survivin expression in rat ischemic preconditioned myocardium. J
Mol Cell Cardiol. 40:138–147. 2006. View Article : Google Scholar
|
|
38
|
Boatright KM and Salvesen GS: Mechanisms
of caspase activation. Curr Opin Cell Biol. 15:725–731. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kumar S: Caspase function in programmed
cell death. Cell Death Differ. 14:32–43. 2007. View Article : Google Scholar
|
|
40
|
Navarro A, Boveris A, Bandez MJ, et al:
Human brain cortex: mitochondrial oxidative damage and adaptive
response in Parkinson disease and in dementia with Lewy bodies.
Free Radic Biol Med. 46:1574–1580. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cassarino DS, Fall CP, Swerdlow RH, et al:
Elevated reactive oxygen species and antioxidant enzyme activities
in animal and cellular models of Parkinson’s disease. Biochim
Biophys Acta. 1362:77–86. 1997.PubMed/NCBI
|
|
42
|
Wallace DC: A mitochondrial paradigm of
metabolic and degenerative diseases, aging, and cancer: a dawn for
evolutionary medicine. Annu Rev Genet. 39:359–407. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Halliwell B: Oxidative stress and
neurodegeneration: where are we now? J Neurochem. 97:1634–1658.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lee M, Cho T, Jantaratnotai N, Wang YT,
McGeer E and McGeer PL: Depletion of GSH in glial cells induces
neurotoxicity: relevance to aging and degenerative neurological
diseases. FASEB J. 24:2533–2545. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hall AG: Review: The role of glutathione
in the regulation of apoptosis. Eur J Clin Invest. 29:238–245.
1999. View Article : Google Scholar : PubMed/NCBI
|