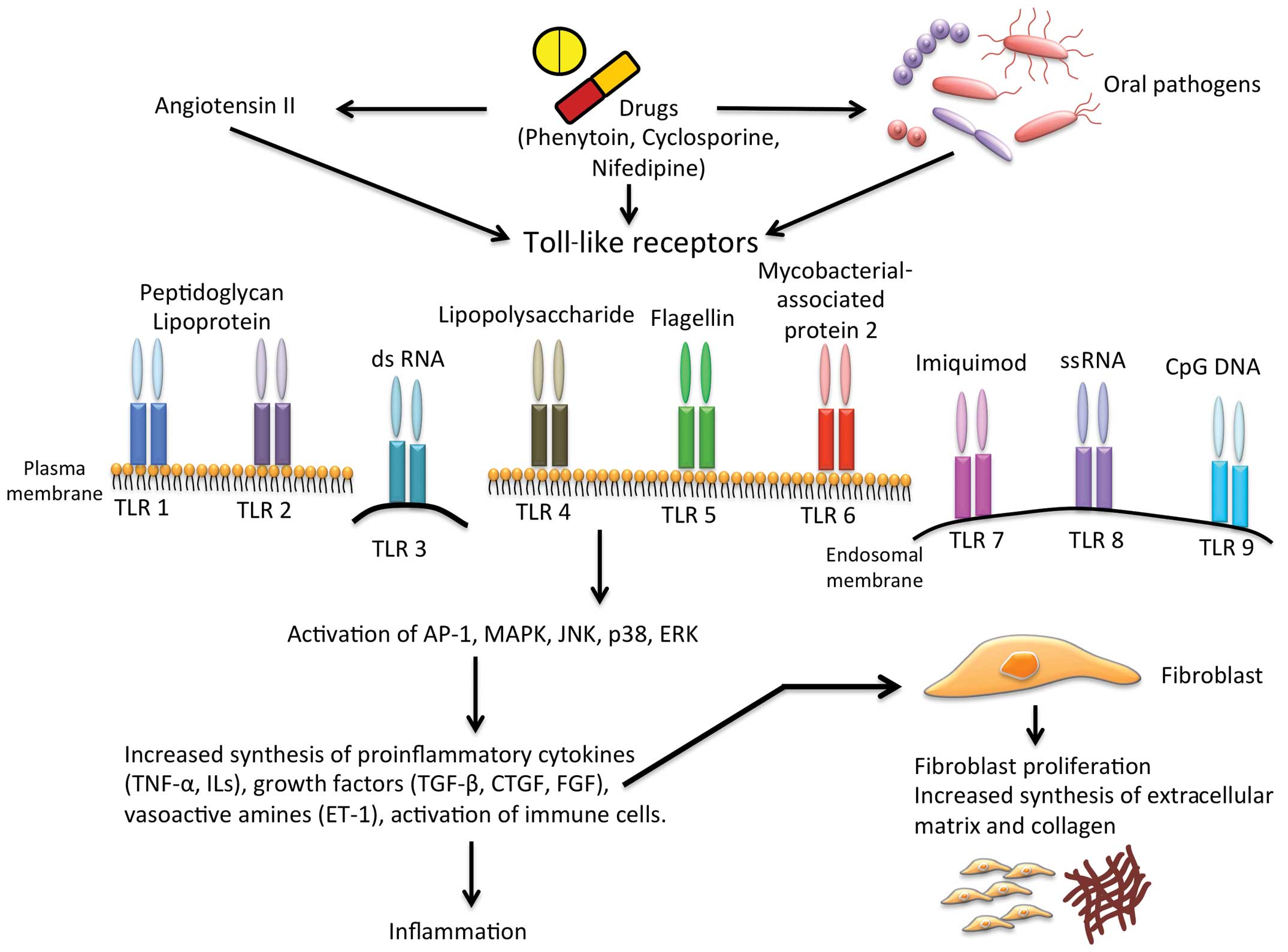
Drug-induced gingival overgrowth is an adverse
reaction mostly associated with three types of regularly
recommended drugs, i.e., immunosuppressants (cyclosporine)
(1), antiepileptic drugs
(phenytoin) (2) and calcium
channel blockers (verapamil, diltiazem and nifedipine) (3–6).
As gingival enlargement develops, regular oral hygiene practice is
disturbed and progressively severe pain develops, often leading to
disfiguration. Several etiological factors triggering gingival
overgrowth have been studied and it was shown that the degrees of
inflammation, fibrosis, dose, duration, quality of oral hygiene and
identity of the drug, local bacterial plaque accumulation,
individual susceptibility and environmental influences contribute
to the progression of gingival overgrowth (7,8).
Bacterial biofilm, one of the primary etiologic factors, harbors
several hundred diverse bacterial species that colonize the sulcus
and periodontal pockets, and initiate inflammation in the gingival
tissues, leading to gingivitis and periodontitis. These bacteria
have a number of structural components that directly destroy the
periodontal tissues, or stimulate host cells to initiate a wide
range of inflammatory responses. These inflammatory responses are
envisioned to overcome the microbial challenge, however, they are
frequently responsible for further tissue destruction. The changes
in the morphology of gingiva during gingival overgrowth lead to the
retention of dental plaque which stimulates the inflammation
(8). At the molecular level, an
excess of mediators are released following gingival inflammation
that contribute to the regulation of fibrogenic and regenerative
signals (8). Out of several
fibrogenic promoters, platelet-derived growth factor (PDGF) and
transforming growth factor-β (TGF-β) play a vital role in the
progression of fibrosis. It has been reported that PDGF and TGF-β
were overexpressed in the promotion of spontaneous fibrosis of the
kidney, liver, lung and gingiva (9–12).
In addition to PDGF and TGF-β, a number of other mediators such as
angiotensin II, endothelin-1, chemokines, leptin, interleukin-4
(IL-4), IL-6 and IL-13 play vital roles in fibrogenesis (13). Despite their protuberant role in
fibrogenesis, recent studies indicate Toll-like receptors (TLRs), a
group of receptors that regulate innate and adaptive immune
responses, as significant modulators of inflammation during
fibrosis (14–16). TLRs recognize pathogen-associated
molecular patterns (PAMPs) and activate innate and adaptive immune
responses to confiscate pathogens. In this review, we analyzed the
role of TLRs in gingival overgrowth and discussed the possibility
that TLRs may reveal innovative targets for the prevention or
treatment of gingival overgrowth.
Microbial biofilm induces the inflammatory process
of gingival tissues and supporting structures, leading to
gingivitis and periodontitis. Oral pathogens present in the biofilm
initiate periodontal diseases by stimulating the host immune
response. Epithelial cells and connective tissues of gingiva
express TLRs, which are crucial in the inflammatory response
(17). TLRs stimulate signaling
via two main pathways. The first one is myeloid differentiation
factor 88 (MyD88)-dependent, while and the second one is
MyD88-independent. Following initiation, pro-inflammatory cytokines
and type Ⅰ interferons (IFNs) are produced. The production of
pro-inflammatory cytokines is dependent on adaptor molecules MyD88
and TIR-associated protein (TIRAP), whereas IFNs are produced by
TIR domain-containing adaptor protein-inducing IFN-β (TRIF) and
TRIF-related adaptor molecules (TRAM) (18–21). Generally, bacterial triacylated
lipopeptides are recognized by heterodimers of the TLR1/TLR2
complex (22). Bacterial
lipoproteins and peptidoglycans derived from Gram-positive
bacteria, are recognized by TLR-2. Endogenous ligands such as heat
shock proteins are also recognized by TLR-2 (23). Double-strand DNA and its synthetic
analogue polyinosine-deoxycytidylic acid are recognized by TLR-3, a
potent stimulator of type I IFNs (24). The extensively studied TLR-4
recognizes a wide range of ligands mainly associated with
Gram-negative bacteria. Apart from microbial pathogens, TLR-4 also
recognizes a series of endogenous ligands in the circulation that
are released during necrosis and cell stress. Following
stimulation, TLR-4 activates a complex downstream signaling pathway
leading to the stimulation of transcription factors, principally
nuclear factor-κB (NF-κB) induces the stimulation of inflammatory
genes, such as tumor necrosis factor-α (TNF-α), IL-1, IL-6 and IL-8
(25,26). The constant domain D1 present in
the monomeric flagellin of bacteria is recognized by TLR-5. The
bacterial triacylated lipopeptides are recognized by the TLR1/TLR6
complex (22) and the bacterial
DNA is recognized by TLR-9 through unmethylated CpG motifs
(27). A summary of TLR ligands,
cellular, gene location and the effector molecules induced are
listed in Table I.
In gingiva, bacteria that inhabit the sulcus and
pockets are attached to the gingival epithelial cells by using
their fimbriae. These epithelial cells protect the organism from
potentially lethal oral pathogens and offer a surface that
tolerates microorganisms and favors the exchange of nutrients. The
gingival epithelial cells express almost all TLRs, such as TLR1-9
(27). The oral epithelial cells
that harbor TLRs have the potential to stimulate pro-inflammatory
cytokines IL-1β, IL-6, IL-8 and TNF-α following ligation with their
respective ligands (28).
Furthermore, IL-8, a well-known chemoattractant enhances the
migration of neutrophils from the circulation (29). Simultaneously, epithelial cells
produced matrix metalloproteinases (MMPs) in response to PAMP,
causing direct damage to periodontal tissues (30). These MMPs are structurally
associated with endopeptidases and capable of degrading virtually
all extracellular matrix (ECM) and basement membrane
components.
Gingival connective tissues predominantly
constituted by gingival fibroblasts that produce ECM are involved
in tissue regeneration by replacing the injured or disrupted
tissue. The expression of TLRs on gingival fibroblasts has
generated substantial interest towards this cell population in
inflammation in addition to their well-documented fibrogenic
effects. It has been demonstrated that the innate immune responses
of gingival fibroblasts produced various inflammatory cytokines,
such as IL-1, IL-6 and IL-8, following stimulation with
lipopolysaccharides from periodontopathic bacteria (31,32). Gingival fibroblast stimulation
with lipopolysaccharides induces the production of chemokines such
as RANTES, IP-10, MCP-1, MIP-1α and MIP-1β (33). Additionally, the stimulated
gingival fibroblasts produce large amounts of collagen and ECM
proteins (Fig. 1). Endothelial
cells also strongly participate in pathogen recognition, which, due
to their blood-exposed location, assigns these cells a key role in
the initial responses to pathogens that have reached the blood.
Accumulating evidence suggests a significant role for gingival
endothelial cell involvement in innate immunity and in controlling
the host responses to pathogens. As indicated in previous
investigations, endothelial cells express functional TLR-2, TLR-4
and TLR-9 (34,35), while the hypothesized contribution
of endothelial cell activities induced via TLRs in pathological
processes such as gingivitis and chronic periodontitis has been
suggested.
The pathogenesis of drug-induced gingival overgrowth
seems complex, especially the involvement of quality of plaque
control. Animal and human models have shown that plaque-mediated
inflammatory changes have a substantial role in the pathogenesis of
drug-induced gingival overgrowth (36–39). The drug may be an important factor
in the pathogenesis of gingival fibrosis by reducing cell
signaling, altering the inflammatory response in gingival tissues
and favoring bacterial invasion and proliferation. Elimination of
the microbial biofilm resulted in reduction of the inflammatory
infiltrate and alteration in connective tissue composition of the
gingival tissues of patients with drug-induced gingival overgrowth
(40). PAMPs are recognized by
pattern recognition receptors (PRRs) presented on several cells,
including fibroblasts (41). The
interaction between PAMPs and PRRs provides a first line of defense
during infection and activates numerous proinflammatory chemokines
as well as cytokine responses that modulate fibroblast
proliferation and ECM synthesis.
Inflammatory and immune mechanisms triggered by
non-infectious as well as, possibly even infectious agents, may be
important in the development of fibrosis (42). TLR ligands induce NF-κB
activation, resulting in the transcription of various types of
genes involved in the inflammatory and proliferative responses of
cells crucial to gingival overgrowth and ultimately leading to the
synthesis and release of inflammatory cytokines which provide a
critical link to adaptive immunity (34,43). These inflammatory mediators can
exert various fibrogenic effects involving the expression of
adhesion molecules on endothelial cells, proliferation of
fibroblasts, activation of immune cells, and stimulation of the
acute-phase response. In addition, TLR ligands can directly
activate fibroblasts and promote fibrogenesis (44–46). TLR-2 and TLR-4 stimulate a series
of events including NF-κB activation following the recognition of
the cell-wall component lipoproteins and peptidoglycan, and
recognition of the outer membrane component lipopolysaccharides,
respectively. NF-κB activation leads to cytokine production and
expression of adhesion molecules in gingival fibroblasts (27). Therefore, considering the role of
TLRs in the recognition of bacterial components and initiation of
host response via the release of several inflammatory mediators,
TLRs have a strong role in the pathogenesis of gingival fibrosis.
An in vitro study using hamster cells treated with phenytoin
and cyclosporine showed that cyclosporine increased signaling by
TLR2 and TLR4, while phenytoin decreased this signaling with a
decreased expression of adhesion molecules such as CD54 (47).
The reduction in cell signaling induced by drugs
such as phenytoin may alter the inflammatory response in gingival
tissues, favoring bacterial invasion and proliferation and,
therefore, may be an important factor in the pathogenesis of
gingival fibrosis (48).
Furthermore, cyclosporine-induced gingival overgrowth patients had
a significantly higher number of TLR-4 expressing cells in the
basal cell layer of the epithelium, as well as in connective tissue
compared to healthy subjects (42). Lim et al (49) demonstrated an association between
cyclosporine-induced renal injury and activation of innate immunity
through TLR-2 and TLR-4 expression in renal tissues of rats and
reported an increased TLR-2 and TLR-4 mRNA and protein expression
in rat kidney. Another study by Suzuki et al (50) showed that TLR-mediated
inflammatory responses were positively regulated by cyclosporine in
human gingival fibroblasts. Moreover, it has been reported that
deficiency of MyD88, the common adaptor for all TLRs except TLR-3,
protects mice from inflammation and fibrosis (51). In contrast to TLR-2 and TLR-4,
TLR-9 appears to promote lung fibrosis (52). Those results were confirmed by
another study, in which TLR-4 deficient mice exhibited a
significant reduction in fibroblast accumulation and renal fibrosis
(53). Together together, those
studies suggest that TLRs, via their common adaptor MyD88,
negatively affect tissue remodeling and fibrotic development.
TLRs activate immune cells including mast cells. The
activated mast cells degranulate and release various mediators such
as ILs, TNF-α and protease enzymes such as chymase and tryptase
(54). Findings of our earlier
studies suggest that an increased expression of mast cells and its
mediators was observed in drug-induced gingival overgrowth compared
to healthy gingival tissues (55). Mast-cell chymase actively
participate in the production of locally expressed angiotensin II
(Ang II) and endothelin-1 in gingiva. Ang II is the effector
peptide of the renin angiotensin system, which acts as a major role
in mediating contractile activity of vascular smooth muscle,
aldosterone release in the adrenal gland, regulation of collagen
synthesis and growth-modulating effects on fibroblasts (54,56,57). Furthermore, Ang II activates TLR4
through AT1 receptor and subsequent extracellular signal-regulated
kinase (ERK)1/2 and mediates NF-κB to initiate the expression of
cytokines involved in the inflammatory and profibrotic response.
Our previous study showed elevated TLR4 expression in gingival
tissues (17) and the significant
increase of Ang II in cyclosporine-induced gingival overgrowth
(58). Consequently, TLR4 is
closely involved in the Ang II-induced inflammatory response and
drug-induced gingival overgrowth. On the other hand, endothelin-1,
a potent vasoconstrictor secreted from endothelial cells (59,60), can induce ECM production (61,62). The source of ET-1 is not
restricted to endothelial cells. Human macrophages have been shown
to produce ET-1 in response to lipopolysaccharide (63), and human monocyte-derived
dendritic cells secrete ET-1 in response to TLR2 and TLR4 agonists
(64). Previously we showed a
significant increase of ET-1 in cyclosporine-treated human gingival
fibroblast cells (65). ET-1
stimulates the synthesis of collagen by gingival fibroblasts from
different species, including humans (66–70). Further profibrotic effects of ET-1
occur at the level of MMPs, with evidence that ET-1, acting via the
ETA receptor, can reduce collagenase activity (71). These findings showed the potential
role of the TLRs-dependent signaling pathway in modulating fibrotic
events in drug-induced gingival overgrowth.
TLR activation also upregulates many growth factors
such as TGF-β, VEGF, CXCR4, and adhesion molecules such as ICAM-1
(70–73). TGF-β has been the most intensively
studied regulator of the ECM and has been associated with the
development of fibrosis in a number of diseases (74–77). Once activated, TGF-β signals
trigger signaling intermediates known as Smad proteins via
transmembrane receptors and modulate the transcription of vital
target genes including procollagen I and III (78). Gingival fibrosis is reduced in
Smad-deficient mice, confirming the significant role for the TGF-β
signaling pathway (79).
Furthermore, it has been reported that loss of TGF-β signaling in
fibroblasts triggers intraepithelial neoplasia, suggesting that
TGF-β1 signaling critically regulates the activity of fibroblasts
as well as the oncogenic potential of neighboring epithelial cells
(80). In pulmonary fibrosis,
alveolar macrophages are thought to produce almost all of the
active TGF-β (81). Nevertheless,
Smad3/TGF-β1-independent mechanisms of fibrosis have been
demonstrated in lung and other tissues (82–84), suggesting that profibrotic
mediators such as IL-4, IL-5, IL-13 and IL-21 can act independently
from the TGFβ/Smad-signaling pathway to stimulate collagen
deposition. The connection between TGF-β and ET-1 has been
established, and several lines of evidence indicate that
transdifferentiation of fibroblasts occurs in response to the
concerted actions of TGF-β, ET-1 and Ang II (85). Activation of AP-1 and the MAPKs
c-jun N-terminal kinase (JNK), p38 and ERK1/2 are other classical
signals regulated by TLR signaling (86,87). AP-1 transcriptional complexes play
a pivotal role in drug-induced gingival overgrowth. AP-1 can be
activated through the TLR Myd88-dependent pathway by a variety of
growth factors and cytokines (88). AP-1 induced the activation of FOS
and Jun, which are observed in many fibrotic conditions (89). Our group previously demonstrated
the elevated expression of Jun and Fos in cyclosporine-induced
gingival overgrowth (90). The
expression of Jun and Fos activates proliferation and ECM synthesis
in gingival fibroblasts. Taken together, there is a significant
amount of evidence for the involvement of TLRs in drug-induced
gingival overgrowth.
Evidence of the involvement of TLRs in gingival
fibrosis largely comes from overexpression in fibrosis and their
activation triggering enhancement in the pathogenesis of diseases.
Inflammatory mediators such as TNF-α and ILs, which are produced as
a consequence of the activation of TLRs have been successfully
targeted in an effort to treat inflammatory diseases. Additionally,
targeting central upstream mediators in the inflammatory cascades
such as the TLRs may modulate pathway activation at an earlier
point and are therefore also likely to be effective in the
manipulation of the immune system to reduce disease severity. A
substantial amount of research has been conducted aiming to develop
TLR-targeted drugs for human use; however, only a few have been
approved thus far. The ubiquitin-modifying enzyme and zinc-finger
protein, A20, has been reported to regulate TLR-4 signaling
(91,92). A20 can suppress both TLR-2- and
TLR-4-induced IL-8 expression in airway epithelial cells (93). OPN305 is a humanized anti-TLR-2
monoclonal antibody that has potential to block TLR-1/2- and
TLR-2/6-mediated signaling and decrease TLR-2-mediated
pro-inflammatory cytokine production (94). NI0101 is a TLR-4 epitope-specific
antibody targeted towards TLR-4 and inhibits TLR-4 dimerization and
decreases pro-inflammatory cytokine production. This antibody
remains in the preclinical developmental stage and has some
potential indications including rheumatoid arthritis, asthma, acute
lung injury and acute respiratory distress syndrome (95). AV411 is another TLR-4-targeted
antagonist that has potential utility for the treatment of
neurological indications (96).
In addition to antibodies, a synthetic analog of lipid A eritoran
is targeted TLR-4, which inhibited the production of LPS-induced
TNF-α and IL-6 (97,98). IMO3100 is a dual TLR-7/TLR-9
antagonist that inhibits TLR ligand-induced gene expression.
Additionally, IMO8400 is a drug capable of antagonising TLR-7,
TLR-8 and TLR-9, that has shown efficacy in mouse models of lupus
(99). Conserning existing
conventional therapies, evidence suggests that many of these
approaches directly or indirectly affect TLR-mediated responses
(100). Although no specific
published data are available on drug-induced gingival overgrowth,
there is a substantial evidence suggesting that TLRs are a good
target for drug-induced gingival overgrowth. By understanding in
more detail the manner in which these modulate the activity of TLRs
to good effect, we can design TLR-directed interventions that
selectively inhibit the inflammatory component of the cascade while
retaining the anti-microbial component.
Evidence of the contribution of TLRs in fibrosis
greatly extends this understanding beyond innate immunity, and
provides important insights into body responses to diseases. As a
major portal of entry for microbes, the gingiva is a key component
of the innate immune system. Oral pathogens encounter a number of
effective defense mechanisms designed to rapidly counteract
potential damage, inhibit colonization and protect against invasion
by pathogens. The existence of TLRs equips gingival tissues with a
uniquely designed mechanism for controlling microbial infection.
However, drug-induced gingival overgrowth is a disease in which the
gingival epithelial cells, endothelial cells and fibroblasts
stimulate inflammatory mediators and profibrotic mediators through
TLRs and facilitate the accumulation of collagen and ECM in
gingiva. Conflicting roles of TLRs in various organs and different
forms of tissue response make it virtually impossible to outline
distinct greater functions for individual TLRs in drug-induced
gingival overgrowth. A possible explanation may be the differential
contribution of endogenous and exogenous ligands to TLR activation,
which likely depends on the anatomical localization and the related
exposure to microbes. Furthermore, drugs such as cyclosporine
stimulate TLR expression in gingival tissues. Thus modulation of
TLRs was important in drug-induced gingival overgrowth. Suppression
of TLRs responses by the use of appropriate inhibitors may reduce
the chronic inflammatory characteristic of this disease. Thus, new
therapeutics designed to selectively activate or inhibit TLR
function specifically and reversibly are powerful tools for the
prevention and treatment of the drug-induced gingival
overgrowth.
|
1
|
McGaw T, Lam S and Coates J:
Cyclosporin-induced gingival overgrowth: correlation with dental
plaque scores, gingivitis scores, and cyclosporin levels in serum
and saliva. Oral Surg Oral Med Oral Pathol Oral Radiol Endod.
64:293–297. 1987. View Article : Google Scholar
|
|
2
|
Perlík F, Kolínová M, Zvárová J and
Patzelová V: Phenytoin as a risk factor in gingival hyperplasia.
Ther Drug Monit. 17:445–448. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Seymour RA: Calcium channel blockers and
gingival overgrowth. Br Dent J. 170:376–379. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Miller CS and Damm DD: Incidence of
verapamil-induced gingival hyperplasia in a dental population. J
Periodontol. 63:453–456. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nishikawa S, Nagata T, Morisaki I, Oka T
and Ishida H: Pathogenesis of drug-induced gingival overgrowth. A
review of studies in the rat model. J Periodontol. 67:463–471.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ellis JS, Seymour RA, Steele JG, Robertson
P, Butler TJ and Thomason JM: Prevalence of gingival overgrowth
induced by calcium channel blockers: a community-based study. J
Periodontol. 70:63–67. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Marshall RI and Bartold PM: A clinical
review of drug-induced gingival overgrowths. Aust Dent J.
44:219–232. 1999. View Article : Google Scholar
|
|
8
|
Seymour RA, Ellis JS and Thomason JM: Risk
factors for drug-induced gingival overgrowth. J Clin Periodontol.
27:217–223. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yoshida M, Sakuma J, Hayashi S, Abe K,
Saito I, Harada S, Sakatani M, Yamamoto S, Matsumoto N, Kaneda Y,
et al: A histologically distinctive interstitial pneumonia induced
by overexpression of the interleukin 6, transforming growth factor
beta 1, or platelet-derived growth factor B gene. Proc Natl Acad
Sci USA. 92:9570–9574. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Campbell JS, Hughes SD, Gilbertson DG,
Palmer TE, Holdren MS, Haran AC, Odell MM, Bauer RL, Ren HP, Haugen
HS, Yeh MM and Fausto N: Platelet-derived growth factor C induces
liver fibrosis, steatosis, and hepatocellular carcinoma. Proc Natl
Acad Sci USA. 102:3389–3394. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Czochra P, Klopcic B, Meyer E, Herkel J,
Garcia-Lazaro JF, Thieringer F, Schirmacher P, Biesterfeld S, Galle
PR, Lohse AW and Kanzler S: Liver fibrosis induced by hepatic
overexpression of PDGF-B in transgenic mice. J Hepatol. 45:419–428.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yoshida T, Nagata J and Yamane A: Growth
factors and prolife-ration of cultured rat gingival cells in
response to cyclosporin A. J Periodontal Res. 40:11–19. 2005.
View Article : Google Scholar
|
|
13
|
Wynn TA: Cellular and molecular mechanisms
of fibrosis. J Pathol. 214:199–210. 2008. View Article : Google Scholar
|
|
14
|
Bataller R and Brenner DA: Liver fibrosis.
J Clin Invest. 115:209–218. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wynn TA: Integrating mechanisms of
pulmonary fibrosis. J Exp Med. 208:1339–1350. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huebener P and Schwabe RF: Regulation of
wound healing and organ fibrosis by toll-like receptors. Biochim
Biophys Acta. 1832:1005–1017. 2013. View Article : Google Scholar
|
|
17
|
Sarah SM, Tamilselvan S, Kamatchiammal S
and Suresh R: Expression of Toll-like receptors 2 and 4 in
gingivitis and chronic periodontitis. Ind J Dent Res. 17:114–116.
2006. View Article : Google Scholar
|
|
18
|
O’Neill LA, Fitzgerald KA and Bowie AG:
The Toll-IL-1 receptor adaptor family grows to five members. Trends
Immunol. 24:286–290. 2003. View Article : Google Scholar
|
|
19
|
Yamamoto M, Sato S, Hemmi H, Hoshino K,
Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K and
Akira S: Role of adaptor TRIF in the MyD88-independent toll-like
receptor signaling pathway. Science. 301:640–643. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yamamoto M, Sato S, Hemmi H, Sanjo H,
Uematsu S, Kaisho T, Hoshino K, Takeuchi O, Kobayashi M, Fujita T,
Takeda K and Akira S: Essential role for TIRAP in activation of the
signaling cascade shared by TLR2 and TLR4. Nature. 420:324–329.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yamamoto M, Sato S, Hemmi H, Uematsu S,
Hoshino K, Kaisho T, Takeuchi O, Takeda K and Akira S: TRAM is
specifically involved in the Toll-like receptor 4-mediated
MyD88-independent signaling pathway. Nat Immunol. 4:1144–1150.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Akira S, Uematsu S and Takeuchi O:
Pathogen recognition and innate immunity. Cell. 124:783–801. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ohashi K, Burkart V, Flohé S and Kolb H:
Cutting edge: heat shock protein 60 is a putative endogenous ligand
of the toll-like receptor-4 complex. J Immunol. 164:558–561. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Oshiumi H, Matsumoto M, Funami K, Akazawa
T and Seya T: TICAM-1, an adaptor molecule that participates in
Toll-like receptor 3-mediated interferon-beta induction. Nat
Immunol. 4:161–167. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fitzgerald KA, Palsson-McDermott EM, Bowie
AG, Jefferies CA, Mansell AS, Brady G, Brint E, Dunne A, Gray P,
Harte MT, McMurray D, Smith DE, Sims JE, Bird TA and O’Neill LA:
Mal (MyD88-adapter-like) is required for Toll-like receptor-4
signal transduction. Nature. 413:78–83. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Medzhitov R: Recognition of microorganisms
and activation of the immune response. Nature. 449:819–826. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Uehara A and Takada H: Functional TLRs and
NODs in human gingival fibroblasts. J Dent Res. 86:249–254. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yan P, Yue J and Jiang H: Expression of
ICAM-1/LFA-1 in the pocket area of adult periodontitis. Zhonghua
Kou Qiang Yi Xue Za Zhi. 34:106–108. 1999.In Chinese.
|
|
29
|
Han YW, Shi W, Huang GT, Kinder Haake S,
Park NH, Kuramitsu H and Genco RJ: Interactions between periodontal
bacteria and human oral epithelial cells: Fusobacterium nucleatum
adheres to and invades epithelial cells. Infect Immun.
68:3140–3146. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Warner RL, Bhagavathula N, Nerusu KC,
Lateef H, Younkin E, Johnson KJ and Varani J: Matrix
metalloproteinases in acute inflammation: induction of MMP-3 and
MMP-9 in fibroblasts and epithelial cells following exposure to
pro-inflammatory mediators in vitro. Exp Mol Pathol. 76:189–195.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takada H, Mihara J, Morisaki I and Hamada
S: Induction of interleukin-1 and -6 in human gingival fibroblast
cultures stimulated with Bacteroides lipopolysaccharides. Infect
Immun. 59:295–301. 1991.PubMed/NCBI
|
|
32
|
Tamura M, Tokuda M, Nagaoka S and Takada
H: Lipopolysaccharides of Bacteroides intermedius (Prevotella
intermedia) and Bacteroides (Porphyromonas) gingivalis induce
interleukin-8 gene expression in human gingival fibroblast
cultures. Infect Immun. 60:4932–4937. 1992.PubMed/NCBI
|
|
33
|
Seki E, De Minicis S, Osterreicher CH,
Kluwe J, Osawa Y, Brenner DA and Schwabe RF: TLR4 enhances TGF-beta
signaling and hepatic fibrosis. Nat Med. 13:1324–1332. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Faure E, Equils O, Sieling PA, Thomas L,
Zhang FX, Kirschning CJ, Polentarutti N, Muzio M and Arditi M:
Bacterial lipopolysaccharide activates NF-kappaB through toll-like
receptor 4 (TLR-4) in cultured human dermal endothelial cells.
Differential expression of TLR-4 and TLR-2 in endothelial cells. J
Biol Chem. 275:11058–11063. 2000. View Article : Google Scholar
|
|
35
|
Li J, Ma Z, Tang ZL, Stevens T, Pitt B and
Li S: CpG DNA-mediated immune response in pulmonary endothelial
cells. Am J Physiol Lung Cell Mol Physiol. 287:L552–L558. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kataoka M, Kido J, Shinohara Y and Nagata
T: Drug-induced gingival overgrowth – a review. Biol Pharm Bull.
28:1817–1821. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Romanos GE, Strub JR and Bernimoulin JP:
Immunohistochemical distribution of extracellular matrix proteins
as a diagnostic parameter in healthy and diseased gingiva. J
Periodontol. 64:110–119. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Seymour RA, Smith DG and Rogers SR: The
comparative effect of azathioprine and cyclosporine on some
gingival health parameters of renal transplant patients. A
longitudinal study. J Clin Periodontol. 14:610–613. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Seymour RA and Jacobs DJ: Cyclosporine and
the gingival tissues. J Clin Periodontol. 19:1–11. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Aimetti M, Romano F, Marsico A and Navone
R: Non-surgical periodontal treatment of cyclosporine A-induced
gingival overgrowth: immunohistochemical results. Oral Dis.
14:244–250. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nurmenniemi PK, Pernu HE, Laukkanen P and
Knuuttila ML: Macrophage subpopulations in gingival overgrowth
induced by nifedipine and immunosuppressive medication. J
Periodontol. 73:1323–1330. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Becerik S, Ozsan N, Gürkan A, Oztürk VÖ,
Atilla G and Eminqil G: Toll like receptor 4 and membrane-bound
CD14 expressions in gingivitis, periodontitis and CsA-induced
gingival overgrowth. Arch Oral Biol. 56:456–465. 2011. View Article : Google Scholar
|
|
43
|
Stoll LL, Denning GM, Li WG, Rice JB,
Harrelson AL, Romig SA, Gunnlaugsson ST, Miller FJ Jr and Weintraub
NL: Regulation of endotoxin-induced proinflammatory activation in
human coronary artery cells: expression of functional
membrane-bound CD14 by human coronary artery smooth muscle cells. J
Immunol. 173:1336–1343. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Meneghin MD and Hogaboam C: Infectious
disease, the innate immune response, and fibrosis. J Clin Invest.
117:530–538. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Otte JM, Rosenberg IM and Podolsky DK:
Intestinal myofibro-blasts in innate immune responses of the
intestine. Gastroenterol. 124:1866–1878. 2003. View Article : Google Scholar
|
|
46
|
Coelho AL, Hogaboam CM and Kunkel SL:
Chemokines provide the sustained inflammatory bridge between innate
and acquired immunity. Cytokine Growth Factor Rev. 16:553–560.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kawai T and Akira S: TLR signaling. Cell
Death Differ. 13:816–825. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Subramani T, Rathnavelu V, Yeap SK and
Alitheen NB: Influence of mast cells in drug-induced gingival
overgrowth. Mediators Inflamm. 2013:2751722013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lim SW, Li C, Ahn KO, Kim J, Moon IS, Ahn
C, Lee JR and Yang CW: Cyclosporine-induced renal injury induces
toll-like receptor and maturation of dendritic cells.
Transplantation. 80:691–699. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Suzuki AM, Yoshimura A, Ozaki Y, Kaneko T
and Hara Y: Cyclosporin A and phenytoin modulate inflammatory
responses. J Dent Res. 88:1131–1136. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gasse P, Mary C, Guenon I, Noulin N,
Charron S, Schnyder-Candrian S, Schnyder B, Akira S, Quesniaux VF,
Lagente V, Ryffel B and Couillin I: IL-1R1/MyD88 signaling and the
inflammasome are essential in pulmonary inflammation and fibrosis
in mice. J Clin Invest. 117:3786–3799. 2007.PubMed/NCBI
|
|
52
|
Trujillo G, Meneghin A, Flaherty KR, Sholl
LM, Myers JL, Kazerooni EA, Gross BH, Oak SR, Coelho AL, Evanoff H,
Day E, Toews GB, Joshi AD, Schaller MA, Waters B, Jarai G, Westwick
J, Kunkel SL, Martinez FJ and Hogaboam CM: TLR9 differentiates
rapidly from slowly progressing forms of idiopathic pulmonary
fibrosis. Sci Tranl Med. 2:57ra822010.
|
|
53
|
Campbell MT, Hile KL, Zhang H, Asanuma H,
Vanderbrink BA, Rink RR and Meldrum KK: Toll-like receptor 4: a
novel signaling pathway during renal fibrogenesis. J Surg Res.
168:e61–e69. 2011. View Article : Google Scholar
|
|
54
|
Mulrow PJ: The intrarenal
renin-angiotensin system. Curr Opin Nephrol Hypertens. 2:41–44.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Dzau VJ: Cell biology and genetics of
angiotensin in cardiovascular disease. J Hypertens Suppl.
12:S3–S10. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Subramani T, Senthilkumar K and Periasamy
S: Histochemical expression of mast cell chymase in chronic
periodontitis and cyclosporine-induced gingival overgrowth. J
Histol. 2013.ID8128422013.
|
|
57
|
Timmermans PB, Benfield P, Chiu AT,
Herblin WF, Wong PC and Smith RD: Angiotensin II receptors and
functional correlates. Am J Hypertens. 5:S221–S235. 1992.
View Article : Google Scholar
|
|
58
|
Subramani T, Senthilkumar K, Periasamy S
and Rao S: Expression of angiotensin II and its receptors in
cyclosporine-induced gingival overgrowth. J Periodontal Res.
48:386–391. 2013. View Article : Google Scholar
|
|
59
|
Inoue A, Yanagisawa M, Kimura S, Kasuya Y,
Miyauchi T, Goto K and Masaki T: The human endothelin family: three
structurally and pharmacologically distinct isopeptides predicted
by three separate genes. Proc Natl Acad Sci USA. 86:2863–2867.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Levin ER: Endothelins. N Engl J Med.
333:356–363. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Leask A: Targeting the TGFbeta,
endothelin-1 and CCN2 axis to combat fibrosis in scleroderma. Cell
Signal. 20:1409–1414. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Leask A: Potential therapeutic targets for
cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF,
partners in fibroblast activation. Circ Res. 106:1675–1680. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ehrenreich H, Anderson RW, Fox CH,
Rieckmann P, Hoffman GS, Travis WD, Coligan JE, Kehrl JH and Fauci
AS: Endothelins, peptides with potent vasoactive properties, are
produced by human macrophages. J Exp Med. 172:1741–1748. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Spirig R, Potapova I, Shaw-Boden J, Tsui
J, Rieben R and Shaw SG: TLR2 and TLR4 agonists induce production
of the vasoactive peptide endothelin-1 by human dendritic cells.
Mol Immunol. 46:3178–3182. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Tamilselvan S, Raju SN, Loganathan D,
Kamatchiammal S, Abraham G and Suresh R: Endothelin-1 and its
receptors ET(A) and ET(B) in drug-induced gingival overgrowth. J
Periodontol. 78:290–295. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kuruvilla L, Nair RR, Umashankar PR, Lal
AV and Kartha CC: Endocardial endothelial cells stimulate
proliferation and collagen synthesis of cardiac fibroblasts. Cell
Biochem Biophyics. 47:65–72. 2007. View Article : Google Scholar
|
|
67
|
Nishida M, Onohara N, Sato Y, Suda R,
Ogushi M, Tanabe S, Inoue R, Mori Y and Kurose H:
Galpha12/13-mediated up-regulation of TRPC6 negatively regulates
endothelin-1-induced cardiac myofibroblast formation and collagen
synthesis through nuclear factor of activated T cells activation. J
Biol Chem. 282:23117–23128. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Katwa LC: Cardiac myofibroblasts isolated
from the site of myocardial infarction express endothelin de novo.
Am J Physiol Heart Circ Physiol. 285:H1132–H1139. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Chintalgattu V and Katwa LC: Role of
protein kinase Cdelta in endothelin-induced type I collagen
expression in cardiac myofibroblasts isolated from the site of
myocardial infarction. J Pharmacol Exp Ther. 311:691–699. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Hafizi S, Wharton J, Chester AH and Yacoub
MH: Profibrotic effects of endothelin-1 via the ETA receptor in
cultured human cardiac fibroblasts. Cell Physiol Biochem.
14:285–292. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Guarda E, Katwa LC, Myers PR, Tyagi SC and
Weber KT: Effects of endothelins on collagen turnover in cardiac
fibroblasts. Cardiovas Res. 27:2130–2134. 1993. View Article : Google Scholar
|
|
72
|
Kelly MG, Alvero AB, Chen R, Silasi DA,
Abrahams VM, Chan S, Visintin I, Rutherford T and Mor G: TLR-4
signaling promotes tumor growth and paclitaxel chemoresistance in
ovarian cancer. Cancer Res. 66:3859–3868. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
He W, Liu Q, Wang L, Chen W, Li N and Cao
X: TLR4 signaling promotes immune escape of human lung cancer cells
by inducing immunosuppressive cytokines and apoptosis resistance.
Mol Immunol. 44:2850–2859. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Ren T, Wen ZK, Liu ZM, Liang YJ, Guo ZL
and Xu L: Functional expression of TLR9 is associated to the
metastatic potential of human lung cancer cell: functional active
role of TLR9 on tumor metastasis. Cancer Biol Ther. 6:1704–1709.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zhou M, McFarland-Mancini MM, Funk HM,
Husseinzadeh N, Mounajjed T and Drew AF: Toll-like receptor
expression in normal ovary and ovarian tumors. Cancer Immunol
Immunother. 58:1375–1385. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Sato M, Muragaki Y, Saika S, Roberts AB
and Ooshima A: Targeted disruption of TGF-beta1/Smad3 signalling
protects against renal tubulointerstitial fibrosis induced by
unilateral ureteral obstruction. J Clin Invest. 112:1486–1494.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Border WA, Noble NA, Yamamoto T, Harper
JR, Yamaguchi Y, Pierschbacher MD and Ruoslahti E: Natural
inhibitor of transforming growth factor-beta protects against
scarring in experimental kidney disease. Nature. 360:361–364. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Clouthier DE, Comerford SA and Hammer RE:
Hepatic fibrosis, glomerulosclerosis, and a lipodystrophy-like
syndrome in PEPCK-TGF-beta1 transgenic mice. J Clin Invest.
100:2697–2713. 1997. View Article : Google Scholar
|
|
79
|
Bonniaud P, Margetts PJ, Ask K, Flanders
K, Gauldie J and Kolb M: TGF-beta and Smad3 signaling link
inflammation to chronic fibrogenesis. J Immunol. 175:5390–5395.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Sime PJ, Xing Z, Graham FL, Csaky KG and
Gauldie J: Adenovector-mediated gene transfer of active
transforming growth factor-beta1 induces prolonged severe fibrosis
in rat lung. J Clin Invest. 100:768–776. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Roberts AB, Russo A, Felici A and Flanders
KC: Smad3: a key player in pathogenetic mechanisms dependent on
TGF-beta. Ann NY Acad Sci. 995:1–10. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Bhowmick NA, Chytil A, Plieth D, Gorska
AE, Dumont N, Shappell S, Washington MK, Neilson EG and Moses HL:
TGF-beta signaling in fibroblasts modulates the oncogenic potential
of adjacent epithelia. Science. 303:848–851. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Khalil N, Corne S, Whitman C and Yacyshyn
H: Plasmin regulates the activation of cell-associated latent
TGF-beta1 secreted by rat alveolar macrophages after in vivo
bleomycin injury. Am J Respir Cell Mol Biol. 15:252–259. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Kaviratne M, Hesse M, Leusink M, Cheever
AW, Davies SJ, McKerrow JM, Wakefield LM, Letterio JJ and Wynn TA:
IL-13 activates a mechanism of tissue fibrosis that is completely
TGF-beta independent. J Immunol. 173:4020–4029. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Ma LJ, Yang H, Gaspert A, Carlesso G,
Barty MM, Davidson JM, Sheppard D and Fogo AB: Transforming growth
factor-beta-dependent and -independent pathways of induction of
tubulointerstitial fibrosis in beta6(−/−) mice. Am J Pathol.
163:1261–1273. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Ashcroft GS, Yang X, Glick AB, Weinstein
M, Letterio JL, Mizel DE, Anzano M, Greenwell-Wild T, Wahl SM, Deng
C and Roberts AB: Mice lacking Smad3 show accelerated wound healing
and an impaired local inflammatory response. Nat Cell Biol.
1:260–266. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
87
|
Chin YT, Liao YW, Fu MM, Tu HP, Shen EC,
Nieh S, Shih KC and Fu E: Nrf-2 regulates cyclosporine-stimulated
HO-1 expression in gingiva. J Dent Res. 90:995–1000. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Schröder NW, Pfeil D, Opitz B, Michelsen
KS, Amberger J, Zähringer U, Göbel UB and Schumann RR: Activation
of mitogen-activated protein kinases p42/44, p38, and
stress-activated protein kinases in myelo-monocytic cells by
Treponema lipoteichoic acid. J Biol Chem. 276:9713–9719. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Kaisho T and Akira S: Toll-like receptor
function and signaling. J Allergy Clin Immunol. 117:979–987. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Subramani T, Rao S, Senthilkumar K,
Periasamy S and Alitheen NB: Angiotensin II stimulates expression
of transcription factors c-Jun and c-Fos in cyclosporine induced
human gingival fibroblasts. Biocell. 37:71–76. 2013.
|
|
91
|
O’Reilly SM and Moynagh PN: Regulation of
Toll-like receptor 4 signalling by A20 zinc finger protein. Biochem
Biophysic Res Commun. 303:586–593. 2003. View Article : Google Scholar
|
|
92
|
Boone DL, Turer EE, Lee EG, Ahmad RC,
Wheeler MT, Tsui C, Hurley P, Chien M, Chai S, Hitotsumatsu O,
McNally E, Pickart C and Ma A: The ubiquitin-modifying enzyme A20
is required for termination of Toll-like receptor responses. Nat
Immunol. 5:1052–1060. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
93
|
Yokota S, Okabayashi T, Yokosawa N and
Fujii N: Measles virus P protein suppresses Toll-like receptor
signal through up-regulation of ubiquitin-modifying enzyme A20.
FASEB J. 22:74–83. 2008. View Article : Google Scholar
|
|
94
|
Arslan F, Houtgraaf JH, Keogh B, Kazemi K,
de Jong R, McCormack WJ, O’Neill LA, McGuirk P, Timmers L, Smeets
MB, Akeroyd L, Reilly M, Pasterkamp G and de Kleijn DP: Treatment
with OPN-305, a humanized anti-Toll-Like receptor-2 antibody,
reduces myocardial ischemia/reperfusion injury in pigs. Circ
Cardiovasc Interv. 5:279–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Mizuno T, Kurotani T, Komatsu Y,
Kawanokuchi J, Kato H, Mitsuma N and Suzumura A: Neuroprotective
role of phosphodiesterase inhibitor ibudilast on neuronal cell
death induced by activated microglia. Neuropharmacology.
46:404–411. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Rolan P, Gibbons JA, He L, Chang E, Jones
D, Gross MI, Davidson JB, Sanftner LM and Johnson KW: Ibudilast in
healthy volunteers: safety, tolerability and pharmacokinetics with
single and multiple doses. Br J Clin Pharmacol. 66:792–801. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Zeisberg EM1, Tarnavski O, Zeisberg M,
Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT,
Roberts AB, Neilson EG, Sayegh MH, Izumo S and Kalluri R:
Endothelial-to-mesenchymal transition contributes to cardiac
fibrosis. Nat Med. 13:952–961. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
98
|
Czeslick E, Struppert A, Simm A and
Sablotzki A: E5564 (Eritoran) inhibits lipopolysaccharide-induced
cytokine production in human blood monocytes. Inflamm Res.
55:511–515. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Hennessy EJ, Parker AE and O’Neill LA:
Targeting Toll-like receptors: emerging therapeutics. Nat Rev Drug
Discov. 9:293–307. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Connolly DJ and O’Neill LA: New
developments in Toll-like receptor targeted therapeutics. Curr Opin
Pharmacol. 12:510–518. 2012. View Article : Google Scholar : PubMed/NCBI
|